

Abstract
Ribonuclease P (RNase P) complexed with external guide sequence (EGS) represents a novel nucleic acid-based gene interference approach to modulate gene expression. We have previously used an in vitro selection procedure to generate EGS variants that efficiently direct human RNase P to cleave a target mRNA in vitro. In this study, a variant was used to target the mRNA encoding the protease of human cytomegalovirus (HCMV), which is essential for viral capsid formation and replication. The EGS variant was about 35-fold more active in inducing human RNase P to cleave the mRNA in vitro than the EGS derived from a natural tRNA. Moreover, a reduction of 95% in the expression of the protease and a reduction of 4,000-fold in viral growth were observed in HCMV-infected cells that expressed the EGS variant, whereas a reduction of 80% in the protease expression and an inhibition of 150-fold in viral growth were detected in cells that expressed the EGS derived from a natural tRNA sequence. No significant reduction in viral protease expression or viral growth was observed in cells that either did not express an EGS or produced a “disabled” EGS, which carried nucleotide mutations that precluded RNase P recognition. Our results provide direct evidence that engineered EGS variant is highly effective in blocking HCMV expression and growth by targeting the viral protease. Furthermore, these results demonstrate the utility of engineered EGS RNAs in gene targeting applications, including the inhibition of HCMV infection by blocking the expression of virus-encoded essential proteins.
Keywords: Antisense RNA, Gene Therapy, RNA, RNA Catalysis, RNA Interference (RNAi), Transfer RNA (tRNA), Virus, Ribonuclease P, Cytomegalovirus
Introduction
Nucleic acid-based gene interference strategies, such as antisense oligonucleotides, ribozymes or DNAzymes, and RNA interference (RNAi), represent powerful research tools and promising therapeutic agents for human diseases (1–4). Ribonuclease P (RNase P)5 has been proposed as a novel RNA-based gene interference strategy for knocking down gene expression (5, 6). This enzyme is a ribonucleoprotein complex found in all organisms examined. It catalyzes a hydrolysis reaction to remove a 5′ leader sequence from tRNA precursors (ptRNA) and is responsible for the maturation of 5′ termini of all tRNAs (5, 7). In Escherichia coli, this enzyme consists of a catalytic RNA subunit (M1 RNA) and a protein subunit (C5 protein) (8). Studies on RNase P substrate recognition revealed that the enzyme recognizes the structure rather than the primary nucleotide sequence of the substrates (9, 10). Thus, RNase P can cleave an mRNA sequence if the mRNA substrate forms a hybrid complex with a custom-designed sequence (external guide sequence or EGS) to resemble a ptRNA molecule (Fig. 1, A and B) (11, 12). EGS RNAs derived from natural tRNA sequences have been shown to be effective in blocking gene expression in bacteria and mammalian cells (12–17). We have previously shown that EGSs that were derived from a natural tRNA effectively induced human RNase P to cleave the mRNA of thymidine kinase (TK) of herpes simplex virus 1 (HSV-1) in vitro (15, 18). A reduction of ∼75% in the expression of TK mRNA and protein was observed in HSV-1-infected cells that expressed these functional EGS RNAs.
FIGURE 1.

Substrates for RNase P. A, a natural substrate (ptRNA). B, a hybridized complex of a target RNA (e.g. mRNA) and an EGS that resembles the structure of a tRNA. C and D, complexes of PR mRNA sequence with EGS PR-SER and PR-C125. The site of cleavage by RNase P is indicated with an arrowhead. The sequence of the PR mRNA around the targeting site is shown in blue and the EGS sequence is shown in green. The sequences of PR-SER that were equivalent to the T-stem and loop, and variable region of a tRNA molecule were derived from tRNASer, whereas those of PR-C125 were from EGS variant C125. The circled positions in the T-loop of EGS PR-SER and PR-C125 represent the nucleotides that are mutated (5′-UUC-3′ → AAG) to generate PR-SER-C and PR-C125-C, respectively.
Increasing the in vitro efficiency of the EGS-induced RNase P cleavage as well as its efficacy in vivo is required to develop EGSs for practical use both as a research tool and as a therapeutic agent for gene-targeting applications. Using an in vitro selection procedure, we have previously isolated novel EGS variants that direct RNase P to cleave TK mRNA in vitro more efficiently than those derived from a natural tRNA sequence (18). Little is currently known about how these EGS RNA variants increase their activity in directing RNase P to cleave a target mRNA. It has not been extensively studied whether selected EGS RNA variants are more effective in blocking gene expression in cultured cells than those derived from a natural tRNA sequence. In this study, one of these EGS variants was used to construct EGS PR-C125 that targets the mRNA encoding the protease (PR) of human cytomegalovirus (HCMV) (19), and the activity of the EGS in blocking viral PR expression and growth in cultured cells was examined.
HCMV, a human herpesvirus, is the leading cause of congenital infections associated with mental retardation in newborns and causes serious clinical manifestations in the immunocompromised population such as AIDS patients (19). Development of effective antiviral compounds and approaches is central for the treatment and prevention of infections by HCMV as well as other herpesviruses. HCMV PR is highly conserved among herpesviruses (20) and is essential for viral capsid formation and replication (21, 22). Thus, PR may serve as a target for novel drug development to combat HCMV and other herpesvirus infections (19).
In this study, we investigated activity of the EGS variant PR-C125 in inducing RNase P to cleave the target PR mRNA and its efficacy in blocking HCMV gene expression and growth in cultured cells. We showed that PR-C125 was about 35-fold more active in directing RNase P to cleave the target mRNA than PR-SER, the EGS derived from a natural tRNA sequence. When expressed in cultured cells that were infected by HCMV, PR-C125 was more effective in inhibiting viral gene expression and growth than PR-SER. A reduction of more than 95% in the PR expression and an inhibition of at least 4,000-fold were observed in cells that expressed PR-C125. In contrast, a reduction of less than 10% in viral gene expression and growth was observed in cells that either did not express an EGS or expressed EGSs that contained point mutations abolishing their ability to induce RNase P-mediated cleavage. Our results provide direct evidence that engineered EGS RNAs are highly effective in blocking HCMV PR expression and growth. These results also demonstrate the feasibility of developing highly active EGSs as a novel class of antiviral agents for treatment of human viral diseases.
EXPERIMENTAL PROCEDURES
Viruses, Cells, and Antibodies
HCMV (strain AD169) was propagated in human foreskin fibroblasts and astrocytoma U373MG cells in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) fetal bovine serum. The anti-rabbit polyclonal antibodies against HCMV protease were kindly provided by Annette Meyer, Pfizer, Inc. (Ann Arbor, MI), and John Wu of Promab, Inc. (Albany, CA). The monoclonal antibodies c1201, c1202, and c1203, which react with HCMV proteins gB, UL44, and IE2, were purchased from the Goodwin Institute for Cancer Research (Plantation, FL). The monoclonal antibodies against gH and human actin were purchased from Biodesign Inc. (Kennebunk, Maine) and Sigma, respectively.
