

Abstract
Vertebrate photoreceptors are thought to adapt to light by a change in Ca2+, which is postulated to mediate modulation of (1) excited rhodopsin (Rh*) by Ca2+-dependent binding of recoverin, (2) guanylyl cyclase activity via Ca2+-dependent GCAP proteins, and (3) cyclic nucleotide-gated channels by binding of Ca2+-calmodulin. Previous experiments genetically deleted recoverin and the GCAPs and showed that significant regulation of sensitivity survives removal of (1) and (2). We genetically deleted the channel Ca2+-calmodulin binding site in the mouse Mus musculus and found that removal of (3) alters response waveform, but removal of (3) or of (2) and (3) together still leaves much of adaptation intact. These experiments demonstrate that an important additional mechanism is required, which other experiments indicate may be regulation of phosphodiesterase 6 (PDE6). We therefore constructed a kinetic model in which light produces a Ca2+-mediated decrease in PDE6 decay rate, with the novel feature that both spontaneously activated and light-activated PDE6 are modulated. This model, together with Ca2+-dependent acceleration of guanylyl cyclase, can successfully account for changes in sensitivity and response waveform in background light.
Introduction
Sensory receptors adapt to constant stimulation. In a vertebrate rod, steady light decreases the concentration of cGMP, which closes cyclic nucleotide-gated channels; this decreases the outer segment free-Ca2+ concentration, which is thought to adapt the rod by modulating one or more steps in the transduction cascade. Three possible mechanisms of adaptation have been identified (Fain et al., 2001): (1) regulation of the lifetime of activated rhodopsin (Rh*) (Matthews et al., 2001; Chen et al., 2010) via a Ca2+-binding protein called S-modulin or recoverin (Kawamura, 1993; Chen et al., 1995, 2010; Erickson et al., 1998); (2) activation of guanylyl cyclase (Koch and Stryer, 1988; Peshenko and Dizhoor, 2004) by Ca2+-binding guanylyl cyclase-activating proteins or GCAPs (see Palczewski et al., 2004); and (3) modulation of the affinity of the cyclic nucleotide-gated channels (CNGs) for cGMP (Hsu and Molday, 1993; Nakatani et al., 1995) by binding of Ca2+-calmodulin to the channel CNGβ1 subunit (Grunwald et al., 1998; Weitz et al., 1998). Previous experiments have studied the roles of (1) and (2) by knocking out recoverin (Makino et al., 2004) and the GCAP proteins (Mendez et al., 2001; Burns et al., 2002). These experiments, together with results we present in this paper, show that recoverin deletion has little or no effect on adaptation of flash sensitivity; removal of GCAPs alters sensitivity regulation, but much of the phenomenology of light adaptation persists in GCAPs−/− rods.
That leaves (3), Ca2+-calmodulin-dependent modulation of the channels. The rod cyclic nucleotide-gated channel is a tetramer composed of CNGA1 and CNGβ1 subunits with a 3:1 stoichiometry (Weitz et al., 2002; Zheng et al., 2002; Zhong et al., 2002); the CNGβ1 subunit is known to contain a binding site for Ca2+-calmodulin (Grunwald et al., 1998; Weitz et al., 1998). Previous experiments (Hsu and Molday, 1993; Gordon et al., 1995; Koutalos et al., 1995; Nakatani et al., 1995) have indicated that Ca2+-calmodulin is bound to the rod cyclic nucleotide-gated channel in the dark, when outer segment free-Ca2+ is high and the affinity of the channel for cGMP is relatively low. As Ca2+ falls in the light, Ca2+ becomes unbound, increasing the affinity of the channel for cGMP and counteracting the fall in cGMP concentration.
To test for a role of channel modulation, we have introduced a targeted mutation into the gene for the CNGβ1 subunit to remove the Ca2+-calmodulin binding site. This should remove all Ca2+-dependent modulation of the channel (Gordon et al., 1995; Warren and Molday, 2002). These CNGB1ΔCaM mice were also crossed with GCAPs−/− mice to remove both channel and cyclase modulation. Our experiments reveal an important contributor to adaptation that none of the proposed mechanisms can explain. We postulate a novel mechanism of modulation of spontaneously activated phosphodiesterase (PDE) 6, and we show with a kinetic model that Ca2+-dependent acceleration of decay of both spontaneous PDE6 and light-activated PDE6, together with GCAP-dependent cyclase regulation, is sufficient to explain the waveform and sensitivity of both wild-type (WT) and GCAPs−/− rod responses during maintained illumination.
Materials and Methods
Generation of CNGB1ΔCaM mice.
Care of experimental animals conformed to procedures approved by the University of Southern California and University of California, Los Angeles Animal Care Committees (Los Angeles, CA). A CNGB1 genomic fragment was obtained by long-range PCR with 129/Sv mouse embryonic stem (ES) cell DNA as template. A targeting vector was constructed whereby 14 aa residues were deleted within the CaM binding site contained within exon 20 (see Fig. 1A). This mutation was flanked by 5′ (1.6 kb) and 3′ (6.2 kb) arms. The neomycin selection cassette was inserted into intron 19, and a thymidine kinase cassette was cloned 5′ to the targeting vector (see Fig. 1B), thus offering both positive and negative selection to the integrated DNA. The construct was electroporated into 129/Sv mouse ES cells. G418- and 2-fluoro-2-deoxy-1β-d-arabino-furanosyl-5-iodo-uracil (FIAU)-resistant colonies were picked, expanded, and analyzed by Southern hybridization. Clones that had undergone homologous recombination were injected into C57BL/6 blastocysts. High degree chimeras (>95%) were bred to C57BL/6 to obtain mice heterozygous for the insertion. These mice were then bred with the Msx2-cre line (Fu et al., 2007) to remove the neomycin cassette.
Figure 1.
