

Abstract
Objectives:
Peroxisome assembly disorders are genetic disorders characterized by biochemical abnormalities, including low docosahexaenoic acid (DHA). The objective was to assess whether treatment with DHA supplementation would improve biochemical abnormalities, visual function, and growth in affected individuals.
Methods:
This was a randomized, double-blind, placebo-controlled trial conducted at a single center. Treatment groups received supplements of DHA (100 mg/kg per day). The primary outcome measures were the change from baseline in the visual function and physical growth during the 1 year follow-up period.
Results:
Fifty individuals were enrolled and randomized. Two were subsequently excluded from study analysis when it was determined that they had a single enzyme disorder of peroxisomal β oxidation. Thirty-four returned for follow-up. Nine patients died during the trial of their disorder, and 5 others were lost to follow-up. DHA supplementation was well tolerated. There was no difference in the outcomes between the treated and untreated groups in biochemical function, electroretinogram, or growth. Improvements were seen in both groups in certain individuals.
Conclusions:
DHA supplementation did not improve the visual function or growth of treated individuals with peroxisome assembly disorders.
Classification of evidence:
This interventional study provides Class II evidence that DHA supplementation did not improve the visual function or growth of treated individuals with peroxisome assembly disorders during an average of 1 year of follow-up in patients aged 1 to 144 months.
GLOSSARY
- AA
= arachidonic acid;
- DHA
= docosahexaenoic acid;
- ERG
= electroretinogram;
- NALD
= neonatal adrenoleukodystrophy;
- RBC
= red blood cell;
- VLCFA
= very long chain fatty acid;
- ZS
= Zellweger syndrome;
- ZSD
= Zellweger spectrum disorder.

LOE Classification
Peroxisome assembly disorders are a genetically heterogeneous group of disorders characterized by the disruption of peroxisomal protein importation.1 The majority of these disorders result from an abnormality of PTS1 targeting and importation and are referred to as Zellweger spectrum disorder (ZSD). This disorder is seen in 1 per 50,000 births.2 Clinically, individuals with ZSD have hypotonia, development delay, retinal degeneration, sensorineural hearing loss, and other organ system involvement. These are serious disorders, and many affected individuals die in infancy or childhood, although survival to adulthood is documented.3
While genetically heterogeneous, the involvement of PTS1-mediated protein importation results in a characteristic set of biochemical abnormalities. The list of involved pathways is extensive but includes plasmalogen synthesis, long chain fatty acid degradation, and the synthesis of docosahexaenoic acid (DHA).4 DHA is a polyunsaturated fatty acid that has been determined to be important in brain and retinal development.5 Individuals with ZSD have been determined to be deficient in DHA,6 but the exact role of this deficiency in the pathogenesis of the disorder is not known.
Preliminary studies7,8 demonstrated the ability to supplement DHA orally and increase the levels in the blood stream. Whereas other investigators8 have reported improvement in the clinical status, we have noted that despite adequate supplementation in all patients studied in an open trial, the variation and severity of preexistent deficits made the experience difficult to interpret.
Our aim in this study was to assess the efficacy of DHA in improving visual function and physical growth through a double-blind, placebo-controlled, randomized trial. We also investigated the relation between DHA supplementation and biochemical peroxisomal measures, including plasma very long chain fatty acid (VLCFA) and red blood cell (RBC) plasmalogen levels.
METHODS
Participants.
Eligible participants were children aged 1 month to 10 years and diagnosed with Zellweger syndrome (ZS), neonatal adrenoleukodystrophy (NALD), and infantile Refsum disease by characteristic biochemical profile, which includes increased VLCFAs, increased plasma phytanic acid, increased plasma pipecolic acid, decreased RBC plasmalogen, and decreased plasma DHA levels.
All participants were admitted to the Pediatric Clinical Research Unit of the Johns Hopkins Hospital, Baltimore, Maryland.
Objectives.
Our primary hypothesis was that DHA supplementation improves visual function and physical growth (Class II evidence) in children affected with peroxisome assembly disorders. Our secondary hypothesis was DHA supplementation normalizes biochemical peroxisomal measures, including plasma VLCFA and RBC plasmalogen levels (Class II evidence).
Standard protocol approvals, registrations, and patient consents.
The study protocol was approved by the institutional review board, and individual written informed consents were obtained from patients' families.
Study design and interventions.
