

Abstract
Neutrophil-dependent reactions catalyzed by myeloperoxidase (MPO) are thought to play important roles in the pulmonary pathobiology of cystic fibrosis (CF). Aerosolized thiol antioxidants such as glutathione (GSH) and N-acetylcysteine (NAC) are currently being utilized as therapeutics to modify CF respiratory tract oxidative processes. We hypothesized that MPO in CF airway lining fluids may be a target of such therapeutics. MPO activity in sputum from 21 adult CF patients was found to be inversely associated with lung function (FEV1). In contrast, systemic inflammation (assessed by plasma C-reactive protein) was not correlated with lung function. Ex vivo studies revealed that GSH and NAC effectively scavenged N-chloramines in sputum, and inhibited sputum MPO activity with potency exquisitely dependent upon MPO activity levels. Detailed kinetic analyses revealed that NAC and GSH inhibit MPO by distinct mechanisms. Activation of the key proinflammatory transcription factor NF-κB in cultured HBE1 cells was inhibited by GSH. Our findings reveal that MPO activity and its reactive products represent useful predictors of the doses of inhaled thiol antioxidants required to ameliorate airway oxidative stress and inflammation in CF patients, and provides mechanistic insight into the antioxidative/anti-inflammatory mechanisms of action of GSH and NAC when administered into the CF lung.
Keywords: myeloperoxidase, cystic fibrosis, sputum, glutathione, N-acetyl cysteine, N-chloramines, antioxidants, individualized medicine
Introduction
Cystic fibrosis (CF) is an autosomal recessive disease caused by mutations in a single gene encoding the transmembrane glycoprotein, CF transmembrane conductance regulator (CFTR) (1). CFTR, a cyclic adenosine monophosphate (cAMP)-activated transmembrane channel protein that primarily conducts chloride ion (2), is also involved in the regulation of other ion channels (e.g., epithelial sodium channel activity) (3, 4). Loss of CFTR function in respiratory tract tissues results in thick, underhydrated mucus due to hyperabsorption of sodium chloride and reduced periciliary water and anion transport, leading to reduced mucociliary clearance, airway bacterial colonization and inflammation characterized by an intense airway neutrophil influx (4–6). The neutrophil influxes and their effector oxidative processes are believed to represent major factors in CF respiratory tract disease (7).
Myeloperoxidase (MPO) is the most abundant protein/enzyme within neutrophils (5% of the dry weight of the cell) (8). MPO catalyzes the formation of hypochlorous acid (HOCl), a potent bactericidal agent, that is capable of oxidizing and chlorinating a broad spectrum of biomolecular species (9–11). Respiratory tract secretions from CF patients are known to contain abundant quantities of active MPO (12, 13). MPO can catalyze protein tyrosine oxidation and produce significant levels of 3-nitrotyrosine, dityrosine and 3-chlorotyrosine in CF (13), thus along with HOCl representing a potent potential source of respiratory tract cytotoxic injury (13, 14).
Not surprisingly, proposed therapeutic strategies for CF have included administrations of oral or aerosol delivered antioxidant substances, especially glutathione (GSH) and N-acetylcysteine (NAC) (15–22). Many of the existing limited therapeutic antioxidant trials have administered a “fixed dose” of oral or aerosolized antioxidant without quantitative consideration of the magnitude of respiratory tract oxidative processes. In the present study we have used MPO activity in CF respiratory tract secretions as a biomarker of respiratory tract “oxidants” and tested various thiol antioxidants utilized as possible therapeutic strategies in CF for their effectiveness in inhibiting MPO activity ex vivo. The primarily focus of this study was on the interactions of GSH and NAC with MPO contained in the complex matrix of CF sputum, as these antioxidant thiols have been widely studied as therapeutic agents (19) and are being used in several clinical trials that are currently in progress in CF patients. We also determined the levels of the long-lived MPO-derived N-chloramines in CF sputum and defined the capacity of NAC and GSH to scavenge these species. Finally, we demonstrate that GSH is capable of modulating the pro-inflammatory effects of CF airway secretions in a respiratory epithelial cell model using NF-κB activation, a key regulator of the inflammatory-immune system (23), as a reporting marker of sputum bioactivity. The results herein provide a biochemical construct to further understand the reactions that GSH and NAC undergo in the CF airway, and provide a platform for the development of novel and more efficacious thiol-based therapeutics for the treatment of CF.
Materials and Methods
Subjects
Studies were performed with approval from the Office of Human Research Protection/Institutional Review Board (IRB) of the University of California, Davis. All subjects gave informed consent. Inclusion criteria included the ability of the patient to spontaneously produce sputum. Exclusion criteria included any use of glutathione (GSH), N-acetyl cysteine (NAC), or ascorbic acid supplementations in the 28 days before recruitment. Freshly expectorated sputum specimens were collected from 21 adult CF patients. Table 1 presents the demographics of the 21 patients (7 male, 14 female; ages 19–58). These patients represent a typical spectrum of the 75 patients being followed in the UC Davis Adult CF clinic.
Table 1.
Adult CF Patient Demographics.
| Patient #, gender, age (yrs) | Sputum bacterial culture (type)† | FEV1 (% predicted) | Mutation |
|---|---|---|---|
| 1, M, 24 | PA (m&n), MRSA, AF, Grp C BHS, Proteus, HI, AX, SP, SM, SA | 38 | ΔF508/Δ508 |
| 2, F, 58 | PA (m&n), MRSA, Ser M, AX, Acaligenes sp. | 43 | ΔF508/Δ508 |
| 3, F, 35 | PA (m&n), MSSA, AX, Acaligenes sp. | 58 | +/ΔF508 |
| 4, M, 22 | PA (m&n), MRSA, | 50 | ΔF508/711+1 (G-T) |
| 5, F, 27 | SM, MSSA, AF | 66 | ΔF508 |
| 6, F, 39 | PA (m&n), MRSA, MSSA | 20 | 3659delC, 2789+5G->A |
| 7, M, 45 | PA (m&n), MSSA, MRSA | 88 | ΔF508 |
| 8, M, 26 | PA (m&n), MRSA, HI, BC, AF, HF | 33 | ΔF508/ΔF508 |
| 9, F, 31 | PA (m&n), MRSA, SA, HI | 29 | ΔF508, 1717-1G->A |
| 10, F, 35 | PA (m&n), HI, AX, MSSA, AF, Mould | 60 | ΔF508 |
| 11, F, 23 | PA (m&n), MSSA | 21 | ΔF508/ΔF508 |
| 12, M, 23 | PA (m&n), MRSA, AF, Mould | 50 | ΔF508/711+1 (G-T) |
| 13, F, 36 | PA (m&n), MSSA, AX, Mould, HI, AD | 38 | ΔF508 |
| 14, M, 26 | PA (m&n) | 54 | ΔF508/ΔF508 |
| 15, F, 36 | PA (m&n), MSSA, HI | 53 | ΔF508 |
| 16, F, 31 | PA (m&n), AF | 31 | ΔF508/ΔF508 |
| 17, F, 48 | PA (m), AF, SM, Ser M, AV, AD, mould | 80 | R117C, 3850-3T->G |
| 18, M, 37 | PA (m&n), MSSA, HI | 29 | ΔF508 |
| 19, F, 19 | PA (m), MSSA, mould | 38 | ΔF508 |
| 20, F, 24 | PA (m), SM, AF, HI | 44 | ΔF508/ΔF508 |
| 21, F, 30 | PA (m), AF, HI | 37 | ΔF508/ΔF508 |
PA (m/n) = Pseudomonas aeruginosa (mucoid/non-mucoid), MSSA = methicillin-susceptible Staphylococcus aureus, SM = Stenotrophomonas maltophilia, AF = Aspergillus flavus, HI = Haemophilus influenzae, MRSA = methicillin-resistant Staphylococcus aureus, Ser M = Serratia marcescens, AX = Achromobacter xylosoxidans, SP = Streptococcus pneumoniae, SA = Streptococcus agalactiae, BC = Burkholderia cepacia, AV = Aspergillus versicolor, AD = Acaligenes denitrificans, Grp C BHS = Grp C Beta Hemolytic Streptococcus
Sputum processing
Freshly obtained sputum specimens were weighed and diluted in equal amounts of phosphate buffered saline (pH 7.0) (w/v). The samples were kept in a shaker for 30 mins at 37°C followed by centrifugation at high speed (14000 rpm, 4°C, 15 mins). The upper clear layer (sol-phase) was aliquoted and stored at −80°C until further analyses. It was not possible to perform all assays on all CF sputum obtained due to limitations of sputum quantity and availability.