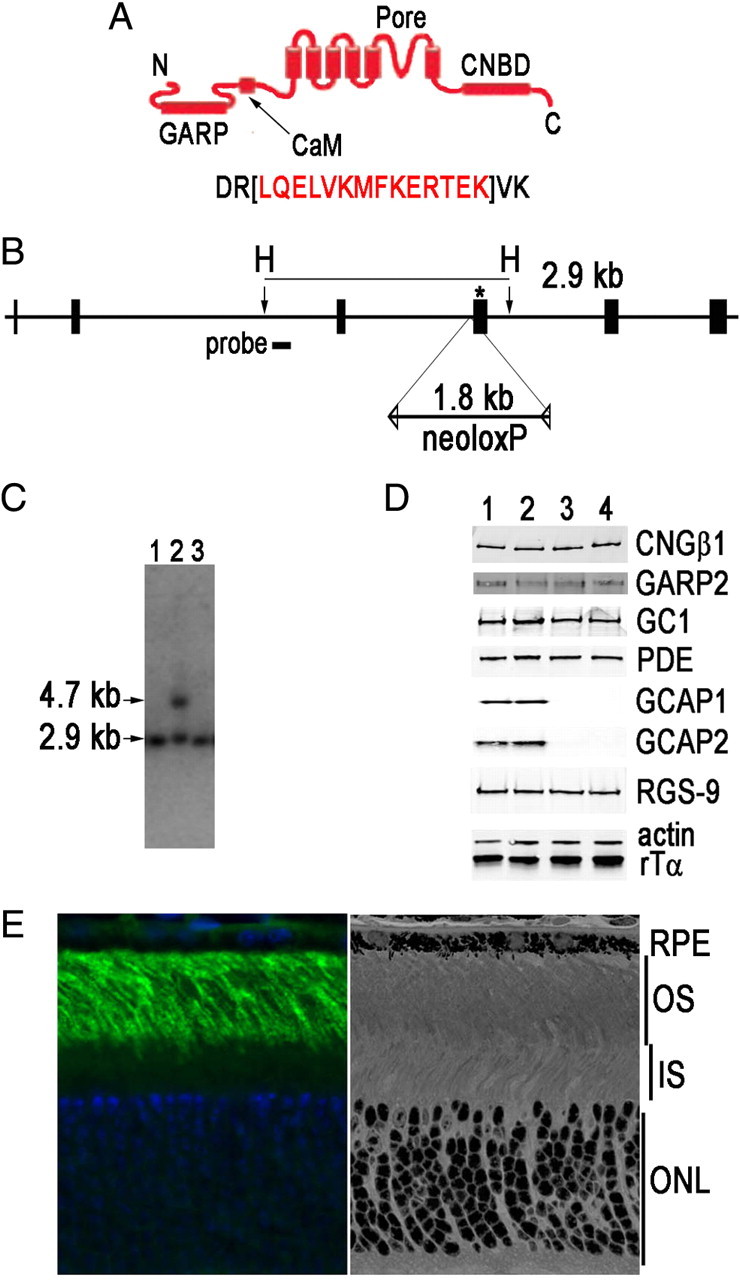
Generation of CNGB1ΔCaM mice. A, Schematic of the domains within the rod CNGβ1 subunit: GARP, Glutamic acid-rich protein; CaM, calmodulin binding domain; CNBD, cyclic nucleotide binding domain. Fourteen amino acid residues (red) were deleted within the CaM binding domain in the targeting construct. B, The calmodulin-binding domain is coded within exon 20 (asterisk). The 1.8 kb neoloxP selection marker was inserted into intron 19. DNA extracted from neomycin-resistant ES cell colonies was digested with HindIII (H). The endogenous fragment is predicted to be 2.9 kb, whereas the correctly targeted homologous recombination event will generate a 4.7 kb fragment (2.9 kb + 1.8 kb from the neomycin cassette) when the indicated probe is used in a Southern blot. C, Southern blot of representative colonies that did not (lanes 1 and 3) and did (lane 2) contain the correct homologous recombination event. D, Representative Western blot (N = 3–5) of retinal homogenates from 1-month-old WT (lane 1), CNGB1ΔCaM (lane 2), CNGB1ΔCaM/GCAPs−/− (lane 3), and GCAPs−/− (lane 4) mice. No changes in levels of indicated retinal proteins were detected except for the absence of GCAP1 and GCAP2 when their genes were knocked out. E, CNGB1ΔCaM is correctly localized to the outer segment (left). The mutation did not affect the retinal morphology (right). RPE, Retinal pigmented epithelium; OS, outer segment; IS, inner segment; ONL, outer nuclear layer.
Western blotting.
Retinas were isolated from 1 month-old mice and homogenized in buffer (80 mm Tris, pH 7.4, 10 mm EDTA, 4 mm MgCl2, 2 mm CaCl2, 0.5 mg/ml Complete Mini protease inhibitors; Roche). For detecting CNGβ1, the retinas were solubilized in radioimmune precipitation assay (RIPA) buffer (50 mm Tris, pH 8.0, 150 mm NaCl, 2 mm EDTA, 1 mm Na3VO4, 2.5 mm NaF, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS, 1 mm PMSF, and protease inhibitor cocktail), followed by DNase I treatment (0.1 mg/ml) at room temperature for 30 min instead of boiling. Equal amounts of retinal homogenates from the indicated mice were fractionated by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were blocked with 5% nonfat dry milk in TBST (20 mm Tris, pH 7.6, 137 mm NaCl, 0.05% Tween 20) followed by incubation with primary antibody (GARP:4B1) (Poetsch et al., 2001), GC1 (H-226, Santa Cruz Biotechnology), actin (MAB 1501; Millipore), PDE6 and rod transducin (MOE and TF-15; Cytosignal), and RGS-9 (Chen et al., 2000). Signals were detected and quantified with the Odyssey infrared imaging system (LI-COR Biosciences). Results from Western blots were normalized to levels of actin.
Histology and immunohistochemistry.
Eyecups were fixed in 2.5% glutaraldehyde/2% paraformaldehyde in 0.1 m cacodylate buffer, pH 7.2, and embedded into epoxy resin or 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.2, and embedded into OCT for frozen sections as described previously (Concepcion et al., 2002).
Suction-electrode recordings.
Methods for recording responses of mouse rods have been given previously (Woodruff et al., 2008; Chen et al., 2010). Animals between 2 and 6 months of age were dark adapted typically for 5 h but for at least 3 h in a light-tight box. Rods were perfused at 37–39°C with DMEM (catalog #D-2902, Sigma) supplemented with 15 mm NaHCO3, 2 mm Na succinate, 0.5 mm Na glutamate, 2 mm Na gluconate, and 5 mm NaCl, bubbled with 5% CO2, pH 7.4. Data were filtered at 30 Hz (8 pole Bessel filter) and sampled at 100 Hz. Flashes of 500 nm light 20 ms in duration were attenuated to different light levels by absorptive neutral density filters. At dim intensities, 10–20 individual responses presented at 5 s intervals were averaged to obtain mean flash responses. At medium intensities, 5–10 responses were averaged and the interflash interval was increased to 10 s. At bright intensities above saturation for the rods, only 3–5 responses were averaged, and the interflash interval was increased to 15–20 s. A 500 nm light was also used for steps of light and backgrounds. Other information about the details of response presentation is given in the figure legends. The values of τD for WT and CNGB1ΔCaM rods were measured as in Woodruff et al. (2008) by giving a series of five flashes at each of between 4 and 7 intensities chosen for each rod to fall within one and a half log units above the flash intensity that just produced saturation of that rod's response amplitude. Flash intensities were in the range of 159–3250 photons μm−2. The time in saturation (Tsat) was measured as the time from the beginning of the flash to the time at which the mean circulating current recovered to 25% of its dark-adapted value. The value of τD was then calculated as the best-fitting slope of Tsat versus the natural logarithm of the flash intensity. Unless otherwise stated, errors are given as standard errors of the mean. Curve fitting and plotting of data were done with the program Origin (OriginLab).
Results
Knocking out Ca2+-dependent modulation of the channel
We investigated the effect of channel regulation on the physiology of the rod by removing the site of calmodulin binding to produce so-called CNGB1ΔCaM mice. To accomplish this, a gene-targeting vector was constructed to delete the amino acids LQELVKMFKERTEK within exon 20 of the CNGB1 gene, which codes for the channel CNGβ1 subunit (Fig. 1A). Removal of these amino acids is known to abolish CaM-mediated inhibition of the channel in vitro (Grunwald et al., 1998) and in vivo for olfactory cyclic nucleotide-gated channels in knock-in mice (Song et al., 2008). The neomycin cassette, flanked by loxP sites, was inserted into intron 19 (Fig. 1B). This construct was electroporated into mouse ES cells, and neomycin-resistant colonies were screened by Southern blots. Digestion of ES cell DNA by the restriction enzyme HindIII created a 2.9 kb fragment from the endogenous CNGB1 locus (Fig. 1C, lanes 1 and 3), while homologous recombination produced a 4.7 kb fragment as the result of insertion of the neomycin cassette (Fig. 1C, lane 2). Knock-in mice were generated with these ES clones. The neoloxP cassette was subsequently removed from the CNGB1 locus by crossing with the Msx2-cre line, which caused excision of neoloxP in the germline of these mice (Fu et al., 2007). Deletion of the calmodulin binding site was confirmed by PCR and restriction mapping. These CNGB1ΔCaM mice were subsequently mated with GCAPs−/− mice lacking the GCAP proteins (Mendez et al., 2001) to produce CNGB1ΔCaM/GCAPs−/− mice.
Expression levels of several transduction proteins were evaluated in retinal homogenates from 1-month-old WT, CNGB1ΔCaM, CNGB1ΔCaM/GCAPs−/−, and GCAPs−/− mice (Fig. 1D, lanes 1, 2, 3, and 4, respectively). Few differences were detected, except for the absence of GCAP1 and GCAP2 in GCAPs−/− mice. These results were quantified and are presented in supplemental Fig. S1, where fluorescent signals from individual proteins were normalized to actin levels and then compared to those of WT. Removing the calmodulin binding site did not lead to expression changes in these transduction proteins.
