

Abstract
The intestinal epithelial barrier represents an important component in the pathogenesis of inflammatory bowel diseases. Interferon (IFN)-γ, a T helper type 1 (Th1) cytokine, regulated by the interleukin (IL)-18/IL-18 binding protein (bp) system, modulates the integrity of this barrier. The aim of this work was to study functionally the consequences of IFN-γ on intestinal epithelial cells (IEC) and to interfere selectively with identified adverse IFN-γ effects. IEC lines were stimulated with IFN-γ. IL-18 and IL-18bp were assessed by enzyme-linked immunosorbent assay. Staining of phosphatidylserine, DNA laddering, lactate dehydrogenase (LDH) release, cleavage of poly-adenosine diphosphate-ribose-polymerase (PARP) and activation of caspase-3 were analysed to determine cell death. Inhibitors of tyrosine kinase, caspase-3 or p38 mitogen-activated kinase ((MAP) activity were used. Cytokines were measured in supernatants of colonic biopsies of healthy controls and inflammatory bowel disease (IBD) patients. In IEC lines, IFN-γ up-regulated IL-18bp selectively. Ex vivo, IFN-γ was present in supernatants from cultured biopsies and up-regulated with inflammation. Contrary to previous reports, IFN-γ alone induced apoptosis in IEC lines, as demonstrated by phosphatidylserin staining, DNA cleavage and LDH release. Further, activation of caspase-3, PARP cleavage and expression of pro-apoptotic Bad were induced. Partial inhibition of caspase-3 and of p38 but not JAK tyrosine kinase, preserved up-regulation of IL-18bp expression. Selective inhibition of IFN-γ mediated apoptosis, while preserving its beneficial consequences on the ratio of IL-18/IL-18bp, could contribute to the integrity of the mucosal barrier in intestinal inflammation.
Keywords: apoptosis, inflammatory bowel disease, interferon-γ, interleukin-18, intestinal epithelial cell
Introduction
The inflammatory bowel diseases (IBD), Crohn's disease (CD) and ulcerative colitis (UC), are believed to be morphological and functional consequences of an interacting genetic disposition, luminal and environmental factors, and an activated immune system [1]. Recently, the intestinal barrier, that is intestinal epithelial cells (IEC) and surrounding constituents, has gained increasing attention in this setting. Impaired barrier integrity is thought to facilitate adhesion and penetration of luminal bacteria and/or bacterial antigens, which then stimulate the mucosal immune system and initiate the chronically relapsing inflammation. A link between a genetic predisposing factor, e.g. mutation of the NOD2 gene, reduced anti-microbial power of the barrier, e.g. insufficient production of defensins, and the occurrence of Crohn's ileitis has been proposed [2,3].
Inflammatory mediators released from activated mucosal immune cells may change the morphology and function of the intestinal epithelial cell layer. IFN-γ is a cytokine that modulates the expression of molecules involved in cell–cell and cell–matrix interactions [4]. Interferons are part of a heterogeneous cytokine family. Interferon (IFN)-γ is produced by natural killer cells and CD4-positive T helper cells. It exerts anti-proliferative and anti-viral effects, regulates major histocompatibility complex (MHC) class II expression of antigen-presenting cells, secretion of chemokines and activation of macrophages as well as lymphocytes [5]. Expression of IFN-γ is increased in IBD, particularly in CD, and the cytokine has therefore been implicated in its pathogenesis [6–8].
Interleukin (IL)-18 is a cytokine formerly called IFN-γ-inducing factor [9,10], and has been implicated in the pathogenesis of IBD. It is expressed by monocytes, macrophages and intestinal epithelial cells [11–13] and functions as a proinflammatory cytokine, thus contributing to differentiation and activation of T cells and inducing production of proinflammatory cytokines such as IFN-γ[14,15]. Blocking IL-18 in animal models, for instance, leads to reduced inflammation [16]. Further, up-regulated systemic levels of IL-18 and increased expression of IL-18 in IEC and mononuclear cells was demonstrated in patients with CD. Additionally, systemic levels of IL-18 are increased in this condition [12,17]. IL-18 binding protein (bp), the natural antagonist of IL-18, is expressed constitutively in the healthy setting of small and large bowel. IL-18bp acts as decoy receptor and binds IL-18 in the extracellular space, thus reducing its biological activity; e.g. binding of IL-18bp to IL-18 reduces expression of IL-8 and IFN-γ, activation of transcription factor nuclear factor (NF)-κB and T cell-mediated immune response [18].
In the past we have investigated cytokine expression in IEC, such as regulation of the IL-1/IL-1 receptor antagonist and the IL-18/IL-18bp system [19–21]. In the present study, we wanted to focus upon IFN-γ as a regulator of the latter. Surprisingly, and in contrast to previous reports, IFN-γ, besides its anti-inflammatory effect via IL-18bp induction, was pro-apoptotic for IEC lines. Using caspase-3 and p38 signalling inhibitors, we reduced the pro-apoptotic effect of IFN-γ successfully, while maintaining its anti-inflammatory function.
Patients and methods
Patients
Intestinal mucosal biopsies were taken from 11 patients with CD [age: 45 ± 9·88 (24–58) years; six female], seven with UC [age: 46 ± 19·66 (19–72) years; three female] and six healthy controls [age: 71 ± 13·62 (52–82) years; three female] during a medically required colonoscopy. All patients had had correctly established disease for more than a year; however, as they were diagnosed primarily outside our own centre, information concerning duration of disease as given by the patients was not reliable. CD patients demonstrated ileocolonic involvement, UC patients displayed left-sided extension of the disease. All IBD patients presented with both macroscopically quiescent and actively inflamed segments, respectively. Concomitant medication was as follows: CD, eight with mesalamine, two with steroids, two with immunosupressants; UC, five with mesalamine, four with steroids, one with immunosuppressants; controls: none. Written informed consent was obtained prior to the procedure, and the study was approved by the local ethics committee. A PCF-160-AL colonoscope (Olympus, Hamburg, Germany) and a biopsy forceps RadialJaw3 (Boston Scientific, Nanterre Cedex, France) were used. Healthy controls underwent colonoscopy in order to exclude gastrointestinal bleeding or malignoma. In healthy controls, biopsies were taken from the ascending colon. In patients diagnosed with IBD and examined for determination of inflammatory activity, biopsies were taken from inflamed and non-inflamed areas. Ulcer grounds were avoided in order to secure epithelial cells in the specimens. Macroscopic activity of the disease was stated in the presence of signs of inflammation such as oedema, reddening, aphthous lesions and ulcerations; however, clinical and endoscopic activity indices were not applied. There was no structured follow-up of the patients.
Tissue culture
Biopsies were washed three times and then cultured for 48 h in 200 µl Rosweli Park Memorial Institute (RPMI)-1640 medium (Gibco, Invitrogen, Karlsruhe, Germany) containing 10% fetal calf serum (FCS) (HyClone Perbio Science, Bonn, Germany), 100 mg/l gentamicin, 2 mM l-glutamine, 100 U/l penicillin, 100 µg/l streptomycin and 100 µg/l fungisone (all obtained from Gibco) in a 5% CO2 atmosphere.
Enzyme-linked immunosorbent assay (ELISA)
The concentrations of IFN-γ were measured using a BD OptEIA set human IFN-γ sandwich-ELISA kit (BD Biosciences-Pharmingen, San Diego, USA), according to the manufacturer's instructions. IL-18bp was measured using a DuoSet Human IL-18bp sandwich-ELISA kit (R&D Systems, Minneapolis, USA), as described in the manufacturer's manual. IL-18 was measured as described previously [22].
Lactate dehydrogenase (LDH) assay
LDH release into the culture medium was assayed according to the manufacturer's instructions (Roche Applied Science, Mannheim, Germany). Triton X-treated cells (set to 100%) served as control of total LDH content, treatment of cells with tumour necrosis factor (TNF)-α (10 ng/ml)/actinomycin D (10 µg/ml) as positive control of apoptosis.