This was a randomized, double-blind, placebo-controlled trial conducted at a single center. Participants were randomly assigned to 2 interventions in a 1:1 ratio using a random number generator. Randomization was performed by the investigational pharmacy of Johns Hopkins Hospital. The treatment group received supplements of DHA and arachidonic acid (AA). Assigned treatments were mailed to patients' homes through the investigational pharmacy at Johns Hopkins Hospital; this pharmacy was the only unmasked entity. Neither the investigators nor the patients were aware of the treatment assignment during the trial period and until the evaluations were completed and analyzed.
It had been previously determined that in otherwise healthy premature infants, supplementation with DHA alone could result in decreased AA levels and decreased growth. We have shown that DHA supplementation with DHA ethyl ester did decrease AA levels significantly in children with peroxisomal disorders (data not shown), so we decided to supplement DHA and AA. This combination is presently used in infant formula supplementation.
The prepared treatment was a microencapsulated powder that contained DHA triglyceride (47% DHA) and AA triglyceride (46% AA). Placebo was similarly prepared soybean oil. Doses of these study medications were 100 mg/kg/d, which were based on previous open study experience.9 This study medication could be mixed with food or infant formula. Both were similar in appearance and comparable in taste and smell. Treatment compliance was monitored by plasma DHA levels. Participants at the time of enrollment were instructed in a low-phytanic diet, mainly restricting whole milk consumption. The registered dietician was in regular contact with families to confirm adequate intake of nutrients. This study was undertaken and completed before the availability of commercial formulas that now routinely contain DHA with AA. Study agents were provided by Martek Biosciences Corporation (Columbia, MD).
Outcome.
Evaluations included neurologic, biochemical, ophthalmologic, growth, and monitoring of hematologic and blood chemistry. Plasma VLCFA, and DHA and plasmalogen levels in RBCs were evaluated at baseline and 3, 6, 9, and 12 months. The remainders of the evaluations (described below) were performed at baseline and 1-year follow-up. The standard ratio of 18:0 dimethylacetals/18:0 in erythrocytes was used as a measure of plasmalogen. Serum VLCFA, DHA and erythrocyte plasmalogen assays were performed in the Peroxisome Diagnostic Laboratories of Kennedy Krieger Institution using standard methodologies.10–12
Visual function was evaluated using electroretinogram (ERG). At baseline, ERGs were graded as a) extinguished, b) very low (<10 μV), c) low (<50 μV), or d) moderate (>50 μV). At follow-up, patients' results were compared with their baseline examination data and were categorized as worse, no change, or improved.
Physical growth was evaluated by following the Z scores of weight and height over the course of the trial.
The primary outcome of the trial was change from baseline in visual function and physical growth during the 1-year follow-up period.
All of our patients received neurologic and neurodevelopmental evaluations as part of the study, but the decision was made to restrict analysis to quantitative outcomes. There was no unusual developmental event on subjective evaluations in any of the participants.
The pretrial anticipated rate of improvement in the visual function was 60% for the treatment group8 and 20% for the control group.2 Power calculations were based on the natural history data at Kennedy Krieger Institute. A sample of 22 subjects per group was required for 80% power and a level of significance of 0.05. Sample size was further inflated by 10% to address missing data or loss to follow-up. Power calculation was also performed for change in weight and height Z scores, before and after treatment with DHA. It was estimated that 25 patients in each group would provide more than 90% power to detect 1 SD change in Z scores of weight and height.
Statistical methods.
Baseline characteristics were compared by randomized groups using t tests for continuous-level variables and χ2 tests for proportions. Primary analyses were performed on 48 randomized participants based on intent-to-treat principle. The χ2 test was used to compare ERG outcomes. Repeated-measures analysis of variance was used to evaluate the difference in aspartate aminotransferase (AST), alanine aminotransferase (ALT), total bilirubin, and change of weight and height from baseline to 1-year follow-up. The criterion for statistical significance was set at ≤0.05.
All analyses were performed using STATA version 8 (STATA Corp LP, College Station, TX).
RESULTS
Because peroxisomal disorders are rare, we informed all centers treating these patients about this study. A total of 51 patients were screened, and 50 of these were enrolled between February 1, 1997, and April 30, 2001 (period of 4 years 3 months). Twenty-five were randomly assigned to receive DHA, and 25 were randomly assigned to receive placebo. Two patients were excluded from study analysis when their diagnosis changed after complete biochemical analysis, which was performed during the baseline visit (both individuals had single enzyme disorders affecting peroxisomal β oxidation). All 48 patients received at least 1 dose of study medication and were included in efficacy and safety analyses. The baseline characteristics of the 2 groups were similar, as shown in table 1. Three patients in the DHA group and 2 patients in the placebo group (10% of the enrolled participants) missed their follow-up visit and could not be contacted. Nine patients died during the course of the trial secondary to the natural progression of the disease. Eight of these 9 patients were diagnosed with ZS, and 1 was diagnosed with NALD. The figure depicts the enrollment and progress of patients throughout the trial.