To localize the mutant channel subunit, retinal sections from CNGB1ΔCaM mice were reacted with an antibody against the GARP domain (Poetsch et al., 2001), which is located near the CNGβ1 N terminus (Fig. 1A). The immunofluorescence signal was restricted to the rod outer segments (Fig. 1E, left panel), similar to the pattern seen in WT controls (data not shown). Retinal morphology was also normal (Fig. 1E, right panel). Therefore the CNGB1ΔCaM mutation did not adversely affect the localization of the CNG channel or the survival of the photoreceptors.
Response properties of CNGB1ΔCaM rods
We compare the responses of WT and CNGB1ΔCaM rods in Figure 2. In Figure 2, A and B, we show mean responses from 10 WT rods and 13 CNGB1ΔCaM rods to the same series of increasing flash intensities, and in Figure 2C we give peak response amplitude as a function of flash intensity. The responses of CNGB1ΔCaM rods are remarkably similar to those of WT rods; there were no significant differences (Student's t test) in the values of circulating current, sensitivity, and integration time (see Table 1). A careful examination of the averaged responses in Figure 2, A and B, however, indicates that the CNGB1ΔCaM rods had a tendency to remain in saturation for a shorter time than WT rods. We examined this property further in Figure 2D, where we have plotted the time the rods remain in saturation, Tsat, as a function of the natural logarithm of the flash intensity (Pepperberg et al., 1992). For both WT (■) and CNGB1ΔCaM rods (□) we give mean values of Tsat measured as the interval from the beginning of the flash to the time at which the circulating current recovered to 25% of its dark value (see Materials and Methods). The best-fitting slopes of the lines gave values for the limiting time constant τD, which from Figure 2D were 178 ms for WT and 153 ms for CNGB1ΔCaM rods. Values of τD measured rod by rod from the two types of photoreceptors gave mean values of 185 ± 11 ms from 21 WT rods and 156 ± 9 from 13 CNGB1ΔCaM rods, which were significantly different at the 0.05 level (Student's t test). In addition, it is quite apparent from the data in Figure 2D that the values of Tsat at each flash intensity are systematically shorter in CNGB1ΔCaM rods, consistent with their more rapid recovery in Figure 2B.
Figure 2.

Comparison of flash responses of WT and CNGB1ΔCaM rods. A, Mean responses averaged from five flash presentations for each of 10 WT rods at the following flash intensities (in photons μm−2): 4, 17, 43, 159, 453, and 1122. B, Mean responses averaged from five flash presentations for each of 13 CNGB1ΔCaM rods at the same flash intensities as those in A. C, Peak response amplitude plotted against flash intensity for 20 WT (■) and 13 CNGB1ΔCaM (□) rods. Curve fitted to WT data is Boltzmann equation with I½ of 26 photons μm−2. D, Time in saturation as a function of the natural logarithm of the flash intensity. Data points give mean with SE from 21 WT rods (■) and 13 CNGB1ΔCaM (□) rods. Straight lines are best linear fits with slopes τD of 178 ms for WT and 153 ms for CNGB1ΔCaM.
Table 1.
Kinetic and sensitivity parameters of rods
| Animal line | rmax (pA) | SFD (pA photon−1 μm2) | I½ (photons μm−2) | ti (ms) |
|---|---|---|---|---|
| WT (20) | 14.5 ± 0.7 | 0.34 ± 0.13 | 27 ± 1 | 262 ± 15 |
| CNGB1ΔCaM (13) | 14.9 ± 0.5 | 0.30 ± 0.03 | 34.2 ± 4.5 | 228 ± 35 |
| GCAPs−/− (15) | 17.6 ± 0.5 | 2.26 ± 0.18 | 4.8 ± 0.5 | 678 ± 46 |
| CNGB1ΔCaM GCAPs−/− (13) | 15.8 ± 1.4 | 1.91 ± 0.18 | 6.4 ± 0.9 | 389 ± 50 |
All values are means ± SE. Values of rmax (maximum response amplitude) were determined cell by cell from responses to saturating flashes; SFD(dark-adapted flash sensitivity), by dividing the peak amplitude of the mean dim-flash response for each cell by the flash intensity; I½ (the intensity required to produce a half-maximal response), from the fit of response-intensity data for each cell to a Boltzmann function in the program Origin; and ti (the integration time), from the time integral of the mean dim-flash response for each cell divided by the peak amplitude of the response.
Changes in flash sensitivity in WT and CNGB1ΔCaM rods
As we and others have shown (Mendez et al., 2001; Makino et al., 2004; Fan et al., 2005; Woodruff et al., 2008), the change in flash response amplitude in the presence of a background light in a WT mouse rod produces a decrease in flash sensitivity, which for dim to moderate intensity backgrounds is in approximate agreement with the Weber–Fechner relation
 |
where SF is the flash sensitivity of the rod in background light, SFD the flash sensitivity of the dark-adapted rod, IB the intensity of the background, and I0 a constant. Flash sensitivity is calculated from the peak amplitude of the small-amplitude response divided by the flash intensity. The data for WT rods (●) have been fitted in Figure 3 with Equation 1 and the best-fitting value of Io for this set of rods of 77 photons μm−2 s−1 (solid black curve).
Figure 3.

Weber–Fechner plot. Sensitivity (SF) in pA photon−1 μm2 was calculated in darkness and in the presence of background light as the peak amplitude of the response in the linear range (for dim flash intensities) divided by the flash intensity. Graph gives mean sensitivity divided by sensitivity in the absence of a background (SFD) as a function of background intensity averaged from 10 WT rods (●), 14 rods lacking the protein recoverin (Rec−/−; ■), 13 CNGB1ΔCaM rods (▴), 5 GCAPs−/− rods (○), and 10 CNGB1ΔCaM/GCAPs−/− rods (□). Solid line is best-fitting Weber–Fechner function for WT rods given by Equation 1 with I0 = 77 photons μm−2 s−1. Black dashed and dotted lines are theoretical predictions of change in sensitivity in the absence of transduction modulation from Equations 2 and 3 (see Results), and red dashed and continuous lines are predictions for GCAPs−/− and WT rods based on a model of light adaptation consisting of two major components: a Ca2+-dependent modulation of guanylyl cyclase via the GCAP molecules, and a Ca2+-dependent regulation of spontaneously activated and light-activated PDE6.
We were unable to observe any effect of removal of the calmodulin binding site on the flash sensitivity of the rods in background light. In Figure 3, we plotted mean values of relative flash sensitivity for 13 CNGB1ΔCaM rods as a function of background intensity (▴). These data nearly superimpose on those of WT rods. We also recorded from rods lacking the Ca2+-binding protein recoverin, which recent experiments show lack the ability to regulate Rh* lifetime (Chen et al., 2010). These measurements (■) are also indistinguishable from those of WT, in confirmation of Makino et al. (2004). Thus, neither channel modulation nor regulation of Rh* seem to make a significant contribution to the control of flash sensitivity during light adaptation.
We then examined the effect of removal of the third of the proposed Ca2+-dependent mechanisms, namely the regulation of guanylyl cyclase by the GCAP proteins. As Mendez et al. (2001) first showed, adaptation is affected by deletion of the GCAPs: flash sensitivity declines in GCAPs−/− rods more rapidly as a function of background intensity than in WT rods. We have repeated their experiments with similar results (Fig. 3, ○). We also measured the flash sensitivity of CNGB1ΔCaM/GCAPs−/− mice lacking both channel and cyclase modulation (Fig. 3, □), and these results are indistinguishable from those of GCAPs−/− rods. There were also no significant differences in maximum response amplitude and dark-adapted sensitivity of GCAPs−/− and CNGB1ΔCaM/GCAPs−/− rods (Table 1, Student's t test); although the integration time appeared to be somewhat shorter, this difference was not significant.
