

Abstract
Retroviral Gag proteins contain short late-domain motifs that recruit cellular ESCRT pathway proteins to facilitate virus budding. ALIX-binding late domains often contain the core consensus sequence YPXnL (where Xn can vary in sequence and length). However, some simian immunodeficiency virus (SIV) Gag proteins lack this consensus sequence, yet still bind ALIX. We mapped divergent, ALIX-binding late domains within the p6Gag proteins of SIVmac239 (40SREKPYKEVTEDLLHLNSLF59) and SIVagmTan-1 (24AAGAYDPARKLLEQYAKK41). Crystal structures revealed that anchoring tyrosines (in lightface) and nearby hydrophobic residues (underlined) contact the ALIX V domain, revealing how lentiviruses employ a diverse family of late-domain sequences to bind ALIX and promote virus budding.
Many enveloped viruses, including retroviruses, recruit proteins of the cellular ESCRT pathway to facilitate budding (reviewed in references 3, 11, and 35). Short sequence motifs, termed late domains, within retroviral Gag polyproteins bind directly to early-acting ESCRT factors, which then recruit and activate the downstream machinery necessary for membrane fission. The three well-characterized late domains are typically denoted by their canonical core amino acid sequences: PTAP late domains bind the ubiquitin E2 variant (UEV) domain of the TSG101 subunit of the ESCRT-I complex, PPXY late domains bind WW domains of NEDD4 family ubiquitin E3 ligases, and YPXnL (where Xn can vary in sequence and length) late domains bind the V domain of ALIX (10, 20, 32, 37). In a number of cases, retroviral Gag proteins have been shown to utilize multiple late domains (e.g., see references 3, 4, 7, 11, 16, 31, 33, 34, and 35). We speculate that this phenomenon may be even more prevalent than is currently appreciated because mutations in auxiliary late domains often produce weak or cell-specific phenotypes and because late domains can be difficult to recognize owing to primary sequence divergence. It is therefore of interest to define the range of different sequences that can function as late domains and to learn how sequence variation is tolerated while late-domain function is retained.
Strack and colleagues initially reported that ALIX binds core sequences of 35LYPLTSL41 and 22LYPDL26 within the late domains of human immunodeficiency virus type 1 (HIV-1) p6Gag and equine infectious anemia virus (EIAV) p9Gag, respectively (32) (anchoring tyrosines are shown in boldface, and nearby hydrophobic residues that contact ALIX are underlined). They also reported that p6Gag proteins from simian immunodeficiency virus SIVmac239 and SIVagmTan-1 can bind and package ALIX into virions, but in those cases the ALIX-binding sites were not fully mapped and were not obvious, because the SIV p6Gag proteins lacked canonical YPXnL ALIX-binding elements. We therefore performed biosensor binding experiments and deletion analyses to quantify and map the ALIX-binding sites. These experiments employed a recombinant ALIX protein that spanned the Bro1 and V domains (residues 1 to 698), here denoted ALIXBro1-V, but lacked the C-terminal proline-rich region (residues 699 to 868). As shown in Fig. 1, ALIXBro1-V bound directly to the full-length p6Gag proteins from SIVmac239 (equilibrium dissociation constant [KD], 66 ± 4 μM) and SIVagmTan-1 (KD, 24 ± 1 μM), with binding affinities that were comparable to those of HIV-1 p6Gag and EIAV p9Gag (KD, 40 and 1.5 μM, respectively) (37).
FIG. 1.