Analysis of sputum MPO activity
The aliquoted CF sputum sol-phase was further diluted and heme peroxidase activity assessed with 3, 3′, 5, 5′-tetramethylbenzidine (TMB) as a substrate (24) against a standard curve using purified human MPO (EMD Biosciences, La Jolla, CA). It should be recognized that this assay is not specific for MPO. However, the vast majority of heme peroxidase activity in CF sputum is due to MPO (13, 25).
Analysis of sputum N-chloramine levels
The basal CF sputum N-chloramine levels were analyzed in 25 μl of sol-phase using iodide to catalyze the oxidation of 3, 3′, 5, 5′-tetramethylbenzidine (TMB) (26). The readings were extrapolated against a standard curve using known concentrations of N-chloro-taurine.
Ex vivo antioxidant treatments
Diluted sol-phase CF sputum was incubated with glutathione (GSH) or N-acetylcysteine (NAC) at different concentrations (1 nM – 100 mM). MPO activities were compared to untreated controls and the half maximal MPO inhibitory concentration (IC50) of each antioxidant was determined.
Similarly, to investigate the effect of GSH and NAC on sputum N-chloramine levels, a known concentration of antioxidants (IC50 obtained from the above experiment) was incubated with 25μl of CF sputum sol-phase and compared to untreated controls.
Cell Culture and treatments
Human bronchial epithelial cells (HBE1), a papilloma virus-immortalized cell line (27), were plated on standard six-well culture plates (BD Falcon, San Jose, CA) at 2 × 104 cells/well in serum-free bronchial epithelial cell basal medium (BEBM)/Dulbecco’s modified Eagle’s medium (DMEM) (1:1) supplemented with various growth factors including bovine pituitary extract (50 μg/ml), gentamycin (50 μg/ml), insulin (5 μg/ml), hydrocortisone (0.1 μM), epidermal growth factor (30 μg/ml) and transferrin (5 μg/ml) (13). When cells attained 80–85% confluency, they were starved for 6 hrs by replacing media with BEBM/DMEM (1:1) medium devoid of growth factors. Following growth factor deprivation, cells were treated with sol-phase of CF sputum (10 μl/ml of BEBM/DMEM (1:1) medium supplemented with growth factors) and harvested at 30 and 60 minutes after treatment for analysis of NF-κB activation. Nuclear and cytosolic protein fractions were separated using Pierce protein-extraction kit as per vendor’s protocol (Thermo Scientific, Rockford, IL) and stored at −80°C until further analyses. In another set of cells, GSH (1mM) was added along with sol-phase of CF sputum (10 μl/ml of BEBM/DMEM (1:1) medium supplemented with growth factors) and processed similarly. For untreated condition, same amount of phosphate buffered saline (PBS), without CF sputum solphase was added to cells. HBE1 cells treated with 20 μg/ml tumor necrosis factor-α (TNF-α) served as positive controls for analysis of NF-κB activation by western blot technique (23).
Western blot for analysis of NF-κB activation
Nuclear and cytosolic fractions of cells were separated on 4–20% gradient precast protein gels (Thermo Scientific, Rockford, IL) (40 μg protein per well) and transferred onto polyvinylidene fluoride (PVDF) membranes. After blocking the membranes in 5% milk (in PBS-Tween 20 solution), they were probed for p65 and IκB-α proteins by incubating with p65 (1:500; Santa Cruz Biotechnology, CA) and IκB-α antibody (1:500; Santa Cruz Biotechnology, CA) respectively. NF-κB activation was monitored by nuclear translocation of p65 and degradation of cytosolic I B-α, subunits of NF-κB (28). Antibodies against β-actin (1:5000; Abcam Inc., Cambridge, MA) and p84 (1 μg/ml; Abcam Inc., Cambridge, MA) proteins were used as loading controls for cytosolic and nuclear proteins respectively. Horseradish peroxidase (HRP) conjugated goat anti-mouse (Abcam Inc., Cambridge, MA) for p84 and goat anti-rabbit secondary antibody (Abcam Inc., Cambridge, MA) for p65, IκB-α and β-actin were used and detected by enhanced chemiluminescence (ECL) western blotting kit (GE Healthcare, Amersham, UK).
Inhibition kinetics of MPO-dependent TMB oxidation by thiol antioxidants
GSH and NAC at different concentrations (0.1–1 μM) were incubated with a known amount of purified human MPO (1.5 nM) and MPO activity assessed with TMB as a substrate at varied concentrations (20–180 μM). From the resulting data, Lineweaver-Burk graphs were plotted between reciprocal values of velocity of reaction (1/V) and TMB substrate concentrations (1/[TMB]).
Statistical analyses
All data are presented as means ± SEM. Spearmann’s correlation coefficient was used to measure correlation coefficient between samples. Statistical tests were performed using GraphPad Prism software (San Diego, CA). P value ≤ 0.05 was considered as significance in each analysis.
Results
Sputum MPO levels inversely correlate with lung function
We observed an inverse correlation between CF sputum MPO levels and lung function as assessed by percent predicted forced expiratory volume in 1 second (FEV1 % predicted) (n=21, r = −0.59, P= 0.004) (Fig 1A), supporting earlier studies in a younger cohort of patients (12). However, we did not observe any significant correlation between plasma CRP levels, a marker of systemic inflammation, and lung function in CF patients (Fig 1B). Sol-fractions of sputum from CF patients contained high levels of MPO activity, consistent with our previous studies (13, 25). No specific investigations were performed that would further characterize outliers and no attempts were made to determine intra-subject variability of the sputum MPO levels. However, although sputum MPO levels in CF can be expected to exhibit sharp rises during acute clinical exacerbations (29), during periods of clinical stability sputum MPO levels in individual CF patients have been reported to be reasonably reproducible (30). Based on the MPO activity, the present group of CF patients could be empirically grouped into low, moderate and high levels of MPO (Fig 2), representing 27, 33 and 38% of the patients, respectively. We believe that this may represent a clinically useful index of the degree of CF respiratory tract inflammation. We did not observe any significant correlation between CF sputum MPO levels and sputum N-chloramine levels (Fig 3).
Figure 1. Sputum MPO but not plasma CRP levels in CF patients correlate with lung function.
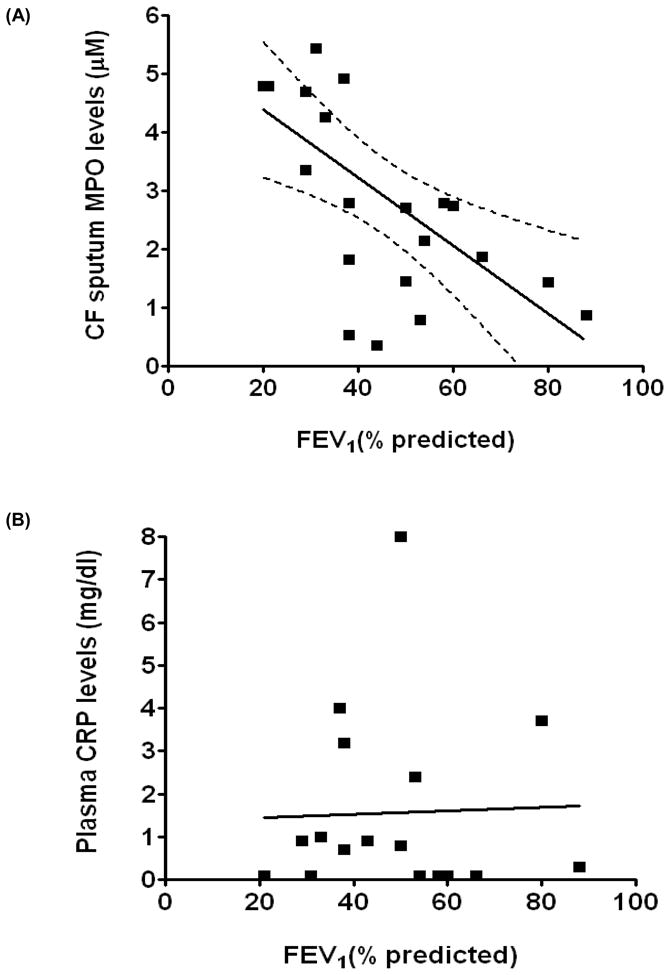
(A) An inverse correlation between CF sputum MPO levels and FEV1 (% predicted) (n=21, r=−0.59, P=0.004) was observed. (B) No correlation was observed between plasma CRP levels and FEV1 (% predicted) (n=17, r=0.033, P=0.9). Spearmann’s correlation coefficient was used to measure correlation coefficient between samples.
