

Abstract
Human inter-alpha-inhibitor proteins (IaIp) are endogenous human plasma proteins that function as serine protease inhibitors. IaIp can block the systemic release of proteases in sepsis and block furin-mediated assembly of protective antigen, an essential stop in the intracellular delivery of the anthrax exotoxins, lethal toxin and edema toxin.
IaIp administered on hour or up to 24 hours after spore challenge with Bacillus anthracis Sterne strain protected mice from lethality if administered with antimicrobial therapy (p<.001). These human plasma proteins possess combined actions against anthrax as general inhibitors of excess serine proteases in sepsis and specific inhibitors of anthrax toxin assembly. IaIp could represent a novel adjuvant therapy for the treatment of established anthrax infection.
Keywords: Anthrax, sepsis, lethal toxin, protective antigen, inter-alpha-inhibitors, protease inhibitor
Introduction
Inter-alpha inhibitor proteins (IαIp) are a family of endogenous serine protease inhibitors found in human plasma (1). Severe sepsis results in reduced IαIp with the loss of protease inhibitory activity (2, 3). Treatment with IαIp is protective in numerous preclinical sepsis models and is an investigational therapy for human sepsis (4, 5).
Inhalation anthrax infection results in a rapidly progressive, high-grade bacteremia and sepsis accompanied by systemic release of two important exotoxins, lethal toxin (LT) and edema toxin (ET) (6). The binding element for these toxins is protective antigen (PA). The assembly of PA requires a host-derived, membrane-associated serine endoprotease known as furin (6, 7). Furin is susceptible to protease inhibition by IαIp in tissue culture systems and in animal models of LT intoxication (8). Thus, IαIp protects against excess protease activation from sepsis and directly limits the assembly of LT and ET. These combined actions of IαIp might represent a unique treatment option in the early phases of systemic anthrax infection.
Bacillus anthracis is a category A biothreat agent causing a highly lethal infection by proliferation and damage to tissues by the exotoxins LT and ET. Both toxins use the same pore-forming binding element formed by PA (6, 9). The 83 kD anthrax PA precursor undergoes extracellular processing by partial proteolysis from the host-derived cellular enzyme furin. A 20 kD soluble fragment is released followed by heptamerization of the 63kD PA monomers to form a membrane pore (6). Once the PA pore is formed within endosomes, LT or ET enter the intracellular space and induce injury or death to susceptible host cells.
Lethal toxin is a metallo-enzyme that inactivates mitogen-activated protein kinase kinase (MAPKK). This event is lethal to monocytes and macrophages and impairs dendritic maturation (6). Edema factor results in excess intracellular levels of cyclic AMP in neutrophils (6, 7). Edema toxin is responsible for the striking edema that surrounds skin lesions and contributes to the pleural effusions and massive fluid shifts seen in patients with systemic anthrax infection (6). Inhibitors of PA assembly, the major epitopes expressed on PA (10), and furin itself (11), have become potential targets for therapeutic intervention against anthrax.
IαIp is a family plasma-derived furin inhibitors that can protect cells from the cytotoxicity of LT (8). IαIp have broad substrate specificity and these protease inhibitors can disrupt an array of plasma proteases implicated in the pathogenesis of septic shock. Some of these proteases include elastase, granzymes, complement components, thrombin, plasmin and other proteases from the coagulation system (8, 12). The IαIp family includes inter-alpha inhibitor, consisting of a light chain (known as bikunin) and two heavy chains linked by chondroitin sulfate, and a related protein known as pre-alpha inhibitor (1, 2). A degradation product found in human urine, known as urinary trypsin inhibitor (UTI), consists of chondroitin sulfate linked to bikunin. The molecule’s active site for serine protease inhibition is located within the two, tightly packed, kunitz domains found on the bikunin light chain.
We hypothesized that the administration of IαIp could be a novel treatment for systemic anthrax infection by serving dual roles: control of excess protease activity from sepsis, and disruption of the final assembly of anthrax toxins by furin inhibition.
