

Abstract
Zinc finger nucleases (ZFNs) have emerged as powerful tools for delivering a targeted genomic double-strand break (DSB) to either stimulate local homologous recombination (HR) with investigator-provided donor DNA or induce gene mutations at the site of cleavage in absence of a donor by non-homologous end joining (NHEJ), both in plant and mammalian cells, including human cells. ZFNs are formed by fusing zinc finger proteins (ZFPs) to the non-specific cleavage domain of FokI restriction enzyme. ZFN-mediated gene targeting yields high gene modification efficiencies (>10%), in a variety of cells and cell types by delivering a recombinogenic DSB to the targeted chromosomal locus, using two designed ZFNs. Mechanism of DSB by ZFNs requires that two ZFN monomers bind to their adjacent cognate sites on DNA and the FokI nuclease domains dimerize to form the active catalytic center for the induction of the DSB. In the case of ZFNs fused to wild-type FokI cleavage domains, homodimers may also form, which could limit the efficacy and safety of the ZFNs by inducing off-target cleavage. In this article, we report further refinements to obligate heterodimer variants of FokI cleavage domain for creating custom ZFNs with minimal cellular toxicity. The efficacy and efficiency of the re-engineered obligate heterodimer variants of FokI cleavage domain were tested using the GFP gene targeting reporter system. The 3- and 4-finger ZFP fusions to REL_DKK pair among the newly generated FokI nuclease domain variants appear to eliminate or greatly reduce the toxicity of designer ZFNs to human cells.
INTRODUCTION
The creation of custom-designed zinc finger nucleases (ZFNs), and hence the development of ZFN-mediated gene targeting, provides molecular biologists with the ability to site-specifically and permanently modify plant and mammalian genomes, including the human genome via homology-directed repair of a targeted genomic DSB (1–5). The ZFNs are inactive as monomers. Mechanism of DSB by ZFNs requires that two different ZFN monomers bind to their adjacent cognate sites on DNA and that the FokI nuclease domains dimerize to form the active catalytic center for the induction of the DSB (6,7). Since dimerization of the FokI cleavage domain is required to produce a DSB, binding of two 3- or 4-finger ZFN monomers (each recognizing a 9- or 12 bp inverted site) to adjacent sites is necessary for delivering a genomic DSB in cells. Such a pair of ZFNs effectively has an 18- or 24-bp recognition site, which is long enough to specify a unique genomic location in plant and mammalian cells, including human cells (8,9). Because the recognition specificities of the ZFPs can be easily manipulated experimentally, designer ZFNs offer a general way for targeted manipulation of the genomes of a variety of cells and cell types (5,14–31).
ZFN-mediated gene modification has been successfully demonstrated in a variety of cells from diverse species like frog oocytes (5), Drosophila (14–16), nematodes (17), zebra fish (18–20), mice (21), rats (22,23), plants (24,25) and humans (21,26–31). High rate of endogenous gene modification efficiencies (>10%) have been achieved using this approach (27). However, in the case of ZFNs fused to wild-type FokI cleavage domains (FokI_WT), homodimers may also form, which could limit the efficacy and safety of the ZFNs by inducing off-target cleavage (32–34). ZFNs toxicity resulting from off-target cleavage, particularly when using 3-finger ZFNs, has been reported to decrease the viability of targeted cells. Two different approaches have been developed to reduce the cytotoxicity of ZFNs to cells: 1) Structure-based redesign of FokI cleavage domains at dimer interface to create obligate heterodimer variants that retained the wild type (WT) catalytic activity of natural FokI enzyme, but show reduced off-target cleavage, which is discussed below (32–34); and 2) Attenuation of ZFN toxicity by small-molecule regulation of protein levels in cells (35). The latter strategy, involves creating ZFNs with shortened half-lives by destabilizing ZFNs either by linking to a ubiquitin moiety to the N-terminus and then regulating ZFN levels by using a small molecule proteosome inhibitor or linking a modified destabilizing FKBP12 domain to the N-terminus and then regulating ZFN levels by using a small molecule that blocks destabilization effect of the N-terminal domain. Thus, it appears that by regulating ZFN levels one could maintain high rates of ZFN-mediated gene targeting while reducing ZFN toxicity.
Here, we report further improvements to obligate heterodimer variants of FokI cleavage domain for creating designer ZFNs with minimal cellular toxicity.
MATERIALS AND METHODS
Construction of ZFNs and the donor plasmid substrate
The design and synthesis of CCR5-specific 3- and 4-finger ZFNs are reported elsewhere (21,28). The obligate heterodimer variants of FokI cleavage domain were constructed using overlapping oligonucleotides as described elsewhere (21). The nucleotide and protein sequences of the various obligate heterodimer variant pairs of FokI cleavage domain are shown in Table S1. Construction of the mutant eGFP genes encoding the desired ZFN target sites and the donor substrate for eGFP gene correction are described elsewhere (21,27). The protocol for generating HEK293 cell lines with an integrated mutant eGFP genes encoding the desired ZFN target sites is reported elsewhere (21,27).
FACS and Microscopy analyses
HEK293 cells carrying a mutated eGFP reporter gene were transiently transfected with a donor plasmid carrying a fragment of wild-type GFP and plasmids expressing various 3- and 4-finger CCR5-specific ZFN constructs using Lipofectamine 2000 as described elsewhere (21,27). Transfections of various obligate heterodimer variants and FokI_WT were performed one after another on the same day. After each transfection, the treated cells were split into 2 flasks. GFP positive cells in about 10,000 treated cells in each flask were determined by FACS and then normalized to one million treated cells. The difference between the two independent FACS readings is shown as error bars. Three, five and seven days post-transfection with ZFNs and donor plasmid, eGFP gene correction was measured by FACS using a BD FACS Canto II. GFP fluorescence (GFP) was measured using BP 530/30 filter. BD FACS Diva Software, v6.1.1 was used for analyses. GFP positive cells were sorted and examined by microscopy to confirm GFP expression. Independent transfections, performed using 3-finger ZFN variant pairs on different days, showed a similar trend for gene correction efficiencies of EL_KK and REL_DKK, respectively.
