

Abstract
BACKGROUND AND PURPOSE
Ca2+ signalling and exocytosis mediated by nicotinic receptor (nAChR) subtypes, especially the α7 nAChR, in bovine chromaffin cells are still matters of debate.
EXPERIMENTAL APPROACH
We have used chromaffin cell cultures loaded with Fluo-4 or transfected with aequorins directed to the cytosol or mitochondria, several nAChR agonists (nicotine, 5-iodo-A-85380, PNU282987 and choline), and the α7 nAChR allosteric modulator PNU120596.
KEY RESULTS
Minimal [Ca2+]c transients, induced by low concentrations of selective α7 nAChR agonists and nicotine, were markedly increased by the α7 nAChR allosteric modulator PNU120596. These potentiated responses were completely blocked by the α7 nAChR antagonist α-bungarotoxin (α7-modulated-response). Conversely, high concentrations of the α7 nAChR agonists, nicotine or 5-iodo-A-85380 induced larger [Ca2+]c transients, that were blocked by mecamylamine but were unaffected by α-bungarotoxin (non-α7 response). [Ca2+]c increases mediated by α7 nAChR were related to Ca2+ entry through non-L-type Ca2+ channels, whereas non-α7 nAChR-mediated signals were related to L-type Ca2+ channels; Ca2+-induced Ca2+-release contributed to both responses. Mitochondrial involvement in the control of [Ca2+]c transients, mediated by either receptor, was minimal. Catecholamine release coupled to α7 nAChRs was more efficient in terms of catecholamine released/[Ca2+]c.
CONCLUSIONS AND IMPLICATIONS
[Ca2+]c and catecholamine release mediated by α7 nAChRs required an allosteric modulator and low doses of the agonist. At higher agonist concentrations, the α7 nAChR response was lost and the non-α7 nAChRs were activated. Catecholamine release might therefore be regulated by different nAChR subtypes, depending on agonist concentrations and the presence of allosteric modulators of α7 nAChRs.
Keywords: chromaffin cells, nicotinic receptors, intracellular calcium
Introduction
Adrenal chromaffin cells derive from the neural crest and are innervated by the preganglionic cholinergic neurons of the splachnic nerve that release acetylcholine, the ligand that induces catecholamine secretion from chromaffin cells. Binding of the physiological neurotransmitter acetylcholine or nicotine to the nicotinic cholinoceptors (nAChR; receptor nomenclature follows Alexander et al., 2009) located in the plasma membrane of the chromaffin cell, opens the nicotinic receptor channel allowing the entry of sodium and calcium and the exit of potassium ions by means of an electrochemical gradient. The resultant depolarization of the membrane opens voltage-operated Ca2+ channels (VOCCs) followed by Ca2+ entry through these channels to trigger the release of catecholamines into the blood stream.
Neuronal nAChRs are pentameric ligand-gated channels, consisting of the same subunit type (homomeric) or different subunit types (heteromeric). In bovine chromaffin cells, α3 (Criado et al., 1992), α5 (Campos-Caro et al., 1997) β4 (Campos-Caro et al., 1997) and α7 (Garcia-Guzman et al., 1995) subunits have been cloned. Furthermore, α3, α5 and β4 transcripts are co-expressed in chromaffin cells and functional receptors have been obtained upon co-injection of these three subunit cRNAs into Xenopus oocytes (Campos-Caro et al., 1997). In primary cultures of bovine chromaffin cells, most studies have shown that the nAChRs are insensitive to α-bungarotoxin (BGT) and there is wide agreement that they are involved in controlling catecholamine release.
As for the α7 nAChR, the presence of BGT-binding sites in the adrenal medullary chromaffin cells was first reported in 1977 (Wilson and Kirshner, 1977). When the α7 nAChRs of the bovine chromaffin cell were cloned, injection in Xenopus oocytes of its cRNA produced BGT binding sites and functional acetylcholine currents sensitive to BGT (Criado et al., 1992). Later on, BGT binding sites were described mainly in adrenergic but not in noradrenergic cells (Criado et al., 1997). More recently, El-Hajj et al. (2007) have reported that the α7 homomeric population of nAChRs in bovine chromaffin cells was greater than the heteromeric nAChR population.
In terms of the control of catecholamine release, it is well established that Ca2+ is required for this process, not only after a depolarizing pulse but also after nicotinic stimulation (Douglas and Rubin, 1961a,b;). After nAChR activation, the cytosolic concentration of Ca2+[Ca2+]c is raised because of Ca2+ entry through VOCCs (Rathouz and Berg, 1994) and by Ca2+-induced Ca2+-release (CICR) from intracellular stores (Verkhratsky, 2002). The VOCCs present in bovine chromaffin cells iclude the L, N and P/Q subtypes (Garcia et al., 2006). Each VOCC subtype has been found to contribute to 45Ca2+ entry (Ballesta et al., 1989; Lopez et al., 1994; Villarroya et al., 1997) and catecholamine release (Lopez et al., 1994; Lara et al., 1998). However, N and P/Q channels are inactivated at depolarizing voltages, in contrast to the L-subtype (Villarroya et al., 1999), and this inactivation blocks Ca2+ entry and catecholamine release (Artalejo et al., 1986; Michelena et al., 1993).
Although Ca2+ entry through the nicotinic receptor itself seems to contribute very little to [Ca2+]c increase after receptor activation (Zhou and Neher, 1993), Ca2+ permeability associated with a particular nAChR subtype represents an important aspect of its physiological role in cellular processes. The homomeric α7 nAChR has the highest Ca2+ : Na+ permeability ratio (Seguela et al., 1993; Fucile et al., 2003) but desensitizes very rapidly (Couturier et al., 1990) in comparison with other nAChR subtypes. Another characteristic of the α7 nAChR is that nicotinic agonists bind to it with low affinity (Pereira et al., 1993), although these receptors show high affinity for the antagonists BGT and methyllycaconitine (MLA).
As indicated before, the existence of α7 nAChRs in bovine chromaffin cells has been well documented by molecular techniques, but its functional relevance in terms of Ca2+ signalling and catecholamine exocytosis is still a matter of debate. We therefore investigated this issue further, using new pharmacological tools such as the α7 nAChR agonist PNU282987 and the α7 nAChR allosteric modulator PNU120596.
Our results showed that activation of α7 nAChRs, in the presence of an allosteric modulator, can mediate BGT-sensitive [Ca2+]c signals and catecholamine release. Furthermore, [Ca2+]c increases mediated by α7 nAChR were related to Ca2+ entry through non-L-VOCCs, whereas non- α7 nAChR-mediated signals were related to L-type VOCCs. CICR also participated in both responses. Our results indicated that functionality of the α7 nAChR in chromaffin cells seems to require the binding of an allosteric modulator together with its agonist and that Ca2+ signalling mediated through α7 and non α7 nAChR stimulation is differentially regulated in bovine chromaffin cells.
Methods
Isolation and culture of bovine chromaffin cells
Bovine adrenal glands were obtained from a local slaughterhouse. Chromaffin cells were isolated by collagenase digestion as previously described (Livett, 1984) with some modifications (Moro et al., 1990); purification of bovine chromaffin cells was achieved by several consecutive centrifugations. Cells were suspended in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% fetal bovine serum, 50 IU·mL−1 penicillin and 50 µg·mL−1 streptomycin. Cells were pre-plated for 30 min and proliferation inhibitors (10 µM cytosine arabinoside, 10 µM fluorodeoxyuridine, and 10 µM leucine methyl ester) were added to the medium to prevent excessive growth of fibroblasts. Cultures were maintained in an incubator at 37°C in a water-saturated atmosphere with 5% CO2. Cells were used between 48 and 72 h post-plating.
Measurement of calcium signals with fluorescent dyes
Fluo-4 measurements in populations of bovine chromaffin cells
Bovine chromaffin cells were plated in black 96 well plates at a density of 200 000 cells per well. Cells were loaded with 4 µM Fluo-4/AM for 1 h at 37°C in DMEM. Then, cells were washed twice with Krebs-HEPES solution (composition (in mM): 140 NaCl, 5.6 KCl, 1.2 MgCl2, 2 CaCl2, 10 HEPES, 11 d-glucose, pH 7.4) and kept at room temperature for 30 min before the beginning of the experiment. When BGT (100 nM), mecamylamine (30 µM) nimodipine (3 µM) or ω-conotoxin MVIIC (2 µM) were employed, these were incubated with the cells for 15 min before injecting the nicotinic agonists.
Fluorescence measurements were carried out for 10 s, as the agonists were injected in a microplate reader (FLUOstar Optima, BMG, Offenburg, Germany). Wavelengths of excitation and emission were 485 and 520 nm respectively.
At the end of the experiment, 75 µL of Triton (5%) was added to each well to calculate maximum fluorescence (Fmax) and then 50 µL of MnCl2 (1 M) to obtain the minimum fluorescence (Fmin). Drug-evoked responses were expressed as percentage of the fluorescence values at each time point (F) minus minimum fluorescence values (F0) divided by Fmax−Fmin as follows:
The maximum value of F520, obtained for each experiment, was considered as the Peak F520 value.
Aequorin measurements
The construction of the aequorin chimeras targeted to either the cytosol or the mitochondria has been described previously (Rizzuto et al., 1992; Brini et al., 1995). Chromaffin cells were plated on coverslips and infected with adenoviruses for expression of both constructs of targeted aequorin. Adenoviruses were prepared as described previously (Santodomingo et al., 2008). Infection was carried out the day after cell isolation and Ca2+ measurements were performed 48–72 h after infection. For aequorin reconstitution, cells expressing either cytosolic or mitochondrial aequorin were incubated for 1–2 h at room temperature with 1 µM of native coelenterazine in standard medium (145 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM glucose, and 10 mM HEPES, pH 7.4). Cells were then placed in the perfusion chamber of a purpose-built luminometer maintained at 37°C. Calibration of the luminescence data into [Ca2+] was made using an algorithm as previously described (Barrero et al., 1997).