Materials and Methods
IαIp (both Inter-alpha Inhibitor and Pre-alpha Inhibitor) were isolated from human fresh frozen plasma (Rhode Island Blood Center, Providence, RI) by cryo-precipitation, solid phase extraction and ion-exchange chromatography as previously described. The PA and LF were purchased from List Biological Laboratory and their activity was confirmed in a cytotoxicity assay (8) in RAW264.7 cells (ATCC # TIB-71). All other reagents used in these experiments were purchased from Sigma (St. Louis, MO).
Male AJ mice were obtained from Jackson Laboratories (Barr Harbor, ME). Animals were housed in an IUCAC- approved facility under Biosafety Level 2 safety conditions. Animal care and protocol adherence were monitored by the Brown University Veterinary staff. Animals were housed in cages with HEPA filter lids and maintained at a constant ambient temperature and humidity with twelve hour day/night cycling.
In-vivo studies of B. anthracis Sterne 34F2 was obtained from Colorado Serum. The Sterne strain has a full complement of pXO1-encoded toxins LF, EF and PA, but lacks the pXO2-encoding anti-phagocytic poly-D-glutamic acid capsule, rendering it non-lethal to humans but still highly lethal in susceptible mouse strains (14). All work was conducted under BSL-2 conditions. B. anthracis spores (103–109/animal) were used in LD50 experiments (n=5/group). IαIp were given (30 mg/kg) ip 1 hour before the spore challenge or PBS control. This dose of IαIp was chosen based upon preliminary dose-finding experiments with recombinant anthrax lethal toxin (8). Moxifloxacin (Schering, Kenilworth, NJ) was given subcutaneously (10mg/kg q24hrX3) beginning 24 hours after spore challenge. In survival experiments, IαIp’s (30mg/kg) were administered ip 1 hour after or 24 hours after the spore challenge with moxifloxacin or PBS control.
Individual parameters between groups were compared using the Mann-Whitney U test. The non-parametric, Kruskal-Wallis one way analysis of variance was used for differences between multiple groups. The survival studies were analyzed using Kaplan-Meier survival plots and differences were assessed by the log-rank test. P values of <.05 were considered significant.
Results
B. anthracis spores were injected intraperitoneally (ip) at increasing doses to determine the LD50 in this mouse strain (<105 spore per animal). A standard antimicrobial agent for B. anthracis, moxifloxacin was given 24 hr after spore challenge and did not significantly improve survival. IαIp (30 mg/kg ip) one hour before the spore challenge did significantly improve the LD50 over 7 days. The combination of IαIp one hour before spore challenge and moxifloxacin therapy daily for 3 days after challenge markedly increased the LD50 by 3 orders of magnitude (figure 1).
Figure 1.
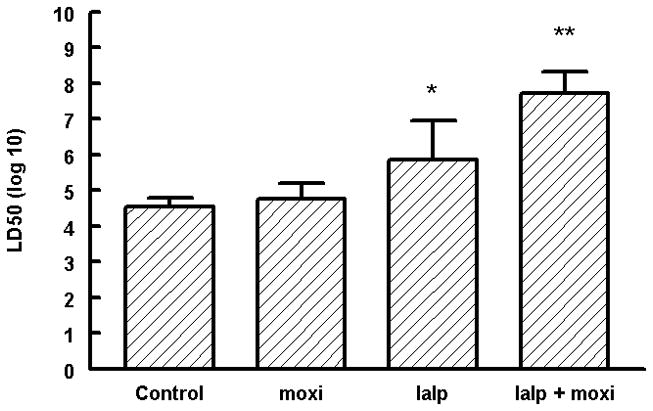
The lethal dose50 of B. anthracis Sterne strain 34F2 after intraperitoneal administration in AJ mice. Moxi –moxifloxacin; IαIp –inter-alpha inhibitor proteins. IαIp + moxifloxacin significantly increased the LD50 compared to the PBS control group.
*p<0.01
**p<0.001
Administration of IαIp and moxifloxacin as a salvage therapy after exposure to an otherwise lethal dose of B. anthracis spores was then investigated (figure 2). No significant improvement in survival was observed with PBS, moxifloxacin alone, or IαIp’s alone. In contrast, the combination of IαIp and moxifloxacin was highly protective, regardless of whether IαIp was given one hour after spore challenge (86% survival; P<.001) or even 24 hours after spore challenge (65% survival; P <.001).
Figure 2.