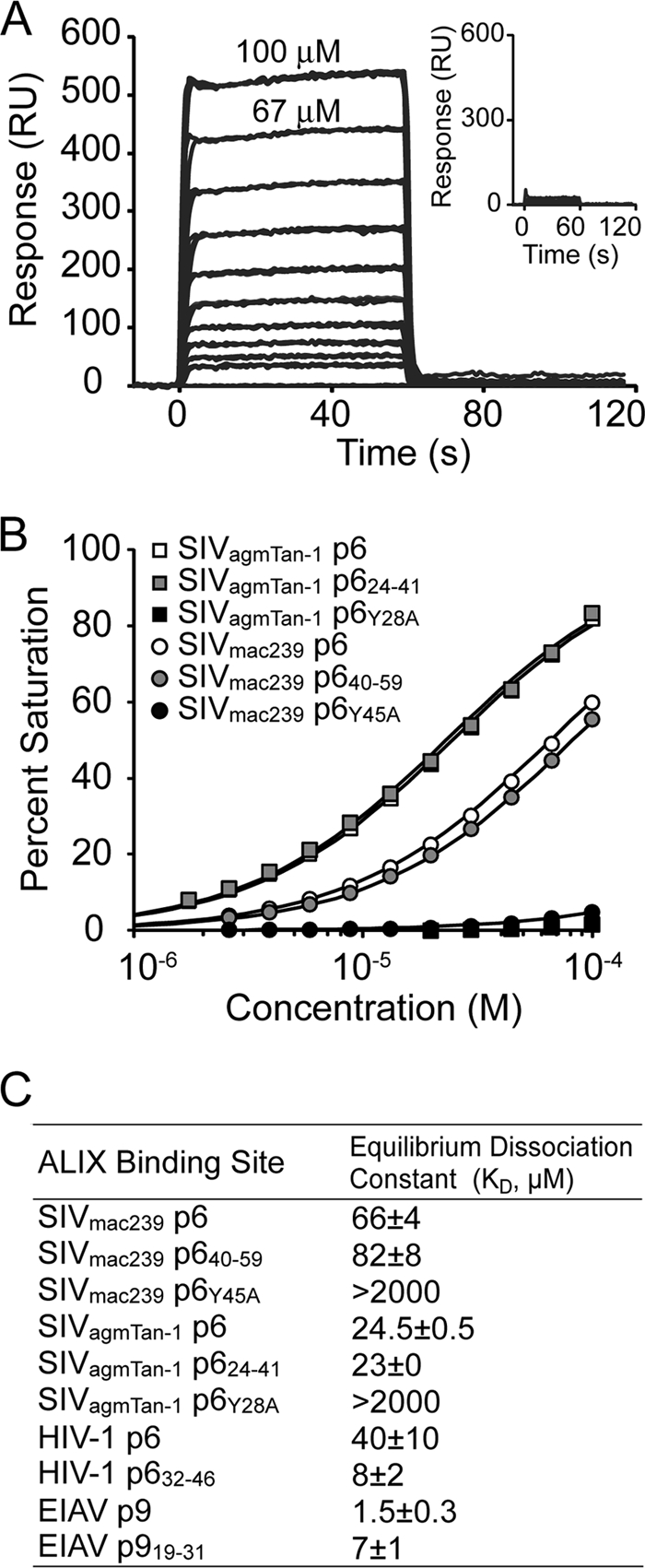
ALIXBro1-V binding to different p6Gag proteins. (A) Representative biosensor sensograms for ALIXBro1-V binding to immobilized wild-type (main graph) or Y45A mutant (inset) SIVmac239 glutathione S-transferase (GST)-p6Gag proteins. Samples were analyzed in triplicate. p6Gag proteins, minimal binding peptides (blocked N and C termini), were created, and the binding studies were performed and analyzed as described previously (37). RU, response units. (B) Representative biosensor binding isotherms for ALIXBro1-V binding to SIVmac239 GST-p6Gag or SIVagmTan-1 p6Gag proteins. (C) Summary of ALIXBro1-V dissociation constants for different retroviral p6/p9 proteins/peptides. Values for SIV peptides are the means from two independent experiments (each measured in triplicate) ± ranges. Dissociation constants for the HIV-1 and EIAV peptides were reported previously (37) but are reprinted here for reference.
Deletion experiments were performed to map the ALIX-binding sites to the following sequences: SIVmac239 p6Gag, 40SREKPYKEVTEDLLHLNSLF59; and SIVagmTan-1 p6Gag, 24AAGAYDPARKLLEQYAKK41 (Fig. 1 and data not shown). In both cases, ALIX bound the full-length SIV p6Gag proteins and the minimal binding sites with comparable affinities, indicating that ALIX binding was not significantly influenced by p6Gag residues beyond the immediate binding site. The late domains of HIV-1 p6Gag and EIAV p9Gag both contain key tyrosine residues that bind in a deep pocket on the second arm of the ALIX V domain (37). The ALIX-binding sites within SIVmac239 and SIVagmTan-1 p6Gag also contained single tyrosine residues (highlighted in boldface), and alanine point mutations in each of these tyrosines eliminated any detectable ALIX binding to the full-length SIV p6Gag proteins (Fig. 1). Thus, these tyrosines are also key determinants of ALIX binding to the SIV p6Gag proteins.