Figure 2. Sputum MPO levels in CF patients.
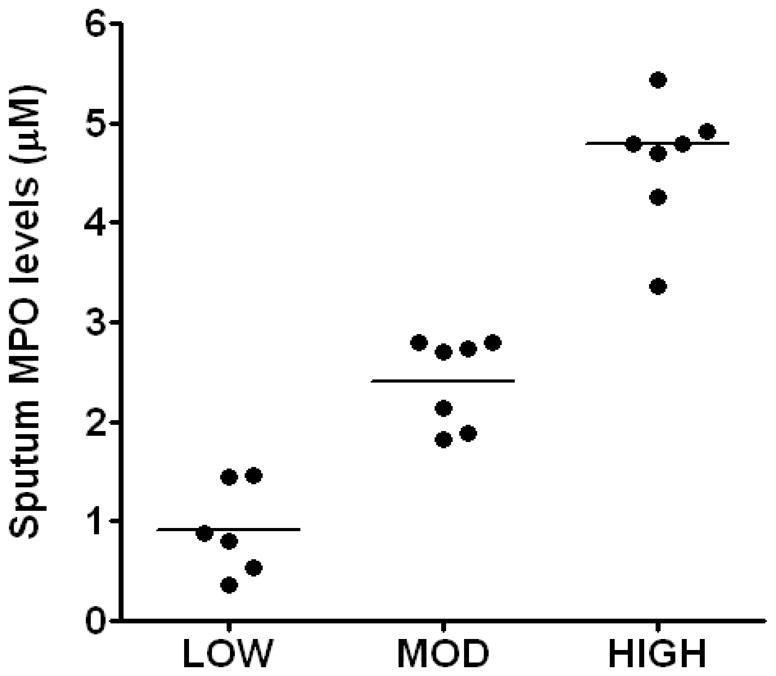
CF patients had varied levels of sputum MPO levels and were categorized into low, moderate and high groups comprising 27%, 33% and 39% of total patients. Values are presented as mean ± SEM.
Figure 3. Correlation between sputum MPO and N-chloramine levels in CF patients.
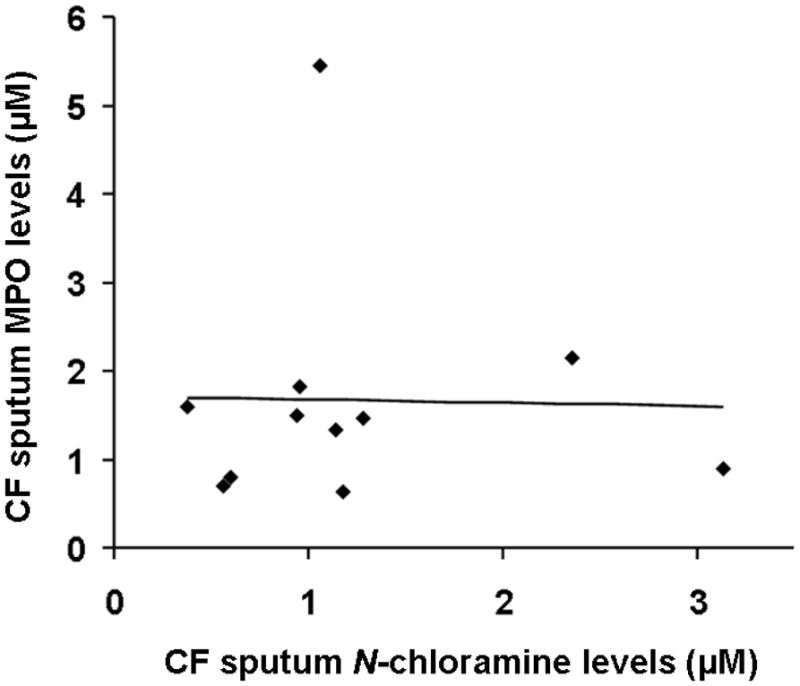
No correlation was observed between sputum MPO and chloramine levels (n=9, r=0.0005). Spearmann’s correlation coefficient was used to measure correlation coefficient between samples.
Inhibition of MPO activity by GSH and NAC is dependent upon sputum MPO concentration
Fig 4A and 4B shows the IC50 of the thiol antioxidants, GSH and NAC respectively, based on their inhibition of sputum MPO activity. Diluted CF sputa having varied ranges of MPO (0.3 nM–2.2 nM) were used. The data illustrate that the higher the MPO activity in CF sputum, the higher the thiol antioxidant concentrations needed to inhibit MPO activity as measured by TMB oxidation.
Figure 4. Inhibition of CF sputum MPO activity by GSH and NAC as represented by IC50 values.
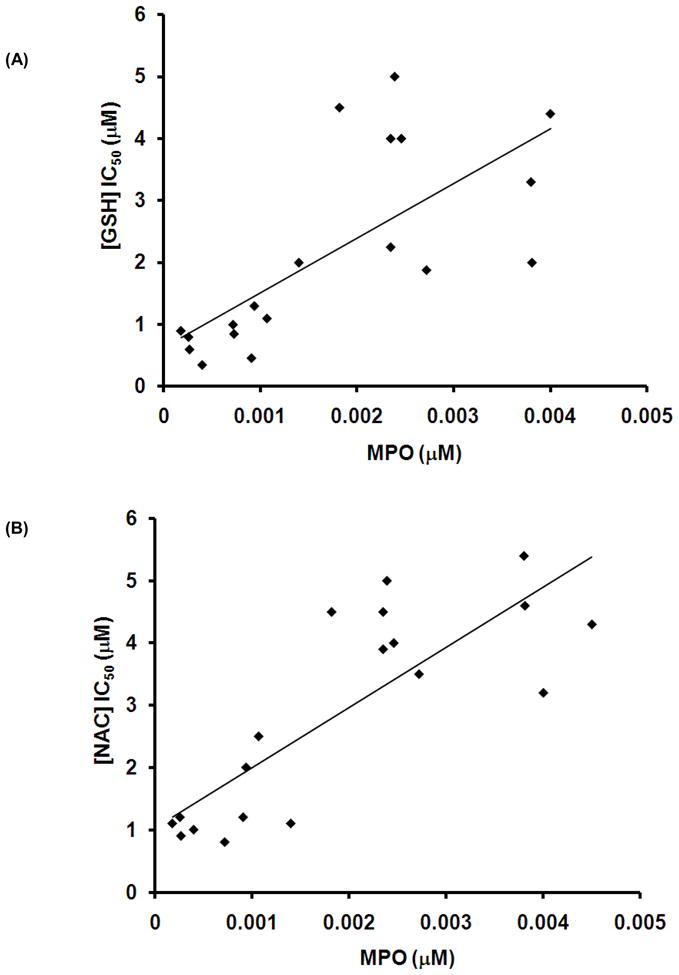
CF sputum was incubated with different concentrations of antioxidants and MPO-activity measured as described in Materials and Methods. MPO levels were determined from the standard graph using purified human MPO. The measurements were compared to untreated samples which served as controls. Antioxidant IC50 values of individual sputum samples were plotted against respective MPO levels. The figure suggests that the higher the MPO levels, the more the antioxidant needed for MPO inhibition.
GSH and NAC inhibit MPO by different mechanisms
To gain insight into the mechanism(s) by which GSH and NAC inhibited the 1-electron oxidation reactions of MPO, we next performed kinetic experiments with varied substrate (TMB) and inhibitor (NAC/GSH) concentrations and generated double reciprocal plots of reaction velocity (1/V) and TMB concentration (1/[TMB]). The results for GSH and NAC are shown in Fig 5. Inhibition of MPO-catalyzed oxidation of TMB by GSH was observed to occur by competitive mode (Fig 5A). However, NAC displays a non-competitive mode of inhibition of MPO as demonstrated by data from reciprocal plots of velocity of reaction (1/V) against TMB concentrations (1/[TMB]). When the slope of each curve from Fig 5A and 5B was plotted against different concentrations of GSH and NAC, a Ki of 0.08 and 0.1 μM was obtained, respectively.