Catecholamine measurements
Bovine chromaffin cells were seeded in 12 well plates at a density of 500 000 cells per well. Catecholamines released from the cells were analyzed by high-performance liquid chromatography (HPLC; Agilent Technologies 1200 series) using electrochemical detection (BioRad, model 1640, Bio-Rad Laboratories, Irvine, CA, USA). A C18 reverse phase column (ZORBAX Eclipse XDB, Agilent Technologies, Santa Clara, CA, USA), eluted with a mobile phase (pH 4; 0.1 M sodium acetate, 0.1 M citric acid, 0.7 mM sodium octyl sulphate and 0.57 mM EDTA containing 10% methanol, v/v) was employed. The system was run continuously with the mobile phase being recycled back into the reservoir and used for up to one week. Flow rate was 1 mL·min−1, at a pressure of 2200 psi. Fixed potentials against H2/H+ reference electrodes were: conditioning electrode 0.4V; pre-oxidation electrode +0.10V; working electrode +0.35V. Adrenaline and noradrenaline concentrations were calculated from the chromatographic peak areas by using external standards. The chromatograms were collected, stored and processed with Clinical Data Management Software (BioRad CDM 1.10).
Data and statistical analysis
Data are given as means ± SEM. Differences between groups were determined by applying a one-way anova followed by Newman–Keuls test. In the aequorin experiments, Student's t-test was used to determine statistical differences between groups. Differences were considered to be statistically significant when P≤ 0.05.
Materials
Nicotine, choline, α-bungarotoxin (BGT), mecamylamine, cadmium, N-methyl glucamine, dimethyl sulfoxide (DMSO), carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) and nimodipine were from Sigma (Madrid, Spain). PNU282987, PNU120596, 5-iodo-A-85380 dihydrochloride and ω-conotoxin MVIIC from Tocris (Biogen Científica, Madrid, Spain). DMEM, fetal bovine serum and penicillin/streptomycin were purchased from Invitrogen (Barcelona, Spain). Fluo-4 AM and coelenterazine were from Molecular Probes (Invitrogen, Madrid, Spain).
Results
Nicotine and 5-iodo-A-85380 induce intracellular calcium signals that are not sensitive to BGT
In Fluo-4 loaded chromaffin cells, addition of increasing concentrations of nicotine (0.03 to 100 µM) showed that this nicotinic agonist caused [Ca2+]c increases in a concentration-dependent manner with an EC50 of 3.73 ± 0.71 µM (Figure 1C). The [Ca2+]c increases induced by nicotine were inhibited by mecamylamine but were unaffected by BGT (see Figure 1A for representative fluorescence tracings performed with 3 µM nicotine).
Figure 1.
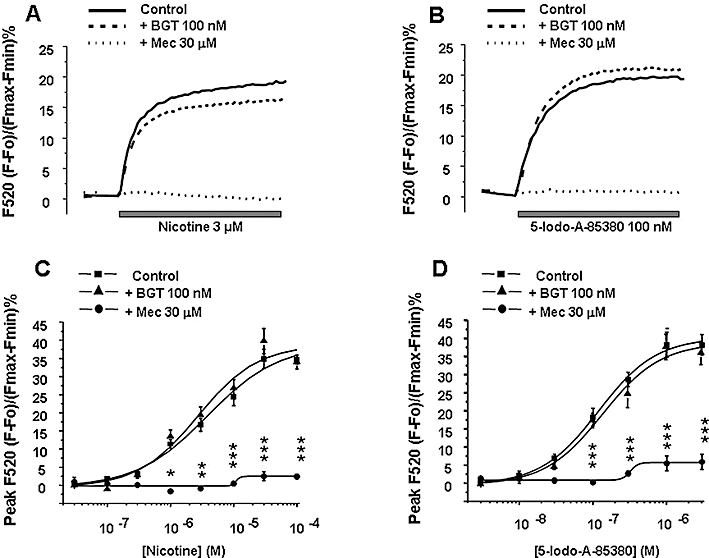
Intracellular calcium signals induced by nicotine and 5-iodo-A-85380 in Fluo-4 loaded bovine chromaffin cells. The upper part of the figure shows representative Fluo-4 fluorescence traces induced by nicotine at 3 µM (A) and 5-iodo-A-85380 at 100 nM (B) measured over 10 s of continuous agonist stimulation alone or in cells pre-treated for 15 min with α-bungarotoxin (BGT; 100 nM) or mecamylamine (Mec; 30 µM). (C) The mean maximum peak fluorescence after increasing concentrations of nicotine (0.03–100 µM) alone or in cells pre-incubated for 15 min with BGT or mecamylamine. (D) Data using the same protocol as used in Figure C, when cells were stimulated by the nicotinic agonist 5-iodo-A-85380 (0.003–3 µM). Bars show the mean ± SEM of five independent experiments; each experiment was performed in triplicate. *P < 0.05, **P < 0.01, ***P < 0.001; significantly different from agonist alone; one-way anovafollowed by a post hoc Newman–Keuls test.
For comparative purposes we used another nAChR agonist, 5-iodo-A-85380, which has been described to have a fivefold higher affinity for α3β4 than for α7 nAChRs (Mukhin et al., 2000). 5-Iodo-A-85380 (0.003 to 3 µM) produced concentration-dependent [Ca2+]c signals with an EC50 of 0.11 ± 0.01 µM (Figure 1D). As in the case of nicotine, the [Ca2+]c increases following 5-iodo-A-85380 were fully blocked by mecamylamine, but were unaffected by BGT (see Figure 1B for representative fluorescence tracings performed with 100 nM 5-iodo-A-85380).
These results suggest that these two agonists, under these experimental conditions, stimulate non-α7 nAChRs and from now on we will refer to these responses as to non-α7.
Cytosolic calcium signals mediated by the α7 nAChR agonists PNU282987 and choline
We first used the selective α7 nAChR agonist PNU282987 (Hajos et al., 2005). At concentrations ranging from 1 to 30 µM, PNU282987 failed to produce any significant [Ca2+]c increase; however, at 100 µM it caused a [Ca2+]c increase that was completely blocked by mecamylamine but unaffected by BGT, suggesting that this calcium increase was due to stimulation of non-α7 nAChRs (Figure 2A).
Figure 2.
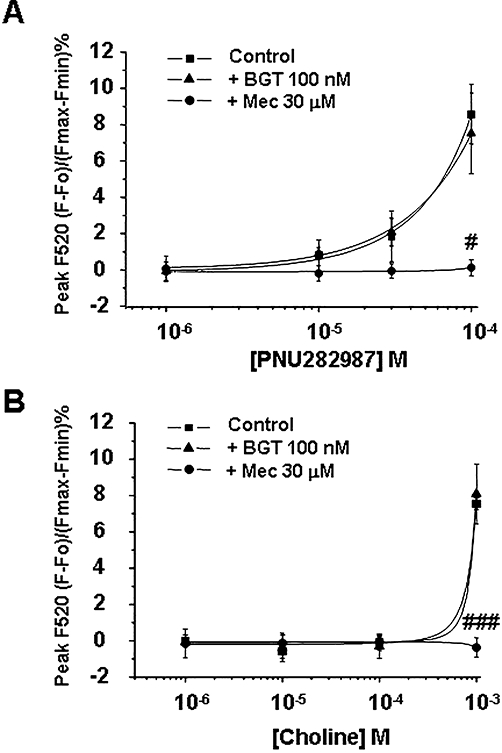
Intracellular Ca2+ responses induced by the α7 nicotinic acetylcholine receptor (nAChR) agonists PNU282987 and choline. Fluo-4 loaded bovine chromaffin cells were stimulated with increasing concentrations of the α7 nAChR agonists (A) PNU282987 (1–100 µM) or (B) choline (1–1000 µM). The antagonists mecamylamine (Mec; 30 µM) or α-bungarotoxin (BGT; 100 nM) were pre-incubated 15 min before injecting the agonists. Data represent the mean ± SEM of four independent experiments; each experiment was performed in triplicate. #P < 0.05, ###P < 0.001, significantly different from agonist in the absence of antagonist. One-way anova followed by a post hoc Newman–Keuls test.
We also used the endogenous α7 nAChR agonist choline (Alkondon et al., 1997). A similar pattern to that observed for PNU282987 was observed for this agonist. Choline did not induce a measurable [Ca2+]c increase at concentrations ranging from 1 to 100 µM; at 1000 µM, choline gave a [Ca2+]c signal that was blocked by mecamylamine but not by BGT (Figure 2B).
The α7 nAChR allosteric modulator PNU120596 potentiates the [Ca2+]c increases induced by low concentrations of PNU282987, choline and nicotine: the modulated α7 nAChR response
We measured the effects of the α7 nAChR positive allosteric modulator PNU120596 (Hurst et al., 2005) on the [Ca2+]c increases mediated by the α7 nAChR agonists PNU282987 and choline. Figure 3A illustrates a typical Fluo-4 recording showing that PNU282987 at 10 µM, a concentration that did not modify basal [Ca2+]c (see Figure 2A), increased by 12-fold the [Ca2+]c signal in cells pre-treated with the α7 nAChR allosteric modulator PNU120596. This potentiated response was completely blocked by BGT. The α7 allosteric modulator potentiated responses to the α7 nAChR agonist PNU282987 (10 µM) in a concentration-dependent manner, with an EC50 of 0.89 ± 0.14 µM and this modulated response was sensitive to BGT (Figure 3B).
Figure 3.