Western Blot Analysis of Obligate Heterodimer Variants Expression in HEK293 Flp-In Cells
Western blot analysis was performed as described elsewhere (32). HEK293 Flp-In cells were grown and transfected as reported elsewhere (21). After 30 h cells were harvested and resuspended in RIPA buffer (Sigma-Aldrich). 50 μg of total protein of cell extract were separated by 10 % SDS-PAGE and transferred to PVDF membrane (Amersham Biosciences). The blot was blocked and incubated with a rabbit anti-FokI antibody (1:200) followed by incubation with horseradish peroxidase-conjugated secondary antibody (Amersham Biosciences, 1:1000) and developed using an ECL chemiluminescence detection system (Thermo Scientific) according to manufacture’s instructions. The expression levels (band intensities) of Fok_WT (171.82), RV_DA (171.54), EL_KK (169.51) and REL_DKK (169.05), respectively, were quantified using the Image J software NIH, Version 1.36b).
Analysis of Genome-wide Double-strand Breaks in ZFN-treated Cells
For cytotoxicity analysis, cells were transfected with 400 ng each of ZFN expression plasmid using Lipofactamine 2000 (Invitrogen) according to manufacture’s instructions. Alternatively, cells were exposed to 10 μM etoposide for 60 minutes 2 h before harvesting. Cells were collected 30 h after transfection and fixed in ice-cold methanol for 15 min at room temperature, permeabilized in 0.5 % Triton X-100 and then blocked using 5 % serum for 45 minutes at room temperature. After blocking, cells were then incubated with anti-53BP1 rabbit polyclonal antibodies (Bethyl Laboratories) overnight at 4º C followed by incubation with AlexaFluor 594-conjugated secondary antibodies (Invitrogen-Molecular Probes) with 2 μg/ml of DAPI (Roche) at room temperature in the dark for one hour. Slides were mounted and analyzed by fluorescence microscope.
RESULTS
Miller et al (2007) strategy for obligate heterodimer development comprised of two key elements: First, ensure that the FokI nuclease domain variants retained the desired catalytic activity by using the functional screen of GFP reporter gene correction in human cells (32). Second, use a stepwise approach to modification of the dimer interface using rational design. This was done through four cycles of variant design and testing, each of which substituted one amino acid at the dimer interface providing incremental improvement in the specificity of heterodimer formation. In each cycle of development, a small panel of amino acid substitutions was generated within one cleavage domain, while its partner was not modified. The choice of substitution was guided by the coordinates of the native crystal structure of FokI restriction endonuclease dimer and mutations were introduced at positions that could contact the unmodified partner with a bias towards charge-charge interactions. Each variant was screened for the ability to stimulate gene correction in two different configurations, namely as a heterodimer with the unmodified partner and as a homodimer, alternating successive development cycles between the two sides of the dimer interface. In each cycle of development, they were able to identify a variant that was efficiently induced gene correction as heterodimer, but showed reduced activity as a homodimer. Through these stringent tests, Sangamo group generated FokI cleavage domain pair with the double mutations, namely Q486E:I499L and E490K:I538K (also known as EL_KK) that promote obligate heterodimer formation. EL_KK pair was shown to possess ~10-fold reduced cytotoxicity as compared to FokI_WT cleavage domains.
Szczepek et al (2007) also used rational design based on the crystal structure of the native FokI endonuclease and protein modeling to identify critical residues involved in the dimerization of ZFNs (33). FokI endonuclease structure has shown that the dimerization is mainly mediated by helices α4 (residues 479–490 through a pair of salt bridge formation between residues D483/R487) and α5 (residues 528–539) of the cleavage domain, which is also supported by functional data (36, 37). The model also predicted the existence of another contact between Q486 and E490. Thus, the FokI crystal structure indicated that an asymmetric dimer interface could be created by rational redesign by swapping a critical pair of interacting residues, like D483R or R487D. By incorporating similarly charged residues D483/D487 or R483/R487 in the same FokI nuclease domain subunit, one could make formation homodimers unlikely, owing to electrostatic repulsion. Similarly, the residues Q486 and E490 were redesigned to generate variants EE (Q486E) and QK (E490K) that favor heterodimerization over homodimerization. Cathomen lab generated a FokI cleavage domain pair with the double mutations, namely D483R:I538V and R487D:I499A (also known as RV_DA) that promote obligate heterodimer formation. RV_DA pair was shown to possess reduced cytotoxicity as compared to FokI_WT cleavage domains. Later, DD_RR pair was also shown to have reduced cytotoxicity (34).
Engineering of Improved Obligate Heterodimer Variants of FokI Cleavage Domain
We hypothesized that one could make further improvements to the obligate heterodimer variants of FokI cleavage domain, to further minimize off-target cleavage and lower ZFN toxicity, and thereby increase the efficacy and efficiency of ZFN-mediated gene targeting of human cells. Such mutants could also greatly increase the viability of gene-modified human cells, especially the sensitive human primary cells, human embryonic stem cells (hESC) and human induced pluripotent stem cells (hiPSC). Here, we have used two different approaches to improve the obligate heterodimer variants of FokI cleavage domain for creating designed ZFNs with minimal toxicity. In the first approach, we reasoned that the mutant residues at the dimer interface identified by Miller et al (2007) (32) and Szczepek et al (2007) (33) using rational design could be combined to form new obligate heterodimer variants of FokI cleavage domain with lowered ZFN toxicity. We generated two such pairs. The monomer of one of the sets contained the mutations, D483R:Q486E:I499L, while its dimer partner contained the mutations, R487D:E490K:I538K. Together the pair is depicted as REL_DKK. The second pair contained one more mutant residue in each of monomer chains in addition to the above: D483R:Q486E:I499L:I538V and R487D:E490K:I499A:I538K (underlined residues), respectively. Together the pair is depicted as RELV_DKAK.