Figure 5. GSH and NAC inhibit MPO by distinct mechanisms.
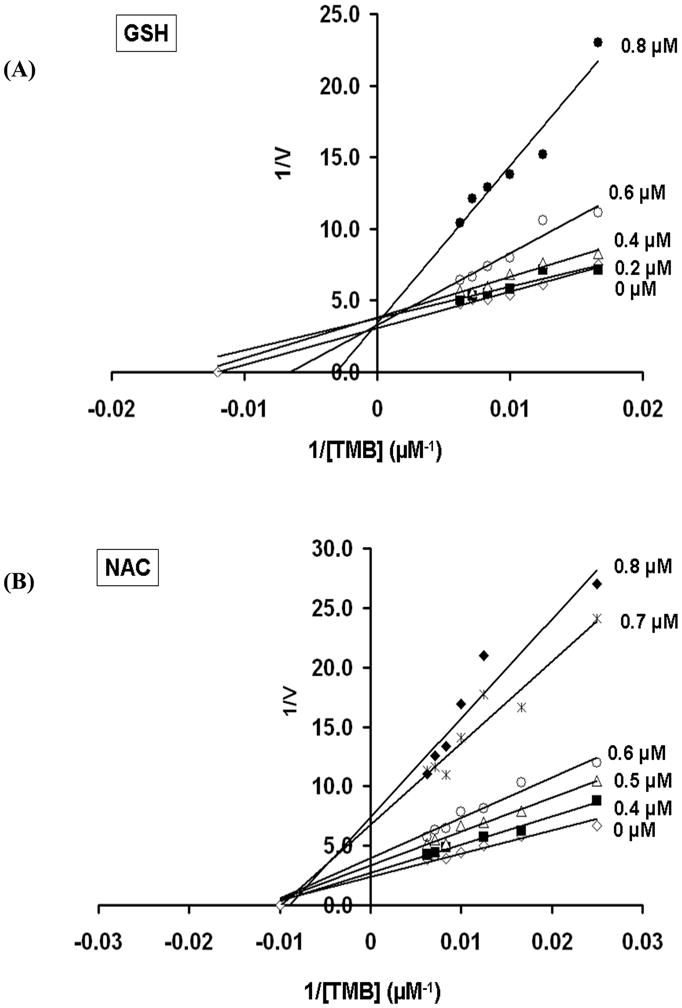
Double reciprocal plot of initial rate of MPO-catalyzed oxidation of TMB vs. its concentration in the presence of (A) 0, 0.2, 0.4, 0.6 or 0.8 μM GSH and (B) 0, 0.4, 0.5, 0.6, 0.7 or 0.8 μM NAC. Inhibition of MPO-catalyzed oxidation of TMB by GSH was observed to occur by competitive mode, whereas NAC showed a non-competitive mode of inhibition as demonstrated by data from reciprocal plots of velocity of reaction (1/V) against TMB concentrations (1/[TMB]).
Sputum N-chloramines are reduced by GSH and NAC
A known concentration of GSH or NAC (2 μM, as derived from data shown in Fig 6A and 6B) was incubated with 25 μl of CF sputum sol-phase for 30 min. Fig 6A and 6B shows CF sputum N-chloramine levels before and after treatment with GSH and NAC respectively. GSH treatment lowered N-chloramine levels by 62 ± 11%, where as NAC treatment reduced N-chloramine levels by 80 ± 11%.
Figure 6. GSH and NAC scavenge N-chloramines in CF sputum.
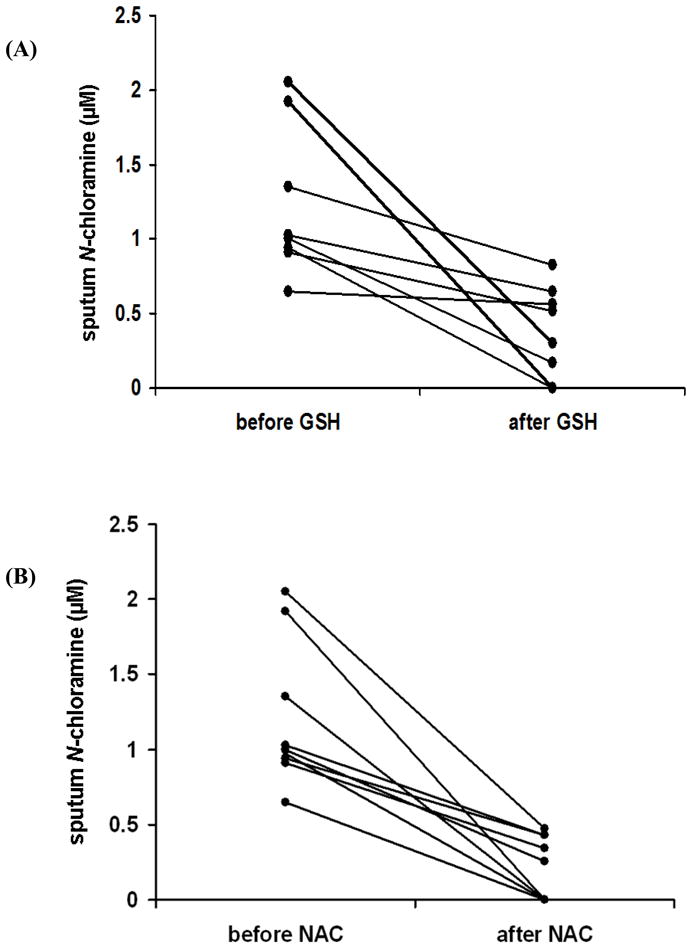
Sol-phase of CF sputum was incubated with/without 2 μM of GSH or NAC for 30 minutes and chloramine levels measured as described in Materials and Methods. N-Chloramine levels were measured by a standard curve using hypochlorous acid. Fig 6A and B shows sputum N-chloramine levels before and after treatment with GSH or NAC, respectively. GSH treatment lowered N-chloramine levels by 62 ± 11 %, whereas NAC treatment lowered N-chloramine levels by 80 ± 11%.
CF sputum mediated NF-κB activation is blocked by GSH in HBE1 cells
As shown in Fig 7A, CF sputum (sol-phase) induces nuclear translocation of NF-κB subunit, p65 and degradation of cytosolic IκB-α suggesting induction of NF-κB pathways in HBE1 cells. Of note, p65 nuclear protein levels in CF sputum treated cells (Fig 7A) were seen more than TNF-a (positive control) treated cells (Fig 7B) at 60 minutes. This suggests a ‘prolonged’ NF-κB activation in response to CF sputum treated cells. It was interesting to note that GSH (1mM) along with CF sputum (solphase) when incubated with HBE1 cells was seen to inhibit p65 nuclear translocation (Fig 7C). Collectively, these results suggest that sol-phase of CF sputum alone can activate NF-κB proteins but this activation can be ameliorated by GSH, suggesting possible scavenging of ‘NF-κB sensitive’ oxidants such as N-chloramines, in CF sputum (31, 32) or by other redox-sensitive mechanisms (33). Due to the complex biochemical composition of CF sputum known to contain a number of well-known inducers of NF-κB, we did not further pursue the identification of the biomolecular species responsible or the upstream mechanisms of NF-κB activation.
Figure 7. GSH inhibits CF sputum-induced NF-κB activation.

HBE1 cells were starved of growth factors and treated with CF sputum (sol-phase) (10 μl/ml of media). Nuclear and cytosolic protein fractions were extracted at 0, 30 and 60 minutes. NF-κB activation was analysed by western blot assay. (A) Nuclear translocation of NF-κB subunit, p65 and degradation of cytosolic IκB-α suggesting CF sputum (sol-phase) mediated NF-κB activation. β-actin and p84 served as loading controls for cytosolic and nuclear protein fractions respectively. (B) Nuclear translocation of p65 in TNF-α treated (20 μg/ml) HBE1 cells which served as controls. (C) HBE1 cells when treated with CF sputum (sol-phase) along with GSH (1 mM) did not show p65 nuclear translocation suggesting inhibition of NF-κB activation. The blots shown are a representative of 3 independent experiments. Densitometric analysis was performed for each blot, and the quantitative data illustrated in graphs to the right of the figure.