Cell culture
Established transformed human colon epithelial cells (HT-29, Caco-2) were obtained from the American Type Culture Collection (Bethesda, MD, USA) and from the German Collection for Micro-organisms and Cell cultures (Braunschweig, Germany). Caco-2 cells were cultured in minimum essential medium (MEM), and parenteral HT-29 cells in Dulbecco's modified Eagle's medium (DMEM), both obtained from Gibco, containing 2 mM l-glutamine, 100 µM non-essential amino-acids, 100 U/ml penicillin and 100 µg/ml streptomycin supplemented with 10% v/v fetal calf serum (FCS) from Gibco. All cells were incubated in non-coated six-well plates (Cellstar, GreinerBioOne, Frickenhausen, Germany) at 37°C in 5% CO2 atmosphere. For the different experiments cells were used reaching confluence of about 50–70% not less than 48 h after passage. Cells were incubated with different concentrations of recombinant human (rh) IFN-γ (10–100 ng/ml) obtained from R&D Systems. To inhibit IFN-γ-mediated apoptosis different concentrations of caspase-3 inhibitor Z-DEVD-FMK (100 and 150 µM) (R&D Systems), JAK tyrosine kinase inhibitor AG490 (40 and 80 µM) (Calbiochem, Merk, Darmstadt, Germany) and p38 inhibitor SB203580 (25 µM), both from Sigma-Aldrich (Steinheim, Germany), were used. For stimulation of cytokine expression the following substances were used: IL-1α 1 ng/ml, IL-1β 1 ng/ml, TNF-α 10 ng/ml, IL-4 100 ng/ml, IL-7 100 ng/ml, IL-9 100 ng/ml, IL-11 30 ng/ml, IL-13 30 ng/ml (all R&D Systems); phorbol myristate acetate (PMA) 10 ng/ml, phytohaemagglutinin 1 µg/ml, lipopolysaccharide 1 µg/ml; butyrate 5 mM, acetate 5 mM and propionate 5 mM (all Sigma-Aldrich). Co-incubation with TNF-α (10 ng/ml) and actinomycin D (5 µg/ml) (Sigma-Aldrich) served as positive control of apoptosis.
Phase-contrast microscopy
Transformed human colon epithelial cells were cultured on uncoated cell culture plates (Cellstar). Morphological features of treated and untreated cells were analysed with the phase-contrast microscope DM IL (Leica, Wetzlar, Germany).
Annexin staining
HT-29 cells were cultured on cover slips (Menzel, Braunschweig, Germany) in 12-well plates (Cellstar). Cells were treated for 48 h with 100 ng/ml IFN-γ. After removing the culture medium, cells were incubated for 5 min in 500 µl/well Hanks' balanced salt solution (BSS) medium [w/o phenol red, w/o Ca (2+), w/o Mg (2+)] (PAA Laboratories, Pasching, Germany) with 2 mM CaCl2 (Sigma-Aldrich). Cell staining was performed by incubation in 300 µl/well Hanks' BSS medium containing 2 mM CaCl2, 5 µg/ml Draq 5 (Hoechst) stock solution (Alexis Corp., Lausen, Switzerland), 5 nM Sytox green nucleic acid stain (Invitrogen, Karlsruhe, Germany) and 0·25 µg/ml annexin V-Cy3 apoptosis detection reagent (Abcam, Cambridge, UK) for 5 min protected from light. After finishing the reaction and mounting the slides the result of the staining was visualized using a confocal microscope DM IRE2 (Leica).
DNA-laddering
After culturing of the cells supernatant was removed and transferred, including the non-adherent cells, into a 2-ml tube (Eppendorf, Hamburg, Germany). Adherent cells from the identical cavity were removed with a cell scraper, transferred into the same tube and then centrifuged at 0·078 g for 3 min at 4°C. The supernatant was then removed and the pellet was lysed in 450 µl lysis buffer containing 10 mM Tris-buffered saline (pH 8·0) (Serva, Heidelberg, Germany), 25 mM ethylenediamine tetraacetic acid (EDTA) (Applichem, Darmstadt, Germany) and 100 mM NaCl (Roth, Karlsruhe, Germany). After removal of cellular protein, DNA extraction and precipitation concentrations of received DNA were measured in a GeneQuant pro photometer (Amersham Biosciences, Freiburg, Germany). Ten µg DNA was loaded in 1·5% v/v agarose gel containing ethidium bromide (Roth). Gel electrophoresis was performed at 80 volts in an effective concentration (EC50) gel electrophoresis apparatus (EC Apparatus Corp., Milford, MA, USA).
Western blot analysis
Transformed human colon epithelial cells were treated under different experimental conditions on non-coated six-well plates (Cellstar). After treatment cells were lysed in RIPA cell lysis buffer (pH 7·2) containing 50 mM Tris buffer (Serva), 250 mM sodium chloride, 2% Nonidet P 40 (Roth), 2·5 mM EDTA, 0·1% sodium dodecyl sulphate, sodium-dideoxycholate, one tablet protease inhibitor (Roche Diagnostics) and 10 µl/ml phosphatase inhibitor cocktail 2 (Sigma-Aldrich). Protein concentration was measured using the DC-protein assay (Bio-Rad, Hercules, CA, USA) following the manufacturer's directions. Then 20–30 µg of cellular protein/well were separated on a NuPAGE sodium dodecyl sulphate-polyacrylamide gel electrophoresis (Invitrogen) and transferred onto nitrocellulose membranes (Protran; Whatman, Dassel, Germany). Blotted membranes were incubated overnight with monoclonal rabbit antibodies for full and cleaved caspase-3, poly-adenosine diphosphate-ribose-polymerase (PARP), total signal transducer and activator of transcription (STAT)-1 and p-STAT-1, all purchased from Cell Signaling (Danvers, MA, USA), and monoclonal mouse antibody for β-actin obtained from Sigma-Aldrich, followed by incubation with the peroxidase-conjugated polyclonal secondary antibodies (Santa Cruz Biotechnology, Heidelberg, Germany). Immunoblots were developed using the enhanced chemiluminescence (ECL) detection system (Amersham Biosciences), following the manufacturer's directions. Immunoreactive bands were detected using a cooled charged couple device camera system LAS-1000 (Fuji, Tokyo, Japan). Band intensity was analysed using the Advanced Image Data Analyzer (AIDA; Raytest GmbH, Straubenhardt, Germany).
Reverse transcription–polymerase chain reaction (RT–PCR)
Human (h)-bcl-xl, h-bad and β-actin were detected by RT–PCR. RNA was isolated from IFN-γ-treated (48 h, 100 ng/ml) or control cultures of HT-29 cells using the RNeasy Mini Kit (Quiagen, Hilden, Germany), as described in the manufacturer's manual. Concentrations of isolated RNA were measured with a GeneQuant pro photometer (Amersham Biosciences). After reverse transcription of 1 µg RNA using the Omniscript RT Kit (Quiagen) as described in the manufacturer's manual, PCR was performed in a PTC-220 DNA Engine Dyad Peltier Thermal Cycler (MJ Research Inc., Waltham, MA, USA) using the following primer pairs and conditions (denaturation, annealing, extension): h-β-actin forward CACCCACACTGTGCCCATC, h-β-actin reverse CTGCTGCTTGCTGATCCAC (94°C, 45 s; 60°C, 45 s; 72°C, 45 s for 25 cycles), h-bcl-xl forward (long) GGTCGCATTGTGGCCTTTTTC, h-bcl-xl reverse (long) TGCTGCATTGTTCCCATAGAG (94°C, 45 s; 62°C, 45 s; 72°C, 45 s for 30 cycles) and h-bad forward CCCAGAGTTTGAGCCGAGTG, h-bad reverse CCCATCCCTTCGTCGTCCT (94°C, 45 s; 62°C, 45 s; 72°C, 45 s for 30 cycles). Amplified products were verified in 1·5% v/v agarose gel by electrophoresis and at predicted sizes for each sample single bands were detectable. There were no products detected in negative controls.