In a second approach, we replaced the FokI segment (IVDTKAYSGGYNLPIGQADEMQRYVEENQTRN KHINPNEWWKVYPSSVTEFKFLFVSGHFKGNYKAQLTRLNHITN) containing the α4 and α5 helices (in italics and shaded yellow) that mediate dimerization between two monomers of the FokI cleavage domains with the corresponding segment from StsI, an isoschizomer of FokI endonuclease. The segment in each of the monomer was replaced with LDSKAYSEGFPLTASHTRAMERYLRQFTERKEELKPTWWDIAPEHLDNTYFAYVSGSSFSGNY KEQLQKFRQDT and ILDSKAYSEGFPLTASHTDAMGDYLKQFTERKEEIKPTWWDIAPEHLDNTYFAY VSGSFSGNYKEQLQKFRQKT respectively, to further decrease the affinity at the dimer interface. Furthermore, based on our results from the first approach, we also replaced three amino acid residues within the StsI segment of each monomer chain (which are shown in bold type and are underlined) to promote obligate heterodimer formation. Together the pair is referred to as FokI_StsI. Since many of the amino acid residues of the StsI segment are quite distinct from the residues within the FokI segment, we reasoned that this would result in further destabilization of the dimer interface and decrease the formation of homodimers, while promoting the heterodimers.
Protein modeling and energy minimization of the obligate heterodimer variant pairs of FokI cleavage domain, EL_KK, RV_DA, REL_DKK and RELV_DKAK respectively, were carried out using the SPDBV software (described in detail in the supplementary material). Visualization of protein structure rendering of images for H-bond interactions were performed with CCP4 MG software. The H-bond interactions that were present between the A and B monomer chains of the different obligate heterodimer variants are shown in Fig. 1. For the REL_DKK pair, the model revealed only one bi-dentate H-bond between residues R487 of chain A and the D483 of chain B, predicting the weakest dimer interface interactions between the two monomers.
Figure 1.
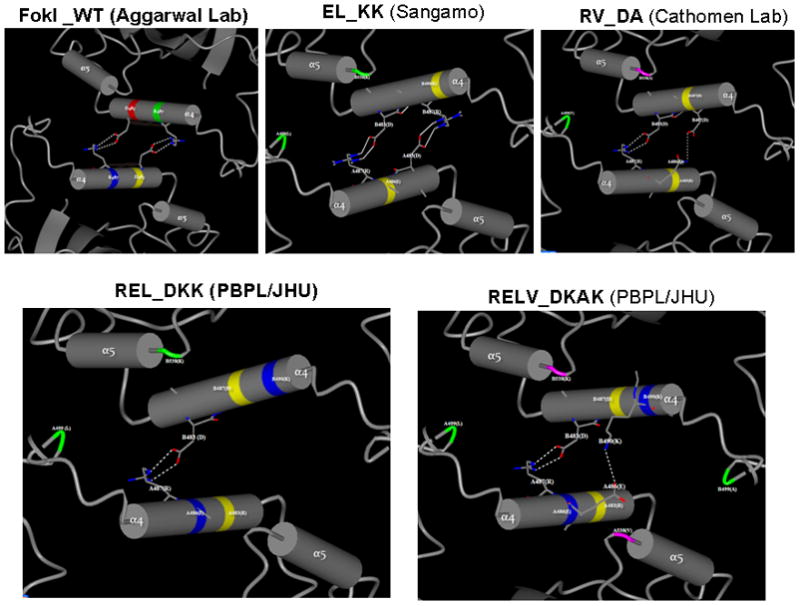
Predicted profiles of H-bond interactions between α4 helices at the dimer interface of obligate heterodimer variants of FokI nuclease domains, based on protein modeling and energy minimization. The 3D structure of the FokI dimer was obtained from RCSB Protein Data Bank, which was generated by X-ray diffraction method at a resolution of 2.3 A by Aggarwaal’s lab (36). The CCP4 Molecular Graphics software (Version 6.1.2) for Macromolecular X-ray Crystallography was used for 3D structure analysis and SPDBV software for protein modeling. Only hydrogen bond interactions between α4 helices at the dimer interface are shown. All potential dimer interface interactions (H-bond interactions and hydrophobic interactions) are listed in Table S2.
Testing the efficacy and efficiency of engineered obligate heterodimer FokI nuclease domain variants using the proxy GFP gene targeting reporter system in HEK293 Cells
We compared the efficiency and efficacy of gene targeting of the various pairs of obligate heterodimer variants (REL_DKK; RELV_DKAK and FokI_StsI) with those of FokI_WT, EL_KK pair from Sangamo and RV_DA pair from Cathoman lab, by making fusions to the previously published pair of 4-finger ZFPs that was shown to target CCR5 in human cells (28,29). ZFN–mediated gene correction at the mutant GFP locus was very efficient in HEK293 Flp-In cells, yielding GFP positive cells upon transduction with the corresponding pairs of ZFNs containing either FokI_WT or the obligate heterodimer variants (Fig. 2). Transfection with donor alone did not yield any GFP positive cells by microscopy. Quantitative FACS analyses of the GFP positive cells at 3, 5 and 7 days post-transfection with designer various ZFN pairs and donor plasmids are shown in Fig. 2A. The number of surviving GFP positive cells using the REL_DKK mutant pair at 3, 5 and 7 days post-transfection was consistently better in gene targeting experiments as compared to the other obligate heterodimer variant pairs, suggesting that they are less toxic to cells, possibly due to reduced off-target cleavage.
Figure 2.