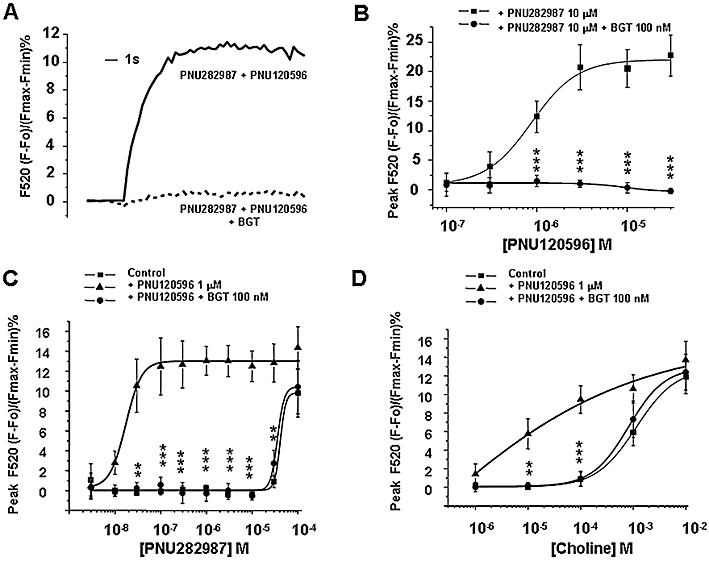
Intracellular Ca2+ responses induced by the α7 nicotinic acetylcholine receptor (nAChR) agonists PNU282987 and choline in the presence of the α7 allosteric modulator PNU120596. (A) Representative Fluo-4 fluorescence traces showing the calcium increase induced by 10 µM PNU282987 plus the allosteric modulator at 1 µM alone or in cells pre-treated with α-bungarotoxin (BGT) 100 nM. (B) The Ca2+ signals induced by 10 µM PNU282987 were potentiated in a concentration-dependent manner by the allosteric modulator (from 0.1 to 30 µM) PNU120596; such potentiation was completely blocked in cells pre-treated with BGT 100 nM. (C,D) Concentration-response curves of PNU282987 (0.003–100 µM) and choline (1 µM–10 mM) in the presence of the α7 allosteric modulator PNU120596 (1 µM). Control cells were pre-incubated for 15 min in the presence of the same volume of DMSO (0.1% (v/v) which was used to assess the maximum concentration of PNU120596. Data represent the mean ± SEM of four independent experiments; each experiment was performed in triplicate. **P < 0.01; ***P < 0.001; significantly different from agonists plus PNU120596: one-way anova followed by a post hoc Newman–Keuls test.
We then performed a concentration response-curve with PNU282987 in bovine chromaffin cells pre-treated with a fixed concentration of the α7 allosteric modulator PNU120596 (1 µM). Under these experimental conditions, we observed a 12-fold potentiation of the PNU282987 response, especially at those concentrations of PNU282987 that per se did not induce detectable [Ca2+]c signals (Figure 3C). The potentiated responses were completely blocked by BGT, suggesting that they were mediated by α7 nAChRs. At higher concentrations of PNU282987 (10–100 µM), the proportion of the response that was sensitive to BGT was reduced (Figure 3C).
We also used the endogenous α7 nAChR agonist choline. In chromaffin cells pre-treated with the α7 nAChR allosteric modulator. The [Ca2+]c signals induced by choline were significantly potentiated, most clearly at low concentrations and this potentiated response was fully blocked by BGT (Figure 3D). These results suggest that α7 nAChRs in chromaffin cells require the binding of an allosteric modulator together with lower concentrations of agonists to produce a measurable [Ca2+]c signal, mediated by α7 nAChRs.
Lastly, we measured the [Ca2+]c increases mediated by the non- specific nAChR agonist nicotine, in cells pre-incubated with PNU120596. The allosteric modulator was able to potentiate the [Ca2+]c signal induced by low concentrations of nicotine (0.3 and 1 µM) (Figure 4A,C); the potentiated response was again fully blocked by BGT. However, concentrations of nicotine above 1 µM were not significantly potentiated by PNU120596 (Figure 5B,C). These results suggest that low concentrations of nicotine, in the presence of an α7 nAChR allosteric modulator, can induce [Ca2+]c increases via α7 nAChRs whereas, at concentrations above 1 µM, the allosteric potentiation is reduced and nicotine seems to induce [Ca2+]c increases, predominantly, via non-α7 nAChRs.
Figure 4.
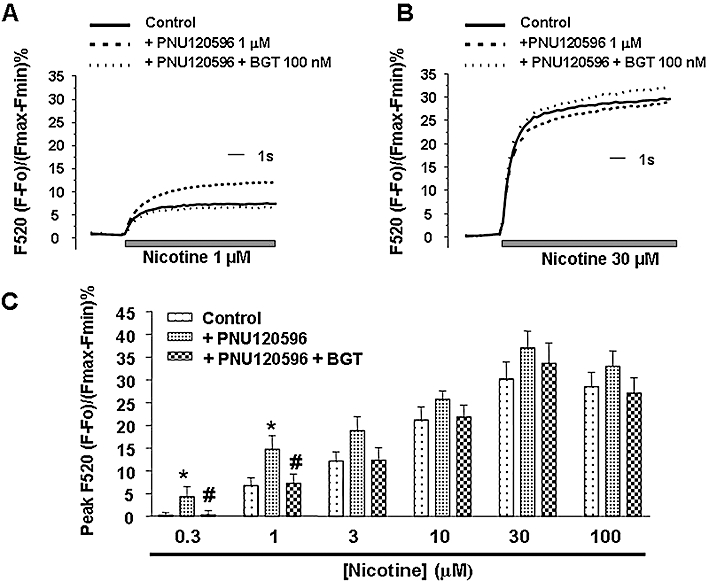
Dual effect of nicotine on the α7 and non-α7 nicotinic acetylcholine receptors (nAChRs). (A) Representative traces of intracellular calcium measured as Fluo-4 fluorescence in cells stimulated with a low concentration of nicotine (1 µM); this response was potentiated by PNU120596 (1 µM) and such potentiation was fully blocked by α-bungarotoxin (BGT) 100 nM, an α7 nAChR response. (B) A similar experiment as earlier but at a higher concentration of nicotine (30 µM). In this case, the nicotinic response was unaffected by PNU120596 or BGT: a non-α7 nAChR response. (C) Concentration-response curve of nicotine alone (control) or in cells pre-incubated with PNU120596 (1 µM) alone or PNU120596 (1 µM) plus BGT (100 nM). The Ca2+ signals induced by low concentrations of nicotine (0.3 and 1 µM) were potentiated by the allosteric modulator; this response was blocked by BGT. Ca2+ increases induced by higher concentrations of nicotine (above 1 µM) were not significantly potentiated by PNU120596 and were not affected by BGT. Data represent the mean ± SEM of five independent experiments; each experiment was performed in triplicate. *P < 0.05, significantly different from control. #P < 0.05, significantly different from nicotine plus PNU120596. One-way anova followed by a post hoc Newman–Keuls test.
Figure 5.
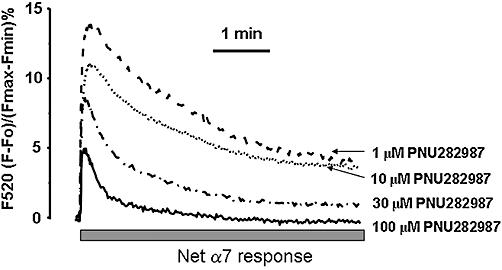
Intracellular Ca2+ traces of pure α7 responses mediated by increasing concentrations of PNU282987 plus PNU120596 (1 µM). Mean traces of the net α7-nicotinic acetylcholine receptor (nAChR)-mediated Ca2+ responses that correspond to increasing concentrations of the α7 nAChR agonist PNU282987 (1–100 µM) plus the α7 nAChR allosteric modulator PNU120596 at 1 µM. To obtain the net α7 nAChR response, we subtracted the non-α7 nAChR response, that is, the fluorescence increases that were not blocked by α-bungarotoxin from the PNU282987 + PNU120596 values. Data correspond to the mean of five independent experiments.
Inactivation of the α7-nAChR-mediated [Ca2+]c signals
To further understand the behavior of the α7 nAChRs in bovine chromaffin cells, we analyzed the kinetics of α7 [Ca2+]c signals, mediated solely by α7 nAChRs, using PNU282987 as the agonist. Cells pre-incubated with a fixed concentration of PNU120596 (1 µM) alone or in the presence of BGT (100 nM), were stimulated with increasing concentrations of PNU282987 (1–100 µM) for 5 min to achieve a plateau in the α7 desensitization state. To estimate the response mediated solely by α7 nAChRs, for a given concentration of the agonist, we subtracted the response that was not blocked by 100 nM BGT (non-α7 response) from the total response (PNU 282987 + PNU 120596) (see Figure S1 for example). After this correction, the [Ca2+]c signals changed as the concentration of the agonist was increased. The maximum peak of the [Ca2+]c signal became lower as the concentration of the α7 nAChR agonist increased (Figure 5). These results correlated with the reduction of the τon (activation time constant) and τoff (inactivation time constant) (see Table 1). Most probably, the lower maximum peak of the [Ca2+]c signals obtained with higher concentrations of the agonist could be related to greater inactivation of the α7 nAChR.
Table 1.
Activation constant (τon), inactivation constant (τoff) and mean maximum peak Fluo-4 fluorescent values of the net α7-nicotinic acetylcholine receptor (nAChR)-mediated Ca2+ response
| PNU 282987 concentration | τon | τoff | Peak max |
|---|---|---|---|
| 1 µM | 7.51 ± 1.18 | 67.5 ± 16.2 | 16.5 ± 2.8 |
| 10 µM | 4.34 ± 0.24 | 64.3 ± 14.3 | 15.6 ± 2.7 |
| 30 µM | 4.00 ± 0.70 | 51.5 ± 13.5 | 12.5 ± 1.8 |
| 100 µM | 2.90 ± 0.68 | 22.2 ± 1.8 | 8.4 ± 2.68 |
To obtain the net α7 nAChR -mediated response, the non-α7 nAChR response, that is, the fluorescence increases that were not blocked by α-bungarotoxin, were subtracted from the PNU282987 + PNU120596 fluorescence value. Once these curves were obtained (see Figure 4), τon, τoff (seconds) and maximum peak were calculated.