Quantitative real-time PCR (qRT–PCR)
One µg of RNA of each sample was reverse-transcribed at 37°C for 1 h in a PTC-220 DNA Engine Dyad Peltier Thermal Cycler (MJ Research Inc., Waltham, MA, USA) using the Omniscript RT KIT (Quiagen), as described in the manufacturer's manual. The qRT–PCR was performed using a TaqMan Universal PCR Master Mix (Applied Biosystems, Foster City, CA, USA) in a sequence detection-system ABI Prism 7700 (Applied Biosystems), as described in the manufacturer's protocol. The detection of human IL-18bp was performed with a TaqMan gene expression assay for human IL-18bp (Applied Biosystems), while glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) (Applied Biosystems) was used as an endogenous loading control. Gene expression analysis was performed using the CT-method (dd CT) for relative quantification of gene expression as described by Livak and co-workers 2001 [23]. The analysis of the PCR products was performed with the ABI Prism 7000 sodium dodecyl sulphate (SDS) software (Applied Biosystems).
Statistical analyses
Data were analysed using the software Statistical Analysis System (sas), release 9·1 (SAS Corp., Cary, NC, USA). Because normal distribution could not be assumed in any case and also because of the small sample sizes, we preferred non-parametric methods. Hence, Wilcoxon's two-sample test or Wilcoxon's signed-rank test was used in order to compare two unpaired or paired samples, respectively. A value of P < 0·05 was considered statistically significant.
Results
IFN-γ up-regulates expression of IL-18bp in IEC lines
IFN-γ increased the IL-18bp expression in IEC dose-dependently, with the most effective concentration of 100 ng/ml, while other stimuli were ineffective (Fig. 1). IL-18bp protein levels were increased 100-fold in culture supernatants and more than 30-fold in cell lysates, respectively, compared to untreated controls (see experiments and figures below). IL-18 protein was not secreted upon exposure to IFN-γ, and intracellular levels remained unchanged. Therefore, IFN-γ shifted the IL-18/IL-18bp ratio towards the latter.
Fig. 1.
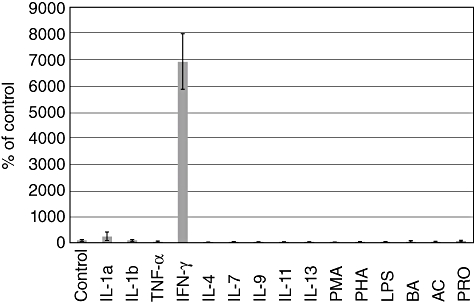
Real-time-polymerase chain reaction (PCR) for detection of interleukin (IL)-18 binding protein (bp) expression in human colon epithelial cells (HT-29) cultured in the presence of stimuli as noted for 24 h. Cells cultured for 24 h in serum-free medium were used as controls. Results of the controls were determined as 100%. PMA: phorbol myristate acetate; PHA: phytohaemagglutinin; LPS: lipopolysaccharide; BA: butyrate, AC: acetate, PRO: propionate.
IFN-γ is released by mucosal biopsies ex vivo
In order to support the concept of a biological significance of IFN-γ in the intestinal mucosa, biopsies were cultured for 48 h and cytokine levels were assessed in supernatants (Fig. 2a–c). In healthy controls the IFN-γ level was 5·11 pg/ml (mean; median: 3·54 pg/ml). The IFN-γ concentration was about fourfold higher in supernatants of biopsies from non-inflamed areas in patients with Crohn's disease [22·14 pg/ml (mean; median: 12·29 pg/ml)], but only slightly increased in according samples of patients with ulcerative colitis [8·33 pg/ml (mean; median: 6·66 pg/ml)]. In Crohn's disease, biopsy cultures from inflamed areas revealed five times increased concentrations of IFN-γ compared to the non-inflamed state [101·67 pg/ml (mean); median: 88·08 pg/ml]. In inflamed areas of patients with ulcerative colitis, the IFN-γ expression in biopsy cultures was significantly higher [33·93 pg/ml (mean); median: 17·82 pg/ml; P < 0·05] than in supernatants of non-inflamed areas. The data supported the concept that IFN-γ is expressed in the intestinal mucosa, up-regulated by inflammation and thereby potentially exerting effector functions on IEC.
Fig. 2.
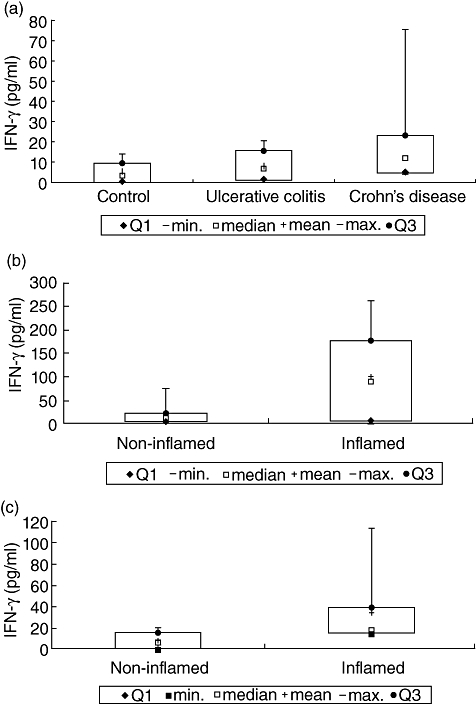
(a) Interferon (IFN)-γ expression in supernatants of biopsies from healthy controls and from non-inflamed areas of patients with inflammatory bowel disease (IBD) using enzyme-linked immunosorbent assay (ELISA). (b) IFN-γ expression in supernatants of biopsies from non-inflamed and inflamed areas of patients with Crohn's disease. (c) IFN-γ expression in supernatants of biopsies from non-inflamed and inflamed areas of patients with ulcerative colitis.
IFN-γ induces apoptosis in IEC
Besides alteration of the IL-18/IL-18bp ratio after treating IEC lines (HT-29, Caco-2) with IFN-γ, morphological changes were visible (Fig. 3). Using phase-contrast microscopy, IFN-γ-treated cells showed typical morphological aspects of apoptotic cells. In contrast to cells cultured in serum-free medium only, IFN-γ exposed cells showed shrinkage and formation of apoptotic bodies. In contrast to untreated cells, these cells lost their cell–cell adherence and were floating in the culture medium (Fig. 3a and b). To confirm these findings, IFN-γ-treated (100 ng/ml) and -untreated HT-29 cells were stained after 48 h to detect the externalization of phosphatidylserine as one central event of the apoptotic cascade. Whereas in untreated cells no externalized phosphatidylserine was detectable, IFN-γ-treated cells showed positive staining for phosphatidylserine by annexin (Fig. 3c and d). Necrosis was excluded by staining the cells using Sytox green (data not shown).
Fig. 3.
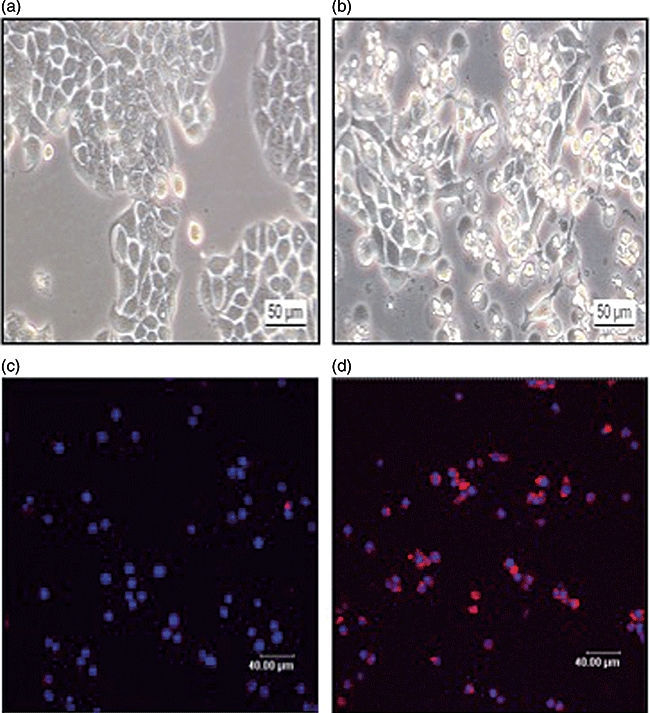
(a) Phase-contrast microscopy of native human colon epithelial cells (HT-29), which were cultured for 48 h in serum-free medium. (b) Phase-contrast microscopy of native HT-29 cells after stimulation with interferon (IFN)-γ (100 ng/ml). (c) Annexin staining of HT-29 cells cultured for 48 h in serum-free medium (coloured blue: staining of intact nuclei with Draq5). (d) Positive detection of externalized phosphatidylserine by annexin (coloured red) as a marker of apoptotic cells after treatment of HT-29 cells with IFN-γ (100 ng/ml) for 48 h.