To learn how these SIV p6Gag proteins recognize and re- cruit ALIX, we crystallized and determined the structures of ALIXBro1-V (KK268,269YY mutant) in complex with binding-site peptides from the SIVagmTan-1 and SIVmac239 p6Gag proteins. Crystallization and data collection were performed as previously described (37). Crystallographic statistics are provided in Table 1. A comparison of the two SIV peptide complex structures with the previously reported HIV and EIAV late-domain complexes reveals that the ALIX protein structure is essentially invariant. In all cases, the dominant interaction is insertion of a tyrosine side chain of the p6 peptide into a deep hydrophobic pocket on ALIX (Fig. 2). The tyrosines of all four late-domain peptides superimpose closely and make the same contacts with ALIX, including a hydrogen bond between the tyrosine phenoxyl and the conserved ALIX Asp506 side chain. As described previously, the EIAV and HIV late-domain interfaces also bury a proline immediately following the tyrosine (the Y + 1 position) and a leucine at Y + 3 (EIAV) or Y + 5 (HIV), with the different leucine positions being accommodated by different conformations of the peptide backbone, either extended (EIAV, designated a type 1 ALIX-binding motif) or helical (HIV, designated type 2) from the Y + 2 position.
TABLE 1.
Crystallographic statistics for ALIX complexesa
| Parameterb | Value for indicated strainc |
|
|---|---|---|
| SIVmac239 | SIVagmTan-1 | |
| Space group | C2 | C2 |
| Cell dimensions | ||
| a (Å) | 145.3 | 145.5 |
| b (Å) | 99.3 | 99.1 |
| c (Å) | 72.5 | 72.6 |
| β (°) | 106.9 | 106.6 |
| Resolution (Å) | 45-2.3 (2.38-2.3) | 45-2.5 (2.59-2.5) |
| Completeness (%) | 97.7 (82.6) | 95.6 (70.8) |
| I/σ(I) | 18.2 (3.9) | 31.5 (3.5) |
| Rsym (%)c | 9.6 (28.2) | 5.5 (32.0) |
| No. of unique reflections | 44,063 | 34,197 |
| Rfactor/Rfree (%)d | 20.5/25.2 | 20.4/26.1 |
| No. of protein atoms | 5,614 | 5,559 |
| No. of water molecules | 58 | 22 |
| Avg B factor (Å2) | ||
| Protein atoms | 77.6 | 86.5 |
| Water molecules | 58 | 63.6 |
| RMSD from ideal geometry | ||
| Bonds (Å) | 0.007 | 0.008 |
| Angles (°) | 0.994 | 1.057 |
Data were collected at beam line X29 of the National Synchrotron Light Source and processed using HKL2000 (23). The structures were determined using rigid-body refinement with the unliganded ALIXBro1-V model and were refined in PHENIX (1) with TLS refinement (24, 25). Model building was performed with O (17) and COOT (9).
Rsym = (|(∑I − 〈I〉)|)/(∑I), where 〈I〉 is the average intensity of multiple measurements. Rfactor = ∑hkl||Fobs(hkl)|| − Fcalc(hkl)||/∑hkl|Fobs(hkl)|. Rfree = the cross-validation R factor for the 5% of reflections against which the model was not refined.
Values in parentheses are for the highest-resolution shell.
FIG. 2.

Structures of SIV late domains bound to the second V-domain arm of human ALIXBro1-V. (A) SIVmac239 p6Gag late-domain peptide (magenta sticks) and human ALIXBro1-V protein, represented as a blue ribbon and surface, with the side chains of binding-site residues shown explicitly and labeled. The p6 tyrosine is indicated with a Y. The p6 peptide is oriented with the N terminus at the bottom of the figure. (B) SIVagmTam-1 p6Gag late-domain peptide and ALIXBro1-V complex. (C) Overlay of the ALIX in complex with late-domain peptides of HIV-1 p6Gag (Protein Data Bank code 2R02; green), EIAV p9Gag (Protein Data Bank code 2R03; turquoise), SIVmac239 p6Gag (magenta), and SIVagmTan-1 p6Gag (salmon). Panels A to C were generated using PyMOL (6). (D) Late-domain peptide sequences used for crystallography and binding studies, aligned on the basis of the crystal structures. Residues modeled in the crystal structures are in boldface, and those lacking electron density are in italics. Residues that are structurally equivalent in all four complexes are highlighted by a gray background. Residues that bury more than 50% of their solvent-accessible surface at the protein interface are underlined. Note that ALIX binds three different types of viral sequence motifs, which we have designated types 1 to 3.