Discussion
MPO and CF disease severity
CF patients demographically described in Table 1 are representative of a typical cross section of adult patients with CF. These CF patients had a wide distribution of sputum MPO levels (Fig 2) that were inversely correlated to their FEV1 levels (Fig 1A). In contrast, although other investigators have observed a negative correlation between plasma C-reactive protein (CRP), a frequently utilized systemic biomarker of inflammation, with lung function (FEV1) in (34, 35), we did not observe a significant correlation in this relatively small cohort of relatively stable adult outpatients with CF (Fig 1B). Our data indicate that systemic inflammatory markers (CRP) do not necessarily reflect the degree of inflammation in the CF airway.
Our observations with MPO were consistent with the reports of other investigators relating MPO levels to pulmonary function decrements in CF patients as a result of increased respiratory tract inflammatory processes (12, 29, 36–40). Of note, the −463GA MPO promoter polymorphism, in which the GG phenotype is found to result in increased MPO gene expression in leukocytes, has been reported to be associated with a more aggressive loss of pulmonary function in CF (41). Interestingly, this latter study also suggests a proinflammatory role of MPO in the early phase of CF, where severity of the disease was associated with higher expressing GG phenotype in the absence of documented bacterial infection (41).
Elevated oxidants in CF respiratory tract lining fluid (42), along with increased amounts of various MPO-derived oxidants and the abundant proteolytic processes accompanying inflammatory processes (13, 43), are important considerations in the pathobiology of CF respiratory tract injury. The results provided herein further support the notion that MPO may play a causative role in CF lung disease, and that current thiol antioxidant therapeutics (GSH and NAC) can influence the activity of MPO by different mechanisms. Additionally, our findings reveal that MPO activity may be an important parameter for determining the dose of thiol antioxidant required to achieve a therapeutic benefit.
Implications for thiol-based antioxidant therapeutic strategies in CF
Aerosolized “antioxidant therapy” represents a current therapeutic approach to counteract oxidative stress in CF airways (42). Several clinical trials using aerosolized antioxidants in CF patients have achieved beneficial effects (15–17, 22), although many others have not been as successful (19). Interestingly, although the positive studies have reported improvements in lung function, few changes have been observed in markers of oxidative stress (16). Surprisingly, most of the aerosolized antioxidant CF clinical trials have not reported the pre-/post-treatment sputum MPO levels/activity, inspite of MPO being one of the most abundant pro-oxidant constituents in CF respiratory tract lining fluids.
While not entirely surprising, data obtained from reacting sputum from CF patients with thiol antioxidants (Fig 4A and B) shows that higher sputum MPO levels require higher concentrations of antioxidants (GSH and NAC) to inhibit MPO activity (Fig 4A and 4B). Of interest, it was necessary to dilute CF sputum (sol-phase) samples approximately 2000 times in order to accurately quantify MPO activity. The ex vivo antioxidant incubations were also carried out in these diluted samples. Hence, using this dilution factor, the actual concentration of antioxidants to be theoretically administered to patients, with respect to MPO inhibition, may require respiratory tract lining fluid levels up to 50 mM to effectively inhibit the highly abundant MPO contained in CF respiratory tract secretions. Griese et al reported an increase in alveolar GSH levels and improved lung function, but not in biomarkers of oxidative state, after administering aerosolized GSH to CF patients (three times daily, doses of 300/450 mg GSH for 14 days) (16). At an inhaled dose of 450 mg, bronchoalveolar lavage (BAL) fluid taken 1 hr after the completion of the 14 day trial, GSH levels were 15.59 ± 7.18 Nmol/L as compared to baseline values of 3.83 ± 1.83 Nmol/L. Interestingly, oxidized GSH levels (GSSG) were 16.32 ± 4.27 Nmol/L as compared to baseline levels of 1.24 ± 0.33 Nmol/L (16). Our current data suggest that to effectively inhibit MPO-catalyzed reactions, GSH may need to be administered at much higher levels to achieve respiratory tract lining fluid concentrations of 1–50 mM (Fig 4A) in order to directly influence MPO activity. This might partially explain why the previous studies did not observe any changes in oxidative stress biomarkers in CF patients after administering aerosolized GSH (16).
A GSH pro-drug, NAC, is occasionally used for inhalation in CF patients but has significant side effects secondary to its acidic nature (pKa 2.2) which can induce bronchospasm. NAC has also been administered orally in CF patients to increase circulating GSH (44). Administration of high doses of NAC orally (0.6–1.0 g, three times daily for 4 weeks) increased neutrophil GSH content, decreased neutrophil influx in CF airways, decreased sputum elastase activity, and decreased sputum IL-8 levels; observations that were attributed to decreased airway inflammation (21). However, there was no improvement in lung function, as assessed by FEV1, possibly due to the relatively short duration of treatment. Of note, there are no clear data on GSH/GSSG levels in CF respiratory tract secretions after a high dose oral NAC administration and/or how NAC might modify the “oxidative stress” of CF airways.
As previously reviewed by Nash et al, either oral or nebulized (aerosolized) thiol derivatives have failed to show significant beneficial effects in CF disease (19). As observed in our ex vivo studies, similar to GSH, the higher the sputum MPO levels, the higher the concentration of NAC required to inhibit MPO activity (Fig 4B), and as for the case of GSH and NAC, a concentration 1–50 mM would appear to be required to inhibit the actions of the high quantities of MPO in the CF airway. Additionally, a significant portion of MPO may be trapped in the gel-phase of the sputum. Hence, one of the major questions still needing to be addressed (and a limitation of the current study) is how administered antioxidants interact with the gel-phase, as most of the proposed therapeutic antioxidants are water soluble and may have difficulty penetrating the highly viscous gel phase of sputum.
Proposed mechanisms of action of thiol antioxidants in CF sputum
N-Chloramines and their scavenging by GSH and NAC
As illustrated in Fig 8A, N-Chloramines are formed by MPO-catalyzed formation of HOCl and subsequent chlorination of amines (45). Somewhat surprisingly, we did not observe a significant correlation between sputum MPO levels and sputum N-chloramine levels (Fig 3). In fact, a negative correlation between N-chloramines and MPO in CF sputum has been previously observed (46). N-Chloramines are selectively reactive with specific molecular targets (ie. sulfur-containing amino acids) that makes them fairly labile in airway secretions and may not necessarily be expected to parallel MPO levels. Ex vivo incubation of CF sputum with GSH and NAC were able to substantially reduce sputum N-chloramine levels (Fig 5). N-chloramines are known to react with GSH, thus leading to GSH depletion (47). Comparing the two antioxidants, NAC appeared to be more potent in reducing N-chloramine levels as compared to GSH (eg. 80 ± 11% vs 62 ± 11%). Peskin and Winterbourn have reported that NAC (k= 46 ± 7 M−1sec−1) was approx. 2.5 times less reactive than GSH (115 ± 14 M−1sec−1) with taurine chloramine (48). However, in our data, NAC seemed to be more effective in scavenging N-chloramines in CF sputum. This potentially could be due to the smaller size of NAC as compared to GSH, that may allow NAC to gain entry to N-chloramines located in sterically-hindered sites within proteins. The reaction of thiol antioxidants with N-chloramines is illustrated in Fig 8A, and may represent one pathway by which thiol antioxidant therapeutics alter the inflammatory and oxidative processes occurring in the CF lung.
Figure 8. Proposed mechanisms by which GSH and NAC inhibit MPO-dependent reactions in the CF airway.

(A) Scavenging of N-chloramines. (B) Scavenging of organic radicals produced by MPO (non-competitive inhibition). (C) GSH-dependent inhibition of substrate binding to MPO (competitive inhibition).
The capacity of GSH to inhibit CF sputum-mediated p65 nuclear translocation in lung epithelial cell culture model (Fig 7) may demonstrate the ‘beneficial effect’ of GSH supplementation in controlling NF-κB mediated inflammatory-immune responses (33). N-chloramines are known to react with GSH (47), and may represent one possible mechanism by which NF-κB was activated by CF sputum (49). In the present study, GSH was able to scavenge CF sputum N-chloramines (Fig 6A) and also block CF sputum mediated p65 nuclear translocation (Fig 7). Of note, a previous study using significantly higher concentrations of N-chloramines has shown inhibition of NF-κB activity (50). Future studies are needed to further interrogate the substances in CF sputum that serve to modulate NF-κB activity in underlying respiratory tract cells, as well as dose-response relationships between the capacity of N-chloramines to modulate NF-κB activation and inhibition.