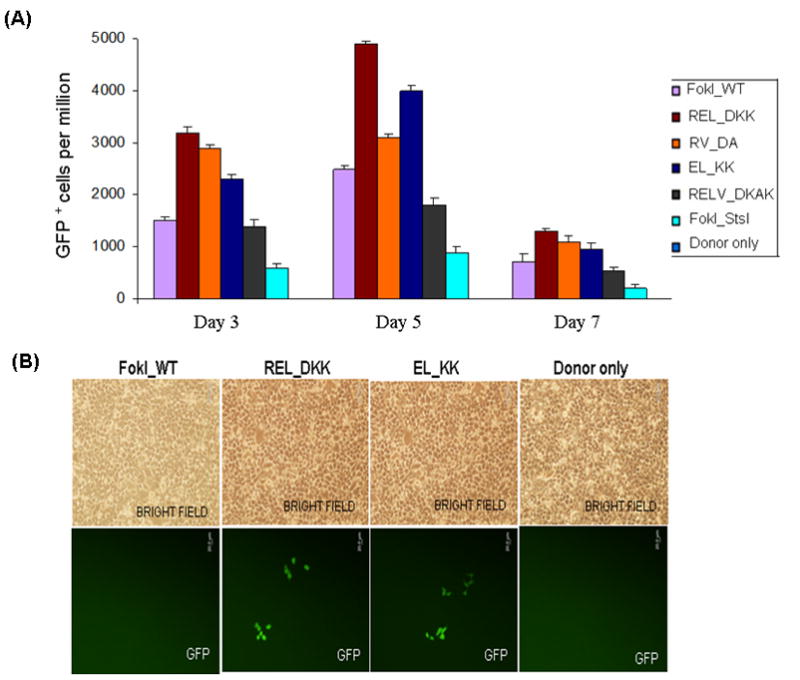
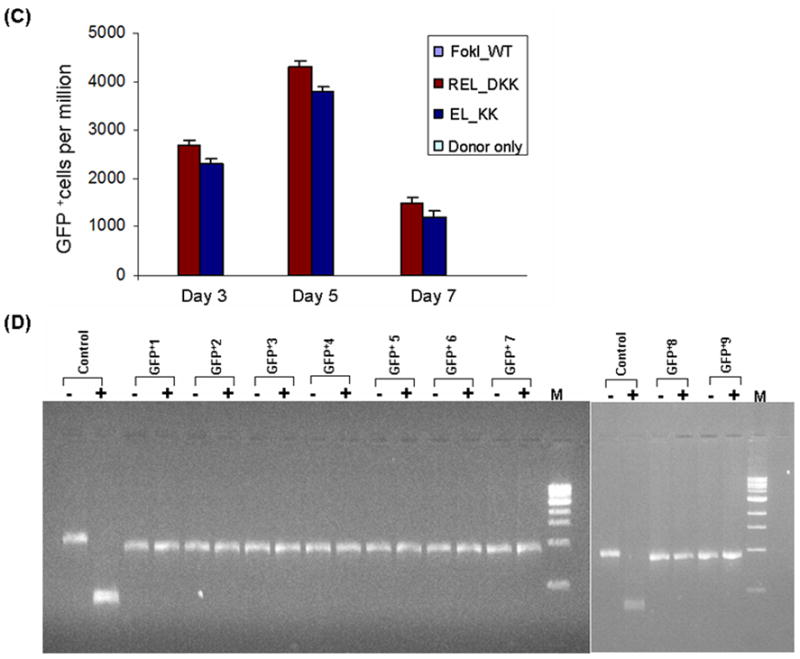
Testing the efficiency and efficacy of CCR5 3- and 4-finger ZFNs (generated by fusing the corresponding ZFPs to various obligate heterodimer variants of FokI nuclease domain) using the GFP gene targeting reporter system. HEK293 cells carrying a mutated eGFP reporter gene were transiently transfected with a donor plasmid carrying a fragment of wild-type GFP and plasmids expressing various 3- or 4-finger CCR5-specific ZFN constructs, using Lipofectamine 2000 as described elsewhere (21,27). Transfections of various obligate heterodimer variants and FokI_WT were performed one after another on the same day. After each transfection, the treated cells were split into 2 flasks. GFP positive cells in about 10,000 treated cells in each flask were determined by FACS and then normalized to one million treated cells. The difference between the two independent FACS readings is shown as error bars. A) Frequency of gene correction in HEK293 Flp-In cells of a chromosomal mutant GFP reporter disabled by insertion of the CCR5 ZFN target sequences using 4-finger ZFNs (28, 29). Quantitative FACS analyses of the GFP positive cells at 3, 5 and 7 days post-transfection with designer CCR5-specific 4-finger ZFNs (constructs carrying the FokI_WT and obligate heterodimer FokI nuclease domain variants) and donor plasmids. WT: wild type; EL_KK, 4-finger CCR5-ZFNs containing the FokI nuclease domain mutants reported by Miller et al., 2007 (32). RV_DA, 4-finger CCR5-ZFN constructs carrying the FokI nuclease domain mutants reported by Szczepek et al., 2007 (33). REL_DKK, RELV_DKAK and FokI_StsI, 4-finger CCR5-ZFN constructs carrying the FokI nuclease domain mutants that were generated at PBPL/JHU. B) Top panel: HEK293 Flp-In cells 5 days post-transfection as seen in brightfield; Bottom panel: GFP positive cells as seen 5 days post-transfection of HEK293 Flp-In cells with 3-finger CCR5-ZFN constructs (carrying Fok_WT, EL_KK or REL_DKK respectively) and donor plasmid. No GFP positive cells were seen with either donor alone or ZFNs containing FokI_WT nuclease domains and donor plasmid. C) FACS analyses of the frequency of gene correction in HEK293 Flp-In cells using 3-finger CCR5 ZFN constructs carrying FokI_WT, EL_KK, REL_DKK or donor alone, respectively. Results from two independent transfections, performed on different days, are shown in Figure S1; both transfections showed a similar trend of gene correction efficiencies for EL_KK and REL_DKK, respectively. The dose response curve using titrations of 3-finger ZFN expression plasmids, EL_KK and REL_DKK respectively, at constant donor plasmid, are shown in Figure S2. D) Analysis of the genotype of nine different individual GFP positive clones. Five days post-transfection with CCR5 3-finger ZFNs and the donor plasmids, GFP positive cells were sorted, serially diluted to get individual clones and grown. The genomic DNA was isolated from the GFP positive clones and the eGFP gene at the Flp-In locus was PCR amplified and digested with BstXI. The mutant eGFP gene has two BstXI sites, where the ZFN binding sites are inserted. Correction of the eGFP gene by homology-directed repair results in the loss of the BstXI sites. The PCR product size of the corrected eGFP gene is 930 bp as compared to 990 bp for the mutant gene. BstXI digestion of the mutant eGFP PCR product generates two bands: 450 bp and 540 bp, respectively. Lanes: Control, PCR product of the mutant eGFP gene from untransfected cells before (-) and after (+) digestion with BstXI; GFP+1–9, PCR products of 9 different individual clones obtained from GFP positive sorted cells before (-) and after (+) digestion with BstXI; M, 1 Kb ladder. All GFP positive cells are resistant to BstXI digestion, confirming ZFN-mediated eGFP gene correction in these cells.