These experiments indicate that, of the three Ca2+-dependent processes postulated to be responsible for regulation of flash sensitivity in vertebrate photoreceptors, only cyclase modulation via the GCAP proteins makes an appreciable contribution. It is of considerable interest, however, as Mendez et al. (2001) first showed, that even in the GCAPs−/− rods the decline of sensitivity is not as great as would be predicted if all of modulation of transduction had been eliminated, and our experiments demonstrate that this is also true for CNGB1ΔCaM/GCAPs−/− receptors. The dashed black curve in Figure 3 is the change in flash sensitivity predicted by the increment saturation relation
 |
where SF is the flash sensitivity of the rod, SFD the flash sensitivity in darkness, IB the intensity of the background, ti the integration time, and rmax the maximum response amplitude. This relation has been used previously to describe the expected change in flash sensitivity if steady illumination simply closed the cyclic nucleotide-gated channels without any regulation of transduction (see for example Mendez et al., 2001). We also show as the dotted black curve in Figure 3 the change in flash sensitivity as a function of background intensity predicted by the model we give later in the paper, again without any feedback regulation of transduction. This relation is as follows:
 |
By either measure, the flash sensitivity in GCAPs−/− or CNGB1ΔCaM/GCAPs−/− rods is much greater than what we would predict if sensitivity regulation were lost completely. It may not seem that this difference is very great, since the horizontal discrepancy between the data in Figure 3 and the black dotted and dashed curves does not seem especially large; but it is the vertical discrepancy that gives a true measure of the adaptation present after the GCAP proteins have been knocked out. By this measure, sensitivity is 20 times greater than Equation 3 of our model would predict at a background of 118 photons μm−2 s−1, and 100 times greater at a background of 438 photons μm−2 s−1. With Equation 2 instead (the exponential saturation equation), these discrepancies are even larger, because that curve falls faster as intensity is increased: measured sensitivity is more than a thousand times greater at a background of 118 photons μm−2 s−1, and the difference at a background of 438 photons μm−2 s−1 is 14 orders of magnitude.
Regulation of response turnoff
A second manifestation of light adaptation also persists in GCAPs−/− and CNGB1ΔCaM/GCAPs−/− rods. In a WT rod, the amplitude of the response to a flash of constant light intensity is decreased in the presence of background light (Fig. 4A). When responses are normalized and superimposed (Fig. 4B), the decay time of the response can be seen to be accelerated, with the exponential time constant of recovery τREC decreasing by as much as a factor of 2 (Woodruff et al., 2008).
Figure 4.

Effect of background light on waveform of decay of rod flash response. A, Mean responses averaged from five presentations each of 10 WT rods to 20 ms flashes, all of flash intensity 453 photons μm−2, given in the presence of backgrounds of the following intensities (in photons μm−2 s−1): 0 (black), 12 (red), 38 (green), 118 (blue), 438 (magenta), and 1354 (dark yellow). B, Responses from A normalized cell by cell to peak amplitude of the response and averaged. C, Mean responses averaged from five presentations each from seven GCAPs−/− rods to 20 ms flashes, all of flash intensity 17 photons μm−2, given in the presence of backgrounds of the following intensities (in photons μm−2 s−1): 0 (black), 4 (red), 13 (green), 38 (blue), and 118 (magenta). D, Responses from C normalized cell by cell to peak amplitude of response and averaged. E, Mean responses averaged from five to seven presentations each from 10 CNGB1ΔCaM/GCAPs−/− rods to 20 ms flashes, all of flash intensity 17 photons μm−2, given in the presence of backgrounds of the following intensities (in photons μm−2 s−1): 0 (black), 4 (red), 13 (green), 38 (blue), and 118 (magenta). F, Responses from E normalized cell by cell to peak amplitude of response and averaged.
The results in Figures 4, C and D, show that a similar phenomenon occurs in GCAPs−/− rods. Response amplitude to a constant intensity flash also decreases in the presence of a background (Fig. 4C), and although responses decay more slowly in the dark in the absence of cyclase regulation and have a much longer integration time (see Table 1) (Mendez et al., 2001), background light accelerates the time course of response decline to an extent that is similar to that in WT rods; single exponential fits to the declining phase of the mean waveform of the response in Figure 4D gave values for τREC of ∼500 ms in dark-adapted rods and ∼250 ms in the presence of the brightest background. Similar data are given for CNGB1ΔCaM/GCAPs−/− rods in Figures 4, E and F. These experiments show that the mechanism responsible for systematic acceleration of response decay in background light can still occur in the absence of Ca2+-dependent modulation of guanylyl cyclase and calmodulin-dependent regulation of the channels.
Responses of GCAPs−/− and CNGB1ΔCaM/GCAPs−/− rods to steps of light
Light adaptation also produces a slow reopening of channels in the presence of background light, evinced by a prominent sag in the response of the rod to steady light. This sag still occurs in rods without recoverin (Makino et al., 2004), and much of it also survives deletion of the GCAPs and the channel Ca2+-calmodulin binding site. In Figure 5, we show averaged responses from WT (A), GCAPs−/− (B), and CNGB1ΔCaM/GCAPs−/− rods (C) to a series of steps of varying duration. As indicated by earlier experiments on amphibian rods (Calvert et al., 2002), the time course of channel reopening in a WT rod is complex, consisting of an initial relaxation of the response (Fig. 5A, inset) with a time constant in mouse of several hundred milliseconds, whose amplitude and time course varied with light intensity but in this experiment restored almost 20% of the dark circulating current. This fast phase was then followed by a slow continuing increase in circulating current over at least 2 min, again variable in amplitude and time course, which at the background intensity of Figure 5A had a time constant of 84 s and could restore an additional 15% of the dark circulating current.
Figure 5.
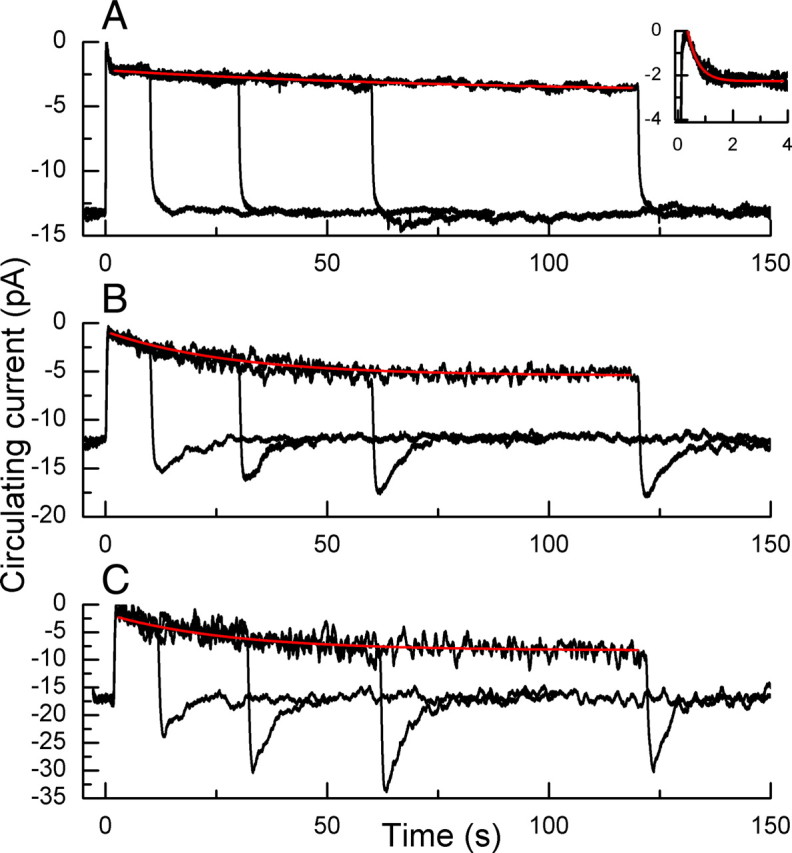
Change in circulating current to steps of light of varying duration. A, WT rods. Data traces are superimposed means of currents of seven rods each exposed three times to steps 10, 30, 60, and 120 s in duration of intensity of 440 photons μm−2 s−1. Red trace is best-fitting single-exponential decay function with a time constant of 84 s. Insert shows first 4 s at higher time resolution; red trace is best-fitting single-exponential decay function with time constant of 380 ms. B, GCAPs−/− rods. Superimposed mean responses of three presentations each from five rods to a step of light of intensity 38 photons μm−2 s−1 for the following durations: 10, 30, 60, and 120 s. Red trace is best-fitting single-exponential decay function with time constant of 28 s. C, CNGB1ΔCaM/GCAPs−/− rods. Superimposed mean responses of three presentations of 2 rods to a step of light of intensity 38 photons μm−2 s−1 for the following durations: 10, 30, 60, and 120 s. Red trace is best-fitting single-exponential decay function with time constant of 28 s.