EGS and PR mRNA Substrate
The DNA sequence coding for EGS PR-SER was generated by PCR using construct pTK112 (15) as the template with 5′ primer oligoPR31 (5′-GGAATTCTAATACGACTCACTATAGGTTAACGCGCCGGGTGCGGTCTCC-3′) and 3′ primer oligoPR32 (5′-AAGCTTTAAATGCTCTCCGCAGGATTTGAACCTGCGCGCG-3′). The DNA sequence coding for PR-C125 was generated by PCR using construct Ptkc125 (18) as the template with the 5′ primer oligoPR41 (5′-GGAATTCTAATACGACTCACTATAGGTTAACGCGCCGGGCAGCTACTAGCAG-3′) and 3′ primer oligoPR42 (5′-AAGCTTTAAATGCTCTCCGCAGGCTCCGAACTGCTAGTAG-3′). The DNA sequences coding for EGS PR-SER-C and PR-C125-C were derived from those for PR-SER and PR-C125, respectively, and contained point mutations (5′-TTC-3′ → AAG) at the three highly conserved positions in the T-loop of these EGSs (Fig. 1, C and D). The DNA sequence that encodes substrate pr39 was constructed by annealing oligonucleotide AF25 (5′-GGAATTCTAATACGACTCACTATAG-3′) and sPR (5′-CGGGATCCGCAGCGCCGGCTGGAGAGCGAGAGGCCGGCCTATAGTGAGTCGTATTA-3′). RNA substrate pr39 and EGS RNAs were synthesized in vitro from the constructed DNA templates using T7 RNA polymerase.
RNase P Assay and in Vitro Studies
Human RNase P was prepared from HeLa cellular extracts as described previously (12, 15, 18). The EGSs and 32P-labeled pr39 were incubated with human RNase P at 37 °C in buffer A (50 mm Tris, pH 7.4, 100 mm NH4Cl, and 10 mm MgCl2). Cleavage products were separated in denaturing gels and analyzed with a STORM840 PhosphorImager (GE Healthcare). Assays to determine kinetic parameters were performed under multiple turnover conditions, as described previously (18, 23). In brief, the cleavage of substrates was assayed in buffer A at various concentrations of substrates, both above and below the Km for the substrate. The amount of substrates was in large excess to that of the enzymes to assure that saturation with the substrate was achieved in the multiple turnover conditions. Aliquots were withdrawn from reaction mixtures at regular intervals and analyzed in polyacrylamide/urea gels, and the values of Km(app) and Vmax(app) were obtained from Lineweaver-Burk double-reciprocal plots (18, 23).
The procedures to measure the equilibrium dissociation constants (Kd) of complexes of the EGSs and the substrates were modified from Pyle et al. (24). In brief, using a gel shift approach, various concentrations of EGSs were preincubated in buffer B (50 mm Tris, pH 7.5, 100 mm NH4Cl, 10 mm MgCl2, 3% glycerol, 0.1% xylene cyanol, 0.1% bromphenol blue) for 10 min before mixing with an equal volume of different concentrations of substrate RNA preheated under identical conditions. The samples were incubated for 10–30 min to allow binding, then loaded on a 5% polyacrylamide gel, and run at 10 watts. The electrophoresis running buffer contained 100 mm Tris/Hepes, pH 7.5, and 10 mm MgCl2 (24). The value of Kd was then extrapolated from a graph plotting the percent of product bound versus EGS concentration (18). The values were the average of three experiments.
Construction of EGS-expressing Cells and Detection of EGS Expression
The protocols for the construction of U373MG cells expressing different EGSs were modified from Miller and Rosman (25) and have been described previously (26). In brief, amphotropic PA317 cells were transfected with retroviral vector DNAs (LXSN-PR-SER, LXSN-PR-SER-C, LXSN-PR-C125, LXSN-PR-C125-C, and LXSN-TK112). Forty-eight hours post-transfection, culture supernatants that contained retroviral vectors were collected and used to infect U373MG cells. At 48–72 h postinfection, cells were incubated in culture medium that contained 600 μg/ml of neomycin. Cells were subsequently selected in the presence of neomycin for 2 weeks and neomycin-resistant cells were cloned.
For Northern analyses of the expression of the EGSs, both nuclear and cytoplasmic RNA fractions from EGS-expressing cells were isolated as described previously (15, 26). The RNA fractions were separated in a 2.5% agarose gel that contained formaldehyde, transferred to a nitrocellulose membrane, hybridized with the 32P-radiolabeled DNA probes that contained the DNA sequences coding for PR-SER, PR-C125, and H1 RNA, and finally analyzed with a STORM840 PhosphorImager.
Viral Infection and Assays for Viral Gene Expression and Growth
Cells (n = 1 × 106) were either mock-infected or infected with HCMV at a multiplicity of infection (m.o.i.) of 0.5–5 in an inoculum of 1.5 ml of DMEM supplemented with 1% fetal calf serum. The inoculum was replaced with DMEM supplemented with 10% (v/v) fetal bovine serum after 2 h incubation with cells. The infected cells were incubated for 4–72 h before harvesting for isolation of viral mRNA or protein. To measure the levels of viral immediate-early (IE) transcripts, some of cells were also treated with 100 μg/ml of cycloheximide prior to and during infection. RNA samples and protein extracts were prepared from the cells as described previously (15, 26).
For detection of viral mRNAs, the RNA fractions were separated in 1% agarose gels that contained formaldehyde, transferred to a nitrocellulose membrane, hybridized with the 32P-radiolabeled DNA probes that contained the HCMV DNA sequences, and analyzed with a STORM840 PhosphorImager. The DNA probes used to detect human H1 RNA, HCMV immediate-early 5-kb RNA transcript, PR mRNA, IE1 mRNA, and US2 mRNA were synthesized from plasmids pH1, pCig27, pPR, pIE1, and pCig38, respectively.
For Western analyses, the polypeptides from cell lysates were separated on either SDS-7.5% polyacrylamide gels or SDS-9% polyacrylamide gels cross-linked with N,N"-methylenebisacylamide, transferred electrically to nitrocellulose membranes, and reacted to the antibodies against HCMV proteins and human actin. The proteins on the membranes were stained subsequently using a Western chemiluminescent substrate kit and quantitated with a STORM840 PhosphorImager, as described previously (15, 26).