Participation of VOCCs in the [Ca2+]c increases mediated by α7 and non-α7 nAChRs
To explore the contribution of the different VOCCs on the α7 and non-α7 nAChR responses, we pre-incubated the cells with a blocker of L-type (nimodipine, 3 µM) or a blocker of non-L-type (ω-conotoxin MVIIC, 2 µM) VOCCs. As illustrated in Figure 6A, the α7 nAChR-mediated [Ca2+]c response (PNU282987 at 10 µM plus PNU120596 at 1 µM) (see Figure 3B,C) was not affected by nimodipine. However, the non-L-type channel blocker, ω-conotoxin MVIIC, decreased the Ca2+ signal by 19.7 ± 7% with respect to control. In contrast, the non-α7 nAChR response to nicotine at 10 µM (see Figure 1C) was decreased by the L-type blocker nimodipine by 16.9 ± 7.7% and was unaffected by ω-conotoxin MVIIC (Figure 6B).
Figure 6.
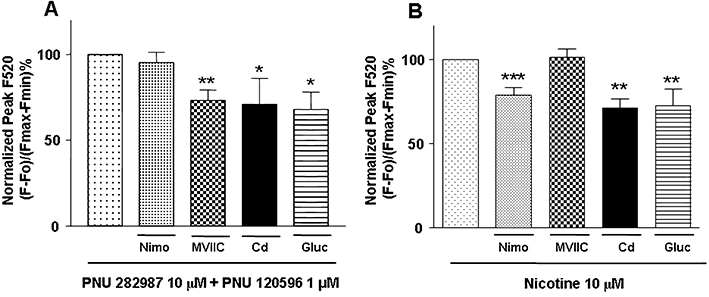
Effect of voltage operated calcium channels (VOCC) blockers on increases in intracellular Ca2+ mediated by α7 and non-α7 nicotinic acetylcholine receptors (nAChRs). Cells were stimulated with PNU282987 10 µM + PNU 120596 1 µM (A), 10 µM nicotine (B), in the presence of 50 µM Cd2+ (Cd), in Krebs-HEPES with N-methyl glucamine in substitution for sodium (gluc) or in the presence of the L-type VOCC blocker nimodipine (nimo 3µM;), or the non-L VOCC blocker ω-conotoxin MVIIC (MVIIC 2 µM) during 10 s. Control cells, to compare with nimodipine, were supplemented with the same percentage of DMSO (0.1%) that was used to dissolve nimodipine. Data represent the mean and SEM of at least four independent experiments performed in triplicate. *P < 0.05, **P < 0.01; ***P < 0.001, significantly different from control: one-way anova followed by a post hoc Newman–Keuls test.
To further test the involvement of VOCCs in the [Ca2+]c increases mediated by stimulation of these different nAChR subtypes, we used two other experimental tools: (i) the non-specific Ca2+ channel blocker cadmium; and (ii) substitution of the extracellular Na+ for N-methyl glucamine to prevent Na+-mediated depolarization. Both cadmium and N-methyl glucamine diminished the responses induced by α7 and non-α7 nAChRs. Blockade obtained by cadmium or Na+ substitution by N-methyl glucamine was not different to that obtained with ω-conotoxin MVIIC in the case of α7 nAChR stimulation (Figure 6A) or nimodipine in the case of non-α7 nAChR stimulation (Figure 6B).
Magnitude and inactivation of α7 and non-α7 nAChRs-mediated [Ca2+]c transients measured with cytosolic targeted aequorins
As the experimental conditions used above did not allow consecutive stimulation of the cells or quantification of the intracellular calcium concentration, we used another experimental approach that allowed superfusion of the cells, consecutive stimulation-washout cycles and estimation of the magnitude of the [Ca2+]c transients. For this purpose we used chromaffin cells transfected with aequorins targeted to the cytosol (Montero et al., 2000).
Figure 7A shows a typical experiment. Addition of PNU282987 plus PNU120596 (at concentrations that gave an α7 nAChR-mediated response) produced a mean [Ca2+]c peak around 200 nM, whereas perfusion of nicotine (at a concentration that gave a non-α7 nAChR response) produced larger peaks of around 600 nM (see statistics in Figure 7C). The nicotine-induced [Ca2+]c peak was also wider than that induced by PNU120596 plus PNU282987. Thus, when the total [Ca2+]c response was measured in terms of area under the curve, the [Ca2+]c response induced by the allosterically modulated α7 nAChRs was only 14 ± 3% (mean ± SEM = 6) of that induced by nicotine. When the cells were pre-treated with 100 nM BGT, an irreversible α7 nAChR antagonist, the [Ca2+]c increase induced by PNU120596 plus PNU282987 was abolished whereas the nicotinic response remained unaltered (Figure 7B,C). These results indicated that with this new experimental approach, we could also distinguish between an α7 and non-α7-nAChR-mediated response.
Figure 7.
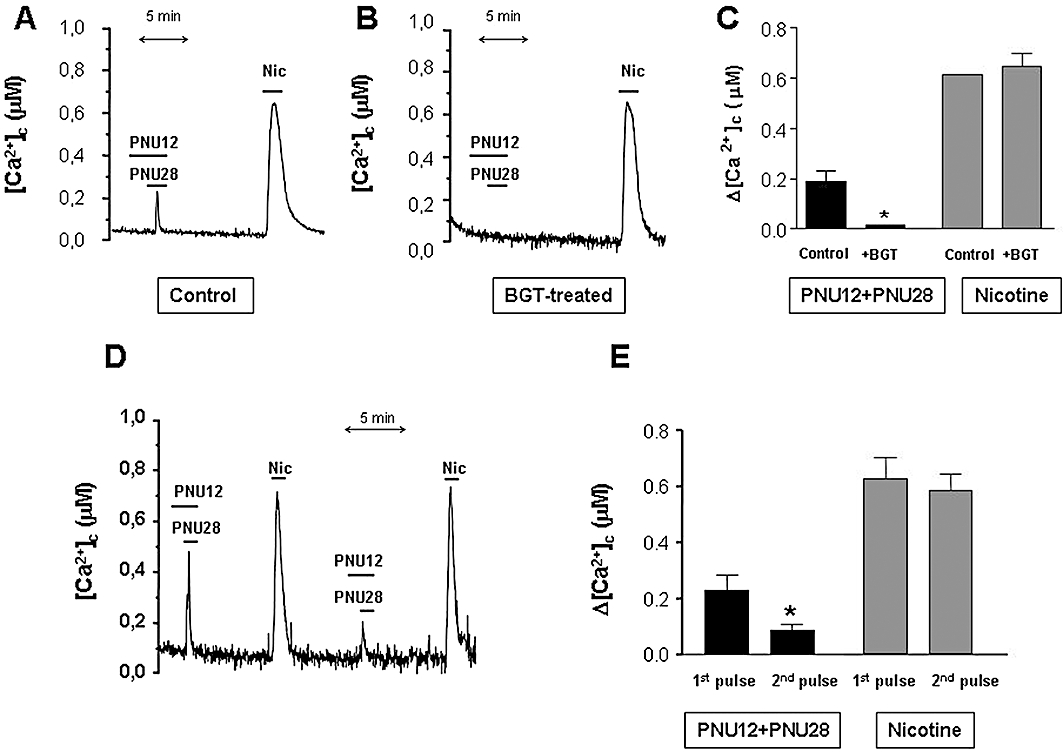
Magnitude and inactivation of [Ca2+]c transients mediated by α7 and non-α7 nicotinic acetylcholine receptors (nAChRs) agonists measured with aequorin. Bovine chromaffin cells expressing cytosolic aequorin were reconstituted with coelenterazine and then luminescence was monitored under continuous perfusion with 2 mM Ca2+-containing external medium. (A) Shows a representative recording illustrating [Ca2+]c transients obtained with submaximal concentrations of PNU282987 10 µM + PNU120596 10 µM and nicotine 30 µM given in the same population of cells. (B) Data from an experiment as in A, but in cells pre-treated with 100 nM α-bungarotoxin (BGT), 15 min prior the experiment, to block the α7 nAChRs. (C) Summary data from several experiments as in A and B, bars correspond to the mean ± SEM of seven individual experiments. *P < 0.01, significantly different from control. (D) Tracing representing the effects of several additions of either PNU282987 10 µM + PNU120596 10 µM or 30 µM nicotine, as indicated. (E) Summary data from seven experiments (mean ± SEM). Note the differences between the first and second stimulation with α7 or non-α7 nAChR stimulation. *P < 0.05, significantly different from the first pulse.
When the cells were stimulated twice with nicotine, this agonist gave two [Ca2+]c peaks of similar magnitude; however, a second stimulation with PNU120596 plus PNU282987 produced a much smaller response, indicating a slower recovery from inactivation (Figure 7D,E). Similar results were given when the order of the agonists was changed.
Contribution of the endoplasmic reticulum (ER) and mitochondria to the [Ca2+]c and [Ca2+]m transients induced by stimulation of α7 and non-α7 nAChR
Figure 8A shows a representative trace of cells consecutively stimulated with PNU120596 plus PNU282987 (α7 nAChR response) and nicotine (non-α7 nAChR response) in control cells expressing cytosolic aequorin. To determine the contribution of the ER to the α7 and non-α7 nAChR-mediated [Ca2+]c increase, we pre-treated the cells with thapsigargin, an irreversible blocker of the ER Ca2+ pump (SERCA), 15 min before nicotinic agonist exposure. Emptying the ER Ca2+ store with thapsigargin reduced, by over 80%, the [Ca2+]c peaks induced by PNU120596 plus PNU282987 and by 60% the [Ca2+]c peaks induced by nicotine (Figure 8B,C). These results were confirmed in chromaffin cells loaded with fluo-4 AM (Figure S2). Contribution of the ER was significantly higher (P < 0.001; n= 3) for the α7 nAChR response, compared with the non-α7 nAChR response.
Figure 8.