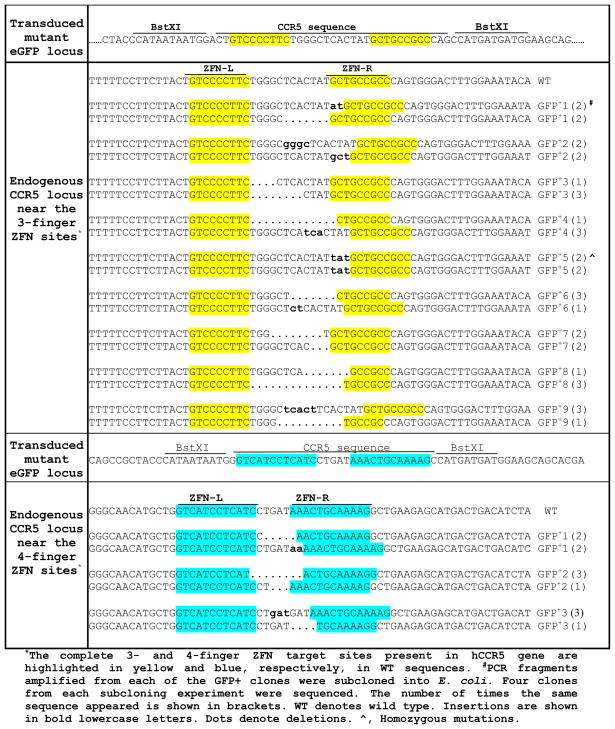
Once established, the gene-altered cells are viable and they continued to increase in number for several weeks. We isolated 3 different individual GFP positive clones from gene targeting experiment REL_DKK mutant pair by serial dilution of FACS sorted GFP positive cells and grew them for genotypic characterization. Three different loci, namely the mutant GFP locus, the endogenous CCR5 locus and the CCR2 locus, were PCR-amplified using the corresponding locus-specific primers from the isolated genomic DNA of the individual GFP positive clones. The PCR DNA from the mutant GFP locus of the gene-altered clones were all resistant to BstXI digestion indicating that gene correction to the wild-type sequence has occurred in the GFP positive clones. The PCR DNA from the three different loci was then cloned in the pGEMT vector for transformation into E. coli. DNA sequence analysis of at least 4 recombinant E. coli clones from each individual GFP positive clone confirmed that gene correction indeed has occurred. DNA sequence analysis of 4 recombinant bacterial clones generated by cloning the PCR-amplified DNA of the endogenous CCR5 locus from each of the three individual GFP positive clones, as expected, showed simple deletion and/or insertion mutations at the targeted 4-finger CCR5 site resulting from NHEJ (Table 1). Extensive genotypic characterization of the GFP positive cells from gene targeting experiments using 4-finger CCR5-specific ZFNs is reported elsewhere (21).
Table 1.
Genotypic analysis of the endogenous CCR5 locus at the 3- and 4-finger ZFN target sites of the GFP+ clones
 |
We then compared the efficiency and efficacy of gene targeting of REL_DKK obligate heterodimer variant pair with those of FokI_WT and EL_KK pair from Sangamo by making fusions to the 3-finger ZFPs that target CCR5 in human cells (13). Unlike the CCR5 4-finger ZFNs which had no linker, the active CCR5-specific 3-finger ZFNs contained the (Gly4S)3 linker between the ZFPs and the FokI nuclease domain variants, since they are designed to target ZFN sites separated by a 12 bp spacer (13). Transfection with donor alone or ZFPs fused to FokI_WT and donor did not yield any GFP positive cells by microscopy (Fig. 2B). ZFN–mediated gene correction at the mutant GFP locus was very efficient in HEK293 Flp-In cells, upon transfection with 3-finger ZFPs fused to REL_DKK or EL_KK obligate heterodimer FokI nuclease domain mutants and donor, yielding GFP positive cells (Fig. 2B). Quantitative FACS analyses of the GFP positive cells at 3, 5 and 7 days post-transfection are shown in Fig. 2C. The GFP positive cells using the REL_DKK mutant pair at 3, 5 and 7 days post-transfection was consistently higher in gene targeting experiments as compared to EL_KK pair, suggesting that REL_DKK variants are less toxic to cells, which is likely due to further reduction in off-target cleavage. Results from two independent transfections, performed on different days, are shown in Fig. S1; both transfections showed a similar trend for gene correction efficiencies of EL_KK and REL_DKK, respectively. We also performed titrations of 3-finger ZFN expression plasmids of obligate heterodimer variants EL_KK and REL_DKK at 0.2, 0.4 and 0.8 μg respectively, with constant donor plasmid (1.0 μg) to obtain a dose response curve to study the differences between the two mutants (Fig. S2). REL_DKK variant consistently yielded more gene corrected cells as compared to EL_KK mutant. The maximal difference of GFP corrected cells was observed 5 days post co-transfection of plasmids (Fig. S2).