To determine the level of the inhibition of viral growth, cells (n = 1 × 105) were infected with HCMV. The m.o.i. is specified under “Results.” The cells and medium were harvested at 1, 2, 3, 4, 5, 6, and 7 days postinfection and viral stocks were prepared by adding an equal volume of 10% (v/v) skim milk, followed by sonication. The titers of the viral stocks were determined by infecting 1 × 105 human foreskin fibroblasts and counting the number of plaques 10–14 days after infection. The values obtained were averages from triplicate experiments.
Assaying the Level of Intracellular HCMV Genome
Viral DNA was detected by PCR amplification of the viral immediate-early IE1 sequence, using the human β-actin sequence as the internal control. The 5′ and 3′ primers for detecting the IE1 sequence were CMV3 (5′-CCAAGCGGCCTCTGATAACCAAGCC-3′) and CMV4 (5′-CAGCACCATCCTCCTCTTCCTCTGG-3′), respectively (27). The 5′ and 3′ primers used to amplify the actin sequence were Actin5 (5′-TGACGGGGTCACCCACACTGTGCCCATCTA-3′) and Actin3 (5′-CTAGAAGCATTGCGGTGGCAGATGGAGGG-3′), respectively (28). PCR cycles and other conditions were optimized to assure that the amplification was within the linear range. To obtain the PCR DNA template, 5 × 105 cells grown on 6-well plates were mock-infected or infected with HCMV. After a 1.5-h incubation at 37 °C, the inoculum was removed and the cells were further incubated and harvested at 72–96 h postinfection. Total and encapsidated (DNase I-treated) DNAs were isolated essentially as described (29) and used as the PCR DNA templates. The PCR consisted of 20 cycles with denaturation at 94 °C for 1 min, followed by primer annealing at 47 °C for 1 min and extension at 72 °C for 1 min. The last cycle was again an extension at 72 °C for 10 min. The amplified HCMV DNA (481 bp) and actin sequence (610 bp) were separated on 4% nondenaturing polyacrylamide gels.
The PCR were carried out in the presence of [α-32P]dCTP. The radiolabeled DNA samples separated on polyacrylamide gels were scanned with a STORM840 PhosphorImager. We also generated a standard (dilution) curve by amplifying different dilutions of the template DNA. The plot of counts for both HCMV and β-actin versus dilutions of DNA did not reach a plateau for the saturation curve (data not shown) under the conditions described above, indicating that quantitation of viral DNA could be accomplished. The PCR results were derived from three independent experiments.
RESULTS
In Vitro RNase P-mediated Cleavage of the PR mRNA Induced by EGSs
Because most intracellular RNAs are associated with proteins and are present in highly organized and folded conformations, choosing a target region of the PR mRNA that is accessible to binding by the EGS is critical to achieve optimal cleavage. We used an in vivo mapping approach with dimethyl sulfate (30, 31) to determine the accessible regions of the PR mRNA. A position 359 nucleotides downstream from the translational initiation codon was chosen as the cleavage site for human RNase P. This site appeared to be one of the regions most accessible to dimethyl sulfate modification and is likely accessible to EGS binding (data not shown).
We have previously employed an in vitro selection procedure to isolate EGS RNA variants that are more efficient in directing human RNase P for cleavage of the TK mRNA sequence than the EGS derived from a natural tRNA sequence (18). The objective of the study was to generate active EGS variants that can be used to target an mRNA, and to study the variants to understand the mechanism of how EGS RNAs efficiently direct RNase P for cleavage of an mRNA substrate. However, little is currently known about how some of these active EGS variants increase their activity in directing RNase P-mediated cleavage in vitro.
We chose variant C125 for this study because the EGS RNAs derived from this variant are among the most active EGSs in inducing RNase P to cleave the TK as well as the PR mRNA sequences in vitro (see below, Table 1). By covalently linking the EGS domain of C125 to the targeting sequences that are complementary to the PR mRNA, we constructed EGS PR-C125, which resembles part of a tRNA structure and contains a T-loop, a T-stem, and a variable region but not the anticodon region that is dispensable for EGS activity (Fig. 1D) (23). Another EGS, PR-SER, which was derived from the natural tRNASer sequence, was also constructed in a similar way and included in the study (Fig. 1C). EGS RNAs were synthesized in vitro from the DNA sequences coding the EGSs by T7 RNA polymerase and subsequently incubated with human RNase P and substrate pr39, which contained a PR mRNA sequence of 39 nucleotides. RNase P-mediated cleavage of the substrate was observed in the presence of PR-SER and PR-C125 (Fig. 2, lanes 1 and 2). In contrast, no cleavage of pr39 by RNase P was observed in the absence of these EGSs (Fig. 2, lane 4). Thus, PR-SER and PR-C125 efficiently directed human RNase P to cleave the PR mRNA sequence in vitro.
TABLE 1.
Kinetic parameters (Vmax(app), Km(app), and Vmax(app)/Km(app)) in the RNase P cleavage of ptRNASer or pr39 in the presence of different EGSs
Multiple turnover kinetic analyses to determine the values of Vmax(app) and Km(app) were carried out in buffer A (50 mm Tris, pH 7.4, 100 mm NH4Cl, and 10 mm MgCl2) at 37 °C, as described previously (18, 23, 45). To determine the binding affinity (Kd) between substrate pr39 and EGSs, binding assays were carried out in the absence of human RNase P in buffer B (50 mm Tris, pH 7.5, 100 mm NH4Cl, 10 mm MgCl2, 3% glycerol, 0.1% xylene cyanol, 0.1% bromophenol blue), using a protocol modified from Pyle et al. (24). The values shown are the average derived from triplicate experiments.
| Substrate | Km | Vmax(app) | Vmax(app)/Km(app) | Kd |
|---|---|---|---|---|
| μm | pmol min−1 | pmol μm−1min−1 | μm | |
| ptRNASer | 0.015 ± 0.003 | 0.045 ± 0.015 | 3.0 ± 0.6 | |
| PR mRNA(pr39) | ||||
| +PR-SER | 0.55 ± 0.10 | 0.030 ± 0.010 | 0.055 ± 0.020 | 1.3 ± 0.3 |
| +PR-SER-C | NDa | ND | <0.001 | 1.2 ± 0.3 |
| +PR-C125 | 0.40 ± 0.09 | 0.80 ± 0.20 | 2.0 ± 0.4 | 0.030 ± 0.005 |
| +PR-C125-C | ND | ND | <0.001 | 0.032 ± 0.006 |
a ND, not determined.
FIGURE 2.

Cleavage of PR mRNA substrate pr39 by human RNase P in the presence of different EGSs. No EGS was added to the reaction mixture in lane 4. 10 (lanes 2 and 3) and 5 nm EGS (lane 1) were incubated with 32P-labeled pr39 RNA substrate (10 nm) and either 5 (lanes 2–4) or 1 unit (lane 1) of human RNase P at 37 °C in a volume of 10 μl for 45 min in buffer A (50 mm Tris, pH 7.0, 100 mm NH4Cl, 10 mm MgCl2). Experimental details can be found under “Experimental Procedures.”