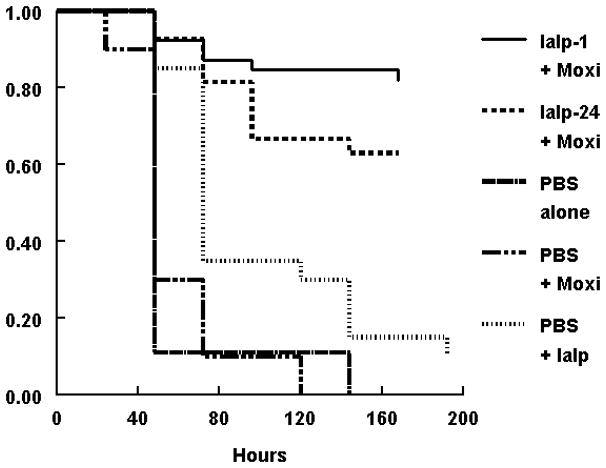
Kaplan-Meier survival plot of A/J mice exposed to 106 B. anthracis Sterne strain spores at time 0. Top line - IαIp at 1 hour + moxifloxacin (10mg/kg) im q24 hrX3 (n=39). Second line - IαIp given at 24 hours plus moxifloxacin (n=27). Third line is IαIp + PBS (n=20). The fourth line is PBS + moxifloxacin (n=10); the bottom line is PBS control (n=9). The combination therapy of IαIp + moxifloxacin significantly improved the outcome compared to the placebo group (P<.001).
Histopathologic findings were strikingly different between groups. Liver tissue and spleen tissue in the control group showed focal areas of necrosis and loss of splenic red pulp with massive quantities of the vegetative forms of B. anthracis found throughout the tissues. Tissue edema, pleural effusions and ascites fluid was often observed. In contrast, the combination therapy group showed preservation of normal tissue architecture and the absence of bacilli. Typical findings observed in lung tissue are displayed on Figure 3A-D.
Figure 3.
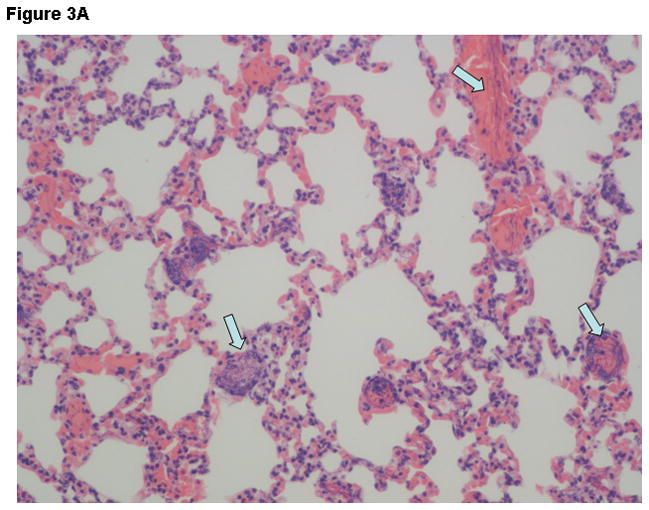
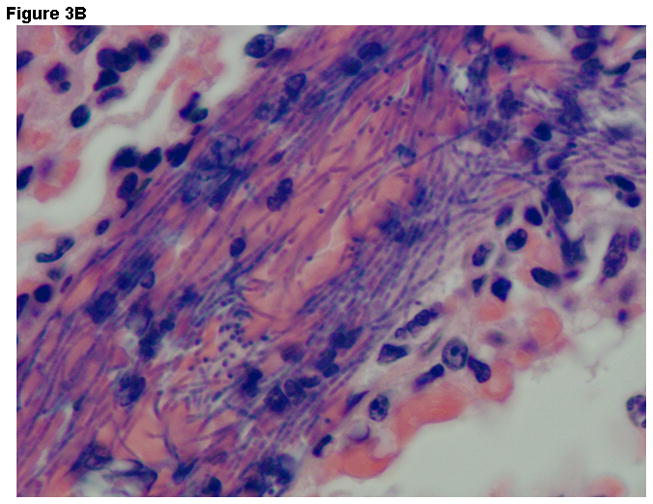
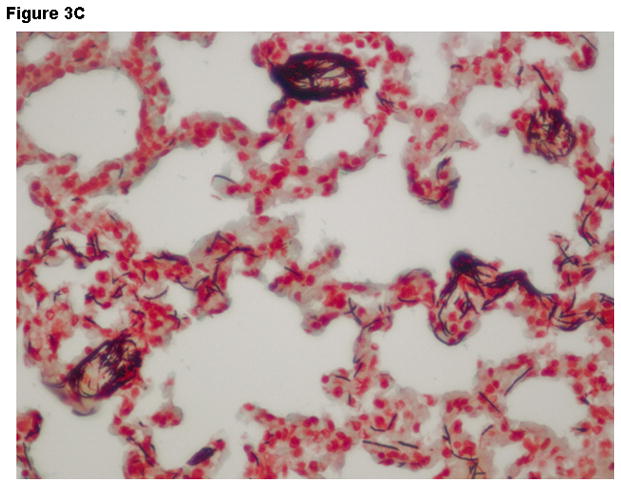
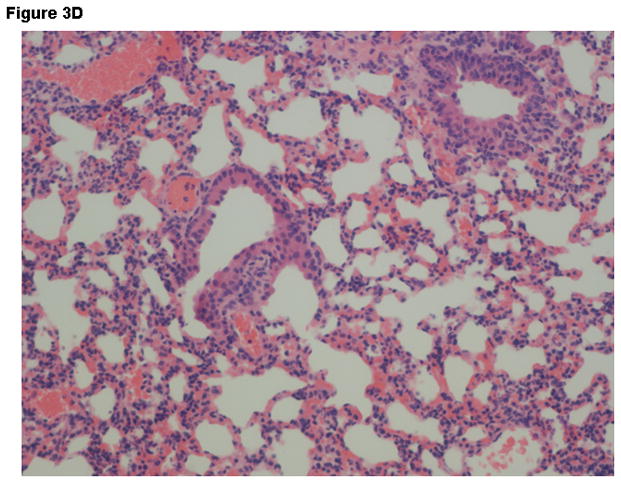
Representative Hematoxylin & Eosin stains lung tissue from animals euthanized 48 hours after the challenge with B. anthracis spores. Panel A –Lung tissue (200X- control group) showing alveolar capillaries and venules clogged with vegetative forms of B. anthracis (arrows point to bacilli in blood vessels); Panel B –Oil immersion view of an alveolar capillary with dense population of intravascular bacteria; Panel C –Tissue gram stain lung tissue (200X) from the control group showing alveolar capillaries with gram-positive bacilli and RBCs. Panel D –lung tissue (200X) in the IαIp + moxifloxacin group showing intact air spaces with minimal cellular infiltrates within the alveolar capillary membranes and the absence of bacterial invasion.
Discussion