Apoptosis-associated cell damage was confirmed by a quantitative LDH assay, demonstrating LDH release upon exposure of IEC to IFN-γ. LDH release of untreated controls (25·96% ± 0·449; n = 3) remained mainly stable over a period of 72 h, while it increased to a maximum of 91·65% ± 2·46 (n = 3) at 72 h in the presence of IFN-γ.
In both IEC lines (HT-29, Caco-2) stimulation with 25 to 500 ng/ml IFN-γ resulted in activation of caspase-3 and cleavage of PARP, two late events in the apoptotic cascade (Fig. 4a and b). Further, HT-29 cells treated with IFN-γ (100 ng/ml) displayed nuclear fragmentation of DNA after 24 h (Fig. 4c) in a DNA-laddering approach.
Fig. 4.
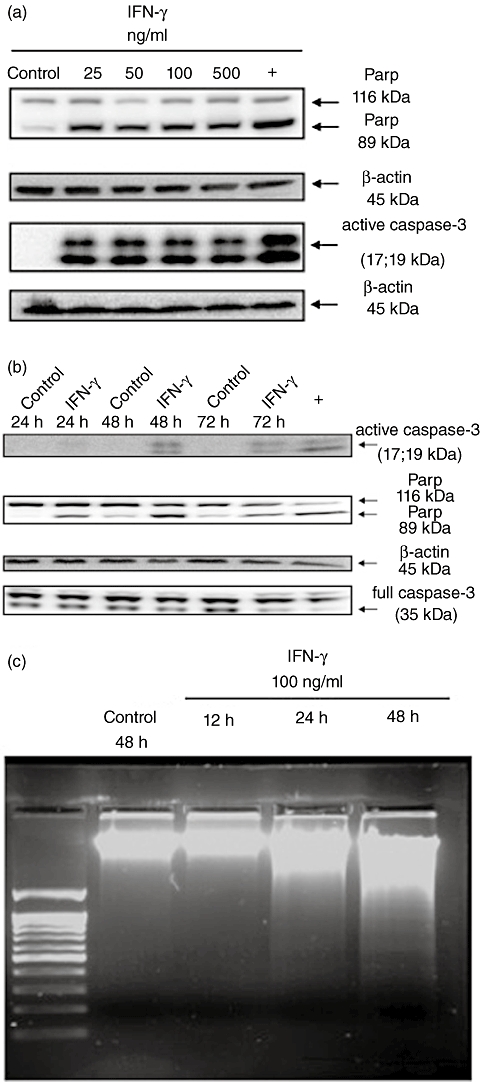
(a) Western blot analysis to detect dose-dependency of caspase-3 activation and consecutive poly-adenosine diphosphate-ribose-polymerase (PARP) cleavage after treatment of human colon epithelial cells (HT-29) with interferon (IFN)-γ for 48 h. Controls were cultured for 48 h in serum-free medium. (b) Time–course for detection of IFN-γ-dependent activation of caspase-3 and consecutive PARP cleavage in HT-29 cells using Western blot analysis. (c) DNA-laddering after stimulation of HT-29 cells for 12–48 h with IFN-γ (100 ng/ml). Controls were cultured for 48 h in serum-free medium.
With regard to the influence of IFN-γ on the expression of apoptosis-related proteins of the bcl2 family, treating HT-29 cells with IFN-γ (100 ng/ml) resulted in up-regulation of the pro-apoptotic Bad after 24 h (Fig. 5), whereas anti-apoptotic bcl-xl remained expressed constitutively.
Fig. 5.
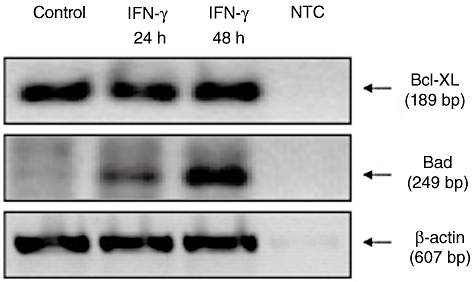
Analysis of interferon (IFN)-γ-dependent regulation of pro- and anti-apoptotic proteins in human colon epithelial cells (HT-29). HT-29 cells were cultured for indicated time with IFN-γ (100 ng/ml) or in serum-free medium. Using reverse transcription–polymerase chain reaction (RT–PCR) the influence of IFN-γ on the pro-apoptotic protein Bad and the anti-apoptotic protein Bcl-XL was examined. NTC: non-template control.
Selective interference with IFN-γ signalling in IEC
Stimulation of HT-29 and Caco-2 cells with IFN-γ (100 ng/ml) led similarly to phosphorylation of the signal transducer and activator of transcription (STAT)-1 (Fig. 6). Pretreatment of IEC with JAK tyrosine kinase inhibitor AG490 (40 and 80 µM) reduced caspase-3 activation (Fig. 7a and c), although STAT-1 phosporylation was not abrogated completely (Fig. 7b).
Fig. 6.
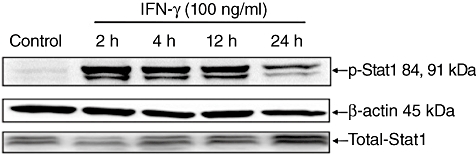
Western blot analysis for detection of interferon (IFN)-γ-mediated signal pathways. Human colon epithelial cells (HT-29) were stimulated with IFN-γ (100 ng/ml) for 2–24 h. Consecutive phosphorylation of signal transducer and activator of transcription (STAT)-1 was detected using Western blot analysis. 24 h in serum-free medium cultured HT-29 cells were used as controls. Similar results were detectable in Caco-2 cells (see Fig. 8b).
Fig. 7.
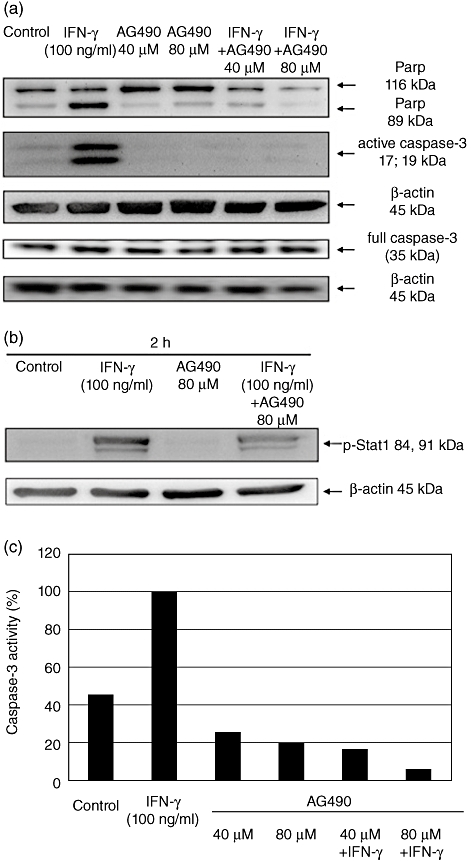
Western blot analysis for detection of interferon (IFN)-γ-mediated caspase-3 activation and consecutive cleavage of poly-adenosine diphosphate-ribose-polymerase (PARP) after inhibition of JAK tyrosine kinase. Caco-2 cells were stimulated for 48 h with IFN-γ (100 ng/ml) and the JAK tyrosine kinase inhibitor alone or in combination. The activation of caspase-3 and consecutive cleavage of PARP were detected using Western blot analysis (48 h in serum-free medium cultured Caco-2 cells were used as controls (a). Caco-2 cells were cultured with IFN-γ and AG490 alone and in combination as indicated. Signal transducer and activator of transcription (STAT)-1 phosphorylation was detected using Western blot analysis (b). After performing Western blotting the caspase-3 activity was also measured semiquantitatively using densitometry and was correlated to β-actin expression. Results after stimulation with IFN-γ were determined as 100% (c).