In GCAPs−/− rods, the value of the circulating current in darkness was somewhat larger than that in WT rods (see Table 1), probably reflecting an increase in basal cyclase activity upon removal of the GCAPs; this difference was significant at the 0.001 level (Student's t test). When we exposed dark-adapted GCAPs−/− rods to prolonged steps of light of varying duration, the initial rapid decrease in circulating current seen in WT rods was absent (Fig. 5B) but a slower current increase was always observed, which for the light intensity in Figure 5B had a time constant of ∼30 s and restored of the order of 40% of the circulating current. The turning off of light produced a transient overshoot, during which the current became larger than its resting value in darkness (see also Burns et al., 2002).
The sag in photocurrent and highly reproducible overshoot following exposure to light steps indicates that some process in the absence of the GCAP proteins is regulating the cGMP concentration and/or the affinity of the outer segment channels for cGMP. This process seems not to be produced by Ca2+-dependent modulation of the channels, since we observed similar declines of current and overshoots in CNGB1ΔCaM/GCAPs−/− rods (Fig. 5C). There was again no detectable rapid phase of response decay after the onset of the light step. The amplitudes of the slow recovery phase and of the overshoot were somewhat larger than in GCAPs−/− rods, consistent with an increase in the affinity of the channels for cGMP upon removal of binding of Ca2+-calmodulin.
Modeling modulation of PDE6
We have shown so far that removal of the Ca2+-calmodulin binding site of the cyclic nucleotide-gated channels produces modest changes in the time of saturation of the light response, consistent with the postulated role of Ca2+-calmodulin in regulating channel affinity, but does not produce any significant alteration of adaptation of flash sensitivity in background light. Furthermore, a substantial part of light adaptation in a mouse rod survives deletion of the GCAPs, with or without the additional removal of the channel Ca2+-calmodulin binding site. Particularly significant are the prominent overshoots of CNGB1ΔCaM/GCAPs−/− rods when steady background light is extinguished, since they cannot be produced by cyclase or calmodulin-dependent channel regulation. Furthermore, modulation of the lifetime of Rh* cannot generate an increase in cGMP at light offset to a concentration greater than the one before the light was turned on. These experiments indicate that some as yet unidentified mechanism must make a large contribution to regulation of current, sensitivity, and response decay in maintained light.
Previous experiments have indicated that PDE6 in mouse rods is directly regulated during (Woodruff et al., 2008) or after steady illumination (Krispel et al., 2003). Furthermore, modulation of the PDE6 could, in principle, explain the large overshoots we have recorded when steady light is extinguished in GCAPs−/− and CNGB1ΔCaM/GCAPs−/− rods. We therefore conjectured that the missing component of adaptation may be produced by direct modulation of the rate of hydrolysis of cGMP through modulation of the lifetime of activated PDE6. Since no molecular mechanisms have been described that produce direct PDE6 modulation, we attempted to test the feasibility of this proposal by constructing a kinetic model. To do this, we built on the biochemical model for rod phototransduction of Hamer et al. (2003; 2005). We removed from this model the modulation of Rh* lifetime mediated by recoverin, and no provision was made for modulation of the channels by Ca2+-calmodulin. Furthermore, to keep the number of fitted parameters in the model to a minimum, we assumed that the fastest reactions in Hamer et al. (2003; 2005) were instantaneous. Further details are given in the supplemental information.
We first simulated our experiments with a model that lacked PDE6 modulation. To simulate recordings from GCAPs−/− mice, we disabled the Ca2+-mediated feedback onto guanylyl cyclase, freezing the rate of synthesis of cGMP by guanylyl cyclase at the dark level (Hamer et al., 2005). We fitted this model to GCAPs−/− rod responses for a family of flash intensities in various background light levels. As expected, we found that this initial model could not account for the GCAPs−/− sensitivity as a function of background light level but is instead in close agreement with Equation 3 and the black dotted curve in Figure 3, which predict much too rapid a decline with increasing background light. This supports the inference from our experiments that there must be a mechanism of adaptation in addition to Ca2+-mediated feedback onto guanylyl cyclase. We found a further dramatic and informative failure of our initial model: at each background there was much more photocurrent that could be blocked by a superimposed bright-light flash in the real GCAPs−/− rods than predicted by our initial model. In other words, at each background light level the steady-state circulating current measured experimentally was much larger than the corresponding current given by the model.
In light of these findings and the experimental evidence for the modulation of PDE6, we embellished the WT and GCAPs−/− models by including a mechanism for modulation of the rate of decay of activated PDE6 (PDE6*). For sake of simplicity, and in agreement with previous evidence (Matthews et al., 1988; Nakatani and Yau, 1988; Tamura et al., 1991), we assumed that this modulation was mediated by the free outer segment Ca2+ concentration; however, we cannot rule out a different unknown intracellular messenger governed by light intensity.
We first tried a version of the model in which only the rate of decay of light-activated PDE6* was subject to this modulation, as in the model of Soo et al. (2008). We found that this assumption alone produced hardly any improvement over the model completely lacking modulation of PDE6* decay. The primary reason for this failure is that the circulating current at each background light level is still far too low in the model compared to that measured experimentally.
Next, we tried a logical and economical extension in which the rate of decay of light-activated and also spontaneously activated PDE6* are both modulated in exactly the same manner, with the spontaneous activation independent of light intensity. We found that this model, with Ca2+-mediated feedback onto guanylyl cyclase disabled, could adequately reproduce the family of mean responses to flashes superimposed on background light levels measured from GCAPs−/− mouse rods (Fig. 6A). The model also adequately predicted the dependence of flash sensitivity on background light level of GCAPs−/− rods; in Figure 3 the dashed red line gives the model calculations and should be compared with the open circles, measured from GCAPs−/− rods. The very same model in WT form, with Ca2+-mediated feedback onto guanylyl cyclase enabled, also accounted satisfactorily for a WT family of responses to flashes superimposed on background light levels (Fig. 6B). The model could in addition predict the dependence of flash sensitivity on background light level of WT rods (Fig. 3, continuous red line). Note that the prediction of the model actually gave a somewhat better fit than Weber's law.
Figure 6.