To further study the activity of the EGS in inducing RNase P to cleave the PR mRNA sequence in vitro, we carried out detailed kinetic analyses for the reactions in the presence of different EGSs, and determined the values of Km(app) and Vmax(app) as well as the overall cleavage efficiency (Vmax(app)/Km(app)) for the cleavage reactions. PR-C125 was highly efficient in directing human RNase P to cleave pr39 and was at least 35-fold more active than PR-SER (Table 1). An increase in the cleavage rate of RNase P may be due to additional tertiary interactions that potentially stabilize the mRNA-EGS complex. If this is the case, the binding affinity of the EGS variant (i.e. PR-C125) to the PR mRNA sequence may be better than that of the EGS (i.e. PR-SER) derived from the natural tRNA sequence. Using gel-shift assays for separating substrate-EGS complexes in polyacrylamide gels under non-denaturing conditions, we measured the dissociation constant (Kd) to determine the binding affinities of EGS PR-C125 and PR-SER to substrate pr39. PR-C125 exhibited about 40 times higher binding affinity to pr39 than PR-SER (Table 1). Because both PR-C125 and PR-SER have the same antisense sequences to pr39 (Fig. 1, C and D), these results suggest that the increased binding affinity and the stability of the substrate-EGS complex in the presence of PR-C125 is probably due to the additional tertiary interactions introduced by this EGS.
Efficient Expression of EGS RNAs in Human Cells
The DNA sequences coding for PR-C125 and PR-SER were subcloned into retroviral vector LXSN and placed under control of the small nuclear U6 RNA promoter (12, 31, 32). This promoter, which has previously been shown to express EGS RNA and other RNAs steadily, is transcribed by RNA polymerase III, and its transcripts are highly expressed and primarily localized in the nucleus (12, 31, 32).
Two additional EGSs, PR-C125-C and PR-SER-C, were also constructed and cloned under the control of the U6 RNA promoter. PR-C125-C and PR-SER-C were derived from PR-C125 and PR-SER, respectively, and contained base substitutions (5′-UUC-3′ → AAG) at the three highly conserved positions in the T-loop of these EGSs (Fig. 1, C and D). Previous studies have shown that these nucleotides were found in most of the known natural tRNA sequences (33, 34) and are believed to be important for the interactions between the tRNA domains and human RNase P (5). EGSs carrying these mutations have been shown to preclude RNase P recognition and exhibit little activity in directing RNase P-mediated cleavage (17, 18, 23, 35). Indeed, cleavage of pr39 by human RNase P in the presence of these two control EGSs was hardly detected (Fig. 2, lanes 3, data not shown) and was at least 2 × 103-fold slower than the cleavage in the presence of PR-C125 (Table 1). PR-C125-C and PR-SER-C contained the same antisense sequence to the PR mRNA sequence as PR-C125 and PR-SER (Fig. 1, C and D), and exhibited similar binding affinities to pr39 as PR-C125 and PR-SER, respectively, when assayed in vitro (Table 1). Therefore, PR-C125-C and PR-SER-C can be used as controls for the antisense effect of these EGSs.
To construct cell lines that express EGS RNAs, amphotropic packaging cells (PA317) were transfected with LXSN-EGS DNAs to produce retroviral vectors that contained the genes for EGS RNAs. Human U373MG cells were then infected with these vectors, and cells expressing the EGSs were cloned. An additional cell line, which expressed EGS TK112 that targeted the HSV-1 TK mRNA (15), was also constructed. No RNase P-mediated cleavage of pr39 in the presence of TK112 was observed in vitro (data not shown). We used this cell line as a control to determine whether EGS RNA with an incorrect guide sequence could target the PR mRNA in tissue culture.
The level of EGS RNA expression in each individual cell clone was determined by Northern analysis, using the expression of human H1 RNA as the internal control. The EGS RNAs were readily expressed in the nuclei as they were detected in the nuclear RNA fractions (Fig. 3, data not shown). This is consistent with previous observations in our laboratory as well as others that the transcripts expressed by the U6 promoter are primarily localized in the nuclei (12, 15, 32). Fig. 3 shows the result from cloned cell lines that expressed PR-SER, PR-C125-C, and PR-C125 (lanes 1–3 and 5–7). Only the cell lines that expressed similar levels of these EGS RNAs were used for further studies in tissue culture. The constructed lines and a control line in which cells were transfected with LXSN vector DNA alone were indistinguishable in terms of their growth and viability for up to 3 months (data not shown), suggesting that the expression of the EGSs did not exhibit significant cytotoxicity.
FIGURE 3.
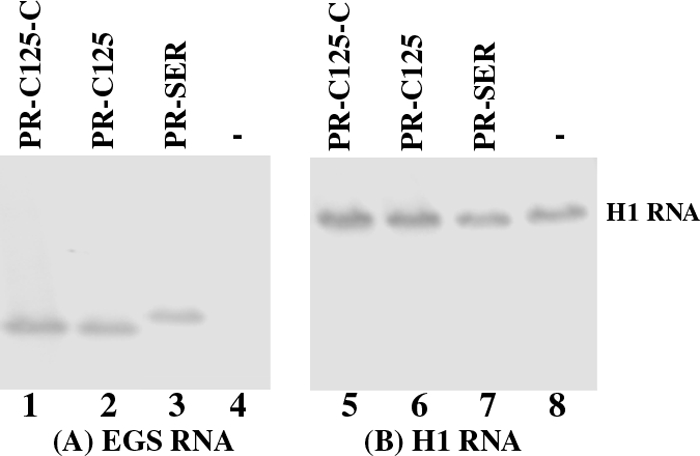
The expression of EGS RNAs in cultured cells. Northern analyses were carried out using nuclear RNA fractions isolated from parental U373MG cells (−, lanes 4 and 8) and a cloned cell line that expressed PR-C125-C (lanes 1 and 5), PR-C125 (lanes 2 and 6), and PR-SER (lanes 3 and 7). Equal amounts of each RNA sample (30 μg) were separated on 2% agarose gels that contained formaldehyde, transferred to nitrocellulose membranes, and hybridized to a 32P-radiolabeled probe that contained the DNA sequence coding for EGS PR-SER/PR-C125 (lanes 1–4) or H1 RNA (lanes 5–8), the RNA subunit of human RNase P and a nuclear RNA (5). The hybridized products corresponding to the full-length retroviral transcripts (∼6 kb), transcribed from the LTR promoter, are at the top of the gel and are not shown.