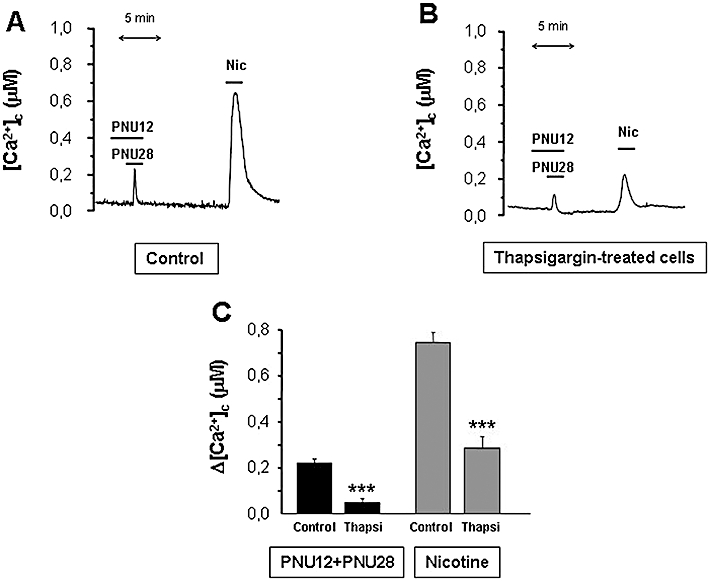
Contribution of the endoplasmic reticulum to the α7 and non-α7 cytosolic Ca2+ transients. Representative tracings of [Ca2+]c induced by α7 (PNU282987 10 µM + PNU120596 10 µM) and non-α 7 (nicotine 30 µM) nicotinic acetylcholine receptors (nAChRs) in control (A) and in cells treated with 1 µM thapsigargin for 15 min prior to the experiment (B). (C) Summary data from several similar experiments as those shown in A and B. Bars represent the mean and SEM of at least six independent experiments from different cultures. ***P < 0.001, significantly different from control.
The other intracellular organelle that participates in the regulation of intracellular calcium homeostasis is the mitochondria. We therefore measured mitochondrial [Ca2+] ([Ca2+]m) with aequorins targeted to this organelle. PNU120596 plus PNU282987 and nicotine elicited larger responses in the mitochondria (Figure 9A,C) than those measured in the cytosol (Figure 9D,F). Stimulation with the α7 agonist and the allosteric modulator, induced [Ca2+]m peaks of 516 ± 119 nM and [Ca2+]c peaks of 218 ± 20 nM (the ratio of [Ca2+]m/[Ca2+]c was 2.4 ± 0.8). Stimulation with nicotine induced [Ca2+]m peaks of 1324 ± 83 nM and [Ca2+]c peaks of 745 ± 44 nM ([Ca2+]m/[Ca2+]c was 1.8 ± 0.2). These ratio values, [Ca2+]m/[Ca2+]c, were not different from each other, indicating that mitochondria captured Ca2+ in the same way, regardless of which nAChR subtype is stimulated. The higher [Ca2+]m measurements are probably a consequence of the ability of mitochondria to amplify the [Ca2+]c variations.
Figure 9.
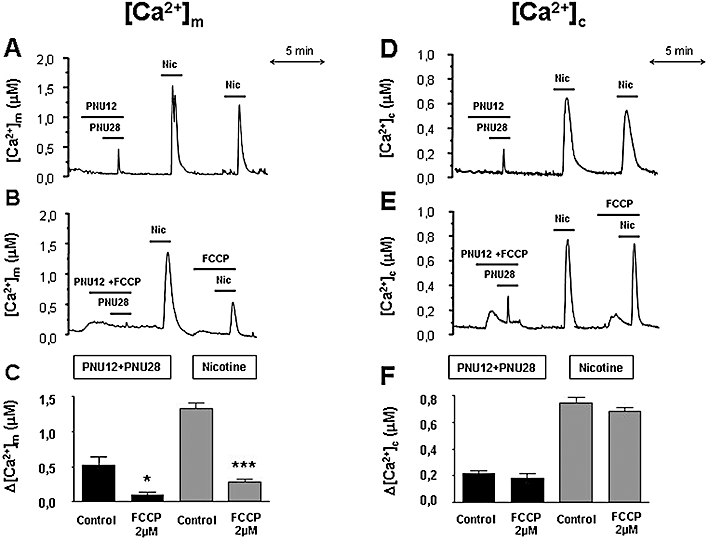
Contribution of the mitochondria to the mitochondrial and cytosolic Ca2+ transients induced by stimulation of α7 or non-α7 nicotinic acetylcholine receptors (nAChRs). Bovine chromaffin cells expressing mitochondrially-or- cytosolic targeted aequorin were reconstituted with coelenterazine and then luminescence was monitored under continuous perfusion with 2 mM Ca2+-containing external medium. Left panels show representative traces of [Ca2+]m transients induced by α7 (PNU282987 10 µM + PNU120596 10 µM) and non-α7 (nicotine 30 µM) nAChRs agonists, in control cells (A) and in cells treated with FCCP (2 µM), 2 min prior to the stimulus (B). (C) Summary data from several similar experiments as those shown in A and B. Right panels show representative traces of [Ca2+]c induced by α7 (PNU282987 10 µM + PNU120596 10 µM) and non-α7 (nicotine 30 µM) nAChR agonists in control cells (D) and in cells treated with FCCP(2 µM), 2 min prior to and during the stimulus (E). (F) Summary data from several similar experiments as those shown in D and E. Data in panels C and F correspond to the mean and SEM of three to five independent experiments. *P < 0.05 and ***P < 0.001, significantly different from control.
In addition, as could be expected, the [Ca2+]m peaks induced by PNU120596 plus PNU282987 and nicotine were markedly reduced by the protonophore FCCP (Figure 9B,C). However, mitochondrial depolarization with FCCP hardly modified [Ca2+]c caused by α7 or non-α7 nAChR activation (Figure 9E,F). Therefore these results indicate that the ER but not the mitochondria participate in the [Ca2+]c transients caused by stimulation of α7 and non-α7 nAChRs.
Catecholamine release induced by α7 and non-α7 nAChR stimulation
To determine if the Ca2+ signals were functional in terms of inducing exocytosis, we measured, by HPLC, secretion of adrenaline and noradrenaline induced by α7 and non-α7 nAChR stimulation. Basal adrenaline release from 106 bovine chromaffin cells was 54 ± 15 ng·mL−1 whereas that of noradrenaline was 84 ± 25 ng·mL−1. The α7 nAChR allosteric modulator PNU120596 at 10 µM did not modify basal levels of adrenaline or noradrenaline. However, in the presence of 10 µM PNU282987, adrenaline release increased to 191 ± 38 ng·mL−1 (354% increase) and noradrenaline to 255 ± 72 ng·mL−1 (305% increase); this catecholamine release was completely blocked by BGT (Figure 10A). Nicotine at 30 µM increased adrenaline release to 364 ± 111 ng·mL−1 (674%) and noradrenaline to 422 ± 116 ng·mL−1 (502%); such secretion was fully blocked by mecamylamine (Figure 10B).
Figure 10.
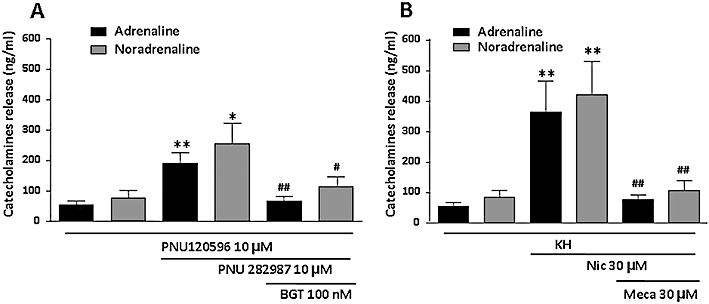
Differential release of adrenaline and noradrenaline induced by α7 (A) and non-α7 (B) nicotinic acetylcholine receptor (nAChR) stimulation. Cells were stimulated for 30 s with both PNUs or nicotine and medium were collected to measure catecholamine release by high-performance liquid chromatography. Antagonists were pre-incubated for 5 min and were present during agonist stimulation. Bars show the mean ± SEM from seven independent experiments. *P < 0.05, **P < 0.01, significantly different from PNU120596 alone for panel A or Krebs-HEPES for panel B. #P < 0.05, ##P < 0.01, significantly different from both PNUs for A or nicotine for B. The statistical analysis used was a one-way anova followed by a post hoc Newman–Keuls test.
When normalized catecholamine release (total amount of catecholamines/basal catecholamines secreted) was divided by the normalized peak [Ca2+]i (total peak [Ca2+]i/basal [Ca2+]i) the proportion obtained for the α7 nAChR response was 1.5 and for the non-α7 nAChR response was 0.6. These results suggest that for a given [Ca2+]i peak, the modulated α7 nAChR stimulus secretes more catecholamines, by a factor of 2.5, than a non-α7 nAChR stimulus.
Discussion
Bovine chromaffin cells are a model extensively used to study calcium signalling and exocytosis, but the contribution of the different subtypes of nAChRs to the control of these parameters are still in debate, especially for the α7 nAChR subtype. By using new pharmacological and selective tools, we have systematically studied how activation of α7 and non-α7 nAChRs of bovine chromaffin cells mediated intracellular Ca2+ signals and catecholamine secretion.
We first characterized the non-α7 nAChR-mediated response. The intracellular Ca2+ transients induced by nicotine and 5-iodo-A-85380 were related to non-α7 nAChRs, as they were fully blocked by mecamylamine but were unaffected by the α7 nAChR antagonist BGT. This non-α7 nAChR response was similar to those previously reported in bovine chromaffin cells and PC12 cells (Afar et al., 1994; Innocent et al., 2008).