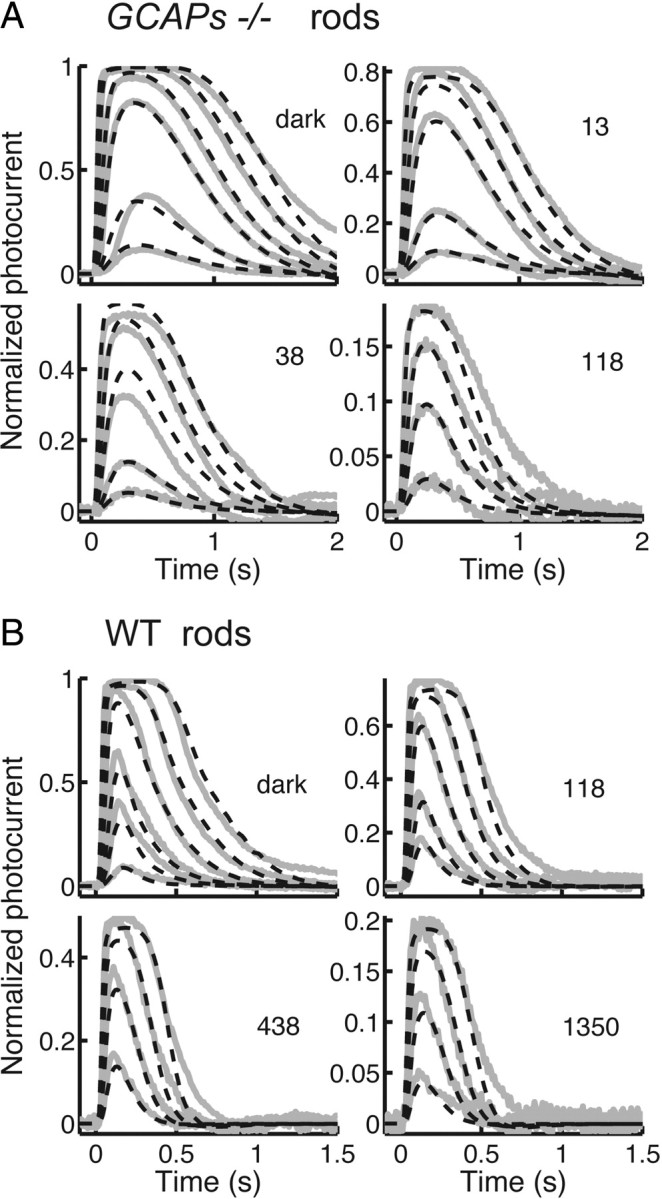
Model predictions of flash–response waveforms. Gray traces give mean responses recorded from the rods, and black dashed curves give model calculations, either with no background (labeled “dark”) or in background light at the indicated intensity in units of photons μm−2 s−1. A, Mean responses of nine GCAPs−/− rods with model containing Ca2+-dependent modulation of decay rate of both light-activated and spontaneous PDE6*, but without GCAP-dependent modulation of cyclase. B, Mean responses of nine WT rods with model containing both PDE6* and cyclase modulation. See text.
The model also predicted the effect of background light on the limiting time constant of the light response. Woodruff et al. (2008) reported that τD decreased by about a factor of 2 from a value in darkness of 168 ms to a value of 78 ms in a background light of 1350 photons μm−2 s−1. On the assumption that τD corresponds to the time constant for the deactivation of spontaneously activated and light-activated PDE6* (τPDE) in our model, we obtained a similar effect: τPDE decreases in the WT model from 184 ms in darkness to 94 ms at the brightest background level (1350 photons photons μm−2 s−1), and it decreases in the GCAPs−/− model from 170 ms in darkness to 70 ms in the brightest background we used (118 photons μm−2 s−1).
Finally, both the WT and GCAPs−/− versions of the model that included modulation of spontaneous and light-activated PDE6* decay gave partial recovery of the photocurrent in response to a prolonged step of light for both WT and GCAPs−/− rods (see supplemental Fig. S2 in the supplemental information), and the kinetics of recovery in the model are similar to those shown in Figure 5, A and B. However, the model gave overshoots upon light offset that were larger than those we have recorded; we think this is because our model lacks Ca2+-calmodulin modulation of cGMP-gated channels and thus behaves more like CNGB1ΔCaM/GCAPs−/− rods, which gave larger overshoots.
Discussion
To explore the possible role of channel modulation in the physiology of the rod, we have removed Ca2+-calmodulin binding by knocking into the genome a CNGB1 construct that lacks the coding region for the calmodulin binding site (Fig. 1A). We showed that the altered channel β subunit is correctly targeted to the rod outer segments (Fig. 1E left), the expression of other outer segment proteins is unaffected (Fig. 1D), and the morphology and survival of the photoreceptors are unaltered (Fig. 1E right). Removal of the Ca2+-calmodulin binding site might be expected to alter rod circulating dark current, but we saw no significant effect on rmax or on sensitivity (Table 1). There were, however, two important consequences for the physiology of the rod. First, there was a decrease in the time in saturation of the rod response manifested by a shift along the intensity axis of the curve relating time in saturation, Tsat, to log light intensity (Fig. 2D). Removal of the Ca2+-calmodulin binding site should produce channels with an increased affinity for cGMP; so, as the rods begin to recover after a bright flash, small increases in cGMP should produce a larger change in current than in WT rods, and current should recover faster. We also observed a small but significant decrease in the value of the limiting time constant τD, for which we presently have no explanation. We detected no change in the expression level of the GAP protein RGS9 in our CNGB1ΔCaM rods (Fig. 1D).
The second effect of removing the Ca2+-calmodulin binding site is revealed by comparing Figure 5B (for GCAPs−/− rods) with Figure 5C (for CNGB1ΔCaM/GCAPs−/− rods): the slow decline of current and the overshoot at the turning off of the light are as large or larger in CNGB1ΔCaM/GCAPs−/− rods as in GCAPs−/− rods. The reason this comparison is important is because it shows that modulation of the channel cannot be responsible for the decrease in current or overshoot in GCAPs−/− rods, since removal of the calmodulin binding site has been shown to eliminate all Ca2+-dependent modulation of the channels (Gordon et al., 1995; Warren and Molday, 2002).
Comparison to olfactory receptor neurons
There is an interesting parallel between our results in rods and the recent observations of Song et al. (2008) on olfactory receptor neurons, which express a CNG channel composed of CNGA2/CNGβ1b/CNGA4 subunits that is gated by cAMP. Song et al. also introduced a mutation that abolishes binding of Ca2+-calmodulin and showed that olfactory responses decay more slowly in mutant receptors, opposite to our observation of faster response recovery, but the reason is the same. In both cell types, the deletion of the channel Ca2+-calmodulin binding site increases the affinity of the channel for cyclic nucleotides, but because stimulation increases cAMP in olfactory receptor neurons but decreases cGMP in rods, the effect on the rate of response recovery is opposite in sign.
Song et al. (2008) also showed that deletion of the Ca2+-calmodulin binding site has little effect on adaptation, in contrast to earlier results (Kurahashi and Menini, 1997). They proposed instead that the primary physiological role of channel modulation in an olfactory receptor neuron is to accelerate termination of the response. We also see no effect on sensitivity regulation in a rod lacking the Ca2+-calmodulin binding site; moreover, our experiments indicate that in a WT rod with Ca2+-calmodulin binding intact, response termination should also be modestly accelerated, particularly for bright flashes, as the free-Ca2+ concentration decreases and Ca2+ falls off the channel. The functional role of channel modulation in the physiology of the rod seems less important than originally supposed (Hsu and Molday, 1993), in substantial agreement with previous observations and modeling from amphibian rods (Koutalos et al., 1995; Nakatani et al., 1995).
Regulation of PDE6 and the mechanism of light adaptation
When a mouse rod is exposed to steady background light, the response adapts; channels first close and then reopen in two temporal phases (Calvert et al., 2002), one with a time constant of a few hundred milliseconds and another with a time constant of tens of seconds (Fig. 5A). In addition, the decay time of the flash response is accelerated (Fig. 4A,B) (see Woodruff et al., 2008), the limiting time constant τD is decreased (Woodruff et al., 2008), and flash sensitivity is reduced (Fig. 3) in approximate agreement with Weber's law (Mendez et al., 2001; Makino et al., 2004; Fan et al., 2005; Woodruff et al., 2008). Three Ca2+-dependent mechanisms have been proposed to explain these phenomena: (1) regulation of the lifetime of Rh* via recoverin, (2) modulation of guanylyl cyclase via the GCAP proteins, and (3) Ca2+-calmodulin-dependent alteration in the affinity of the channels for cGMP. Our results, together with those of previous investigators, show that neither removal of recoverin (Makino et al., 2004) nor deletion of the channel Ca2+-calmodulin binding site has any detectable effect on regulation of flash sensitivity (Fig. 3). We therefore conclude that mechanisms (1) and (3) make little contribution to light adaptation.