As expected, AG490 abrogated IFN-γ-induced IL-18bp expression in supernatants and cell lysates. Stimulation of Caco-2 cells with IFN-γ alone for 48 h resulted in a significantly higher IL-18bp expression in supernatants and in lysates compared to controls cultivated in serum-free medium only (P < 0·05) (Fig. 8a and b). AG490 in 40 µM and 80 µM concentrations in the presence of IFN-γ resulted in a significant reduction of the IL-18bp expression in supernatants (P < 0·05) and in cell lysates (P < 0·05).
Fig. 8.
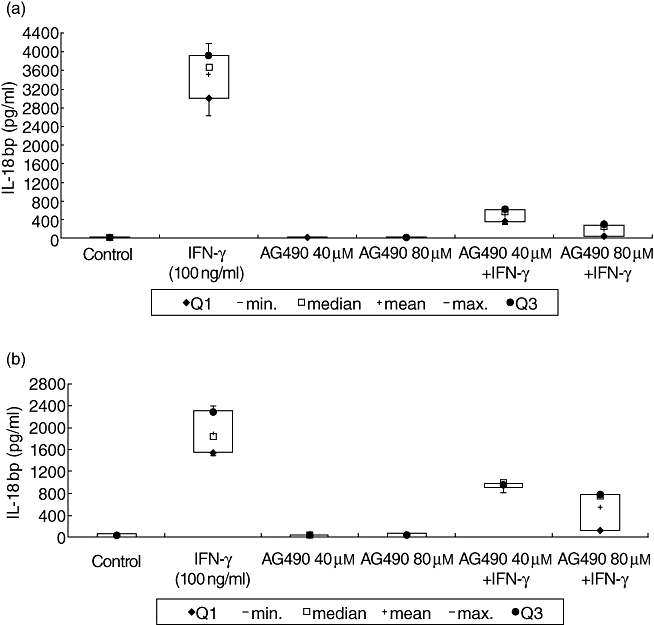
Interferon (IFN)-γ-mediated interleukin (IL)-18 binding protein (bp) expression after inhibition of JAK tyrosine kinase. Caco-2 cells were cultured for 48 h with IFN-γ (100 ng/ml) and JAK tyrosine kinase inhibitor AG490 (40 and 80 µM) alone and in combination. Resulting IL-18bp expression was measured by enzyme-linked immunosorbent assay in cell supernatants (a) as well as in cell lysates (b). Caco-2 cells cultured for 48 h in serum-free medium were used as controls. Data represent the results from three independent experiments.
In contrast to inhibition of IFN-γ-dependent JAK tyrosine kinase activation, inhibition of caspase-3 preserved the positive influence of IFN-γ on IL-18bp expression (Fig. 9). However, as expected, the inhibition of caspase-3 resulted in reduced activation of the pro-apoptotic cascade (Fig. 9). Compared to treatment of Caco-2 cells with IFN-γ (100 ng/ml) alone, treatment of cells in the presence of the caspase-3 inhibitor Z-DEVD-FMK (150 µM) reduced caspase-3 activity substantially.
Fig. 9.
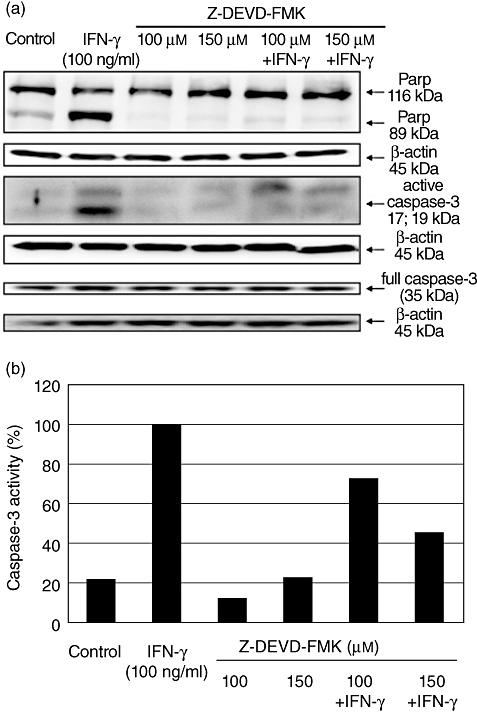
Western blot analysis for detection of interferon (IFN)-γ-mediated caspase-3 activation and consecutive cleavage of poly-adenosine diphosphate-ribose-polymerase (PARP) after caspase inhibition. Caco-2 cells were stimulated for 48 h with IFN-γ (100 ng/ml) and the caspase-3 inhibitor Z-DEVD-FMK alone or in combination. One representative experiment of three is shown. The activation of caspase-3 and consecutive cleavage of PARP were detected using Western blot analysis (a). Caco-2 cells cultured for 48 h in serum-free medium were used as controls. After performing Western blotting the caspase-3 activity was also measured semiquantitatively using densitometry and was correlated with β-actin expression (b). Results after stimulation with IFN-γ were determined as 100%.
Here, significant lower concentrations of IL-18bp were detectable in untreated controls, compared with IFN-γ-treated cells (P < 0·05). Stimulation of Caco-2 cells with IFN-γ alone for 48 h resulted in a significantly higher IL-18bp expression in supernatants and in lysates compared to cells cultured in serum-free medium after 48 h (P < 0·05). IFN-γ in the presence of 100 µM Z-DEVD-FMK was followed by similar IL-18bp expression, as in cells cultured only in the presence of IFN-γ, which was 3% higher in supernatant and about 5% higher in lysates (Fig. 10). Furthermore, IFN-γ-induced stimulation after pretreatment with 150 µM Z-DEVD-FMK resulted in an expression of IL-18bp which was one-third higher in supernatants and at about 25% higher in lysates compared to IFN-γ treatment without inhibitor. Surprisingly, similar results were obtained in the presence of the p38 MAP kinase inhibitor SB203580. We were able to reduce IFN-γ-mediated activity of the pro-apoptotic cascade, while not altering the positive effects of IFN-γ on the IL-18/IL-18bp ratio (Figs 11, 12).
Fig. 10.
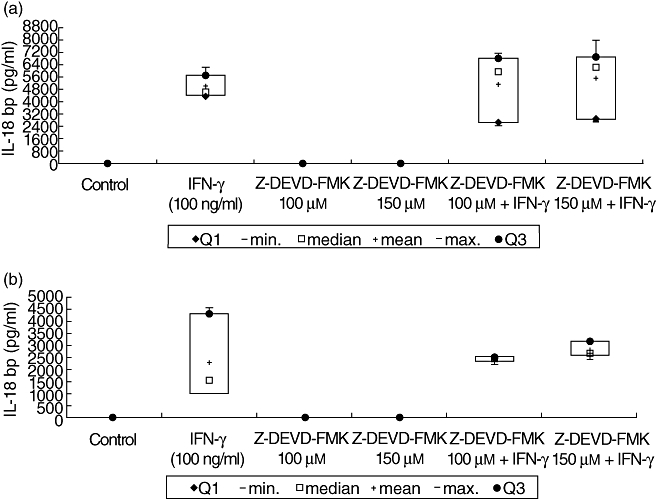
Interferon (IFN)-γ-mediated interleukin (IL)-18 binding protein (bp) expression after inhibition of caspase-3. Caco-2 cells were cultured for 48 h with IFN-γ (100 ng/ml) and caspase-3 inhibitor Z-DEVD-FMK (100 and 150 µM) alone and in combination. Resulting IL-18bp expression was measured by enzyme-linked immunosorbent assay in cell supernatants (a) as well as in cell lysates (b). Cultured Caco-2 cells were used as controls for 48 h in serum-free medium. Data represent the results from three independent experiments.