The results indicate that the administration of IαIp can salvage animals after a potentially lethal dose of B. anthracis spores, even up to 24 hours after infection has been initiated. The beneficial effects of IaIp’s therapy are most readily observed when administered in combination with appropriate antimicrobial therapy. This rapidly replicating bacterial pathogen must be inhibited by the use of antimicrobial agents without delay. The capsular poly-glutamate outer membrane around B. anthracis possesses anti-phagocytic properties that also contribute to hepatic uptake and proliferation within micro colonies within the liver (15, 16).
Current therapeutic options for inhalational anthrax in patients are limited to antibiotics and supportive care, and the resulting mortality rate, even in the best of critical care settings, is unacceptably high (17). B. anthracis is a rapidly proliferating gram-positive bacillus that overwhelms host defenses resulting in high-grade bacteremia and severe sepsis. As the population of B. anthracis expands within the host, progressive release of anthrax exotoxins results in fatal anthrax infection (6, 18, 19).
Treatment will necessitate early intervention with antibacterial agents to limit bacterial growth. In addition, adjuvant therapies that inhibit exotoxin-mediated tissue injury and ameliorate the ongoing systemic inflammatory state might improve outcome. A combination of antimicrobial agents plus IαIp provides a particularly potent combination therapy for anthrax. IαIp are direct furin inhibitors with the capacity to limit the final assembly of the PA pore for both LT and ET (8, 9). IαIp also functions as a broad spectrum protease inhibitor, reducing the excessive systemic protease activity that results from the host’s acute phase response during systemic infection and sepsis (8, 12). Anthrax exotoxins do not induce significant cytokine responses (20, 21), but experimental infection by B. anthracis spores induces a robust, acute phase, pro-inflammatory cytokine response that might be amenable to IaIp inhibitory action (22).