Inhibition of Viral PR Expression in Cells by EGS-directed RNase P Cleavage
To determine the efficacy of the EGS RNAs in inhibiting the expression of their target PR mRNA, cells were infected with HCMV at a m.o.i. of 0.5–1. Total RNAs were isolated from the infected cells at 8–72 h postinfection. The expression levels of PR mRNA were determined by Northern analyses. The level of the 5-kb long viral immediate-early transcript (5-kb RNA), which expression is not regulated by PR under the assay conditions (19), was used as an internal control for the quantitation of expression of PR mRNA (Fig. 4). In addition, the expression level of human H1 RNA was also used as a control and was similar in each lane in Fig. 4C, suggesting that an equal amount of cellular RNAs was present in each lane of the gel. A reduction of ∼95 and 80% (average of three experiments) in the levels of PR mRNA expression was observed in cells that expressed EGS PR-C125 and PR-SER, respectively (Fig. 4, lanes 8 and 10). In contrast, cells that expressed PR-C125-C and PR-SER-C RNAs exhibited a reduction of less than 10% (Fig. 4, lane 9) (Table 2). We detected no products of the cleavage of the PR mRNA in the Northern analyses, presumably because these RNAs, which lacked either a cap structure or a poly(A) sequence, were rapidly degraded by intracellular RNases. The low level of inhibition found in cells that expressed PR-C125-C and PR-SER-C RNAs was probably due to an antisense effect as PR-C125-C and PR-SER-C, which contained the point mutations at the T-loop (Fig. 1, C and D), exhibited little targeting activity but bound to the PR mRNA sequence as well as PR-C125 and PR-SER (Table 1). These observations suggest that the significant reduction of PR mRNA expression in cells expressing PR-C125 and PR-SER was due to RNase P-mediated cleavage of the target mRNA directed by these EGSs.
FIGURE 4.

Levels of HCMV mRNAs (A and B) and human H1 RNA (C) as determined by Northern analysis. 1 × 106 cells were either mock-infected (lanes 1, 6, and 11) or infected with HCMV (m.o.i. = 1) (lanes 2–5, 7–10, and 12–15) and were harvested at 36 h postinfection. Northern analyses were carried out using RNA isolated from parental U373MG cells (−, lanes 1, 2, 6, 7, and 11, 12) and cell lines that expressed PR-SER (lanes 3, 8, and 13), PR-C125-C (lanes 4, 9, and 14), and PR-C125 (lanes 5, 10, and 15). Equal amounts of each RNA sample (30 μg) were separated on agarose gels that contained formaldehyde, transferred to a nitrocellulose membrane, and hybridized to a 32P-radiolabeled probe that contained the cDNA sequence of the HCMV 5-kb transcript (lanes 1–5), PR mRNA (lanes 6–10), and human H1 RNA (lanes 11–15).
TABLE 2.
Levels of inhibition of viral gene expression in the cells expressing EGS PR-C125, PR-C125-C, PR-SER, PR-SER-C, and TK112, as compared to parental U373MG cells that did not express an EGS (U373MG)
The values are the means derived from triplicate experiments and values for the standard deviation that were less than 5% are not shown.
| Viral gene class | EGS RNA |
||||||
|---|---|---|---|---|---|---|---|
| U373-MG | TK112 | PR-SER-C | PR-C125-C | PR-SER | PR-C125 | ||
| IE1 mRNA | α | 0% | 0% | 1% | 0% | 1% | 0% |
| US2 mRNA | β | 0% | 0% | 0% | 1% | 0% | 2% |
| PR mRNA | γ | 0% | 1% | 7% | 8% | 80 ± 7% | 95 ± 9% |
| IE2 protein | α | 0% | 0% | 0% | 1% | 1% | 2% |
| UL44 protein | β,γ | 0% | 0% | 2% | 1% | 2% | 2% |
| PR protein | γ | 0% | 0% | 7% | 6% | 79 ± 7% | 95 ± 7% |
| gB | γ | 0% | 0% | 0% | 1% | 2% | 0% |
| gH | γ | 0% | 0% | 0% | 1% | 0% | 0% |
The protein levels of PR in EGS-expressing cells are expected to decline due to decreased levels of their mRNAs. Proteins were isolated from cells at 24–72 h postinfection, separated in SDS-polyacrylamide gels, and transferred to identical membranes. One membrane was stained with an anti-PR antibody (Fig. 5B), whereas another membrane was stained with a monoclonal antibody against human actin (Fig. 5A). The latter serves as an internal control for the quantitation of PR protein expression. The results of three independent experiments are summarized in Table 2: a reduction of about 95 and 79% in the level of PR protein was observed in cells that expressed PR-C125 and PR-SER, respectively. In contrast, a reduction of less than 10% was found in cells that expressed PR-C125-C, PR-SER-C, or TK112 RNAs.
FIGURE 5.

Levels of human actin and HCMV proteins as determined by Western blot analysis. Protein samples were isolated from the parental U373MG cells or EGS-expressing cells that were either mock-infected (lanes 5 and 10) or infected with HCMV (m.o.i. = 0.5–1) (lanes 1–4 and 6–9) for 48 h, separated in SDS-polyacrylamide gels, and then transferred to membranes. One membrane was allowed to react with a monoclonal antibody (anti-actin) against human actin (A), whereas the other was stained with the monoclonal antibody (anti-PR) against HCMV PR (B).
Inhibition of HCMV Growth by EGS-directed RNase P Cleavage of PR mRNA
To determine whether viral growth was inhibited in the EGS-expressing cells, cells were infected with HCMV at a m.o.i. of 0.5–2. We prepared virus stocks from the infected cultures (cells and culture medium together) at 1-day intervals through 7 days postinfection and determined the number of plaque forming units by measuring the viral titer on human foreskin fibroblasts. After 5 days postinfection, a reduction of about 4,000 and 150-fold in viral yield was observed in cells that expressed PR-C125 and PR-SER, respectively (Fig. 6). No significant reduction was found in those that expressed the control EGSs PR-C125-C, PR-SER-C, or TK112 (Fig. 6, data not shown).
FIGURE 6.

Growth of HCMV in U373MG cells and cell lines expressing EGS RNAs. 5 × 105 cells were infected with HCMV at m.o.i. of 2. Virus stocks were prepared from the infected cells at 1-day intervals through 7 days postinfection and the plaque forming unit count was determined by measurement of the viral titer on human foreskin fibroblasts. These values are derived from the means of triplicate experiments. The standard deviation is indicated by the error bars.