Because nicotine has been reported to act on different nAChR subtypes in PC12 cells (Nakayama et al., 2001) and in chick ciliary ganglion neurons (Vijayaraghavan et al., 1992), we used PNU120596, an α7 nAChR allosteric modulator that is known to potentiate the α7 nAChR agonist response in PC12 and SHSY5Y cells (Dickinson et al., 2007; Innocent et al., 2008). When we used PNU120596, which does not display any effect on α3β4 nAChRs expressed in Xenopus oocytes (Gronlien et al., 2007), we observed that the minimal [Ca2+]c transients induced by low concentrations of nicotine (1 µM or below) were markedly increased and that this potentiated response was fully blocked by the selective α7 nAChR antagonist BGT. However, nicotine at concentrations above 3 µM, provided concentration-dependent [Ca2+]c transients in the absence of the allosteric modulator and this response was blocked by mecamylamine but not by BGT. Therefore, low concentrations of nicotine, in the presence of an α7 nAChR allosteric modulator, gave an α7 nAChR-modulated response, whereas high concentrations of nicotine alone give a non-α7 nAChR-mediated signal.
To further investigate how activation of α7 nAChRs could raise intracellular Ca2+ and catecholamine secretion we used the selective α7 nAChR agonists like PNU282987 and choline. In spite of PNU282987 being highly selective for α7 nAChRs (Bodnar et al., 2005; Hajos et al., 2005) and showing almost negligible blockade by α3β4 nAChR antagonists (Wishka et al., 2006), we found that this agonist, at concentrations ranging from 1 to 10 µM, did not induce measurable [Ca2+]c increases. However at concentrations above 30 µM, it did induce [Ca2+]c increases that were fully blocked by mecamylamine but were unaffected by BGT. We also found a similar pattern with the endogenous α7 nAChR agonist choline. In rat superior cervical ganglion neurones, such BGT-insensitive responses to choline have also been reported (Alkondon et al., 1997; Seddik et al., 2003). However, in the presence of the α7 nAChR allosteric modulator PNU120596, the α7 nAChR agonist and choline, at concentrations that did not elicit measurable [Ca2+]c increases by themselves, were markedly increased. These potentiated responses were completely blocked by BGT, indicating that it was an α7-nAChR-mediated response.
The activation and inactivation constants as well as the maximum peak of the α7 nAChR-mediated [Ca2+]c responses decreased, as the concentration of the α7 nAChR agonist increased (Fatt, 1950; Katz and Thesleff, 1957), suggesting a rapid desensitization of the receptor (Couturier et al., 1990). The rapid inactivation of this receptor could also explain the loss of the α7 nAChR response at higher concentrations of the agonists.
The faster inactivating profile of the α7 nAChR-mediated response was corroborated in the aequorin experiments, where a second stimulation with PNU282987 plus PNU120596 gave practically no response. However, two consecutive nicotine pulses, a non-α7 nAChR-mediated response, gave [Ca2+]c transients of similar amplitude, i.e. there was hardly any inactivation. Although it has been reported that PNU120596 increases the mean opening time of the α7 nAChR channel even in the desensitized state (Bodnar et al., 2005; Gronlien et al., 2007), our results (Figure 4) suggest a time-dependent decrease of the Ca2+ signal after prolonged exposure to the agonist.
Cell depolarization and participation of VOCCs in the [Ca2+]c transients observed with PNU282987 plus PNU120596 (α7 response) and nicotine (non-α7 response) were corroborated with the N-methyl glucamine and cadmium experiments. Substitution of Na+ by N-methyl glucamine, which prevents membrane depolarization mediating Na+ entry through its channels, reduced the α7 and non-α7 nAChR-mediated increases in [Ca2+]c to levels that were not statistically different to those obtained with ω-conotoxin MVIIC in the case of the α7 response or nimodipine in the case of the non-α7 nAChR response. Moreover, the observation that the non-specific Ca2+ channel blocker cadmium did not further increase the blockade of the [Ca2+]c responses induced by nimodipine or ω-conototoxin MVIIC in the α7 or non-α7-mediated responses, respectively, further supports the idea that non-L-type Ca2+ channels were coupled to the α7 nAChRs, whereas L-type Ca2+channels were coupled to non-α7 nAChRs.
Differential coupling of L and non-L-type VOCCs to a specific nAChR subtype can also contribute to the interpretation of the kinetics of the Ca2+ signals mediated by these receptor subtypes, and the dual activation pattern of a given agonist. For example, Ca2+ entry through L-type VOCCs favoured the inhibition of N and P/Q channels in voltage-clamped bovine adrenal chromaffin cells (Rosa et al., 2009). Therefore, when a nicotinic agonist acts on non-α7 nAChRs, coupled to L-type Ca2+ channels, the α7 nAChRs are inactivated because they are coupled to non-L Ca2+ channels that inactivate more rapidly (Villarroya et al., 1999). Also, depolarization itself can inactivate N-and-P/Q-type-Ca2+ channels (Villarroya et al., 1999) that are more prone to inactivation than L-type Ca2+ channels; this action translates into a reduction of [Ca2+]c and catecholamine release (Artalejo et al., 1986; Michelena et al., 1993). Therefore, rapid inactivation of α7 nAChRs could be related to the fact that they seem to be coupled to non-L-type Ca2+ channels that inactivate faster, whereas non-α7 nAChRs, which are coupled to the slower inactivating L-type Ca2+ channels, show less inactivation. However, in other systems like GH3 rat pituitary cells expressing homomeric α7 nAChR, [Ca2+]c transients induced by ACh were blocked by isradipine, an L-type Ca2+ channel antagonist (Feuerbach et al., 2005).
Besides Ca2+ entry through VOCCs, CICR from the ER can also contribute to the nAChR- mediated [Ca2+]c transients. Our results in bovine chromaffin cells indicated that CICR made a major contribution to [Ca2+]c transients upon nAChR stimulation, in comparison with Ca2+ entry through VOCC. We also found, as previously reported in PC12 cells (Dickinson et al., 2007), that CICR was a major contributor during α7 nAChR stimulation, when compared with non-α7 nAChR stimulation.
As to the participation of the mitochondria, the [Ca2+]c transients induced by both α7 and non-α7 nAChRs were slightly and similarly amplified in this organelle, resulting in [Ca2+]m transients of about twice the magnitude compared with the corresponding cytosolic transients. In spite of this observation, we did not observe significant differences between mitochondrial Ca2+ uptake, relative to cytosolic Ca2+ levels, which could be related to the nAChR subtype producing Ca2+ entry. Moreover, blockade of mitochondrial Ca2+ uptake with the protonophore FCCP produced no changes in the [Ca2+]c peaks induced by either α7 or non-α7 nAChR responses, indicating that under these experimental conditions, mitochondrial Ca2+ sequestration was relatively small and could not significantly modify the overall [Ca2+]c transient. As shown before, massive Ca2+ accumulation into mitochondria and its implication in catecholamine modulation requires the application of much more potent stimuli (Herrington et al., 1996; Montero et al., 2000; 2001;). Therefore in this case, the physiological significance of the observed increases in the [Ca2+]m would be more related with the activation of energy production than with the buffering of cytosolic [Ca2+].
Activation of postsynaptic nAChRs located in chromaffin cells, by acetylcholine, is responsible of catecholamine secretion under stressful conditions. The results of this study show that calcium entry through the modulated α7 nAChR seems to be more effective in inducing exocytosis, in comparison with the α3β4/ α3β4α5 receptor (Tachikawa et al., 2001), in terms of secretion/[Ca2+]c ratios. However, the magnitude of catecholamine release induced by nicotine, at concentrations related to non-α7 nAChRs was much higher compared with the α7nAChR -modulated response. The fact that α7 nAChRs seem to be coupled to non-L type VOCC, which have been described to be located closer to the exocytotic sites (Lopez et al., 1994; Lara et al., 1998), could explain why Ca2+ entry through this receptor seems to be more efficient in inducing exocytosis. Although it had been previously reported (Criado et al., 1997) that BGT-labelled cells were mainly adrenergic in adrenal slices, we have not found statistical differences between adrenaline and noradrenaline release when secretion was stimulated via α7 or non-α7 nAChRs. This finding could be probably attributed to differences in the preparation used; in this study we have employed isolated bovine chromaffin cells in culture instead of adrenal slices.
In conclusion, although the functional role of the α7 nAChR in the CNS has been well characterized (Barrantes et al., 1995; Meyer et al., 1998; Martin et al., 2004; Ulloa, 2005), its functional properties in the peripheral nervous system have been less well defined (Fucile et al., 2005). The expression density of this subtype of nAChR in plasma membranes from bovine chromaffin cells is similar ([125I]αBGT 49.9 ± 7.6 fmol·mg−1 protein) (El-Hajj et al., 2007) to those reported for rat hippocampal membranes ([125I]αBGT 51.1 fmol·mg−1 protein) (Garcia-Guzman et al., 1995). So, the question is why if bovine chromaffin cells express α7 nAChRs, most of the attempts to establish their functional role have produced negative results? (Afar et al., 1994; Tachikawa et al., 2001). Nevertheless, we (Lopez et al., 1998) were able to partially block ACh-induced calcium signals and catecholamine secretion with a selective α7 nAChR antagonist, which was an indirect indication to its participation in these processes. However, detection of calcium signals or exocytosis by direct activation of α7 nAChRs in these cells have yet not been clearly demonstrated. Our data offer at least three reasons why a functional role for the chromaffin α7 nAChRs, in terms of increasing cytosolic Ca2+ and evoking catecholamine release, has been difficult to find: (i) the α7 nAChRs of bovine chromaffin cells need the presence of an allosteric modulator and low concentrations of agonists to achieve a measurable [Ca2+]c transient; (ii) the magnitude of the [Ca2+]c signal, compared with the non-α7nAChR -mediated response, is about 25%, in terms of maximum peak response, and around 15%, in terms of total calcium entry; and (iii) the α7 nAChR inactivates very rapidly.
In summary, the results of this study indicate that α7 nAChRs, in bovine chromaffin cells, are able to mediate measurable calcium signals and catecholamine exocytosis under circumstances that require an allosteric modulator and low concentrations of agonists. Notably, these requirements are not necessary for the effects mediated by the non-α7 nAChRs.