The two SIV peptides form equivalent ALIX interfaces but do so by adopting yet another conformation (termed a type 3 ALIX-binding motif). In both cases, they are helical from the Y + 1 residue, which results in the Y + 3 Val/Ala and Y + 7 Leu occupying the same locations as the Pro and Leu of EIAV and HIV (Fig. 2). The most notable difference between the two SIV peptides is that their helices project at an angle of 15° with respect to each other. This presumably results from differences in residues that contact ALIX, especially Val versus Ala at position Y + 3, and results in a 2.5-Å displacement of the SIVagmTan-1 Y + 7 Leu compared to the structurally equivalent Leu of SIVmac239, HIV, and EIAV. Thus, late-domain sequences adopt a range of conformations in order to preserve the interaction motif: ΦYX0/2ΦX1/3L, with the alternative 0/2 and 1/3 spacings of the intervening X residues accommodated by extended versus helical backbone conformations.
Sequence analyses revealed that nearly all primate lentiviruses carry one of the three different types of ALIX-binding motifs, supporting the idea that type 3 ALIX-binding motifs can function as late domains that enhance virus budding (Fig. 3A). SIVmac239 p6Gag has been analyzed by deletion analysis (26), but unfortunately the functional importance of the key tyrosine residue in the ALIX-binding site was not tested. We therefore used a SIVmac251-based system (21), for which a vector was available, to test whether noncanonical ALIX-binding sites within SIV p6Gag proteins can function as late domains. SIVmac251 and SIVmac239 are closely related and have identical p6Gag ALIX-binding sites. Both isolates also contain PTAP elements within p6Gag that presumably bind TSG101 and function as late domains.
FIG. 3.

Primary sequences and functional analyses of the late domains within primate lentiviral p6Gag proteins. (A) Maximum likelihood phylogenetic tree showing the different primate lentiviral lineages and their p6Gag proteins (drawn to scale as white boxes). TSG101-binding P(S/T)AP late domains are shown in blue, and putative ALIX-binding late domains are shown in red (predicted ALIX contact residues) or black (solvent exposed residues). ALIX-binding-site types are designated at right (see text for explanation). Sequences that most closely match the crystallographically characterized ALIX complexes are denoted with stars. Consensus sequences for the designated lineage(s) were derived from reference 19, and residues conserved at >85% identity are shown in capital letters. p6Gag proteins from the HIV-1/SIVcpz lineage fall into three different classes that either have type 1 or 2 ALIX-binding sites or lack apparent ALIX-binding sites (and also lack tyrosines). Putative ALIX-binding sites within SIVwrc proteins can have either a Trp or Tyr residue in the second position, and the effect of the Trp substitution on ALIX binding has not been tested. The scale bar represents 0.1 substitution per site, and the tree was adapted from reference 1a. (B) Mutations in the SIVmac251 p6Gag 11PTAP14 (ΔPTAP) and 38EKPYKEVTEDLLHL51 (ΔYL) sequences inhibited virion release (Western blot, panel 1) and reduced viral titers (bottom graph, single-cycle infectivity assays), and ALIX overexpression stimulated release of the SIVmac251 ΔPTAP construct. Cells were transfected with the designated SIVmac251 vectors (WISP10-480-484), and cotransfected with either an empty pCI-neo vector control (lanes 1 to 4) or vectors expressing either wild-type (WT) FLAG-ALIX (lanes 5 to 8) or the designated mutant (lanes 9 and 10) FLAG-ALIX proteins. Vector transduction titers shown in the graph were measured in single-cycle infectivity assays (n = 5 assays; values are shown plus standard deviations). Western blots showing the levels of cell-associated CA and ALIX are also shown (CELL), with endogenous ALIX expression levels (lanes 1 to 4) enhanced 20-fold relative to exogenous ALIX expression levels (lanes 5 to 10) for ease of visualization.
Constructs were designed to mutate key residues in both candidate late domains within the SIVmac251 p6Gag proteins encoded by the pSIV3+ helper vector without altering the underlying Pol reading frame (11PTAP14 to 11LIAL14, termed ΔPTAP; and 38EKPYKEVTEDLLHL51 to 38EKPSKEVTEDSLHL51, termed ΔYL; mutated residues are italicized). Virions were produced in 293T cells and analyzed as described previously (30), with Western blotting used to detect virion-associated and cellular CA levels (anti-SIVmac CA mouse monoclonal antibody, 1:3,000 [14]) and cellular ALIX levels (anti-ALIX rabbit polyclonal antibody, 1:5,000 [10]). Viral titers were measured using flow cytometry to detect green fluorescent protein (GFP) expression from the packaged pSIV-gaMES4sin vector in transduced 293T cells.