Fig. 11.
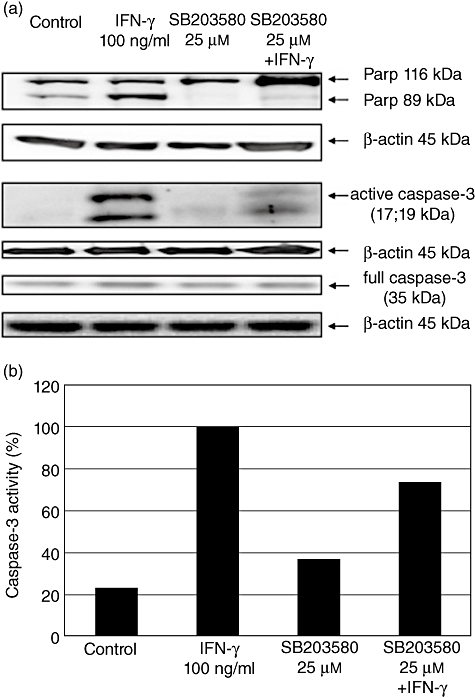
Western blot analysis for detection of interferon (IFN)-γ-mediated caspase-3 activation and consecutive cleavage of poly-adenosine diphosphate-ribose-polymerase (PARP) after inhibition of mitogen-activated protein (MAP) kinase p38. Caco-2 cells were stimulated for 48 h with IFN-γ (100 ng/ml) and the p38 inhibitor SB203580 alone or in combination. One representative experiment of three is shown. The activation of caspase-3 and consecutive cleavage of PARP were detected using Western blot analysis (a). Caco-2 cells cultured for 48 h in serum-free medium were used as controls. After performing Western blotting the caspase-3 activity was also measured semiquantitatively using densitometry and was correlated to β-actin expression (b). Results after stimulation with IFN-γ were determined as 100%.
Fig. 12.
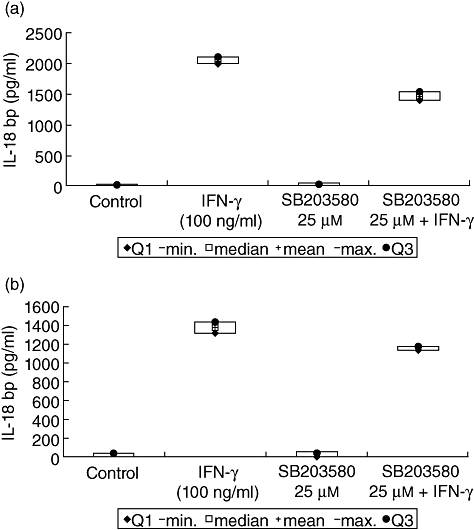
Interferon (IFN)-γ-mediated interleukin (IL)-18 binding protein (bp) expression after inhibition of p38. Caco-2 cells were cultured for 48 h with IFN-γ (100 ng/ml) and p38 mitogen-activated protein (MAP) kinase inhibitor SB203580 (25 µM) alone and in combination. Resulting IL-18bp expression was measured by enzyme-linked immunosorbent assay in cell supernatants (a) as well as in cell lysates (b). Caco-2 cells cultured for 48 h in serum-free medium were used as controls. Data represent the results from two independent experiments.
Discussion
IFN-γ is a cytokine with pleiotropic effects and it exerts an important role in the pathogenesis of chronic inflammatory bowel diseases. The data from this work demonstrate, first, the ability of IFN-γ alone to induce apoptosis in IEC. Secondly, while confirming a previous report that IFN-γ is a potent inducer of IL-18bp [24], we are able to broaden the knowledge by investigating involved signalling pathways and the association with the occurrence of apoptosis. Thirdly, we disclose an approach that, in the presence of IFN-γ, maintains induction of anti-inflammatory IL-18bp while reducing activity of the pro-apoptotic cascade.
IL-18bp is the natural antagonist of IL-18, which potentially perpetuates chronic inflammation in patients with IBD, mainly through induction of IFN-γ expression. IL-18bp exists in humans in several isoforms, especially in the form of IL-18bpa and IL-18bpc, which bind IL-18 in the extracellular space and thereby antagonize its proinflammatory effect – other isoforms do not have this ability [25]. An increased expression of IL-18bp, mainly of the functioning isoforms, which bind IL-18 effectively, could therefore decrease the degree of inflammation in patients with IBD.
Inhibition of early signalling events (i.e. STAT-1 phosphorylation) prevents IFN-γ-mediated apoptosis, but also expression of IL-18bp. Further downstream, in the presence of inhibitors of p38 MAP kinase and caspase-3 IL-18bp expression and secretion is maintained.
Apoptosis is induced by IFN-γ due potentially to an alteration of the stoichiometry of pro- and anti-apoptotic proteins. We found an induction of pro-apoptotic Bad after treatment with IFN-γ in HT-29 cells. Bad is a direct antagonist of anti-apoptotic proteins such as Bcl-2 and Bcl-XL, which inhibit apoptosis by preventing the release of cytochrome c from mitochondria [26,27]. Bad forms a heterodimer with Bcl-2 and Bcl-XL and thereby enhances the liberation of cytochrome c [28]. In our hands, Bcl-XL was expressed constitutively, whereas Bcl-2 was not detectable reproducibly in HT-29 cells; neither was altered in the presence of IFN-γ (data not shown). Treatment of intestinal epithelial cells with parathormone has been shown to cause translocation of the cytosolic Bad in the mitochondria and, in the end, also results in apoptosis of the cell by release of cytochrome c [28]. Several studies indicate that the ratio of pro- to anti-apoptotic proteins influences the cell volume of the intestinal epithelium physiologically. Proliferation of epithelial cells dominates in the epithelial crypts, whereas surface cells undergo degradation, e.g. by apoptosis, in order to guarantee a permanent renewal. In crypts anti-apoptotic proteins such as Bcl-2 dominate [29], whereas pro-apoptotic proteins such as Bax dominate in surface cells [30].
Binding of IFN-γ to its receptor leads to phosphorylation of the transcription factor STAT-1 by activation of the Janus kinases JAK1/JAK2 [31]. We used AG490, a JAK tyrosine kinase inhibitor, in order to analyse blockade of an early event of the IFN-γ signalling cascade on apoptosis and IL-18bp expression and release. Decreased phosphorylation of STAT-1 and significantly decreased expression of IL-18bp were noted. Similarly, almost complete inhibition of caspase-3 activity and consecutive cleavage of PARP occurred. Kwak and colleagues were able to show that AG490 can, potentially, avert programmed cell death by inhibition of JAK2 in osteoclasts, associated with decreased cytochrome c release and suppression of the activity of pro-apoptotic proteins Bad and Bim [32]. Similar results were found in glioma cells [33]. Here, AG490 caused an inhibition of apoptosis by suppressing the activation of pro-apoptotic proteins Bad and Bax. Interestingly, inhibition of apoptosis by AG490 occurred only in the presence of p53, a protein responsible for cell cycle control. AG490 reinforced the effect of pro-apoptotic stimuli in cells, which had a mutation or deletion of the p53 gene. AG490 was cytostatic due to its pro-apoptotic effects in acute lymphatic leukaemia, both in vitro and in vivo. We were unable to detect this pro-apoptotic effect in intestinal epithelial cells. This also leads to the assumption that other signalling pathways besides STAT-1 might be involved. It is known that the JAK2 Janus kinase, which is activated after binding of IFN-γ to its receptor, also phosphorylates not only STAT-1 but also other STAT molecules [34]. In fact, the inhibitor AG490 does not block JAK2 selectively, but is also able to inhibit other tyrosine kinases such as JAK3 [35].