Specific Inhibition of HCMV Capsid Formation but Not the Expression of Other Viral Genes or Viral Genome Replication in the EGS-expressing Cells
HCMV protease plays an essential role in DNA encapsidation during capsid assembly and is required for viral growth (19–22). Meanwhile, it is possible that the observed reduction of viral growth may be due to other effects of the EGS on viral lytic replication and may not necessarily be due to the specific RNase P cleavage of PR mRNA. To determine the antiviral mechanism resulting from the EGS-directed cleavage, we carried out a series of experiments to examine the effects of inhibition of viral PR expression on several key steps of the HCMV lytic replication cycle. In the first set of experiments, we investigated whether the expression of other viral genes is affected in EGS-expressing cells. Inhibition of PR expression is not expected to affect the expression of other viral genes, including α, β, and γ genes, which are not regulated by the protease (19). Northern analyses were carried out to determine the expression levels of the IE1 mRNA (an immediate early (α) transcript) and the US2 mRNA (a early (β) transcript). Moreover, Western analyses were performed to determine the expression level of viral protein UL44, a viral late (βγ) protein and gH, a viral late (γ) protein. No significant reduction in the expression levels of these genes was observed in cells that expressed PR-C125, PR-125-C, PR-SER, PR-SER-C, and TK112 RNA (Table 2). Similar results were also found in the expression of HCMV IE2 (an immediate early (α) gene) and gB (a late (γ) gene) (Table 2). These results suggest that expression of the EGS RNAs specifically inhibits the expression of PR and does not affect overall viral gene expression.
The second set of experiments was carried out to investigate whether EGS-directed inhibition of PR expression affects viral genomic DNA replication as well as viral DNA encapsidation. Total DNA was isolated from lysates of the mock-infected or HCMV-infected cells that were either treated with DNase I or not. The encapsidated viral DNAs would be resistant to DNase I digestion, whereas those that are not packaged in the capsid would be susceptible. The level of intracellular viral genomic DNA was determined by PCR detection of the sequence of viral immediate-early gene IE1 (Fig. 7). The level of human β-actin DNA was used as the internal control for the quantitation of HCMV DNA. When assaying the DNA samples from cell lysates that were not treated with DNase I, no significant difference in the level of intracellular HCMV DNA was observed in cells that do not express an EGS or express PR-C125, PR-SER, PR-C125-C, PR-SER-C, or TK112 (Fig. 7, lanes 1–4). These results suggest that the total levels (both encapsidated and unencapsidated DNAs) of intracellular viral DNAs are similar among these cells and that replication of the viral genome is not affected by the EGS-mediated reduction of PR expression. However, when the DNase I-treated samples were assayed, the levels of “encapsidated” DNA in cells expressing functional EGS PR-C125 and PR-SER were significantly less than those in cells expressing no EGS RNAs or control EGSs PR-C125-C, PR-SER-C, or TK112 (Fig. 7, lanes 5–8). Indeed, the encapsidated DNA was hardly detected in cells that expressed PR-C125 compared with the PR-SER-expressing cells (Fig. 7, compare lane 7 with lane 8). These observations suggest that EGS-directed inhibition of PR expression has no effect on HCMV genome replication but blocks viral capsid formation. Moreover, these results indicate that PR-C125, which was derived from a highly active EGS variant, was more effective in blocking encapsidation of the HCMV genome than PR-SER, an EGS derived from a natural tRNA sequence.
FIGURE 7.
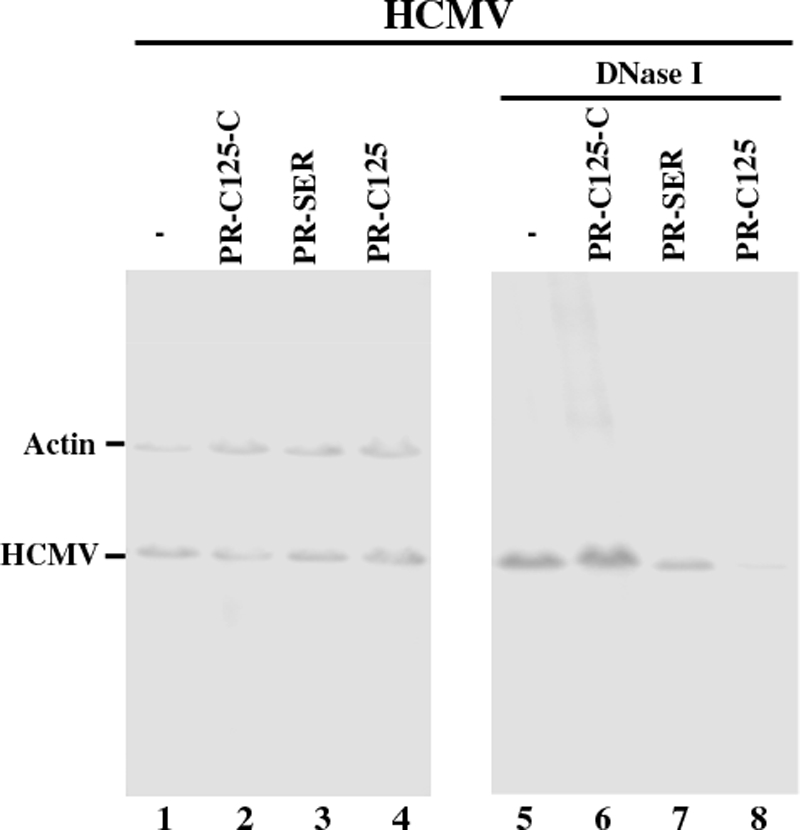
Level of total intracellular (left) and encapsidated (right) viral DNA as determined by semi-quantitative PCR. Total DNA (lanes 1–4) or DNase I-treated DNA samples (lanes 5–8) were isolated from cells that either did not express an EGS (−, lanes 1 and 5) or express EGS PR-C125-C (lanes 2 and 6), PR-SER (lanes 3 and 7), or PR-C125 (lanes 4 and 8). Cells were infected with HCMV at m.o.i. of 1. The levels of viral IE1 sequence was determined by PCR using human actin DNA sequence as the internal controls.
DISCUSSION
The EGS-based technology represents an attractive approach for gene inactivation because it utilizes endogenous RNase P to generate highly efficient and specific cleavage of the target RNA (5, 6). Thus, EGS molecules represent promising general gene-targeting agents that can be used in both basic research and clinical applications. To develop the EGS technology for practical gene-targeting applications, further studies are needed to study how to improve the efficacy of the EGSs in inhibiting gene expression.
Studies have suggested that substrate binding represents one of the most important steps affecting the efficacy of small ribozymes in mammalian cells (1). Furthermore, Fedor and colleagues (36) have shown that substrate binding is the rate-determining step in the intracellular cleavage pathway of the hairpin ribozyme in yeast. However, little is known about the rate-limiting step of EGS targeting in cultured cells. Equally unclear is whether the efficacy of the EGSs can be improved, and if so, how it can be improved. In the present study, EGS RNAs were constructed to target an accessible region of the HCMV PR mRNA. Moreover, EGSs were expressed by the small nuclear U6 RNA promoter. This design would increase the probability for the EGS RNAs to bind to their target mRNA sequence and co-localize with human RNase P, which is exclusively localized in the nuclei (5). Under the described settings, we hypothesized that the efficacy of EGS technology in cultured cells is dictated by the catalytic efficiency (Vmax/Km) of RNase P-mediated cleavage directed by the EGS. If this is the case, increasing the activity of EGS in directing RNase P-mediated cleavage may lead to more effective inhibition of the target mRNA expression in vivo.