Deletion of the GCAP proteins eliminates the rapid phase of current recovery in steady light but not the slower phase (Fig. 5B), and a similar result is produced by removing both the GCAPs and the channel Ca2+-calmodulin binding site (Fig. 5C). Thus, the rapid phase of current recovery depends in some way on GCAP-dependent modulation of guanylyl cyclase (Calvert et al., 2002), but the slower phase must be produced by some other mechanism. Removal of the GCAPs or of both the GCAPs and channel modulation has no effect on the acceleration of flash recovery decay time in background light (Figs. 4C–F) and modifies but does not eliminate the regulation of sensitivity (Fig. 3). Thus, GCAP-dependent activation of guanylyl cyclase contributes to light adaptation, but a considerable part of the phenomenology of sensitivity and waveform adjustment persists when the GCAPs and other proposed Ca2+-dependent regulatory mechanisms have all been eliminated.
This remaining component of adaptation is unlikely to be produced by regulation of any of the initial steps in transduction, because background light has no effect on the initial rising phase (or gain of activation) of the light response (Nikonov et al., 2000; Soo et al., 2008). Furthermore, modulation of the lifetime of Rh* could not produce overshoots like those observed from GCAPs−/− and CNGB1ΔCaM/GCAPs−/− rods at the turning off of steady illumination. Since previous experiments indicate that the time constant of response decay τREC and the limiting time constant τD are both decreased in mouse by steady background light (Woodruff et al., 2008), and because both τREC and τD at least in WT rods are thought to reflect the time course of decay of the PDE6 (Krispel et al., 2006; Tsang et al., 2006; Woodruff et al., 2008; Chen et al., 2010; Gross and Burns, 2010; but see Doan et al., 2009;), we have previously proposed that modulation of this enzyme is an essential component of adaptation of mammalian photoreceptors to background light (Woodruff et al., 2008; see also Soo et al., 2008).
We therefore tested the explanatory power of a hypothetical PDE modulation mechanism by constructing a kinetic model based on the previously published model of Hamer et al. (2003, 2005). In contrast to an earlier model (Soo et al., 2008), we found it essential to hypothesize that background light increases the rate of decay of spontaneous PDE6* as well as of light-activated PDE6*. With this model we could satisfactorily account for the changes in response waveform and sensitivity in GCAPs−/− and WT rods (Figs. 3, 6). There were small discrepancies between model predictions and recorded waveforms, particularly in bright background light (Fig. 6), which we believe are the result of an inadequate simulation of outer segment Ca2+ buffering; we made no attempt to refine the model further to eliminate these differences. Modulation of only light-activated PDE6* gave a poor fit to our data and was not substantially better than a model with no PDE6* modulation at all. The main reason is that without modulation of spontaneously activated PDE6*, the steady circulating current declined far too rapidly with background light.
Our model calculations show that modulation of PDE6* can in principle explain the missing component of light adaptation and are consistent with other evidence indicating that the rate of decay of activated PDE6* is modulated by light. Modulation of spontaneously activated PDE6 is a novel proposed mechanism of light adaptation and is the simplest of the possibilities we can envisage. Identical modulation of spontaneously activated and light-activated PDE6* would be expected if PDE6 was modulated directly or if modulation occurred at the level of transducin. This latter possibility seems less likely, however, in view of the evidence that spontaneous PDE6 activation does not occur via transducin in amphibian rods (Rieke and Baylor, 1996). We cannot exclude the contribution of other possible regulatory processes that have not as yet been described. In particular, a model that includes modulation of light-activated PDE6 and a hypothetical regulation of channel open probability by a mechanism not dependent on calmodulin can also adequately account for our results (see supplemental information). The challenge for the future is to identify the molecular mechanisms of the missing components of modulation, which at present are completely unknown, and show how, together with cyclase regulation, they can produce the changes in signal transduction that are responsible for light adaptation in mammalian rods.
Footnotes
This work was supported by National Institutes of Health Grants EY01844 (G.L.F.) and EY12703 and EY12155 (J.C.). We thank Dr. Robert Molday for providing the 4B1 antibody that recognizes the GARP domain on the CNGβ1 subunit.
References
- Burns ME, Mendez A, Chen J, Baylor DA. Dynamics of cyclic GMP synthesis in retinal rods. Neuron. 2002;36:81–91. doi: 10.1016/s0896-6273(02)00911-x. [DOI] [PubMed] [Google Scholar]
- Calvert PD, Govardovskii VI, Arshavsky VY, Makino CL. Two temporal phases of light adaptation in retinal rods. J Gen Physiol. 2002;119:129–145. doi: 10.1085/jgp.119.2.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CK, Inglese J, Lefkowitz RJ, Hurley JB. Ca2+-dependent interaction of recoverin with rhodopsin kinase. J Biol Chem. 1995;270:18060–18066. doi: 10.1074/jbc.270.30.18060. [DOI] [PubMed] [Google Scholar]
- Chen CK, Burns ME, He W, Wensel TG, Baylor DA, Simon MI. Slowed recovery of rod photoresponse in mice lacking the GTPase accelerating protein RGS9–1. Nature. 2000;403:557–560. doi: 10.1038/35000601. [DOI] [PubMed] [Google Scholar]
- Chen CK, Woodruff ML, Chen FS, Chen D, Fain GL. Background light produces a recoverin-dependent modulation of activated-rhodopsin lifetime in mouse rods. J Neurosci. 2010;30:1213–1220. doi: 10.1523/JNEUROSCI.4353-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concepcion F, Mendez A, Chen J. The carboxyl-terminal domain is essential for rhodopsin transport in rod photoreceptors. Vision Res. 2002;42:417–426. doi: 10.1016/s0042-6989(01)00195-x. [DOI] [PubMed] [Google Scholar]
- Doan T, Azevedo AW, Hurley JB, Rieke F. Arrestin competition influences the kinetics and variability of the single-photon responses of mammalian rod photoreceptors. J Neurosci. 2009;29:11867–11879. doi: 10.1523/JNEUROSCI.0819-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson MA, Lagnado L, Zozulya S, Neubert TA, Stryer L, Baylor DA. The effect of recombinant recoverin on the photoresponse of truncated rod photoreceptors. Proc Natl Acad Sci U S A. 1998;95:6474–6479. doi: 10.1073/pnas.95.11.6474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fain GL, Matthews HR, Cornwall MC, Koutalos Y. Adaptation in vertebrate photoreceptors. Physiol Rev. 2001;81:117–151. doi: 10.1152/physrev.2001.81.1.117. [DOI] [PubMed] [Google Scholar]
- Fan J, Woodruff ML, Cilluffo MC, Crouch RK, Fain GL. Opsin activation of transduction in the rods of dark-reared Rpe65 knockout mice. J Physiol. 2005;568:83–95. doi: 10.1113/jphysiol.2005.091942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, Ishii M, Gu Y, Maxson R. Conditional alleles of Msx1 and Msx2. Genesis. 2007;45:477–481. doi: 10.1002/dvg.20316. [DOI] [PubMed] [Google Scholar]
- Gordon SE, Downing-Park J, Zimmerman AL. Modulation of the cGMP-gated ion channel in frog rods by calmodulin and an endogenous inhibitory factor. J Physiol. 1995;486:533–546. doi: 10.1113/jphysiol.1995.sp020832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross OP, Burns ME. Control of rhodopsin's active lifetime by arrestin-1 expression in mammalian rods. J Neurosci. 2010;30:3450–3457. doi: 10.1523/JNEUROSCI.5391-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunwald ME, Yu WP, Yu HH, Yau KW. Identification of a domain on the β-subunit of the rod cGMP-gated cation channel that mediates inhibition by calcium-calmodulin. J Biol Chem. 1998;273:9148–9157. doi: 10.1074/jbc.273.15.9148. [DOI] [PubMed] [Google Scholar]