As expected, the ΔPTAP mutation inhibited SIVmac251 release, as measured by reductions in virion-associated CA (p27) protein levels (Fig. 3B, VIRION Western blot, compare lanes 1 and 2), without altering cellular CA expression levels (CELL blot). Vector titers were also dramatically reduced, essentially to background levels [from 1.06 (±0.06) × 106/ml to 6 (±2) × 103/ml; 180-fold reduction] (Fig. 3B, bottom panel, compare lanes 1 and 2). SIVmac251 release and infectivity were also reduced by the ΔYL mutation in the ALIX-binding site, although the reduction was much less dramatic than for the ΔPTAP mutation (Fig. 3B, compare lanes 1 and 3; 3-fold infectivity reduction). The ΔPTAP/ΔYL double mutation inhibited virus release to an even greater extent than either single mutation alone (Fig. 3B, compare lanes 2 and 4), with viral titers again near background levels. Mutations in either (or both) late domains led to accumulation of the CA-SP1 processing intermediate within cells (Fig. 3B, CELL blot, compare lane 1 to lanes 2 to 4). This phenotype is also seen for HIV-1 late-domain mutants and is indicative of budding defects (13). Thus, both the 11PTAP14 and 38EKPYKEVTEDLLHL51 sequences within SIVmac251 p6Gag promote Gag processing, virion release, and viral infectivity, and the PTAP sequence serves as the dominant late domain under these experimental conditions. The situation is similar for HIV-1; both the ALIX- and TSG101-binding late domains are functional, but mutations in the PTAP element are more detrimental in most cell types (12).
To confirm that the SIVmac251 p6Gag 38EKPYKEVTEDLLHL51late domain was ALIX responsive, we tested whether ALIX overexpression stimulated virus release via this sequence (10, 33). ALIX overexpression did not alter the release and infectivity of wild-type SIVmac251, presumably because the 11PTAP14 late domain was already highly active (Fig. 3B, compare lanes 1 and 5). In contrast, ALIX overexpression substantially stimulated the release and infectivity of an SIVmac251 ΔPTAP construct [to 1.4 (±0.2) × 105; 23-fold infectivity increase] (Fig. 3B, compare lanes 2 and 6). This stimulation was dependent upon the ALIX-binding site within SIVmac251 p6Gag, because ALIX overexpression failed to stimulate either the ΔYL or the ΔPTAP/ΔYL mutant constructs significantly (compare lanes 3 to 7 and 4 to 8). Stimulation also required the YPXnL-binding site of ALIX, because an inactivating point mutation within this site (F676D) blocked the ability of ALIX to stimulate release of the ΔPTAP construct (compare lanes 6 and 9). Thus, the 38EKPYKEVTEDLLHL51 site within SIVmac251 p6Gag functions as an ALIX-dependent late domain.
As shown in Fig. 3A, p6Gag proteins from nearly every known primate lentiviral lineage contain a type 1, type 2, or type 3 ALIX-binding site, implying that (i) the ability to bind ALIX must provide primate lentiviruses with a strong selective advantage and (ii) these three types probably account for all (or nearly all) of the different ALIX-binding modes. The only exceptions are a subset of viruses within the HIV-1/SIVcpz lineage, which lack identifiable ALIX-binding sites, and possibly also a subset of SIVwrc viruses whose type 1 ALIX-binding sites have Trp in place of Tyr. Type 3 ALIX-binding sites are widespread throughout primate lentiviruses, and type 1 and 3 sites are more common than type 2 sites (which predominate only in HIV-1 strains). Interestingly, p6Gag proteins that lack the ability to bind TSG101 typically have type 1 ALIX-binding sites (2). Type 1 sites bind ALIX with relatively high affinities, at least in the cases examined to date (Fig. 1C), and this correlation may therefore reflect a need to recruit ALIX more efficiently when TSG101 cannot be recruited directly.