Table 1 Baseline demographic and clinical characteristics of each group
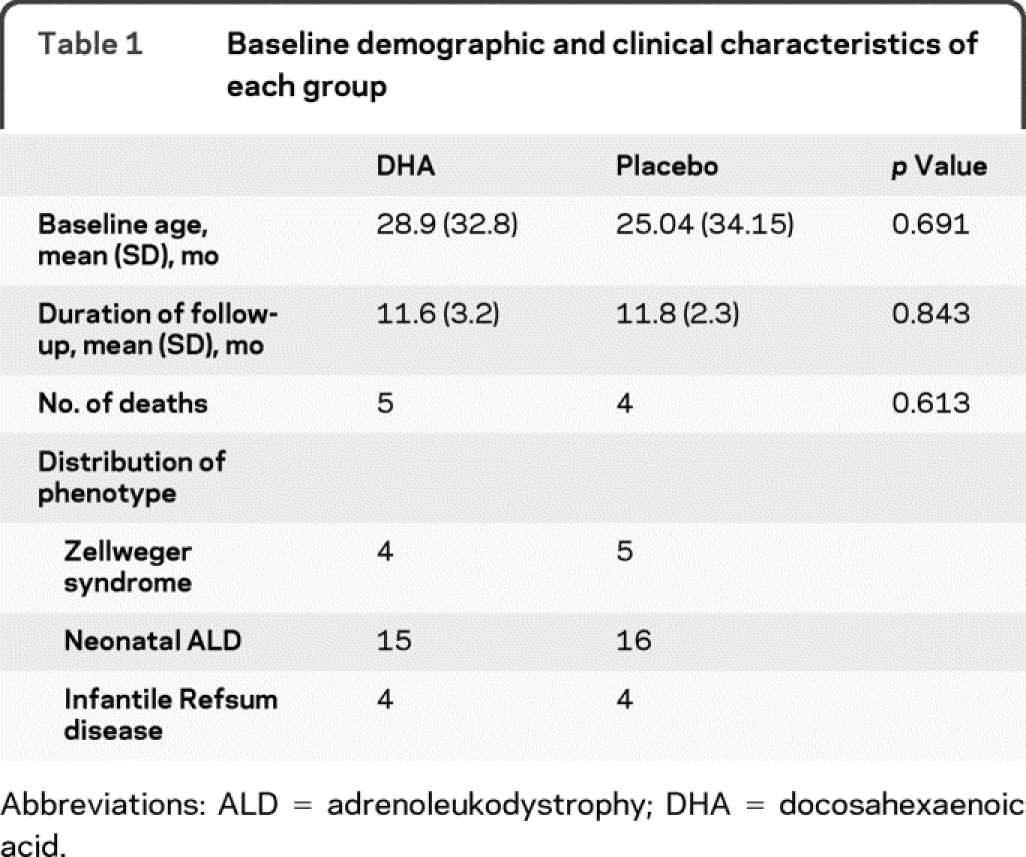

Figure Participant flow through the trial
DHA = docosahexaenoic acid.