Scavenging of MPO-derived radicals by NAC and GSH
The present results indicate that the thiol antioxidants NAC and GSH can inhibit the MPO system in CF respiratory tract secretions. This could occur at a number of potential mechanistic points. First, it is theoretically possible that GSH and NAC could decrease MPO activity by simple scavenging of H2O2; that is, decreasing the substrate available to MPO. However, at pH 7.4, the second-order rate constants for reaction of NAC and GSH with H2O2 are very slow (0.16 and 0.87 M−1s−1, respectively) (51) and can’t compete with the very fast reaction of H2O2 with ground state ferric MPO (k = 2.6 × 107 M−1s−1) (52). Thus, it is unlikely this mechanism can explain the present results.
Another possible mechanism by which NAC and GSH could modulate MPO one-electron peroxidase activity is by direct reaction with either compound I or compound II of MPO. However, because of their structure and charge state, NAC and GSH are poor substrates for reaction with both compound I and II of MPO (k = 101–102 M−1s−1) (53). In fact, the reaction of our substrate molecule TMB is 4–5 orders of magnitude faster with MPO compound I and II (3.6 × 106 and 9.4 × 105 M−1s−1) (54), compared to NAC and GSH. Given the vast excess of TMB substrate in the biochemical assays, and its much faster reaction with MPO, it is unlikely that NAC and GSH inhibit MPO activity by directly reacting with compounds I and II of MPO.
Having excluded scavenging of H2O2 and largely negating the direct reactions with MPO, one reasonable explanation is that NAC and GSH exert their ‘inhibitory’ effects on MPO by scavenging radical species produced by MPO (in the present case TMB substrate radicals) (Fig 8B). There is precedence for such reactions (55, 56), and conjugates of GSH with TMB have been previously identified in horseradish peroxidase-catalyzed reactions (57). Taken together, scavenging of intermediate TMB radicals, or adduction of benzoquinonediimine intermediates, is one likely mechanism by which NAC and GSH alter MPO reactions occurring in CF sputum.
Competitive inhibition of MPO by GSH: A previously unrecognized interaction
Detailed kinetic analyses in our study have revealed that NAC and GSH inhibit MPO by two independent mechanisms. On the one hand, NAC inhibits MPO by a non-competitive mechanism (Fig 5B), suggesting an interaction/reaction of NAC with the MPO-TMB radical complex not at the TMB binding site (ie. consistent with the radical scavenging mechanism in Fig 8B). In contrast, GSH is a competitive inhibitor of MPO. Kinetic analyses shown in Fig 5A reveal that GSH increases the apparent Km for TMB as substrate for MPO, and this strongly suggests that GSH combines reversibly with MPO at or near the active site. Previous studies have revealed that GSH is a poor substrate for MPO (58), and that the oxidation of GSH by MPO is largely dependent upon the formation of HOCl (59). This, taken together with our data presented herein, suggests that GSH serves to inhibit binding and oxidation of MPO substrates without actually participating in the catalytic peroxidase cycle. While there are no data that we are aware of in the literature to document a binding site for GSH on MPO, there is precedence for a binding interaction between GSH and lactoperoxidase (60). Additionally, the microsomal heme protein prostaglandin E synthase-1 binds GSH and utilizes it as a cofactor in its catalytic cycle (61). It remains possible that a specific binding site for GSH exists at, or near, the substrate binding site/active site in MPO. Structural interrogation of the active site of MPO, combined with computational docking models, and binding experiments are needed to further understand the previously unrecognized interaction of MPO with GSH. Lastly, structure-activity relationships of diverse GSH derivatives remain future goals for potentially developing novel inhibitors of MPO for use in the treatment of CF.
Conclusions
The major aim of this investigation was to explore the effect of ex vivo antioxidants on MPO activity in CF respiratory tract secretions. It must be noted that CF sputum is a rich source of inflammatory biomarkers, containing abundant amounts of oxidants, proteases, bacterial products, chemokines, cytokines, etc (43, 62). It is thus very challenging to ‘single-out’ a molecule in CF sputum that could be contributing to the detrimental effect in respiratory tract epithelial cell NF-κB activation (63). As reported here, possible inhibition of NF- B activation processes in HBE1 cells by GSH-mediated consumption of N-chloramines remains speculative as other mechanisms cannot be excluded. Equally likely would be the GSH scavenging of other endogenously produced oxidants in CF sputum or even bacterial-derived oxidants such as pyocyanin (64). Different methods of CF sputum processing also significantly influence several parameters analyzed (62, 65). Based on the present study, we suggest that using fresh respiratory tract secretions collected from CF patients and using these fluids to characterize interactions of these secretions on airway epithelial cells represents a useful simulation of pathobiological events occurring in the airways of CF patients. Finally, we suggest that strategies designed to therapeutically address “oxidative stress” in CF patients may need to take into consideration the overall magnitude of MPO in CF respiratory tract secretions.
Acknowledgments
The authors would like to thank the patient volunteers who participated in the study. This work was supported by a Fellowship from the Cystic Fibrosis Research, Inc. (VTV), the Cystic Fibrosis Foundation support of the UCD Adult CF Program (BMM and CEC), the NIH (HL092506; CEC and JPE), pre-doctoral Training Grants in Comparative Respiratory Biology and Medicine (NHLBI HL07013; JSH) and Advanced Training in Environmental Health Sciences (T32 ES07059; KAH). We also wish to recognize the generous support of Pam Fair and Glen Sullivan.
References
- 1.Davis PB, Drumm M, Konstan MW. Cystic fibrosis. Am J Respir Crit Care Med. 1996;154:1229–1256. doi: 10.1164/ajrccm.154.5.8912731. [DOI] [PubMed] [Google Scholar]
- 2.Anderson MP, Gregory RJ, Thompson S, Souza DW, Paul S, Mulligan RC, Smith AE, Welsh MJ. Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science. 1991;253:202–205. doi: 10.1126/science.1712984. [DOI] [PubMed] [Google Scholar]
- 3.Donaldson SH, Boucher RC. Sodium channels and cystic fibrosis. Chest. 2007;132:1631–1636. doi: 10.1378/chest.07-0288. [DOI] [PubMed] [Google Scholar]
- 4.Tummler B, Puchelle E. CFTR:a multifaceted epithelial molecule. Trends Cell Biol. 1997;7:250–251. doi: 10.1016/S0962-8924(97)01062-3. [DOI] [PubMed] [Google Scholar]
- 5.Wine JJ. The genesis of cystic fibrosis lung disease. J Clin Invest. 1999;103:309–312. doi: 10.1172/JCI6222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Rose V. Mechanisms and markers of airway inflammation in cystic fibrosis. Eur Respir J. 2002;19:333–340. doi: 10.1183/09031936.02.00229202. [DOI] [PubMed] [Google Scholar]
- 7.Meyer KC. Neutrophils, myeloperoxidase, and bronchiectasis in cystic fibrosis: green is not good. J Lab Clin Med. 2004;144:124–126. doi: 10.1016/j.lab.2004.05.014. [DOI] [PubMed] [Google Scholar]