Activation of apoptosis ultimately results, besides other cellular and subcellular changes, in the activation of effector caspase-3 and cleavage of the caspase-3-specific substrate PARP [36]. Z-DEVD-FMK is a potential inhibitor of caspase-3 [37]. Usage of this inhibitor, although incomplete, enabled us to inhibit the terminal pathway of apoptosis almost completely. The inhibitor reduced caspase-3 activation and decreased cleavage of PARP. Fortunately, inhibition of caspase-3 by Z-DEVD-FMK did not suppress IFN-γ-induced expression of IL-18bp. Rather, the opposite was the case: in the presence of the inhibitor, IL-18bp levels increased. Obviously, inhibition of apoptosis and maintained cellular integrity permitted increased expression of IL-18bp. It is known that different pro-apoptotic cytokines in intestinal epithelial cells might alter cell–cell connections [38]. Anoikis describes the induction of a programmed cell death of cells which lose contact with each other or with the extracellular matrix [39]. In this context, apoptosis as well as changes of zonulae occludentes after treatment of IEC with IL-13 have been reported [38]. Scaife and colleagues postulated that the apoptosis in HT-29 and Caco-2 cells was caused not only by inhibitors of NF-κB but also by loss of contact of these cells with their united cell structure [40].
Western blot analyses showed a constitutive phosphorylation of p38 and JNK, which was not altered by IFN-γ (data not shown). Whereas in one report phosphorylated p38 and JNK was not found in HT-29 and Caco-2 cells, which were cultivated in serum-free medium [41], Honore and colleagues confirmed our observation of a constitutive phosphorylation of these signalling molecules in Caco-2 cells [42]. Even though IFN-γ does not influence the phosphorylation level of these kinases, they seem to play a role in the IFN-γ-induced IL-18bp expression and programmed cell death. SB203580 leads to a considerable decrease of caspase 3 activity and consecutive cleavage of PARP; p38 activation in connection with induction of apoptosis is also thought to play an important role in the pathogenesis of the necrotizing enterocolitis in newborns [43]. In this context, the authors showed phosphorylation of p38 – caused by oxidative stress, with consecutive occurrence of apoptosis – in intestinal epithelial cells, which were isolated from rats. H2O2-induced cell death was reduced by the inhibitor SB203580. Data generated in Caco-2 cells demonstrate that inhibition of p38 prevents apoptosis in these cells [44]. In an experimental model of colitis in mice it was possible to inhibit cytokine-mediated apoptosis by inhibition of MAP kinases [45]. According to the study, inhibition of stress-dependent activation of MAP kinase could have a positive effect on the inflammatory reaction and preservation of the intestinal barrier in patients with IBD in vivo. As already mentioned, SB203580 inhibited IFN-γ-dependent apoptosis in Caco-2 cells. However, the positive influence of IFN-γ on IL-18bp expression could be conserved. These findings, which are shown here for the first time, support the notion that signalling pathways which are induced by p38 are not part of the IL-18bp regulation by IFN-γ in Caco-2 cells. It would be worthwhile to clarify in detail the molecular mechanism of these signalling cascades regarding IFN-γ-mediated apoptosis as well as IL-18bp expression. Therefore, strategies that follow the inhibition of MAP kinases such as p38 in vivo could be interesting, even though a clinical study failed to show efficacy in patients with Crohn's disease [46].
The data presented here do not implicate immediate therapeutic consequences for patients with IBD. However, the contribution of the epithelial barrier to the pathogenesis of IBD is accepted widely. Therefore, better knowledge of the balance of pro- and anti-inflammatory cytokines expressed by epithelial cells may help to interfere earlier in the disease process. In the above-discussed context, enhancement of secretion of the anti-inflammatory IL-18bp could, potentially, stabilize remission of the disease, but concomitantly, epithelial cell death would need to be evaded by selective interference with pro-apoptotic cascades.
Acknowledgments
The authors would like to thank Carolin Stump, Frank Herweck and Jutta Gundt for expert technical assistance as well as Mrs S. Hugues for help with the manuscript.
Disclosure
The authors declare that there is no conflict of interest.
References
- 1.Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008;134:577–94. doi: 10.1053/j.gastro.2007.11.059. [DOI] [PubMed] [Google Scholar]
- 2.Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem. 2001;276:4812–18. doi: 10.1074/jbc.M008072200. [DOI] [PubMed] [Google Scholar]
- 3.Ramasundara M, Leach ST, Lemberg DA, Day AS. Defensins and inflammation: the role of defensins in inflammatory bowel disease. J Gastroenterol Hepatol. 2009;24:202–8. doi: 10.1111/j.1440-1746.2008.05772.x. [DOI] [PubMed] [Google Scholar]
- 4.Colgan SP, Parkos CA, Delp C, Arnaout MA, Madara JL. Neutrophil migration across cultured intestinal epithelial monolayers is modulated by epithelial exposure to IFN-gamma in a highly polarized fashion. J Cell Biol. 1993;120:785–98. doi: 10.1083/jcb.120.3.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-gamma. Annu Rev Immunol. 1997;15:749–95. doi: 10.1146/annurev.immunol.15.1.749. [DOI] [PubMed] [Google Scholar]
- 6.Breese E, Braegger CP, Corrigan CJ, Walker-Smith JA, MacDonald TT. Interleukin-2- and interferon-gamma-secreting T cells in normal and diseased human intestinal mucosa. Immunology. 1993;78:127–31. [PMC free article] [PubMed] [Google Scholar]
- 7.Fuss IJ, Neurath M, Boirivant M, et al. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn's disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol. 1996;157:1261–70. [PubMed] [Google Scholar]
- 8.Niessner M, Volk BA. Altered Th1/Th2 cytokine profiles in the intestinal mucosa of patients with inflammatory bowel disease as assessed by quantitative reversed transcribed polymerase chain reaction (RT–PCR) Clin Exp Immunol. 1995;101:428–35. doi: 10.1111/j.1365-2249.1995.tb03130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Okamura H, Tsutsi H, Komatsu T, et al. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature. 1995;378:88–91. doi: 10.1038/378088a0. [DOI] [PubMed] [Google Scholar]
- 10.Ushio S, Namba M, Okura T, et al. Cloning of the cDNA for human IFN-gamma-inducing factor, expression in Escherichia coli, and studies on the biologic activities of the protein. J Immunol. 1996;156:4274–9. [PubMed] [Google Scholar]
- 11.Stoll S, Jonuleit H, Schmitt E, et al. Production of functional IL-18 by different subtypes of murine and human dendritic cells (DC): DC-derived IL-18 enhances IL-12-dependent Th1 development. Eur J Immunol. 1998;28:3231–9. doi: 10.1002/(SICI)1521-4141(199810)28:10<3231::AID-IMMU3231>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 12.Pizarro TT, Michie MH, Bentz M, et al. IL-18, a novel immunoregulatory cytokine, is up-regulated in Crohn's disease: expression and localization in intestinal mucosal cells. J Immunol. 1999;162:6829–35. [PubMed] [Google Scholar]
- 13.Okamura H, Nagata K, Komatsu T, et al. A novel costimulatory factor for gamma interferon induction found in the livers of mice causes endotoxic shock. Infect Immun. 1995;63:3966–72. doi: 10.1128/iai.63.10.3966-3972.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Micallef MJ, Ohtsuki T, Kohno K, et al. Interferon-gamma-inducing factor enhances T helper 1 cytokine production by stimulated human T cells: synergism with interleukin-12 for interferon-gamma production. Eur J Immunol. 1996;26:1647–51. doi: 10.1002/eji.1830260736. [DOI] [PubMed] [Google Scholar]