IαIp are endogenous human proteins that are likely to be non-immunogenic when administered in therapeutic doses to patients. Related IaIp compounds such as UTI have already been demonstrated to be safe in human clinical studies in pancreatitis and sepsis (4, 5). The combined, anti-inflammatory and antitoxic activities of IaIp against systemic anthrax infection could be a potential salvage therapy, when administered with antimicrobial agents, in the treatment of this highly lethal biohazardous infection. We now hope to extend these investigations to large animal models of inhalation anthrax under high-level, bio-containment laboratory conditions.
Acknowledgments
This work was supported by a US National Institutes of a Health Grant R41AI062095-01AI. The authors wish to thank James Harper and Gordon Hankinson and their excellent veterinary assistance.
Footnotes
Presented in part at the 48th Annual Meeting of the Interscience Conference of Antimicrobial Agents and Chemotherapy/Infectious Diseases Society of America Meeting, Washington, DC. October 25, 2008. (abstract # B-1912)
Author Contributions. SMO developed the protocols, performed in vitro and in vivo experiments and generated the manuscript. AWA generated the protocols and reviewed the manuscript. PC and DD assisted in the animal experiments and laboratory assays. NK performed the histologic evaluations in a blinded fashion and reviewed the manuscript. JEP and NP preformed the in vitro and in vivo experiments, and Y-PL purified the IαIp, developed the study protocols and reviewed the manuscript.
Competing Interest Statements.
Yow-Pin Lim has equity in ProThera Biologics which is developing IαIp for commercial use. The other authors claim no conflict of interest in the completion of these studies.
References
- 1.Bost F, Diarra-Mehrpour M, Martin J. Inter-alpha-inhibitor-trypsin inhibitor proteoglycan family; a group of proteins binding and stabilizing the extracellular matrix. Eur J Biochem. 1998;252:339–349. doi: 10.1046/j.1432-1327.1998.2520339.x. [DOI] [PubMed] [Google Scholar]
- 2.Opal SM, Lim Y-P, Siryaporn E, et al. Longitudinal studies of inter-alpha inhibitor proteins in severely septic patients: A potential clinical marker and mediator of severe sepsis. Crit Care Med. 2007;35(2):387–392. doi: 10.1097/01.CCM.0000253810.08230.83. [DOI] [PubMed] [Google Scholar]
- 3.Lim Y-P, Bendelja K, Opal SM, et al. Correlation between mortality and the levels of inter-alpha inhibitors in the plasma of patients with severe sepsis. J Infect Dis. 2003;188:919–926. doi: 10.1086/377642. [DOI] [PubMed] [Google Scholar]
- 4.Ying Z, Chen H, Li Y-M, et al. Thymosin α1- and ulinastatin-based immunomodulatory strategy for sepsis arising from intra-abdominal infection due to carbapenem-resistant bacteria. J Infect Dis. 2008;198:723–730. doi: 10.1086/590500. [DOI] [PubMed] [Google Scholar]
- 5.Yumin L, Hao C, Xun L, et al. A new immunomodulatory therapy for severe sepsis: ulinastatin plus thymosin α1. J Intensive Care Med. 2009;24:47–53. doi: 10.1177/0885066608326970. [DOI] [PubMed] [Google Scholar]
- 6.John AT, Collier J. Anthrax toxin: Receptor binding, internalization, pore formation, and translocation. Ann Rev Biochem. 2007;76:243–265. doi: 10.1146/annurev.biochem.75.103004.142728. [DOI] [PubMed] [Google Scholar]
- 7.Molloy S, Bresnahan P, Leppla S, et al. Human furin is a calcium-dependent serine endoprotease that recognizes the sequence Argx-x-arg and efficiently cleaves anthrax toxin protective antigen. J Biol Chem. 2002;267:16396–16402. [PubMed] [Google Scholar]