Although ALIX binds rather weakly to most isolated late domains, several factors likely enhance ALIX recruitment in vivo. First, p6-ALIX V domain interactions of the type studied here can be augmented by upstream interactions between the ALIX Bro1 domain and the HIV-1 Gag NC domain (8, 28, 29). Analogous SIV NC-ALIX Bro1 interactions are also possible, although such interactions alone are apparently not sufficient to stimulate virus release because ALIX overexpression does not substantially rescue SIVmac251 release or infectivity in the absence of the p6Gag ALIX-binding site. Second, activated ALIX is dimeric (27), which should enhance binding avidity to oligomeric Gag assemblies. Third, ALIX can associate with ubiquitin, and ubiquitylation of Gag (or associated proteins) could therefore enhance ALIX recruitment (18). Finally, both Gag and ALIX can associate with membranes, which may increase the effective local ALIX concentrations at budding sites. Thus, relatively weak ALIX-p6Gag interactions of the type described here are apparently sufficient to ensure that ALIX is recruited to function in virus budding.
Once recruited, ALIX can stimulate virus budding by recruiting the downstream ESCRT-III membrane fission machinery via direct interactions with CHMP4 subunits (10, 33) and also via additional stimulatory activities of the N-terminal Bro1 domain that may involve membrane deformation (28). CHMP4 recruitment appears to be important in the case of SIVmac251, because the ALIXI212D mutant, which cannot bind CHMP4 (22), also failed to stimulate release of the ΔPTAP construct (Fig. 3B, compare lanes 6 and 10). Thus, the ALIX-binding site in SIVmac251 p6Gag functions, at least in part, to provide access to the membrane fission activity of the downstream ESCRT-III proteins.
Our results also have important implications for the identification of cellular ALIX-binding partners. The YPXnL-binding site within the ALIX V domain presumably evolved to bind cellular partners, rather than viral late domains. To date, however, only one cellular interaction of this type has been identified: that between the Aspergillus ALIX homolog PalA and its binding partner PacC (36). PalA binds tandem YPXL/I motifs within PacC that match canonical EIAV late domains, and this interaction facilitates the pH-regulated cleavage of the PacC transcription factor. Such pH-sensing pathways are not conserved outside of fungi, however, suggesting that additional ALIX-binding partners have yet to be identified. Our studies show that ALIX can bind a broader range of sequences than previously appreciated, requiring only an anchoring tyrosine interaction and downstream hydrophobic residues that can vary in both identity and spacing. Cellular ALIX-binding partners (and possibly also other viruses) can presumably also employ this very loose consensus motif, which may help explain why the mammalian ALIX-binding partner(s) has thus far escaped detection.
Protein structure accession numbers.
Coordinates and diffraction data for ALIXBro1-V (KK268,269YY mutant) in complex with the SIVmac239 and SIVagmTan-1 peptides have been deposited in the Protein Data Bank (PDB codes 2XS1 and 2XS8, respectively).
Acknowledgments
We thank Niels Pedersem for supplying the SIVmac p27 monoclonal antibody (55-2F12) through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH, Jean-Luc Darlix for the gift of the SIVmac251 vector system, David Myszka and Rebecca Rich at the University of Utah HSC Protein Interactions Core Research Facility for assistance with the biosensor binding experiments, and Martine Peeters, Vanessa Hirsch, and Beatrice Hahn for helpful direction on primate lentivirus lineages.
Operations of the National Synchrotron Light Source (NSLS) are supported by the Office of Basic Energy Sciences at the U.S. Department of Energy and by the NIH. Data collection at the NSLS was funded by the National Center for Research Resources. This work was supported by NIH grants AI051174 (to W.I.S.) and P50 082545 (W.I.S. and C.P.H.).
Footnotes
Published ahead of print on 20 October 2010.
REFERENCES
- 1.Adams, P. D., P. V. Afonine, G. Bunkóezi, V. B. Chen, I. W. Davis, N. Echols, J. J. Headd, L.-W. Hung, G. J. Kapral, R. W. Grosse-Kunstleve, A. J. McCoy, N. W. Moriarty, R. Oeffner, R. J. Read, D. C. Richardson, J. S. Richardson, T. C. Terwilliger, and P. H. Zwart. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66:213-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1a.Ahuka-Mundeke, S., F. Liegeois, A. Ayouba, Y. Foupouopouognini, E. Nerriennet, E. Delaporte, and M. Peeters. 25 August 2010. Full-length genome sequence of a simian immunodeficiency virus infecting a captive agile mangabey (Cercocebus agilis) is closely related to SIVrcm infecting wild red-capped mangabeys (Cercocebus torquatus) in Cameroon. J. Gen. Virol. [Epub ahead of print.] [DOI] [PMC free article] [PubMed]
- 2.Bibollet-Ruche, F., E. Bailes, F. Gao, X. Pourrut, K. L. Barlow, J. P. Clewley, J. M. Mwenda, D. K. Langat, G. K. Chege, H. M. McClure, E. Mpoudi-Ngole, E. Delaporte, M. Peeters, G. M. Shaw, P. M. Sharp, and B. H. Hahn. 2004. New simian immunodeficiency virus infecting De Brazza's monkeys (Cercopithecus neglectus): evidence for a cercopithecus monkey virus clade. J. Virol. 78:7748-7762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bieniasz, P. D. 2009. The cell biology of HIV-1 virion genesis. Cell Host Microbe 5:550-558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chung, H. Y., E. Morita, U. von Schwedler, B. Muller, H. G. Krausslich, and W. I. Sundquist. 2008. NEDD4L overexpression rescues the release and infectivity of human immunodeficiency virus type 1 constructs lacking PTAP and YPXL late domains. J. Virol. 82:4884-4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reference deleted.