- 15.Kohno K, Kataoka J, Ohtsuki T, et al. IFN-gamma-inducing factor (IGIF) is a costimulatory factor on the activation of Th1 but not Th2 cells and exerts its effect independently of IL-12. J Immunol. 1997;158:1541–50. [PubMed] [Google Scholar]
- 16.Lochner M, Forster I. Anti-interleukin-18 therapy in murine models of inflammatory bowel disease. Pathobiology. 2002;70:164–9. doi: 10.1159/000068149. [DOI] [PubMed] [Google Scholar]
- 17.Monteleone G, Trapasso F, Parrello T, et al. Bioactive IL-18 expression is up-regulated in Crohn's disease. J Immunol. 1999;163:143–7. [PubMed] [Google Scholar]
- 18.Novick D, Kim SH, Fantuzzi G, Reznikov LL, Dinarello CA, Rubinstein M. Interleukin-18 binding protein: a novel modulator of the Th1 cytokine response. Immunity. 1999;10:127–36. doi: 10.1016/s1074-7613(00)80013-8. [DOI] [PubMed] [Google Scholar]
- 19.Böcker U, Damiao A, Holt L, et al. Differential expression of interleukin 1 receptor antagonist isoforms in human intestinal epithelial cells. Gastroenterology. 1998;115:1426–38. doi: 10.1016/s0016-5085(98)70021-6. [DOI] [PubMed] [Google Scholar]
- 20.Kalina U, Koyama N, Hosoda T, et al. Enhanced production of IL-18 in butyrate-treated intestinal epithelium by stimulation of the proximal promoter region. Eur J Immunol. 2002;32:2635–43. doi: 10.1002/1521-4141(200209)32:9<2635::AID-IMMU2635>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 21.Böcker U, Schottelius A, Watson JM, et al. Cellular differentiation causes a selective down-regulation of interleukin (IL)-1beta-mediated NF-kappaB activation and IL-8 gene expression in intestinal epithelial cells. J Biol Chem. 2000;275:12207–13. doi: 10.1074/jbc.275.16.12207. [DOI] [PubMed] [Google Scholar]
- 22.Manigold T, Böcker U, Traber P, et al. Lipopolysaccharide/endotoxin induces IL-18 via CD14 in human peripheral blood mononuclear cells in vitro. Cytokine. 2000;12:1788–92. doi: 10.1006/cyto.2000.0783. [DOI] [PubMed] [Google Scholar]
- 23.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 24.Paulukat J, Bosmann M, Nold M, et al. Expression and release of IL-18 binding protein in response to IFN-gamma. J Immunol. 2001;167:7038–43. doi: 10.4049/jimmunol.167.12.7038. [DOI] [PubMed] [Google Scholar]
- 25.Kim SH, Eisenstein M, Reznikov L, et al. Structural requirements of six naturally occurring isoforms of the IL-18 binding protein to inhibit IL-18. Proc Natl Acad Sci USA. 2000;97:1190–5. doi: 10.1073/pnas.97.3.1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang J, Liu X, Bhalla K, et al. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–32. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 27.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–6. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 28.Schurmann A, Mooney AF, Sanders LC, et al. p21-activated kinase 1 phosphorylates the death agonist bad and protects cells from apoptosis. Mol Cell Biol. 2000;20:453–61. doi: 10.1128/mcb.20.2.453-461.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hockenbery DM, Zutter M, Hickey W, Nahm M, Korsmeyer SJ. BCL2 protein is topographically restricted in tissues characterized by apoptotic cell death. Proc Natl Acad Sci USA. 1991;88:6961–5. doi: 10.1073/pnas.88.16.6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krajewski S, Krajewska M, Shabaik A, Miyashita T, Wang HG, Reed JC. Immunohistochemical determination of in vivo distribution of Bax, a dominant inhibitor of Bcl-2. Am J Pathol. 1994;145:1323–36. [PMC free article] [PubMed] [Google Scholar]
- 31.Ghosh S, Chaudhary R, Carpani M, Playford R. Interfering with interferons in inflammatory bowel disease. Gut. 2006;55:1071–3. doi: 10.1136/gut.2005.090134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kwak HB, Sun HM, Ha H, Lee JH, Kim HN, Lee ZH. AG490, a Jak2-specific inhibitor, induces osteoclast survival by activating the akt and ERK signaling pathway. Mol Cell. 2008;26:436–42. [PubMed] [Google Scholar]
- 33.Jane EP, Premkumar DR, Pollack IF. AG490 influences UCN-01-induced cytotoxicity in glioma cells in a p53-dependent fashion, correlating with effects on BAX cleavage and BAD phosphorylation. Cancer Lett. 2007;257:36–46. doi: 10.1016/j.canlet.2007.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bunting KD. STAT5 signaling in normal and pathologic hematopoiesis. Front Biosci. 2007;12:2807–20. doi: 10.2741/2274. [DOI] [PubMed] [Google Scholar]
- 35.Kirken RA, Erwin RA, Wang L, Wang Y, Rui H, Farrar WL. Functional uncoupling of the Janus kinase 3-Stat5 pathway in malignant growth of human T cell leukemia virus type 1-transformed human T cells. J Immunol. 2000;165:5097–104. doi: 10.4049/jimmunol.165.9.5097. [DOI] [PubMed] [Google Scholar]
- 36.Ramachandran A, Madesh M, Balasubramanian KA. Apoptosis in the intestinal epithelium: its relevance in normal and pathophysiological conditions. J Gastroenterol Hepatol. 2000;15:109–20. doi: 10.1046/j.1440-1746.2000.02059.x. [DOI] [PubMed] [Google Scholar]
- 37.Wu JN, Huang J, Yang J, Tashiro S, Onodera S, Ikejima T. Caspase inhibition augmented oridonin-induced cell death in murine fibrosarcoma l929 by enhancing reactive oxygen species generation. J Pharmacol Sci. 2008;108:32–9. doi: 10.1254/jphs.fp0072079. [DOI] [PubMed] [Google Scholar]
- 38.Heller F, Florian P, Bojarski C, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology. 2005;129:550–64. doi: 10.1016/j.gastro.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 39.Frisch SM, Francis H. Disruption of epithelial cell–matrix interactions induces apoptosis. J Cell Biol. 1994;124:619–26. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scaife CL, Kuang J, Wills JC, et al. Nuclear factor kappaB inhibitors induce adhesion-dependent colon cancer apoptosis: implications for metastasis. Cancer Res. 2002;62:6870–8. [PubMed] [Google Scholar]
- 41.Barry OP, Mullan B, Sheehan D, et al. Constitutive ERK1/2 activation in esophagogastric rib bone marrow micrometastatic cells is MEK-independent. J Biol Chem. 2001;276:15537–46. doi: 10.1074/jbc.M010847200. [DOI] [PubMed] [Google Scholar]
- 42.Honore S, Kovacic H, Pichard V, Briand C, Rognoni JB. Alpha2beta1-integrin signaling by itself controls G1/S transition in a human adenocarcinoma cell line (Caco-2): implication of NADPH oxidase-dependent production of ROS. Exp Cell Res. 2003;285:59–71. doi: 10.1016/s0014-4827(02)00038-1. [DOI] [PubMed] [Google Scholar]
- 43.Zhou Y, Wang Q, Mark Evers B, Chung DH. Oxidative stress-induced intestinal epithelial cell apoptosis is mediated by p38 MAPK. Biochem Biophys Res Commun. 2006;350:860–5. doi: 10.1016/j.bbrc.2006.09.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leone V, di Palma A, Ricchi P, et al. PGE2 inhibits apoptosis in human adenocarcinoma Caco-2 cell line through Ras-PI3K association and cAMP-dependent kinase A activation. Am J Physiol Gastrointest Liver Physiol. 2007;293:G673–81. doi: 10.1152/ajpgi.00584.2006. [DOI] [PubMed] [Google Scholar]
- 45.Assi K, Pillai R, Gomez-Munoz A, Owen D, Salh B. The specific JNK inhibitor SP600125 targets tumour necrosis factor-alpha production and epithelial cell apoptosis in acute murine colitis. Immunology. 2006;118:112–21. doi: 10.1111/j.1365-2567.2006.02349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schreiber S, Feagan B, D'Haens G, et al. Oral p38 mitogen-activated protein kinase inhibition with BIRB 796 for active Crohn's disease: a randomized, double-blind, placebo-controlled trial. Clin Gastroenterol Hepatol. 2006;4:325–34. doi: 10.1016/j.cgh.2005.11.013. [DOI] [PubMed] [Google Scholar]