Once established, the gene-corrected cells are viable and they continued to increase in number for several weeks. We isolated 9 different individual GFP positive clones from gene targeting experiment using REL_DKK mutant pair by serial dilution of FACS sorted GFP positive cells and grew them for genotypic characterization. Three different loci, namely the mutant GFP locus, the endogenous CCR5 locus and the CCR2 locus, were PCR-amplified using the corresponding locus-specific primers from the isolated genomic DNA of the individual GFP positive clones. The PCR DNA from the mutant GFP locus of the gene-altered clones were all resistant to BstXI digestion indicating that gene correction to the wild-type sequence has occurred in the GFP positive clones (Fig. 2D). The PCR DNA from the three different loci was then cloned in the pGEMT vector for transformation into E. coli. DNA sequence analysis of at least 4 recombinant E. coli clones from each individual GFP positive clone confirmed that gene correction indeed has occurred. DNA sequence analysis of 4 recombinant bacterial clones generated by cloning the PCR-amplified DNA of the endogenous CCR5 locus from each of the individual GFP positive clones, as expected, showed simple deletion and/or insertion mutations at the targeted 3-finger CCR5 site resulting from NHEJ (Table 1). No change in the nucleotide sequence of the CCR2 locus was observed in the limited number of GFP positive clones that were sequenced suggesting that the designed pair of 3-finger CCR5 ZFNs did not cleave at a distantly related site (data not shown).
Reduced levels of DNA Damage by REL_DKK Heterodimer Variant Pair
As a direct measurement for ZFNs’ cytotoxicity, we then monitored whether REL_DKK heterodimer variant pair reduced genome-wide DNA cleavage levels when expressed in human HEK293 Flp-In cells using the well-established assay for visualizing DNA double-strand breaks as described elsewhere (32). We used antibody-mediated detection of the protein 53BP1, which localizes to sites of DNA damage and forms foci that are visualized by immunofluoroscence. Antibody-based detection of 53BP1 revealed that substitution of either EL_KK or REL_KK heterodimer variant pair for FokI_WT cleavage domain showed marked reduction in the number of 53BP1-stained foci in HEK293 Flp-In cells (Fig. 3A). Moreover, the fraction of 53BP1-stained cells with multiple foci (>3 foci) for REL_DKK heterodimer variants was lower than that of EL-KK variants (Fig. 3A). We confirmed that the observed results were not due to poor protein expressions of REL_DKK variants in HEK293 Flp-In cells, since western blot analysis showed comparable levels of ZFN expressions for FokI_WT, EL_KK and REL_DKK, respectively (Fig. 3B).
Figure 3.
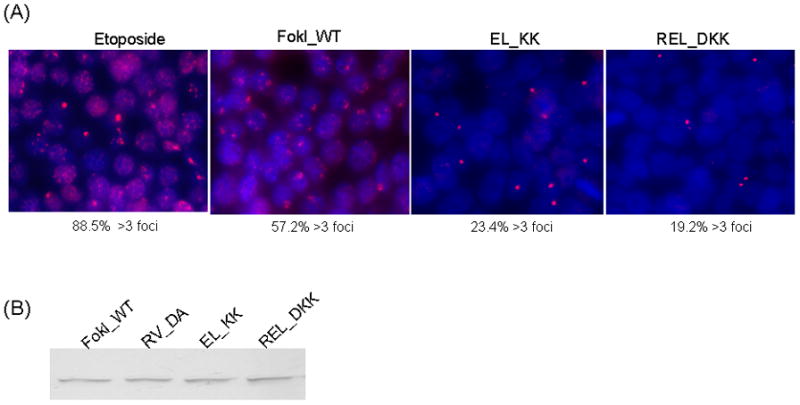
Reduced genome-wide DNA damage levels by REL_DKK obligate heterodimer variant pair of FokI nuclease domain. A), Representative images of cells treated with the DNA cleavage agent etoposide or transfected with the indicated ZFN expression plasmids. Cells were fixed after 30 h and stained with antibodies against 53BP1 (red) and then with DAPI (blue). The fraction of cells containing more than 3 foci is indicated under each panel. The total number of cells analyzed is: etoposide, 152; FokI_WT, 245; EL_KK, 282; REL_DKK, 289. B), ZFN expression levels were examined by anti-FokI immunoblot analysis. HEK293 Flp-In cells were transfected with indicated ZFN expression plasmids and cells were harvested after 30 h. Equal amounts of total cellular protein was separated by 10% SDS-PAGE gel and transferred to PVDF membrane. The blot was probed with anti-FokI antibody. The ZFNs migrate as a single band on the gel. Western blot analysis shows comparable levels ZFN expression for various obligate heterodimer variants in HEK293 Flp-In cells.
DISCUSSION
This report details further improvements to obligate heterodimer variants, by incorporating multiple mutations at the dimer interface of FokI cleavage domains, to eliminate or greatly reduce ZFN toxicity, while increasing the efficacy and efficiency of ZFN-mediated gene targeting in cells. We hypothesized that incorporation of different combinations of mutations at the ZFN dimer interface based on previous studies, would eliminate or further reduce toxicity associated with off-target cleavage. We expected RELV_DKAK pair, which incorporates almost all of the mutations from previous studies, to perform better than that of REL_DKK in the GFP gene targeting reporter assays in HEK293 cells. On the contrary, our results indicate that the REL-DKK pair fused to 3- and 4-finger ZFPs consistently performed better as compared to RELV_DKAK or EL_KK or RV_DA, suggesting this pair exhibits the lowest toxicity to human cells, while retaining catalytic efficiency of the FokI_WT cleavage domains. Using 3D protein modeling based on FokI crystal structure and energy minimization calculations, we analyzed the H-bond and hydrophobic interactions present at the dimer interface of the newly generated mutant pairs. The RELV_DKAK model predicts the existence of one additional H-bond interaction including those that are present in REL_DKK pair, which likely provides added stability to the dimer interface. The FokI_StsI pair generated by replacing the FokI segment (encoding the α4 and α5 helices that are involved in the dimer interface interactions) with the StsI segment in FokI nuclease domain, although active, appears to have much diminished catalytic activity, which is probably due to the destabilizing effect of the StsI segment on protein folding of the FokI nuclease domains.