- Hamer RD, Nicholas SC, Tranchina D, Liebman PA, Lamb TD. Multiple steps of phosphorylation of activated rhodopsin can account for the reproducibility of vertebrate rod single-photon responses. J Gen Physiol. 2003;122:419–444. doi: 10.1085/jgp.200308832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamer RD, Nicholas SC, Tranchina D, Lamb TD, Jarvinen JL. Toward a unified model of vertebrate rod phototransduction. Vis Neurosci. 2005;22:417–436. doi: 10.1017/S0952523805224045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YT, Molday RS. Modulation of the cGMP-gated channel of rod photoreceptor cells by calmodulin. Nature. 1993;361:76–79. doi: 10.1038/361076a0. [DOI] [PubMed] [Google Scholar]
- Kawamura S. Rhodopsin phosphorylation as a mechanism of cyclic GMP phosphodiesterase regulation by S-modulin. Nature. 1993;362:855–857. doi: 10.1038/362855a0. [DOI] [PubMed] [Google Scholar]
- Koch KW, Stryer L. Highly cooperative feedback control of retinal rod guanylate cyclase by calcium ions. Nature. 1988;334:64–66. doi: 10.1038/334064a0. [DOI] [PubMed] [Google Scholar]
- Koutalos Y, Nakatani K, Yau KW. The cGMP-phosphodiesterase and its contribution to sensitivity regulation in retinal rods. J Gen Physiol. 1995;106:891–921. doi: 10.1085/jgp.106.5.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krispel CM, Chen CK, Simon MI, Burns ME. Novel form of adaptation in mouse retinal rods speeds recovery of phototransduction. J Gen Physiol. 2003;122:703–712. doi: 10.1085/jgp.200308938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krispel CM, Chen D, Melling N, Chen YJ, Martemyanov KA, Quillinan N, Arshavsky VY, Wensel TG, Chen CK, Burns ME. RGS expression rate-limits recovery of rod photoresponses. Neuron. 2006;51:409–416. doi: 10.1016/j.neuron.2006.07.010. [DOI] [PubMed] [Google Scholar]
- Kurahashi T, Menini A. Mechanism of odorant adaptation in the olfactory receptor cell. Nature. 1997;385:725–729. doi: 10.1038/385725a0. [DOI] [PubMed] [Google Scholar]
- Makino CL, Dodd RL, Chen J, Burns ME, Roca A, Simon MI, Baylor DA. Recoverin regulates light-dependent phosphodiesterase activity in retinal rods. J Gen Physiol. 2004;123:729–741. doi: 10.1085/jgp.200308994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews HR, Murphy RL, Fain GL, Lamb TD. Photoreceptor light adaptation is mediated by cytoplasmic calcium concentration. Nature. 1988;334:67–69. doi: 10.1038/334067a0. [DOI] [PubMed] [Google Scholar]
- Matthews HR, Cornwall MC, Crouch RK. Prolongation of actions of Ca2+ early in phototransduction by 9-demethylretinal. J Gen Physiol. 2001;118:377–390. doi: 10.1085/jgp.118.4.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez A, Burns ME, Sokal I, Dizhoor AM, Baehr W, Palczewski K, Baylor DA, Chen J. Role of guanylate cyclase-activating proteins (GCAPs) in setting the flash sensitivity of rod photoreceptors. Proc Natl Acad Sci U S A. 2001;98:9948–9953. doi: 10.1073/pnas.171308998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatani K, Yau KW. Calcium and light adaptation in retinal rods and cones. Nature. 1988;334:69–71. doi: 10.1038/334069a0. [DOI] [PubMed] [Google Scholar]
- Nakatani K, Koutalos Y, Yau KW. Ca2+ modulation of the cGMP-gated channel of bullfrog retinal rod photoreceptors. J Physiol. 1995;484:69–76. doi: 10.1113/jphysiol.1995.sp020648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikonov S, Lamb TD, Pugh EN., Jr The role of steady phosphodiesterase activity in the kinetics and sensitivity of the light-adapted salamander rod photoresponse. J Gen Physiol. 2000;116:795–824. doi: 10.1085/jgp.116.6.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K, Sokal I, Baehr W. Guanylate cyclase-activating proteins: structure, function, and diversity. Biochem Biophys Res Commun. 2004;322:1123–1130. doi: 10.1016/j.bbrc.2004.07.122. [DOI] [PubMed] [Google Scholar]
- Pepperberg DR, Cornwall MC, Kahlert M, Hofmann KP, Jin J, Jones GJ, Ripps H. Light-dependent delay in the falling phase of the retinal rod photoresponse. Vis Neurosci. 1992;8:9–18. doi: 10.1017/s0952523800006441. [DOI] [PubMed] [Google Scholar]
- Peshenko IV, Dizhoor AM. Guanylyl cyclase-activating proteins (GCAPs) are Ca2+/Mg2+ sensors: implications for photoreceptor guanylyl cyclase (RetGC) regulation in mammalian photoreceptors. J Biol Chem. 2004;279:16903–16906. doi: 10.1074/jbc.C400065200. [DOI] [PubMed] [Google Scholar]
- Poetsch A, Molday LL, Molday RS. The cGMP-gated channel and related glutamic acid-rich proteins interact with peripherin-2 at the rim region of rod photoreceptor disc membranes. J Biol Chem. 2001;276:48009–48016. doi: 10.1074/jbc.M108941200. [DOI] [PubMed] [Google Scholar]
- Rieke F, Baylor DA. Molecular origin of continuous dark noise in rod photoreceptors. Biophys J. 1996;71:2553–2572. doi: 10.1016/S0006-3495(96)79448-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Cygnar KD, Sagdullaev B, Valley M, Hirsh S, Stephan A, Reisert J, Zhao H. Olfactory CNG channel desensitization by Ca2+/CaM via the B1b subunit affects response termination but not sensitivity to recurring stimulation. Neuron. 2008;58:374–386. doi: 10.1016/j.neuron.2008.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soo FS, Detwiler PB, Rieke F. Light adaptation in salamander L-cone photoreceptors. J Neurosci. 2008;28:1331–1342. doi: 10.1523/JNEUROSCI.4121-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura T, Nakatani K, Yau KW. Calcium feedback and sensitivity regulation in primate rods. J Gen Physiol. 1991;98:95–130. doi: 10.1085/jgp.98.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang SH, Woodruff ML, Chen CK, Yamashita CY, Cilluffo MC, Rao AL, Farber DB, Fain GL. GAP-Independent termination of photoreceptor light response by excess gamma subunit of the c-GMP-phosphodiesterase. J Neurosci. 2006;26:4472–4480. doi: 10.1523/JNEUROSCI.4775-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren R, Molday RS. Regulation of the rod photoreceptor cyclic nucleotide-gated channel. In: Baehr W, Palczewski K, editors. Photoreceptors and calcium, Vol 514, Advances in experimental medicine and biology. New York: Springer; 2002. pp. 205–223. [DOI] [PubMed] [Google Scholar]
- Weitz D, Zoche M, Müller F, Beyermann M, Körschen HG, Kaupp UB, Koch KW. Calmodulin controls the rod photoreceptor CNG channel through an unconventional binding site in the N-terminus of the beta-subunit. EMBO J. 1998;17:2273–2284. doi: 10.1093/emboj/17.8.2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitz D, Ficek N, Kremmer E, Bauer PJ, Kaupp UB. Subunit stoichiometry of the CNG channel of rod photoreceptors. Neuron. 2002;36:881–889. doi: 10.1016/s0896-6273(02)01098-x. [DOI] [PubMed] [Google Scholar]
- Woodruff ML, Janisch KM, Peshenko IV, Dizhoor AM, Tsang SH, Fain GL. Modulation of phosphodiesterase6 turnoff during background illumination in mouse rod photoreceptors. J Neurosci. 2008;28:2064–2074. doi: 10.1523/JNEUROSCI.2973-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Trudeau MC, Zagotta WN. Rod cyclic nucleotide-gated channels have a stoichiometry of three CNGA1 subunits and one CNGB1 subunit. Neuron. 2002;36:891–896. doi: 10.1016/s0896-6273(02)01099-1. [DOI] [PubMed] [Google Scholar]
- Zhong H, Molday LL, Molday RS, Yau KW. The heteromeric cyclic nucleotide-gated channel adopts a 3A:1B stoichiometry. Nature. 2002;420:193–198. doi: 10.1038/nature01201. [DOI] [PMC free article] [PubMed] [Google Scholar]