- 8.Schultz J, Kaminker K. Myeloperoxidase of the leucocyte of normal human blood. I. Content and localization. Arch Biochem Biophys. 1962;96:465–467. doi: 10.1016/0003-9861(62)90321-1. [DOI] [PubMed] [Google Scholar]
- 9.Eiserich JP, Hristova M, Cross CE, Jones AD, Freeman BA, Halliwell B, van der Vliet A. Formation of nitric oxide-derived inflammatory oxidants by myeloperoxidase in neutrophils. Nature. 1998;391:393–397. doi: 10.1038/34923. [DOI] [PubMed] [Google Scholar]
- 10.Hazen SL, Hsu FF, Gaut JP, Crowley JR, Heinecke JW. Modification of proteins and lipids by myeloperoxidase. Methods Enzymol. 1999;300:88–105. doi: 10.1016/s0076-6879(99)00117-2. [DOI] [PubMed] [Google Scholar]
- 11.Hawkins CL. The role of hypothiocyanous acid (HOSCN) in biological systems. Free Radic Res. 2009;43:1147–1158. doi: 10.3109/10715760903214462. [DOI] [PubMed] [Google Scholar]
- 12.Regelmann WE, Siefferman CM, Herron JM, Elliott GR, Clawson CC, Gray BH. Sputum peroxidase activity correlates with the severity of lung disease in cystic fibrosis. Pediatr Pulmonol. 1995;19:1–9. doi: 10.1002/ppul.1950190102. [DOI] [PubMed] [Google Scholar]
- 13.Van Der Vliet A, Nguyen MN, Shigenaga MK, Eiserich JP, Marelich GP, Cross CE. Myeloperoxidase and protein oxidation in cystic fibrosis. Am J Physiol Lung Cell Mol Physiol. 2000;279:L537–546. doi: 10.1152/ajplung.2000.279.3.L537. [DOI] [PubMed] [Google Scholar]
- 14.Cantin AM. Taurine modulation of hypochlorous acid-induced lung epithelial cell injury in vitro. Role of anion transport. J Clin Invest. 1994;93:606–614. doi: 10.1172/JCI117013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bishop C, Hudson VM, Hilton SC, Wilde C. A pilot study of the effect of inhaled buffered reduced glutathione on the clinical status of patients with cystic fibrosis. Chest. 2005;127:308–317. doi: 10.1378/chest.127.1.308. [DOI] [PubMed] [Google Scholar]
- 16.Griese M, Ramakers J, Krasselt A, Starosta V, Van Koningsbruggen S, Fischer R, Ratjen F, Mullinger B, Huber RM, Maier K, Rietschel E, Scheuch G. Improvement of alveolar glutathione and lung function but not oxidative state in cystic fibrosis. Am J Respir Crit Care Med. 2004;169:822–828. doi: 10.1164/rccm.200308-1104OC. [DOI] [PubMed] [Google Scholar]
- 17.Hartl D, Starosta V, Maier K, Beck-Speier I, Rebhan C, Becker BF, Latzin P, Fischer R, Ratjen F, Huber RM, Rietschel E, Krauss-Etschmann S, Griese M. Inhaled glutathione decreases PGE2 and increases lymphocytes in cystic fibrosis lungs. Free Radic Biol Med. 2005;39:463–472. doi: 10.1016/j.freeradbiomed.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 18.Jaques A, Daviskas E, Turton JA, McKay K, Cooper P, Stirling RG, Robertson CF, Bye PT, Lesouef PN, Shadbolt B, Anderson SD, Charlton B. Inhaled mannitol improves lung function in cystic fibrosis. Chest. 2008;133:1388–1396. doi: 10.1378/chest.07-2294. [DOI] [PubMed] [Google Scholar]
- 19.Nash EF, Stephenson A, Ratjen F, Tullis E. Nebulized and oral thiol derivatives for pulmonary disease in cystic fibrosis. Cochrane Database Syst Rev. 2009:CD007168. doi: 10.1002/14651858.CD007168.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roum JH, Borok Z, McElvaney NG, Grimes GJ, Bokser AD, Buhl R, Crystal RG. Glutathione aerosol suppresses lung epithelial surface inflammatory cell-derived oxidants in cystic fibrosis. J Appl Physiol. 1999;87:438–443. doi: 10.1152/jappl.1999.87.1.438. [DOI] [PubMed] [Google Scholar]
- 21.Tirouvanziam R, Conrad CK, Bottiglieri T, Herzenberg LA, Moss RB. High-dose oral N-acetylcysteine, a glutathione prodrug, modulates inflammation in cystic fibrosis. Proc Natl Acad Sci U S A. 2006;103:4628–4633. doi: 10.1073/pnas.0511304103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Visca A, Bishop CT, Hilton SC, Hudson VM. Improvement in clinical markers in CF patients using a reduced glutathione regimen: An uncontrolled, observational study. J Cyst Fibros. 2008 doi: 10.1016/j.jcf.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 23.de Oliveira-Marques V, Cyrne L, Marinho HS, Antunes F. A quantitative study of NF-kappaB activation by H2O2: relevance in inflammation and synergy with TNF-alpha. J Immunol. 2007;178:3893–3902. doi: 10.4049/jimmunol.178.6.3893. [DOI] [PubMed] [Google Scholar]
- 24.Thomas EL, Jefferson MM, Joyner RE, Cook GS, King CC. Leukocyte myeloperoxidase and salivary lactoperoxidase: identification and quantitation in human mixed saliva. J Dent Res. 1994;73:544–555. doi: 10.1177/00220345940730021001. [DOI] [PubMed] [Google Scholar]
- 25.Chapman AL, Morrissey BM, Vasu VT, Juarez MM, Houghton JS, Li CS, Cross CE, Eiserich JP. Myeloperoxidase-dependent oxidative metabolism of nitric oxide in the cystic fibrosis airway. J Cyst Fibros. 2010;9:84–92. doi: 10.1016/j.jcf.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dypbukt JM, Bishop C, Brooks WM, Thong B, Eriksson H, Kettle AJ. A sensitive and selective assay for chloramine production by myeloperoxidase. Free Radic Biol Med. 2005;39:1468–1477. doi: 10.1016/j.freeradbiomed.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 27.Yankaskas JR, Haizlip JE, Conrad M, Koval D, Lazarowski E, Paradiso AM, Rinehart CA, Jr, Sarkadi B, Schlegel R, Boucher RC. Papilloma virus immortalized tracheal epithelial cells retain a well-differentiated phenotype. Am J Physiol. 1993;264:C1219–1230. doi: 10.1152/ajpcell.1993.264.5.C1219. [DOI] [PubMed] [Google Scholar]
- 28.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 29.Kohlhoff SA, Kohler E, Sollich V. Serum myeloperoxidase in cystic fibrosis patients receiving systemic antibiotics. Scand J Infect Dis. 2007;39:163–166. doi: 10.1080/00365540600951259. [DOI] [PubMed] [Google Scholar]
- 30.Ordonez CL, Kartashov AI, Wohl ME. Variability of markers of inflammation and infection in induced sputum in children with cystic fibrosis. J Pediatr. 2004;145:689–692. doi: 10.1016/j.jpeds.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 31.Kaul N, Forman HJ. Activation of NF kappa B by the respiratory burst of macrophages. Free Radic Biol Med. 1996;21:401–405. doi: 10.1016/0891-5849(96)00178-5. [DOI] [PubMed] [Google Scholar]
- 32.Kaul N, Choi J, Forman HJ. Transmembrane redox signaling activates NF-kappaB in macrophages. Free Radic Biol Med. 1998;24:202–207. doi: 10.1016/s0891-5849(97)00209-8. [DOI] [PubMed] [Google Scholar]
- 33.Staal FJ, Roederer M, Herzenberg LA. Intracellular thiols regulate activation of nuclear factor kappa B and transcription of human immunodeficiency virus. Proc Natl Acad Sci USA. 1990;87:9943–9947. doi: 10.1073/pnas.87.24.9943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith JW, Colombo JL, McDonald TL. Comparison of serum amyloid A and C-reactive protein as indicators of lung inflammation in corticosteroid treated and non-corticosteroid treated cystic fibrosis patients. J Clin Lab Anal. 1992;6:219–224. doi: 10.1002/jcla.1860060410. [DOI] [PubMed] [Google Scholar]
- 35.Levy H, Kalish LA, Huntington I, Weller N, Gerard C, Silverman EK, Celedon JC, Pier GB, Weiss ST. Inflammatory markers of lung disease in adult patients with cystic fibrosis. Pediatr Pulmonol. 2007;42:256–262. doi: 10.1002/ppul.20563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mohammed JR, Mohammed BS, Pawluk LJ, Bucci DM, Baker NR, Davis WB. Purification and cytotoxic potential of myeloperoxidase in cystic fibrosis sputum. J Lab Clin Med. 1988;112:711–720. [PubMed] [Google Scholar]