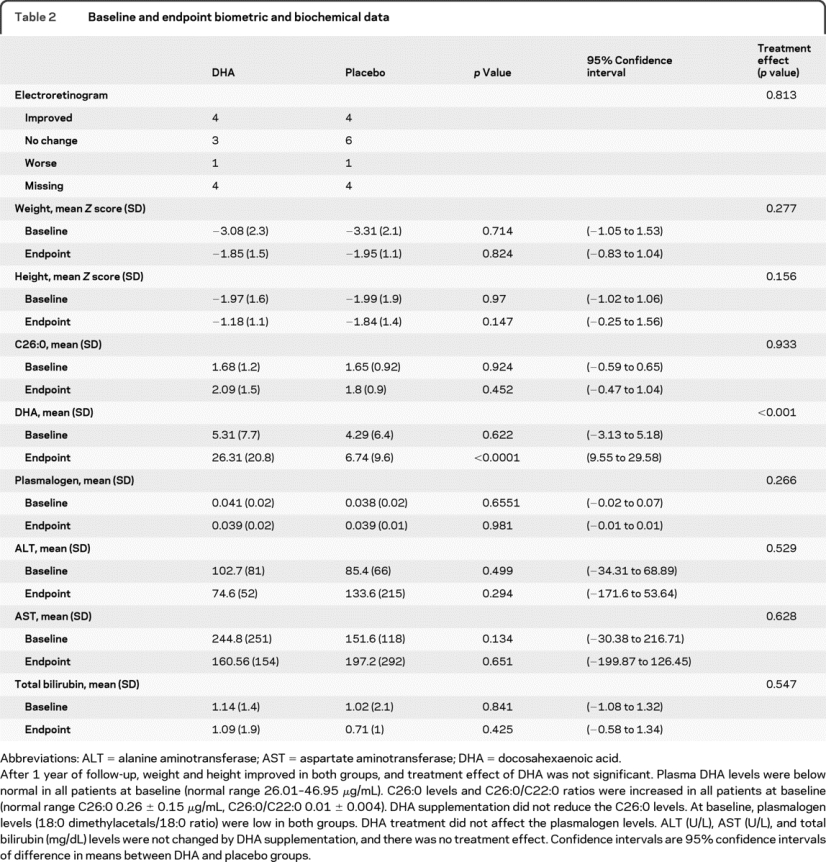
The frequency of patients with improved ERG was similar in 2 groups, as shown in table 2. The treatment effect of DHA supplementation on other outcomes is also shown in table 2. DHA was well tolerated, and there was no serious adverse event reported that was conclusively related to interventions.
Table 2 Baseline and endpoint biometric and biochemical data
DISCUSSION
In this placebo-controlled, randomized trial among children with peroxisome assembly disorders, we found no overall effects of oral DHA supplementation on visual function, growth, or biochemical measures. We noted that oral supplementation increases DHA blood levels without any adverse effects. Surprisingly, visual function deteriorated in only 1 patient of each group who was able to return for follow-up; the rest either improved or stayed the same. Similarly, weight and height improved over time regardless of intervention. DHA supplementation had no significant impact on VLCFA or plasmalogen levels, which were characteristic peroxisomal biochemical abnormalities in our patients.
In previous open studies,7,8 it had been stated that DHA supplementation was associated with improvements in vision and growth of these children. In a similar fashion, we also noticed improvement in vision, weight, and height in both groups, but these effects were not related to DHA supplementation because they were present also in the untreated group. Additionally, unexplained biochemical improvements were also reported. Most likely these changes in VLCFAs and plasmalogens may reflect mild variations with age and survival. In light of these findings of maturational effects in untreated individuals, other alleged benefits, including changes in myelin, are suspect without a comparison group.13
This study, in comparison with previous open trials, highlights the limitations of presuming to know the natural history of a rare disorder. It is apparent that the children who did not die during the study period maintained stable features and in some instances improved.
The limits of our study are that there was no uniformity in the genetic etiology of the affected children. It is possible that certain mild genotypes of particular PEX genes may receive more benefit from postnatal supplementation with DHA, but given the rarity of these conditions, such a limited study is not feasible. Similarly, the restricted sample size and variability in age of participants may have missed a small treatment effect, but prior open case reports did not limit their claims of efficacy, which were not shown to be valid here.
Unfortunately, therapies for peroxisomal disorders remain limited. Peroxisome assembly disorders involve multiple systems and have their onset during fetal development. Many of the pathologic features are set in motion at this developmental stage, and any therapy instituted after birth is unlikely to offset this. Although supplementation with DHA is without significant adverse effect in this population, its therapeutic use cannot be endorsed at this time.
AUTHOR CONTRIBUTIONS
Statistical analyses were performed by Asif M. Paker, MD, MPH.
ACKNOWLEDGMENT
The authors thank Dr. Irene Maumenee and the fellows, residents, and staff who assisted in the evaluations. The authors also are indebted to Polly Green, Willie Foreman, and Dr. Hugo Moser (all deceased), who were instrumental in the design and carrying out of this project.