Acknowledgments
This work was supported in part by grants from: Dirección General de Investigación Científica y Técnica SAF2006-08540 and SAF2009-12150, the Spanish Ministry of Health (Instituto de Salud Carlos III) RETICS-RD06/0026 and Comunidad Autónoma de Madrid SAL2006/0275 to MGL. Dirección General de Investigación Científica y Técnica BFU2008-01871 and Junta de Castilla y León (VA103A08 and GR105) to JA. ARN is supported by FIS n°: PI052124. We also thank Ms Lorena Cortés and Inés Colmena for preparing cell cultures and technical assistance, Prof Antonio G. García for useful comments on the paper and the continuous support of Fundación Teófilo Hernando.
Glossary
Abbreviations
- [Ca2+]c
cytosolic concentration of calcium
- BGT
α-bungarotoxin
- DMEM
Dulbecco's modified Eagle's medium
- ER
endoplasmic reticulum
- nAChR
nicotinic acetylcholine receptor
- VOCC
voltage operated calcium channel
Conflicts of interest
None to declare.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 To determine the pure α7 response (Trace C), we subtracted the response that was not blocked by 100 nM a-bungarotoxin (non-α7 response- Trace B) from the total response (PNU 282987 + PNU 120596- Trace A). Variables of Trace A and Trace B were performed in parallel in the same batches of cell cultures.
{kind=link}
Figure S2 Comparison of the contribution of the ER to the α7 and non-α7 nACh-mediated responses. Intracellular Ca2+ was measured in bovine chromaffin cells loaded with Fluo-4AM using a fluorescence microplate reader as described in the Materials and Methods section. Blockade of the ER was achieved by treating the cells for 15 min with thapsigargin 1 mM prior to the addition of the nicotinic agonists. Data correspond to the mean and S.E.M of five experiments. ***P < 0.001 in comparison to control response. $$$P < 0.001 comparing the α7 and non-α7 response in thapsigargin-treated cells.
{kind=link}
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Afar R, Trifaro JM, Quik M. Nicotine-induced intracellular calcium changes are not antagonized by alpha-bungarotoxin in adrenal medullary cells. Brain Res. 1994;641:127–131. doi: 10.1016/0006-8993(94)91824-4. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Cortes WS, Maelicke A, Albuquerque EX. Choline is a selective agonist of alpha7 nicotinic acetylcholine receptors in the rat brain neurons. Eur J Neurosci. 1997;9:2734–2742. doi: 10.1111/j.1460-9568.1997.tb01702.x. [DOI] [PubMed] [Google Scholar]
- Artalejo CR, Bader MF, Aunis D, Garcia AG. Inactivation of the early calcium uptake and noradrenaline release evoked by potassium in cultured chromaffin cells. Biochem Biophys Res Commun. 1986;134:1–7. doi: 10.1016/0006-291x(86)90518-8. [DOI] [PubMed] [Google Scholar]
- Ballesta JJ, Palmero M, Hidalgo MJ, Gutierrez LM, Reig JA, Viniegra S, et al. Separate binding and functional sites for omega-conotoxin and nitrendipine suggest two types of calcium channels in bovine chromaffin cells. J Neurochem. 1989;53:1050–1056. doi: 10.1111/j.1471-4159.1989.tb07394.x. [DOI] [PubMed] [Google Scholar]
- Barrantes GE, Rogers AT, Lindstrom J, Wonnacott S. alpha-Bungarotoxin binding sites in rat hippocampal and cortical cultures: initial characterisation, colocalisation with alpha 7 subunits and up-regulation by chronic nicotine treatment. Brain Res. 1995;672:228–236. doi: 10.1016/0006-8993(94)01386-v. [DOI] [PubMed] [Google Scholar]
- Barrero MJ, Montero M, Alvarez J. Dynamics of [Ca2+] in the endoplasmic reticulum and cytoplasm of intact HeLa cells. A comparative study. J Biol Chem. 1997;272:27694–27699. doi: 10.1074/jbc.272.44.27694. [DOI] [PubMed] [Google Scholar]
- Bodnar AL, Cortes-Burgos LA, Cook KK, Dinh DM, Groppi VE, Hajos M, et al. Discovery and structure-activity relationship of quinuclidine benzamides as agonists of alpha7 nicotinic acetylcholine receptors. J Med Chem. 2005;48:905–908. doi: 10.1021/jm049363q. [DOI] [PubMed] [Google Scholar]
- Brini M, Marsault R, Bastianutto C, Alvarez J, Pozzan T, Rizzuto R. Transfected aequorin in the measurement of cytosolic Ca2+ concentration ([Ca2+]c). A critical evaluation. J Biol Chem. 1995;270:9896–9903. doi: 10.1074/jbc.270.17.9896. [DOI] [PubMed] [Google Scholar]
- Campos-Caro A, Smillie FI, Dominguez del Toro E, Rovira JC, Vicente-Agullo F, Chapuli J, et al. Neuronal nicotinic acetylcholine receptors on bovine chromaffin cells: cloning, expression, and genomic organization of receptor subunits. J Neurochem. 1997;68:488–497. doi: 10.1046/j.1471-4159.1997.68020488.x. [DOI] [PubMed] [Google Scholar]
- Couturier S, Bertrand D, Matter JM, Hernandez MC, Bertrand S, Millar N, et al. A neuronal nicotinic acetylcholine receptor subunit (alpha 7) is developmentally regulated and forms a homo-oligomeric channel blocked by alpha-BTX. Neuron. 1990;5:847–856. doi: 10.1016/0896-6273(90)90344-f. [DOI] [PubMed] [Google Scholar]
- Criado M, Alamo L, Navarro A. Primary structure of an agonist binding subunit of the nicotinic acetylcholine receptor from bovine adrenal chromaffin cells. Neurochem Res. 1992;17:281–287. doi: 10.1007/BF00966671. [DOI] [PubMed] [Google Scholar]
- Criado M, Dominguez del Toro E, Carrasco-Serrano C, Smillie FI, Juiz JM, Viniegra S, et al. Differential expression of alpha-bungarotoxin-sensitive neuronal nicotinic receptors in adrenergic chromaffin cells: a role for transcription factor Egr-1. J Neurosci. 1997;17:6554–6564. doi: 10.1523/JNEUROSCI.17-17-06554.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson JA, Hanrott KE, Mok MH, Kew JN, Wonnacott S. Differential coupling of alpha7 and non-alpha7 nicotinic acetylcholine receptors to calcium-induced calcium release and voltage-operated calcium channels in PC12 cells. J Neurochem. 2007;100:1089–1096. doi: 10.1111/j.1471-4159.2006.04273.x. [DOI] [PubMed] [Google Scholar]
- Douglas WW, Rubin RP. Mechanism of nicotinic action at the adrenal medulla: calcium as a link in stimulus-secretion coupling. Nature. 1961a;192:1087–1089. doi: 10.1038/1921087b0. [DOI] [PubMed] [Google Scholar]
- Douglas WW, Rubin RP. The role of calcium in the secretory response of the adrenal medulla to acetylcholine. J Physiol. 1961b;159:40–57. doi: 10.1113/jphysiol.1961.sp006791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hajj RA, McKay SB, McKay DB. Pharmacological and immunological identification of native alpha7 nicotinic receptors: evidence for homomeric and heteromeric alpha7 receptors. Life Sci. 2007;81:1317–1322. doi: 10.1016/j.lfs.2007.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatt P. The electromotive action of acetylcholine at the motor end-plate. J Physiol. 1950;111:408–422. doi: 10.1113/jphysiol.1950.sp004492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerbach D, Lingenhohl K, Dobbins P, Mosbacher J, Corbett N, Nozulak J, et al. Coupling of human nicotinic acetylcholine receptors alpha 7 to calcium channels in GH3 cells. Neuropharmacology. 2005;48:215–227. doi: 10.1016/j.neuropharm.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Fucile S, Renzi M, Lax P, Eusebi F. Fractional Ca(2+) current through human neuronal alpha7 nicotinic acetylcholine receptors. Cell Calcium. 2003;34:205–209. doi: 10.1016/s0143-4160(03)00071-x. [DOI] [PubMed] [Google Scholar]
- Fucile S, Sucapane A, Eusebi F. Ca2+ permeability of nicotinic acetylcholine receptors from rat dorsal root ganglion neurones. J Physiol. 2005;565:219–228. doi: 10.1113/jphysiol.2005.084871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia AG, Garcia-De-Diego AM, Gandia L, Borges R, Garcia-Sancho J. Calcium signaling and exocytosis in adrenal chromaffin cells. Physiol Rev. 2006;86:1093–1131. doi: 10.1152/physrev.00039.2005. [DOI] [PubMed] [Google Scholar]
- Garcia-Guzman M, Sala F, Sala S, Campos-Caro A, Stuhmer W, Gutierrez LM, et al. alpha-Bungarotoxin-sensitive nicotinic receptors on bovine chromaffin cells: molecular cloning, functional expression and alternative splicing of the alpha 7 subunit. Eur J Neurosci. 1995;7:647–655. doi: 10.1111/j.1460-9568.1995.tb00668.x. [DOI] [PubMed] [Google Scholar]
- Gronlien JH, Hakerud M, Ween H, Thorin-Hagene K, Briggs CA, Gopalakrishnan M, et al. Distinct profiles of alpha7 nAChR positive allosteric modulation revealed by structurally diverse chemotypes. Mol Pharmacol. 2007;72:715–724. doi: 10.1124/mol.107.035410. [DOI] [PubMed] [Google Scholar]
- Hajos M, Hurst RS, Hoffmann WE, Krause M, Wall TM, Higdon NR, et al. The selective alpha7 nicotinic acetylcholine receptor agonist PNU-282987 [N-[(3R)-1-Azabicyclo[2.2.2]oct-3-yl]-4-chlorobenzamide hydrochloride] enhances GABAergic synaptic activity in brain slices and restores auditory gating deficits in anesthetized rats. J Pharmacol Exp Ther. 2005;312:1213–1222. doi: 10.1124/jpet.104.076968. [DOI] [PubMed] [Google Scholar]