While the 4-finger CCR5 ZFNs (created by fusing highly specific 4-finger ZFPs to FokI_WT cleavage domains) were efficient in GFP gene correction assays, the 3-finger CCR5-specific ZFNs (created by fusing modular 3-finger ZFPs to FokI_WT cleavage domains) did not yield any GFP positive cells, suggesting that sequence-specificity of the designed ZFNs is the major determinant of ZFN activity for efficient gene targeting and for greatly reduced cellular toxicity. The toxicity resulting from off-target cleavage could be attributed to: (i) Homodimer formation by the individual ZFN species; and (ii) Relaxed specificity of the ZF modules (used to generate ZFPs by modular assembly), which results in degenerate or off-target binding by ZFNs (10). However, when the corresponding CCR5-specific ZFPs were fused to the obligate heterodimer variants of FokI nuclease domains, the ZFNs were active in the GFP gene targeting reporter system indicating that the off-target cleavage could be eliminated or greatly reduced by fusing 3-finger ZFPs to obligate heterodimer variant pair (REL_DKK).
Our results from GFP gene correction experiments are consistent with the mechanism of double-strand cleavage by natural FokI enzyme (36–39) and by ZFNs (7). Bitinaite et al (1998) reported that although FokI enzyme binds DNA as a monomer, dimerization of the nuclease domains is required to form active sites in order to cleave DNA (36, 37). Later studies have shown that an active dimer could form with just one subunit bound to its recognition site, albeit through a weak protein–protein interaction of the nuclease domains, particularly at high enzyme concentrations (38). Studies on the mechanism of cleavage by ZFNs suggest that binding of two ZFN monomers to two binding sites is required for effective double-strand DNA cleavage (7). We speculate that ZFN monomers bound to a single site are unlikely to form an active dimer by associating with another ZFN monomer that is not bound to DNA, since this has to occur through the weak protein-protein interaction. It is more likely that the off-target cleavage results, when one ZFN monomer is bound to its cognate site while the other is bound to a nearby degenerate site (since all ZF modules do not always contact all the three bases within their cognate triplets, which could result in relaxed specificities) or bound non-specifically to DNA, especially at high protein concentrations. Interactions at the dimer interface could provide additional stability to the off-target cleavage site complexes, inducing DSB at these sites, resulting in ZFNs’ toxicity to human cells. In such instances, destabilization of the dimer interface would greatly diminish ZFNs off-target cleavage, leading to lowered toxicity to cells. Our results lend support to this idea.
In summary, ZFN technology applications in human therapeutics depend on the ability to create custom-designed ZFNs that cleave the target sequences with exquisite sequence-specificity and high affinity (27). Highly specific ZFPs fused to the re-engineered obligate heterodimer variant pair (like REL_DKK described in this article) will be critical for eliminating or greatly reducing ZFN toxicity to human cells. This will allow one to deliver a targeted genomic DSB to human cells, while leaving the rest of the genome unchanged. Such engineered highly specific ZFNs will enable wider application of ZFN technology in human therapeutics, by further increasing the viability of the gene-modified cells, especially the sensitive human primary cells, human embryonic stem cells (hESC) and human induced pluripotent stem cells (hiPSC).
Supplementary Material
Acknowledgments
This work was supported by a grant to K.K. from the Bill & Melinda Gates Foundation through the Grand Challenge Explorations Initiative (Grant Id: 52067). S.C. is supported by a grant from National Institute of General Medical Sciences (GM077291). We thank Mr. S. Gunasegaran (President and CEO of PBPL, India) for his constant support and encouragement; Dr. Jithesh Narayanan at PBPL for helpful suggestions and Dr. Margolick’s lab at JHU for assistance with the flow cytometry and FACS studies. S.R. and K.K. contributed equally to this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kim YG, Cha J, Chandrasegaran S. Proc Natl Acad Sci USA. 1996;93:1156–1160. doi: 10.1073/pnas.93.3.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li L, Wu LP, Chandrasegaran S. Proc Natl Acad Sci USA. 1992;89:4275–4279. doi: 10.1073/pnas.89.10.4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin L, Chandrasegaran S. Proc Natl Acad Sci USA. 1993;90:2764–2768. doi: 10.1073/pnas.90.7.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim YG, Chandrasegaran S. Proc Natl Acad Sci USA. 1994;91:883–887. doi: 10.1073/pnas.91.3.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bibikova M, Carroll D, Segal DJ, Trautman JK, Smith J, Kim Y–G, Chandrasegaran S. Mol Cell Biol. 2001;21:289–297. doi: 10.1128/MCB.21.1.289-297.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith J, Bibikova M, Whitby FG, Reddy AR, Chandrasegaran S, Carroll D. Nucleic Acids Res. 2000;28:3361–3369. doi: 10.1093/nar/28.17.3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mani M, Smith J, Kandavelou K, Berg JM, Chandrasegaran S. Biochem Biophyl Res Commun. 2005;334:1191–1197. doi: 10.1016/j.bbrc.2005.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu J, Kandavelou K, Chandrasegaran S. Cellular and Molecular Life Sciences. 2007;64:2933 –2944. doi: 10.1007/s00018-007-7206-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kandavelou K, Mani M, Durai S, Chandrasegaran S. Nat Biotechnol. 2005;23:686–687. doi: 10.1038/nbt0605-686. [DOI] [PubMed] [Google Scholar]