- 37.Brown RK, Wyatt H, Price JF, Kelly FJ. Pulmonary dysfunction in cystic fibrosis is associated with oxidative stress. Eur Respir J. 1996;9:334–339. doi: 10.1183/09031936.96.09020334. [DOI] [PubMed] [Google Scholar]
- 38.Dominguez C, Gartner S, Linan S, Cobos N, Moreno A. Enhanced oxidative damage in cystic fibrosis patients. Biofactors. 1998;8:149–153. doi: 10.1002/biof.5520080125. [DOI] [PubMed] [Google Scholar]
- 39.Starosta V, Rietschel E, Paul K, Baumann U, Griese M. Oxidative changes of bronchoalveolar proteins in cystic fibrosis. Chest. 2006;129:431–437. doi: 10.1378/chest.129.2.431. [DOI] [PubMed] [Google Scholar]
- 40.Kim JS, Okamoto K, Rubin BK. Pulmonary function is negatively correlated with sputum inflammatory markers and cough clearability in subjects with cystic fibrosis but not those with chronic bronchitis. Chest. 2006;129:1148–1154. doi: 10.1378/chest.129.5.1148. [DOI] [PubMed] [Google Scholar]
- 41.Reynolds WF, Sermet-Gaudelus I, Gausson V, Feuillet MN, Bonnefont JP, Lenoir G, Descamps-Latscha B, Witko-Sarsat V. Myeloperoxidase promoter polymorphism -463G is associated with more severe clinical expression of cystic fibrosis pulmonary disease. Mediators Inflamm. 2006;2006:36735. doi: 10.1155/MI/2006/36735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cantin AM, White TB, Cross CE, Forman HJ, Sokol RJ, Borowitz D. Antioxidants in cystic fibrosis. Conclusions from the CF antioxidant workshop, Bethesda, Maryland, November 11–12, 2003. Free Radic Biol Med. 2007;42:15–31. doi: 10.1016/j.freeradbiomed.2006.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Downey DG, Bell SC, Elborn JS. Neutrophils in cystic fibrosis. Thorax. 2009;64:81–88. doi: 10.1136/thx.2007.082388. [DOI] [PubMed] [Google Scholar]
- 44.Cantin AM. Potential for antioxidant therapy of cystic fibrosis. Curr Opin Pulm Med. 2004;10:531–536. doi: 10.1097/01.mcp.0000138997.29276.a1. [DOI] [PubMed] [Google Scholar]
- 45.Thomas EL, Grisham MB, Jefferson MM. Myeloperoxidase-dependent effect of amines on functions of isolated neutrophils. J Clin Invest. 1983;72:441–454. doi: 10.1172/JCI110992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Witko-Sarsat V, Delacourt C, Rabier D, Bardet J, Nguyen AT, Descamps-Latscha B. Neutrophil-derived long-lived oxidants in cystic fibrosis sputum. Am J Respir Crit Care Med. 1995;152:1910–1916. doi: 10.1164/ajrccm.152.6.8520754. [DOI] [PubMed] [Google Scholar]
- 47.Witko-Sarsat V, Nguyen AT, Descamps-Latscha B. Immunomodulatory role of phagocyte-derived chloramines involving lymphocyte glutathione. Mediators Inflamm. 1993;2:235–241. doi: 10.1155/S0962935193000328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peskin AV, Winterbourn CC. Kinetics of the reactions of hypochlorous acid and amino acid chloramines with thiols, methionine, and ascorbate. Free Radic Biol Med. 2001;30:572–579. doi: 10.1016/s0891-5849(00)00506-2. [DOI] [PubMed] [Google Scholar]
- 49.Rahman I, Biswas SK, Jimenez LA, Torres M, Forman HJ. Glutathione, stress responses, and redox signaling in lung inflammation. Antioxid Redox Signal. 2005;7:42–59. doi: 10.1089/ars.2005.7.42. [DOI] [PubMed] [Google Scholar]
- 50.Midwinter RG, Cheah FC, Moskovitz J, Vissers MC, Winterbourn CC. IkappaB is a sensitive target for oxidation by cell-permeable chloramines: inhibition of NF-kappaB activity by glycine chloramine through methionine oxidation. Biochem J. 2006;396:71–78. doi: 10.1042/BJ20052026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Winterbourn CC, Metodiewa D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic Biol Med. 1999;27:322–328. doi: 10.1016/s0891-5849(99)00051-9. [DOI] [PubMed] [Google Scholar]
- 52.Bolscher BG, Wever R. A kinetic study of the reaction between human myeloperoxidase, hydroperoxides and cyanide. Inhibition by chloride and thiocyanate. Biochim Biophys Acta. 1984;788:1–10. doi: 10.1016/0167-4838(84)90290-5. [DOI] [PubMed] [Google Scholar]
- 53.Burner U, Jantschko W, Obinger C. Kinetics of oxidation of aliphatic and aromatic thiols by myeloperoxidase compounds I and II. FEBS Lett. 1999;443:290–296. doi: 10.1016/s0014-5793(98)01727-x. [DOI] [PubMed] [Google Scholar]
- 54.Marquez LA, Dunford HB. Mechanism of the oxidation of 3, 5, 3′, 5′-tetramethylbenzidine by myeloperoxidase determined by transient- and steady-state kinetics. Biochemistry. 1997;36:9349–9355. doi: 10.1021/bi970595j. [DOI] [PubMed] [Google Scholar]
- 55.Naumchik IV, Karasyova EI, Metelitza DI, Edimecheva IP, Sorokin VL, Shadyro OI. Inhibition of peroxidase-catalyzed oxidation of 3, 3, 5, 5 -tetramethylbenzidine by aminophenols. Biochemistry (Mosc) 2005;70:322–329. doi: 10.1007/s10541-005-0118-z. [DOI] [PubMed] [Google Scholar]
- 56.Karaseva EI, Losev Iu P, Metelitsa DI. Inhibition of peroxidase oxidation of 3, 3′, 5, 5′-tetramethylbenzidine and o-phenylenediamine by 1-amino-2-naphthol-4-sulfonic acid and its polysulfide. Prikl Biokhim Mikrobiol. 2001;37:713–721. [PubMed] [Google Scholar]
- 57.McGirr LG, O’Brien PJ. Glutathione conjugate formation without N-demethylation during the peroxidase catalysed N-oxidation of N, N′, N, N′-tetramethylbenzidine. Chem Biol Interact. 1987;61:61–74. doi: 10.1016/0009-2797(87)90019-6. [DOI] [PubMed] [Google Scholar]
- 58.Svensson BE, Graslund A, Strom G, Moldeus P. Thiols as peroxidase substrates. Free Radic Biol Med. 1993;14:167–175. doi: 10.1016/0891-5849(93)90007-h. [DOI] [PubMed] [Google Scholar]
- 59.Turkall RM, Tsan MF. Oxidation of glutathione by the myeloperoxidase system. J Reticuloendothel Soc. 1982;31:353–360. [PubMed] [Google Scholar]
- 60.Pommier J, Cahnmann HJ. Interaction of lactoperoxidase with thiols and diiodotyrosine. J Biol Chem. 1979;254:3006–3010. [PubMed] [Google Scholar]
- 61.Hamza A, Tong M, AbdulHameed MD, Liu J, Goren AC, Tai HH, Zhan CG. Understanding microscopic binding of human microsomal prostaglandin E synthase-1 (mPGES-1) trimer with substrate PGH2 and cofactor GSH: insights from computational alanine scanning and site-directed mutagenesis. J Phys Chem B. 114:5605–5616. doi: 10.1021/jp100668y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim JS, Hackley GH, Okamoto K, Rubin BK. Sputum processing for evaluation of inflammatory mediators. Pediatr Pulmonol. 2001;32:152–158. doi: 10.1002/ppul.1101. [DOI] [PubMed] [Google Scholar]
- 63.Blackwell TS, Stecenko AA, Christman JW. Dysregulated NF-kappaB activation in cystic fibrosis: evidence for a primary inflammatory disorder. Am J Physiol Lung Cell Mol Physiol. 2001;281:L69–70. doi: 10.1152/ajplung.2001.281.1.L69. [DOI] [PubMed] [Google Scholar]
- 64.Schwarzer C, Fu Z, Fischer H, Machen TE. Redox-independent activation of NF-kappaB by Pseudomonas aeruginosa pyocyanin in a cystic fibrosis airway epithelial cell line. J Biol Chem. 2008;283:27144–27153. doi: 10.1074/jbc.M709693200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dauletbaev N, Rickmann J, Viel K, Diegel H, von Mallinckrodt C, Stein J, Wagner TO, Bargon J. Antioxidant properties of cystic fibrosis sputum. Am J Physiol Lung Cell Mol Physiol. 2005;288:L903–909. doi: 10.1152/ajplung.00349.2004. [DOI] [PubMed] [Google Scholar]