- Herrington J, Park YB, Babcock DF, Hille B. Dominant role of mitochondria in clearance of large Ca2+ loads from rat adrenal chromaffin cells. Neuron. 1996;16:219–228. doi: 10.1016/s0896-6273(00)80038-0. [DOI] [PubMed] [Google Scholar]
- Hurst RS, Hajos M, Raggenbass M, Wall TM, Higdon NR, Lawson JA, et al. A novel positive allosteric modulator of the alpha7 neuronal nicotinic acetylcholine receptor: in vitro and in vivo characterization. J Neurosci. 2005;25:4396–4405. doi: 10.1523/JNEUROSCI.5269-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innocent N, Livingstone PD, Hone A, Kimura A, Young T, Whiteaker P, et al. Alpha-conotoxin Arenatus IB[V11L,V16D][corrected] is a potent and selective antagonist at rat and human native alpha7 nicotinic acetylcholine receptors. J Pharmacol Exp Ther. 2008;327:529–537. doi: 10.1124/jpet.108.142943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B, Thesleff S. A study of the desensitization produced by acetylcholine at the motor end-plate. J Physiol. 1957;138:63–80. doi: 10.1113/jphysiol.1957.sp005838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara B, Gandia L, Martinez-Sierra R, Torres A, Garcia AG. Q-type Ca2+ channels are located closer to secretory sites than L-type channels: functional evidence in chromaffin cells. Pflugers Arch. 1998;435:472–478. doi: 10.1007/s004240050541. [DOI] [PubMed] [Google Scholar]
- Livett BG. Adrenal medullary chromaffin cells in vitro. Physiol Rev. 1984;64:1103–1161. doi: 10.1152/physrev.1984.64.4.1103. [DOI] [PubMed] [Google Scholar]
- Lopez MG, Montiel C, Herrero CJ, Garcia-Palomero E, Mayorgas I, Hernandez-Guijo JM, et al. Unmasking the functions of the chromaffin cell alpha7 nicotinic receptor by using short pulses of acetylcholine and selective blockers. Proc Natl Acad Sci USA. 1998;95:14184–14189. doi: 10.1073/pnas.95.24.14184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez MG, Villarroya M, Lara B, Martinez Sierra R, Albillos A, Garcia AG, et al. Q- and L-type Ca2+ channels dominate the control of secretion in bovine chromaffin cells. FEBS Lett. 1994;349:331–337. doi: 10.1016/0014-5793(94)00696-2. [DOI] [PubMed] [Google Scholar]
- Martin LF, Kem WR, Freedman R. Alpha-7 nicotinic receptor agonists: potential new candidates for the treatment of schizophrenia. Psychopharmacology (Berl) 2004;174:54–64. doi: 10.1007/s00213-003-1750-1. [DOI] [PubMed] [Google Scholar]
- Meyer EM, Tay ET, Zoltewicz JA, Meyers C, King MA, Papke RL, et al. Neuroprotective and memory-related actions of novel alpha-7 nicotinic agents with different mixed agonist/antagonist properties. J Pharmacol Exp Ther. 1998;284:1026–1032. [PubMed] [Google Scholar]
- Michelena P, Garcia-Perez LE, Artalejo AR, Garcia AG. Separation between cytosolic calcium and secretion in chromaffin cells superfused with calcium ramps. Proc Natl Acad Sci USA. 1993;90:3284–3288. doi: 10.1073/pnas.90.8.3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero M, Alonso MT, Albillos A, Cuchillo-Ibanez I, Olivares R, García AG, et al. Control of secretion by mitochondria depends on the size of the local [Ca2+] after chromaffin cell stimulation. Eur J Neurosci. 2001;13:2247–2254. doi: 10.1046/j.0953-816x.2001.01602.x. [DOI] [PubMed] [Google Scholar]
- Montero M, Alonso MT, Carnicero E, Cuchillo-Ibanez I, Albillos A, Garcia AG, et al. Chromaffin-cell stimulation triggers fast millimolar mitochondrial Ca2+ transients that modulate secretion. Nat Cell Biol. 2000;2:57–61. doi: 10.1038/35000001. [DOI] [PubMed] [Google Scholar]
- Moro MA, Lopez MG, Gandia L, Michelena P, Garcia AG. Separation and culture of living adrenaline- and noradrenaline-containing cells from bovine adrenal medullae. Anal Biochem. 1990;185:243–248. doi: 10.1016/0003-2697(90)90287-j. [DOI] [PubMed] [Google Scholar]
- Mukhin AG, Gundisch D, Horti AG, Koren AO, Tamagnan G, Kimes AS, et al. 5-Iodo-A-85380, an alpha4beta2 subtype-selective ligand for nicotinic acetylcholine receptors. Mol Pharmacol. 2000;57:642–649. doi: 10.1124/mol.57.3.642. [DOI] [PubMed] [Google Scholar]
- Nakayama H, Numakawa T, Ikeuchi T, Hatanaka H. Nicotine-induced phosphorylation of extracellular signal-regulated protein kinase and CREB in PC12h cells. J Neurochem. 2001;79:489–498. doi: 10.1046/j.1471-4159.2001.00602.x. [DOI] [PubMed] [Google Scholar]
- Pereira EF, Alkondon M, Tano T, Castro NG, Froes-Ferrao MM, Rozental R, et al. A novel agonist binding site on nicotinic acetylcholine receptors. J Recept Res. 1993;13:413–436. doi: 10.3109/10799899309073670. [DOI] [PubMed] [Google Scholar]
- Rathouz MM, Berg DK. Synaptic-type acetylcholine receptors raise intracellular calcium levels in neurons by two mechanisms. J Neurosci. 1994;14:6935–6945. doi: 10.1523/JNEUROSCI.14-11-06935.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Simpson AW, Brini M, Pozzan T. Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature. 1992;358:325–327. doi: 10.1038/358325a0. [DOI] [PubMed] [Google Scholar]
- Rosa JM, Gandia L, Garcia AG. Inhibition of N and PQ calcium channels by calcium entry through L channels in chromaffin cells. Pflugers Arch. 2009;458:795–807. doi: 10.1007/s00424-009-0662-2. [DOI] [PubMed] [Google Scholar]
- Santodomingo J, Vay L, Camacho M, Hernandez-Sanmiguel E, Fonteriz RI, Lobaton CD, et al. Calcium dynamics in bovine adrenal medulla chromaffin cell secretory granules. Eur J Neurosci. 2008;28:1265–1274. doi: 10.1111/j.1460-9568.2008.06440.x. [DOI] [PubMed] [Google Scholar]
- Seddik R, Bradaia A, Trouslard J. Choline induces Ca2+ entry in cultured sympathetic neurones isolated from rat superior cervical ganglion. Eur J Pharmacol. 2003;471:165–176. doi: 10.1016/s0014-2999(03)01860-0. [DOI] [PubMed] [Google Scholar]
- Seguela P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW. Molecular cloning, functional properties, and distribution of rat brain alpha 7: a nicotinic cation channel highly permeable to calcium. J Neurosci. 1993;13:596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachikawa E, Mizuma K, Kudo K, Kashimoto T, Yamato S, Ohta S. Characterization of the functional subunit combination of nicotinic acetylcholine receptors in bovine adrenal chromaffin cells. Neurosci Lett. 2001;312:161–164. doi: 10.1016/s0304-3940(01)02211-x. [DOI] [PubMed] [Google Scholar]
- Ulloa L. The vagus nerve and the nicotinic anti-inflammatory pathway. Nat Rev Drug Discov. 2005;4:673–684. doi: 10.1038/nrd1797. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A. The endoplasmic reticulum and neuronal calcium signalling. Cell Calcium. 2002;32:393–404. doi: 10.1016/s0143416002001896. [DOI] [PubMed] [Google Scholar]
- Vijayaraghavan S, Pugh PC, Zhang ZW, Rathouz MM, Berg DK. Nicotinic receptors that bind alpha-bungarotoxin on neurons raise intracellular free Ca2+ Neuron. 1992;8:353–362. doi: 10.1016/0896-6273(92)90301-s. [DOI] [PubMed] [Google Scholar]
- Villarroya M, De la Fuente MT, Lopez MG, Gandia L, Garcia AG. Distinct effects of omega-toxins and various groups of Ca(2+)-entry inhibitors on nicotinic acetylcholine receptor and Ca2+ channels of chromaffin cells. Eur J Pharmacol. 1997;320:249–257. doi: 10.1016/s0014-2999(96)00902-8. [DOI] [PubMed] [Google Scholar]
- Villarroya M, Olivares R, Ruiz A, Cano-Abad MF, de Pascual R, Lomax RB, et al. Voltage inactivation of Ca2+ entry and secretion associated with N- and P/Q-type but not L-type Ca2+ channels of bovine chromaffin cells. J Physiol. 1999;516(Pt 2):421–432. doi: 10.1111/j.1469-7793.1999.0421v.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SP, Kirshner N. The acetylcholine receptor of the adrenal medulla. J Neurochem. 1977;28:687–695. doi: 10.1111/j.1471-4159.1977.tb10615.x. [DOI] [PubMed] [Google Scholar]
- Wishka DG, Walker DP, Yates KM, Reitz SC, Jia S, Myers JK, et al. Discovery of N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]furo[2,3-c]pyridine-5-carboxamide, an agonist of the alpha7 nicotinic acetylcholine receptor, for the potential treatment of cognitive deficits in schizophrenia: synthesis and structure – activity relationship. J Med Chem. 2006;49:4425–4436. doi: 10.1021/jm0602413. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Neher E. Calcium permeability of nicotinic acetylcholine receptor channels in bovine adrenal chromaffin cells. Pflugers Arch. 1993;425:511–517. doi: 10.1007/BF00374879. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.