Our results indicated that an EGS variant, PR-C125, is about 35 times more active (Vmax(app)/Km(app)) in directing RNase P to cleave the PR mRNA sequence in vitro than PR-SER, an EGS derived from the natural tRNASer sequence. Moreover, PR-C125 inhibited PR expression in cultured cells by more than 95% and was more effective than PR-SER, which reduced PR expression by about 80%. A reduction of about 4,000-fold in viral growth was observed in the PR-C125-expressing cells, whereas a reduction of about 150-fold was observed in cells expressing PR-SER. In contrast, a reduction of less than 10% in the PR expression level and viral growth was observed in cells that expressed PR-C125-C, PR-SER-C, or TK112. PR-C125-C and PR-SER-C exhibited similar binding affinity to pr39 as PR-C125 and PR-SER, respectively, but were inactive in directing RNase P-mediated cleavage due to the presence of the mutations at the T-loop that precluded RNase P recognition (Figs. 1 and 2, Table 1). Our results suggest that the observed reduction in viral gene expression and inhibition of viral growth with PR-C125 and PR-SER is primarily attributed to the specific targeted RNase P-mediated cleavage induced by these two EGSs as opposed to the antisense effect or other nonspecific effects of the EGSs. Moreover, the EGS (i.e. PR-C125) that is more active (Vmax(app)/Km(app)) in inducing RNase P to cleave the PR mRNA sequence in vitro is also more effective in inhibiting HCMV gene expression and growth in cultured cells. These results support our hypothesis that increasing the activity of EGS in directing RNase P cleavage in vitro may lead to improved efficacy in inhibiting gene expression in cultured cells. The difference between the in vivo efficacies of PR-C125 and PR-SER (e.g. 95% versus 80%) appeared to be more limited than that of the in vitro cleavage efficiencies (∼35-fold difference). One of the possible explanations is that about 5% of the target mRNA may not be accessible to EGS binding or RNase P cleavage, possibly due to its specific nuclear localization and its rapid transport to the cytoplasm. Another possibility is that the efficacy of the EGS-based approach, in contrast to its activity in vitro, may be influenced by the intrinsic intracellular decay of the target RNA, because hairpin ribozymes effectively eliminate mRNAs in cells only when cleavage is much faster than normal degradation (37, 38). Moreover, proper folding of the mRNA-EGS complexes in cells may also affect the efficacy of the EGS approach. Further studies will be needed to address these issues.
The targeting activity of EGSs appears to be highly specific. First, expression of the EGSs did not exhibit significant cytotoxicity as cells expressing EGSs are indistinguishable from the parental cells in terms of cell growth and viability for up to 3 months (data not shown). Second, the antiviral effect (inhibition of viral growth and encapsidation of viral genomic DNA) associated with the expression of PR-C125 and PR-SER appears to be due to a reduction in the expression of PR. This is because the level of DNA encapsidation, viral growth, and the expression of PR were found to be significantly reduced in cells that expressed PR-C125 and PR-SER but not in those that expressed control PR-C125-C, PR-SER-C, or TK112 (Figs. 4–7, Table 2). The extent of the observed inhibition of DNA encapsidation and viral growth correlates with that of the inhibition of the PR expression. Third, expression of the EGS only inhibits the expression of the PR mRNA and protein. We found no reduction in the expression levels of other viral genes examined (e.g. IE1, IE2, US2, UL44, gB, and gH) in the EGS-expressing cells (Figs. 4 and 5, Table 2). Moreover, the expression of EGSs and the reduction of PR expression do not appear to affect the replication of viral genomic DNA (Fig. 7). These results are consistent with previous observations that the protease is required for viral capsid maturation and does not play a role in regulating viral gene expression and genome replication (19–22). Thus, the EGS-based technology is highly specific in inhibiting the expression of its target mRNA.
Delivery of the EGS into the nuclear compartment is essential to the success of EGS technology because RNase P is exclusively localized in the nuclei (5, 6). The expression cassette used to produce the EGSs is the promoter for small nuclear U6 RNA (12, 15, 32). This promoter has been used extensively to express functional RNAs and ribozymes for gene targeting applications and the transcript from this promoter is quite stable and primarily localized in the nuclei (12, 15, 32). In addition, the efficient delivery and proper localization of the EGS may be mediated by cellular tRNA-binding proteins. Viral and non-viral vectors can be used to deliver the EGS expression cassette sequence to specific cells and tissues in vivo (39–41). Further studies on EGS delivery will facilitate the development of EGS-based technology for both basic research and clinical therapeutic applications.
In vitro selection has been widely used to generate highly active ribozymes and functional RNA molecules with increased activity (42–44). For example, Yuan and Altman (23) have used this procedure to generate active EGS molecules that direct human RNase P to cleave the mRNA encoding chloramphenicol acetyltransferase in vitro. However, whether these selected EGSs exhibit higher efficacies in targeting chloramphenicol acetyltransferase mRNA in tissue culture has not been extensively studied. Using an EGS RNA variant selected from a pool of EGS molecules containing randomized sequences, we, in this study, provide direct evidence that an EGS selected in vitro with increased targeting activity also exhibited improved efficacy in inhibiting HCMV gene expression and growth in cultured cells. Our results suggest that the enhanced stability of the mRNA-EGS complexes may possibly contribute to the increased targeting activity of EGS PR-C125. PR-C125 bound to substrate pr39 at least 40-fold greater than PR-SER (Table 1). Previous studies on tRNA molecules indicated that tertiary interactions between the variable region and D-loop are important for maintaining the tRNA conformation and RNase P cleavage (5, 7). Given the fact that, in the pr39-PR-C125 complex, the 3′ region of pr39 can be considered equivalent to the D-loop in a tRNA (Fig. 1, A and D), it is conceivable that the additional interactions between pr39 and PR-C125 stabilize the mRNA-EGS complex and result in an enhanced binding affinity and increased targeting activity of the EGS. Thus, our study may provide a direction for the engineering and generation of highly active and effective EGS molecules by carrying out selection procedures and manipulation of the EGS domain to interact with the mRNA substrates. Further investigation of engineered EGSs should provide insights into the mechanism of how an EGS RNA efficiently directs RNase P to cleave an mRNA and facilitate the development of novel and effective EGSs for gene targeting applications.
Acknowledgments
We thank Tianhong Zhou, Phong Trang, Jiaming Zhu, Naresh Sunkara, and Ed Yang for critical comments and technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants AI041927 and DE014842.