- 10.Durai S, Mani M, Kandavelou K, Wu J, Porteus M, Chandrasegaran S. Nucleic Acids Research. 2005;33:5978–5990. doi: 10.1093/nar/gki912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Durai S, Bosley A, Abulencia AB, Chandrasegaran S, Ostermeier M. Journal of Combinatorial Chemistry & High Throughput Screening. 2006;9:301–311. doi: 10.2174/138620706776843147. [DOI] [PubMed] [Google Scholar]
- 12.Maeder ML, Thibodeau-Beganny S, Osiak A, Wright DA, Anthony RM, Eichtinger M, Jiang T, Foley JE, Winfrey RJ, Townsend JA, et al. Mol Cell. 2008;31:294–301. doi: 10.1016/j.molcel.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mani M, Kandavelou K, Dy JF, Durai S, Chandrasegaran S. Biochem Biophys Res Commun. 2005;335:447– 457. doi: 10.1016/j.bbrc.2005.07.089. [DOI] [PubMed] [Google Scholar]
- 14.Beumer K, Bhattacharyya G, Bibikova M, Trautman JK, Carroll D. Genetics. 2006;172:2391–2403. doi: 10.1534/genetics.105.052829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bibikova M, Beumer K, Trautman JK, Carroll D. Science. 2003;300:764. doi: 10.1126/science.1079512. [DOI] [PubMed] [Google Scholar]
- 16.Bibikova M, Golic M, Golic KG, Carroll D. Genetics. 2002;161:1169–1175. doi: 10.1093/genetics/161.3.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morton J, Davis MW, Jorgensen EM, Carroll D. Proc Natl Acad Sci USA. 2006;103:16370–16375. doi: 10.1073/pnas.0605633103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meng X, Noyes MB, Zhu LJ, Lawson ND, Wolfe SA. Nat Biotechnol. 2008;26:695– 701. doi: 10.1038/nbt1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doyon Y, McCammon JM, Miller JC, Faraji F, Ngo C, Katibah GE, Amora R, Hocking TD, Zhang L, Rebar EJ, et al. Nat Biotechnol. 2008;26:702–708. doi: 10.1038/nbt1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foley JE, Yeh JR, Maeder ML, Reyon D, Sander JD, Peterson RT, Joung JK. PLoS ONE. 2009;4:e4348. doi: 10.1371/journal.pone.0004348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kandavelou K, Ramalingam S, London V, Mani M, Wu J, Alexeev V, Civin CI, Chandrasegaran S. Biochem BiophyRes Commun. 2009;388:56–61. doi: 10.1016/j.bbrc.2009.07.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Geurts AM, Cost GJ, Freyvert Y, Zeitler B, Miller JC, et al. Science. 2009;325:433. doi: 10.1126/science.1172447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mashimo T, Takizawa A, Voigt B, Yoshimi K, Hiai H, Kuramoto T, Serikawa T. PLoS ONE. 2010;5:e8870. doi: 10.1371/journal.pone.0008870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Townsend JA, Wright DA, Winfrey RJ, Fu F, Maeder ML, Joung JK, Voytas DF. Nature. 2009;459:442–445. doi: 10.1038/nature07845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shukla VK, Doyon Y, Miller JC, DeKelver RC, Moehle EA, Worden SE, Mitchell JC, Arnold NL, Gopalan S, Meng X, et al. Nature. 2009;459:437–441. doi: 10.1038/nature07992. [DOI] [PubMed] [Google Scholar]
- 26.Porteus MH, Baltimore D. Science. 2003;300:763. doi: 10.1126/science.1078395. [DOI] [PubMed] [Google Scholar]
- 27.Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S, Jamieson AC, Porteus MH, Gregory PD, Holmes MC. Nature. 2005;435:646–651. doi: 10.1038/nature03556. [DOI] [PubMed] [Google Scholar]
- 28.Lombardo A, Genovese P, Beausejour CM, Colleoni S, Lee YL, Kim KA, Ando D, Urnov FD, Galli C, Gregory PD, et al. Nat Biotechnol. 2007;25:1298–1306. doi: 10.1038/nbt1353. [DOI] [PubMed] [Google Scholar]
- 29.Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, Wang N, Lee G, Bartsevich VV, Lee YL, et al. Nat Biotechnol. 2008;26:808–816. doi: 10.1038/nbt1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hockemeyer D, Soldner F, Beard C, Gao Q, Mitalipova M, DeKelver RC, Katibah GE, Amora R, Boydston EA, Zeitler B, et al. Nat Biotechnol. 2009;27:851–857. doi: 10.1038/nbt.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim HJ, Lee HJ, Kim H, Cho SW, Kim JS. Genome Res. 2009;19:1279–1288. doi: 10.1101/gr.089417.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller JC, Holmes MC, Wang J, Guschin DY, Lee YL, et al. Nat Biotechnol. 2007;25:778–785. doi: 10.1038/nbt1319. [DOI] [PubMed] [Google Scholar]
- 33.Szczepek M, Brondani V, Buchel J, Serrano L, Segal DJ, Cathomen T. Nat Biotechnol. 2007;25:786–793. doi: 10.1038/nbt1317. [DOI] [PubMed] [Google Scholar]
- 34.Pruett-Miller SM, Connelly JP, Maeder ML, Joung JK, Porteus MH. Molecular Therapy. 2008;16:707–717. doi: 10.1038/mt.2008.20. [DOI] [PubMed] [Google Scholar]
- 35.Pruett–Miller SM, Reading DW, Porter SN, Porteus MH. PLoS Genet. 2009;5:e1000376. doi: 10.1371/journal.pgen.1000376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wah DA, Hirsch JA, Dorner LF, Schildkraut I, Aggarwal AK. Proc Natl Acad Sci USA. 1998;95:10564–10569. doi: 10.1073/pnas.95.18.10564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bitinaite J, Wah DA, Aggarwal AA, Schildkraut I. Proc Natl Acad Sci USA. 1998;95:10570–10575. doi: 10.1073/pnas.95.18.10570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vanamee ES, Santagata S, Aggarwal AK. J Mol Biol. 2001;309:69–78. doi: 10.1006/jmbi.2001.4635. [DOI] [PubMed] [Google Scholar]
- 39.Catto LE, Ganguly S, Milsom SE, Welsh AJ, Halford SE. Nucleic Acids Research. 2006;34:1711–1720. doi: 10.1093/nar/gkl076. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.