- 6.DeLano, W. L. 2008. The PyMOL molecular graphics system. DeLano Scientific LLC, Palo Alto, CA.
- 7.Dilley, K. A., D. Gregory, M. C. Johnson, and V. M. Vogt. 2010. An LYPSL late domain in the gag protein contributes to the efficient release and replication of Rous sarcoma virus. J. Virol. 84:6276-6287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dussupt, V., M. P. Javid, G. Abou-Jaoude, J. A. Jadwin, J. de La Cruz, K. Nagashima, and F. Bouamr. 2009. The nucleocapsid region of HIV-1 Gag cooperates with the PTAP and LYPXnL late domains to recruit the cellular machinery necessary for viral budding. PLoS Pathog. 5:e1000339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Emsley, P., and K. Cowtan. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126-2132. [DOI] [PubMed] [Google Scholar]
- 10.Fisher, R. D., H. Y. Chung, Q. Zhai, H. Robinson, W. I. Sundquist, and C. P. Hill. 2007. Structural and biochemical studies of ALIX/AIP1 and its role in retrovirus budding. Cell 128:841-852. [DOI] [PubMed] [Google Scholar]
- 11.Fujii, K., J. H. Hurley, and E. O. Freed. 2007. Beyond Tsg101: the role of Alix in ‘ESCRTing’ HIV-1. Nat. Rev. Microbiol. 5:912-916. [DOI] [PubMed] [Google Scholar]
- 12.Fujii, K., U. M. Munshi, S. D. Ablan, D. G. Demirov, F. Soheilian, K. Nagashima, A. G. Stephen, R. J. Fisher, and E. O. Freed. 2009. Functional role of Alix in HIV-1 replication. Virology 391:284-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Göttlinger, H. G., T. Dorfman, J. G. Sodroski, and W. A. Haseltine. 1991. Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc. Natl. Acad. Sci. U. S. A. 88:3195-3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Higgins, J. R., S. Sutjipto, P. A. Marx, and N. C. Pedersen. 1992. Shared antigenic epitopes of the major core proteins of human and simian immunodeficiency virus isolates. J. Med. Primatol. 21:265-269. [PubMed] [Google Scholar]
- 15.Reference deleted.
- 16.Jadwin, J. A., V. Rudd, P. Sette, S. Challa, and F. Bouamr. 2010. Late domain-independent rescue of a release-deficient Moloney murine leukemia virus by the ubiquitin ligase itch. J. Virol. 84:704-715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones, T. A., J. Y. Zou, S. W. Cowan, and Kjeldgaard. 1991. Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47:110-119. [DOI] [PubMed] [Google Scholar]
- 18.Joshi, A., U. Munshi, S. D. Ablan, K. Nagashima, and E. O. Freed. 2008. Functional replacement of a retroviral late domain by ubiquitin fusion. Traffic 9:1972-1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuiken, C., B. Foley, T. Leitner, C. Apetrei, B. Hahn, I. Mizrachi, J. Mullins, A. Rambaut, S. Wolinsky, and B. Korber (ed.). 2010. HIV sequence compendium 2010. Los Alamos National Laboratory, Theoretical Biology and Biophysics Division, Los Alamos, NM.