- 8.Opal SM, Artenstein AW, Cristofaro PA, et al. Inter-alpha-inhibitor proteins are endogenous furin inhibitors and provide protection against experimental anthrax intoxication. Infect Immun. 2005;73(8):5101–5105. doi: 10.1128/IAI.73.8.5101-5105.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petosa C, Collier RJ, Klimpel KR, et al. Crystal structure of the anthrax toxin protective antigen. Nature. 1997;385:833–838. doi: 10.1038/385833a0. [DOI] [PubMed] [Google Scholar]
- 10.Chen Z, Moayeri M, Zhou Y-H, et al. Efficient neutralization of anthrax toxin by chimpanzee monoclonal antibodies against protective antigen. J Infect Dis. 2006;193:625–633. doi: 10.1086/500148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jean F, Stella K, Thomas L, et al. Alpha antitrypsin Portland, a bioengineered serpin highly selective for furin:application as an antipathogenic agent. Proc Natl Acad Sci USA. 1998;95:7293–7298. doi: 10.1073/pnas.95.13.7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garatzoitis S, Hollingsworth JW, Ghanayem RB, et al. Inter-α-trypsin inhibitor attenuates complement activation and complement-induced lung injury. J Immunol. 1997;179:4187–4192. doi: 10.4049/jimmunol.179.6.4187. [DOI] [PubMed] [Google Scholar]
- 13.Josic D, Brown MK, Huang F, et al. Proteomic characterization on inter-alpha inhibitor proteins from human plasma. Proteomics. 2006;6(9):2874–2885. doi: 10.1002/pmic.200500563. [DOI] [PubMed] [Google Scholar]
- 14.Lyons CR, Lovchik J, Hutt J, et al. Murine model of pulmonary anthrax: Kinetics of dissemination, histopathology, and mouse strain susceptibility. Infect Immun. 2004;72(8):4801–4809. doi: 10.1128/IAI.72.8.4801-4809.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Piris-Gimenez A, Corre J-P, Jouvion G, Candela T, Khun H, Goossens PL. Encapsulated Bacillus anthracis interacts closely with live endothelium. J Infect Dis. 2009;200:1381–1389. doi: 10.1086/644506. [DOI] [PubMed] [Google Scholar]
- 16.Gregory SH, Sagnimeni AJ, Wing EJ. Bacteria in the bloodstream are trapped in the liver and killed by immigrating neutrophils. J Immunol. 1996;157:2514–2520. [PubMed] [Google Scholar]
- 17.Inglesby TV, O’Tolle T, Henderson DA, et al. Anthrax as a biological weapon: medical and public health management. JAMA. 2002;287(17):2236–2252. doi: 10.1001/jama.287.17.2236. [DOI] [PubMed] [Google Scholar]
- 18.Mock M, Fouet A. Anthrax. Annu Rev Microbiol. 2001;55:647–671. doi: 10.1146/annurev.micro.55.1.647. [DOI] [PubMed] [Google Scholar]
- 19.Tournier JN, Quesnel-Hellmann A, Cleret A, Vidal DR. Contribution of toxins to the pathogenesis of inhalational anthrax. Cell Microbiol. 2007;9:555–565. doi: 10.1111/j.1462-5822.2006.00866.x. [DOI] [PubMed] [Google Scholar]
- 20.Erwin JL, DaSilva LM, Bavari S, Little SF, Friedlander AM, Chanh TC. Macrophase-derived cell lines do not express proinflammatory cytokines after exposure to Bacillus anthracis lethal toxin. Infect Immun. 2001;69(2):1175–1177. doi: 10.1128/IAI.69.2.1175-1177.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Agrawal A, Lingappa J, Leppla SH, Agrawal S, Jabbar A, Quinn C, Pulendran B. Impairment of dendritic cells and adaptive immunity by anthrax lethal toxin. Nature. 2003;424:329–334. doi: 10.1038/nature01794. [DOI] [PubMed] [Google Scholar]
- 22.Pickering AK, Osorio M, Lee GM, Grippe VK, Bray M, Merkel TJ. Cytokine response to infection with Bacillus anthracis spores. Infect Immun. 2004;72(11):6382–6389. doi: 10.1128/IAI.72.11.6382-6389.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]