DISCLOSURE
Dr. Paker reports no disclosures. Dr. Sunness serves on scientific advisory boards for Potentia Pharmaceuticals and Acucela Inc.; has received funding for travel from Acucela Inc., Merck Serono, Jerini AG, and Genentech, Inc.; has received publishing royalties from Ryan Retina Online (low vision course); serves as a consultant for SYTERA, LLC, Eyetec GmbH, Alcon, Merck Serono, Johnson & Johnson, Othera Pharmaceuticals, Inc., Acucela Inc., Jerini AG, Taligen Therapeutics, Genentech, Inc., Pfizer Inc, Potentia Pharmaceuticals, Avnet, Inc., Health Advances, LLC, Advanced Vision Therapies, Inc., Ophthotech Corporation, Shire plc, and Neurotech; has received research support from the NIH (R03 EY14148 [PI]); and holds stock options in Potentia Pharmaceuticals. Ms. Brereton is a research nutritionist of the Institute for Clinical and Translational Research, which is funded by the NIH. Dr. Speedie, Dr. Albanna, Dr. Dharmaraj, Ms. Moser, and Dr. Jones report no disclosures. Dr. Raymond received a speaker honorarium from Garrod Association of Canada; receives research support from the NIH (NICHD R01 HD39276 [PI], R01 FD003030 [PI], and NICHD R01 HD057136 [PI]), the European Leukodystrophy Association, and the Myelin Project; and has served as a consultant for the Department of Health and Human Services in the Division of Vaccine Injury compensation.
Address correspondence and reprint requests to Dr. Gerald V. Raymond, Department of Neurogenetics, Kennedy Krieger Institute, 707 N. Broadway, Baltimore, MD 21205 raymond@kennedykrieger.org
Study funding: Supported by the Food and Drug FD-R-001289 (G.V.) and the Johns Hopkins University School of Medicine General Clinical Research Center M01 RR00052 from the National Center for Research Resources. Supply of agent and placebo were provided by Martek Biosciences Corp, Columbia, Maryland.
Disclosure: Author disclosures are provided at the end of the article.
Received April 3, 2009. Accepted in final form May 13, 2010.
REFERENCES
- 1.Goldfischer S, Moore CL, Johnson AB, et al. Peroxisomal and mitochondrial defects in the cerebro-hepato-renal syndrome. Science 1973;182:62–64. [DOI] [PubMed] [Google Scholar]
- 2.Gould S, Raymond G, Valle D. The peroxisome biogenesis disorders. In: Scriver C, Beaudet A, Sly W, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw Hill; 2001:3181–3217. [Google Scholar]
- 3.Wilson GN, Holmes RG, Custer J, et al. Zellweger syndrome: diagnostic assays, syndrome delineation, and potential therapy. Am J Med Genet 1986;24:69–82. [DOI] [PubMed] [Google Scholar]
- 4.Wanders RJ, Tager JM. Lipid metabolism in peroxisomes in relation to human disease. Mol Aspects Med 1998;19:69–154. [DOI] [PubMed] [Google Scholar]
- 5.Neuringer M, Connor WE, Lin DS, Barstad L, Luck S. Biochemical and functional effects of prenatal and postnatal omega 3 fatty acid deficiency on retina and brain in rhesus monkeys. Proc Natl Acad Sci USA 1986;83:4021–4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinez M. Abnormal profiles of polyunsaturated fatty acids in the brain, liver, kidney and retina of patients with peroxisomal disorders. Brain Res 1992;583:171–182. [DOI] [PubMed] [Google Scholar]
- 7.Martinez M, Pineda M, Vidal R, Conill J, Martin B. Docosahexaenoic acid: a new therapeutic approach to peroxisomal-disorder patients—experience with two cases. Neurology 1993;43:1389–1397. [DOI] [PubMed] [Google Scholar]
- 8.Martinez M. Restoring the DHA levels in the brains of Zellweger patients. J Mol Neurosci 2001;16:309–316. [DOI] [PubMed] [Google Scholar]
- 9.Raymond GV, Moser A, Moser H. Docosahexaenoic acid therapy in peroxisome biogenesis disorders. Ann Neurol 1997;42(suppl):P41. Abstract.
- 10.Moser AB, Kreiter N, Bezman L, et al. Plasma very long chain fatty acids in 3,000 peroxisome disease patients and 29,000 controls. Ann Neurol 1999;45:100–110. [DOI] [PubMed] [Google Scholar]
- 11.Moser AE, Singh I, Brown FR III, et al. The cerebrohepatorenal (Zellweger) syndrome: increased levels and impaired degradation of very-long-chain fatty acids and their use in prenatal diagnosis. N Engl J Med 1984;310:1141–1146. [DOI] [PubMed] [Google Scholar]
- 12.Moser AB, Jones DS, Raymond GV, Moser HW. Plasma and red blood cell fatty acids in peroxisomal disorders. Neurochem Res 1999;24:187–197. [DOI] [PubMed] [Google Scholar]
- 13.Martinez M, Vazquez E. MRI evidence that docosahexaenoic acid ethyl ester improves myelination in generalized peroxisomal disorders. Neurology 1998;51:26–32. [DOI] [PubMed] [Google Scholar]