- 20.Lee, S., A. Joshi, K. Nagashima, E. O. Freed, and J. H. Hurley. 2007. Structural basis for viral late-domain binding to Alix. Nat. Struct. Mol. Biol. 14:194-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mangeot, P. E., D. Negre, B. Dubois, A. J. Winter, P. Leissner, M. Mehtali, D. Kaiserlian, F. L. Cosset, and J. L. Darlix. 2000. Development of minimal lentivirus vectors derived from simian immunodeficiency virus (SIVmac251) and their use for gene transfer into human dendritic cells. J. Virol. 74:8307-8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCullough, J., R. D. Fisher, F. G. Whitby, W. I. Sundquist, and C. P. Hill. 2008. ALIX-CHMP4 interactions in the human ESCRT pathway. Proc. Natl. Acad. Sci. U. S. A. 105:7687-7691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Otwinowski, Z., and W. Minor. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307-326. [DOI] [PubMed] [Google Scholar]
- 24.Painter, J., and E. A. Merritt. 2005. A molecular viewer for the analysis of TLS rigid-body motion in macromolecules. Acta Crystallogr. D Biol. Crystallogr. 61:465-471. [DOI] [PubMed] [Google Scholar]
- 25.Painter, J., and E. A. Merritt. 2006. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D Biol. Crystallogr. 62:439-450. [DOI] [PubMed] [Google Scholar]
- 26.Pikora, C. A., C. Wittish, and R. C. Desrosiers. 2006. p6gag of human and simian immunodeficiency viruses is tolerant to small in-frame deletions downstream of the late domain. Virology 346:479-489. [DOI] [PubMed] [Google Scholar]
- 27.Pires, R., B. Hartlieb, L. Signor, G. Schoehn, S. Lata, M. Roessle, C. Moriscot, S. Popov, A. Hinz, M. Jamin, V. Boyer, R. Sadoul, E. Forest, D. I. Svergun, H. G. Göttlinger, and W. Weissenhorn. 2009. A crescent-shaped ALIX dimer targets ESCRT-III CHMP4 filaments. Structure 17:843-856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Popov, S., E. Popova, M. Inoue, and H. G. Göttlinger. 2009. Divergent Bro1 domains share the capacity to bind human immunodeficiency virus type 1 nucleocapsid and to enhance virus-like particle production. J. Virol. 83:7185-7193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Popov, S., E. Popova, M. Inoue, and H. G. Göttlinger. 2008. Human immunodeficiency virus type 1 Gag engages the Bro1 domain of ALIX/AIP1 through the nucleocapsid. J. Virol. 82:1389-1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sandrin, V., D. Muriaux, J. L. Darlix, and F. L. Cosset. 2004. Intracellular trafficking of Gag and Env proteins and their interactions modulate pseudotyping of retroviruses. J. Virol. 78:7153-7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Segura-Morales, C., C. Pescia, C. Chatellard-Causse, R. Sadoul, E. Bertrand, and E. Basyuk. 2005. Tsg101 and Alix interact with murine leukemia virus Gag and cooperate with Nedd4 ubiquitin ligases during budding. J. Biol. Chem. 280:27004-27012. [DOI] [PubMed] [Google Scholar]
- 32.Strack, B., A. Calistri, S. Craig, E. Popova, and H. G. Göttlinger. 2003. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 114:689-699. [DOI] [PubMed] [Google Scholar]
- 33.Usami, Y., S. Popov, and H. G. Göttlinger. 2007. Potent rescue of human immunodeficiency virus type 1 late domain mutants by ALIX/AIP1 depends on its CHMP4 binding site. J. Virol. 81:6614-6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Usami, Y., S. Popov, E. Popova, and H. G. Göttlinger. 2008. Efficient and specific rescue of human immunodeficiency virus type 1 budding defects by a Nedd4-like ubiquitin ligase. J. Virol. 82:4898-4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Usami, Y., S. Popov, E. Popova, M. Inoue, W. Weissenhorn, and, G. G. H. 2009. The ESCRT pathway and HIV-1 budding. Biochem. Soc. Trans. 37:181-184. [DOI] [PubMed] [Google Scholar]
- 36.Vincent, O., L. Rainbow, J. Tilburn, H. N. Arst, Jr., and M. A. Penalva. 2003. YPXL/I is a protein interaction motif recognized by Aspergillus PalA and its human homologue, AIP1/Alix. Mol. Cell. Biol. 23:1647-1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhai, Q., R. D. Fisher, H. Y. Chung, D. G. Myszka, W. I. Sundquist, and C. P. Hill. 2008. Structural and functional studies of ALIX interactions with YPX(n)L late domains of HIV-1 and EIAV. Nat. Struct. Mol. Biol. 15:43-49. [DOI] [PubMed] [Google Scholar]