- RNase P
- ribonuclease P
- EGS
- external guide sequence
- HCMV
- human cytomegalovirus
- TK
- thymidine kinase
- IE
- immediate-early.
REFERENCES
- 1. Scherer L. J., Rossi J. J. (2003) Nat. Biotechnol. 21, 1457–1465 [DOI] [PubMed] [Google Scholar]
- 2. Santoro S. W., Joyce G. F. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 4262–4266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Castanotto D., Rossi J. J. (2009) Nature 457, 426–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fedor M. J., Williamson J. R. (2005) Nat. Rev. Mol. Cell. Biol. 6, 399–412 [DOI] [PubMed] [Google Scholar]
- 5. Gopalan V., Altman S. (2006) in The RNA World (Gesteland R., Cech T., Atkins J. eds) Vol. 277, Chapter 6.1 (online only at rna.cshl.edu), Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 6. Liu F. (2010) in Ribonuclease P (Liu F., Altman S. eds) pp. 257–276, Springer, New York [Google Scholar]
- 7. Kazantsev A. V., Pace N. R. (2006) Nat. Rev. Microbiol. 4, 729–740 [DOI] [PubMed] [Google Scholar]
- 8. Guerrier-Takada C., Gardiner K., Marsh T., Pace N., Altman S. (1983) Cell 35, 849–857 [DOI] [PubMed] [Google Scholar]
- 9. Gopalan V., Vioque A., Altman S. (2002) J. Biol. Chem. 277, 6759–6762 [DOI] [PubMed] [Google Scholar]
- 10. Marvin M. C., Engelke D. R. (2009) J. Cell. Biochem. 108, 1244–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Forster A. C., Altman S. (1990) Science 249, 783–786 [DOI] [PubMed] [Google Scholar]
- 12. Yuan Y., Hwang E. S., Altman S. (1992) Proc. Natl. Acad. Sci. U.S.A. 89, 8006–8010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guerrier-Takada C., Li Y., Altman S. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 11115–11119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Plehn-Dujowich D., Altman S. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 7327–7332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kawa D., Wang J., Yuan Y., Liu F. (1998) RNA 4, 1397–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kraus G., Geffin R., Spruill G., Young A. K., Seivright R., Cardona D., Burzawa J., Hnatyszyn H. J. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 3406–3411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhu J., Trang P., Kim K., Zhou T., Deng H., Liu F. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 9073–9078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou T., Kim J., Kilani A. F., Kim K., Dunn W., Jo S., Nepomuceno E., Liu F. (2002) J. Biol. Chem. 277, 30112–30120 [DOI] [PubMed] [Google Scholar]
- 19. Mocarski E. S., Shenk T., Pass R. F. (2007) in Fields Virology (Knipe D. M., Howley P. M., Griffin D. E., Martin M. A., Lamb R. A., Roizman B., Straus S. E. eds) pp. 2701–2772, Lippincott-Williams and Wilkins, Philadelphia, PA [Google Scholar]
- 20. Welch A. R., Woods A. S., McNally L. M., Cotter R. J., Gibson W. (1991) Proc. Natl. Acad. Sci. U.S.A. 88, 10792–10796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dunn W., Chou C., Li H., Hai R., Patterson D., Stolc V., Zhu H., Liu F. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 14223–14228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yu X., Trang P., Shah S., Atanasov I., Kim Y. H., Bai Y., Zhou Z. H., Liu F. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 7103–7108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yuan Y., Altman S. (1994) Science 263, 1269–1273 [DOI] [PubMed] [Google Scholar]
- 24. Pyle A. M., McSwiggen J. A., Cech T. R. (1990) Proc. Natl. Acad. Sci. U.S.A. 87, 8187–8191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Miller A. D., Rosman G. J. (1989) BioTechniques 7, 980–990 [PMC free article] [PubMed] [Google Scholar]
- 26. Trang P., Lee M., Nepomuceno E., Kim J., Zhu H., Liu F. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 5812–5817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang J., Jiang H., Liu F. (2000) RNA 6, 571–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Daftarian P. M., Kumar A., Kryworuchko M., Diaz-Mitoma F. (1996) J. Immunol. 157, 12–20 [PubMed] [Google Scholar]
- 29. Matusick-Kumar L., Hurlburt W., Weinheimer S. P., Newcomb W. W., Brown J. C., Gao M. (1994) J. Virol. 68, 5384–5394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zaug A. J., Cech T. R. (1995) RNA 1, 363–374 [PMC free article] [PubMed] [Google Scholar]
- 31. Liu F., Altman S. (1995) Genes Dev. 9, 471–480 [DOI] [PubMed] [Google Scholar]
- 32. Bertrand E., Castanotto D., Zhou C., Carbonnelle C., Lee N. S., Good P., Chatterjee S., Grange T., Pictet R., Kohn D., Engelke D., Rossi J. J. (1997) RNA 3, 75–88 [PMC free article] [PubMed] [Google Scholar]
- 33. Jühling F., Mörl M., Hartmann R. K., Sprinzl M., Stadler P. F., Pütz J. (2009) Nucleic Acids Res. 37, D159–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sprinzl M., Vassilenko K. S. (2005) Nucleic Acids Res. 33, D139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li H., Trang P., Kim K., Zhou T., Umamoto S., Liu F. (2006) RNA 12, 63–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yadava R. S., Mahen E. M., Fedor M. J. (2004) RNA 10, 863–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Donahue C. P., Fedor M. J. (1997) RNA 3, 961–973 [PMC free article] [PubMed] [Google Scholar]
- 38. Fedor M. J. (2000) J. Mol. Biol. 297, 269–291 [DOI] [PubMed] [Google Scholar]
- 39. Bai Y., Li H., Vu G. P., Gong H., Umamoto S., Zhou T., Lu S., Liu F. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 7269–7274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Robbins P. D., Ghivizzani S. C. (1998) Pharmacol. Ther. 80, 35–47 [PubMed] [Google Scholar]
- 41. Vassaux G., Nitcheu J., Jezzard S., Lemoine N. R. (2006) J. Pathol. 208, 290–298 [DOI] [PubMed] [Google Scholar]
- 42. Gold L., Polisky B., Uhlenbeck O., Yarus M. (1995) Annu. Rev. Biochem. 64, 763–797 [DOI] [PubMed] [Google Scholar]
- 43. Szostak J. W. (1992) Trends Biochem. Sci. 17, 89–93 [DOI] [PubMed] [Google Scholar]
- 44. Joyce G. F. (1992) Sci. Am. 267, 90–97 [DOI] [PubMed] [Google Scholar]
- 45. Liu F., Altman S. (1994) Cell 77, 1093–1100 [DOI] [PubMed] [Google Scholar]