

The ability to modify and directly target nanoparticulate carriers has greatly increased their applicability in diagnostic and therapeutic studies. Generally essential to the targeting of nanoparticles is the bioconjugation of targeting ligands to the agent’s surface. While bioconjugation techniques have steadily improved in recent years, the field is still plagued with inefficient conjugations reactions and/or the lack of site-specific coupling. To overcome these limitations, click chemistry and expressed protein ligation (EPL) were combined to produce a highly efficient, site-specific reaction. This new EPL-Click conjugation strategy was applied to create superparamagnetic iron oxide nanoparticles (SPIO) labeled with HER2/neu affibodies. These HER2-SPIO nanoparticles proved to be highly potent and receptor specific in both in vitro cell studies and murine tumor models. Moreover, when EPL-Click derived HER2-SPIO were compared with SPIO that had been labeled with HER2 affibodies using other popular bioconjugation methods, they produced a statistically significant improvement in contrast enhancement upon cell binding. The EPL-Click system was also successfully extended to other nanoparticle platforms, i.e. liposomes and dendrimers, highlighting the versatility of the approach.
1. Introduction
Superparamagnetic iron oxide (SPIO) nanoparticles (NPs) have emerged as an attractive class of magnetic resonance (MR) contrast agents, providing T2*-weighted contrast enhancement in MR imaging applications.[1] Due to their strong contrast-enhancing capabilities, SPIO have recently been evaluated as molecular imaging agents, whereby they are used to report the expression level of target cell-surface receptors to improve the specificity of disease detection. Generally essential to the effectiveness of any NP-based molecular imaging contrast agent is the successful bioconjugation of targeting molecules to the nanoparticle platform. Maleimide-, N-hydroxysuccinimide,[2, 3] and carbodiimide-based chemistries[4, 5] have traditionally been applied for this purpose, but their utility is hindered by low reaction efficiencies. This has recently led to a great deal of interest in bioconjugation strategies based on click chemistry, which offer stereospecificity and high reaction efficiencies.[6] The Cu(I)-catalyzed terminal azide-alkyne cycloaddition (CuAAC) has perhaps been the most widely adopted click chemistry reaction, with improved reaction efficiencies being reported in several SPIO functionalization studies.[7, 8] Click chemistry reactions, however, are also subject to some limitations. For example, the indiscriminate labeling of protein-based targeting molecules (e.g. antibodies) with click-reactive groups has the potential to render them nonfunctional. Moreover, it is generally not possible to control the orientation of the targeting molecule on the nanoparticle. Clearly, it would be beneficial to implement a technique that allows for the site-specific conjugation of targeting molecules to nanoparticles, with the targeting domains uniformly available for binding.
Recently, expressed protein ligation (EPL) has garnered some interest as a chemoselective bioconjugation method that allows for site-specific coupling reactions.[9] EPL refers to a native chemical ligation between a recombinant protein with a C-terminal thioester and a second agent with an N-terminal cysteine. The C-terminal thioester can readily be introduced onto any recombinant protein (i.e the targeting ligand) through the use of auto-processing proteins called inteins. Specifically, when an intein is cloned downstream of the targeting ligand, thiols (using 2-mercaptoethanesulfonic acid, MESNA) can be used to induce the site-specific cleavage of the intein, resulting in the formation of a reactive thioester. The thioester will then react with any agent that has an N-terminal cysteine.[10] EPL operates in a site-specific manner, and the reaction is known to be very efficient if both functional groups are in high concentrations.[11] Several studies have shown the efficiency of the reaction between small, unbound molecules to be at or near 100%. However, when EPL was applied to nanoparticle systems the reaction efficiencies were much lower,[12, 13] likely due to the limited nanoparticle concentrations that were obtainable.
While neither click chemistry nor EPL offer a perfect solution for bioconjugation, we investigated whether these methods could be combined to create a highly efficient, site-specific coupling strategy (Figure 1). In our approach, bacterially expressed HER2/neu-targeted Affibodies (HER2-Affibodies) were ligated to an alkynated-fluorescent peptide (AFP) via EPL. The product of this reaction, which we refer to as HER2-AFP, was then clicked to azide-labeled SPIO nanoparticles via the CuAAC reaction (HER2-SPIO). The targeting specificity of HER2-SPIO was assessed in vitro as well as in a murine tumor model. Moreover, HER2-SPIO prepared via the EPL-click approach was compared with conjugates prepared using carbodiimide chemistry and conventional CuAAC. In all of the studies, HER2-Affibodies were used as the targeting agent. Affibodies comprise a new class of high affinity ligands based on a protein scaffold derived from the IgG-binding domains of staphylococcal protein A.[14] These small (6.5 kDa), robust molecules have been shown to exhibit remarkable specificity and affinity (pM range) for the HER2/neu receptor.[15]
Figure 1.

Schematic of EPL-Click conjugation strategy. Expressed protein ligation between a HER2-Affibody containing a C-terminal thioester and an alkynated fluorescent peptide (AFP) containing an N-terminal cysteine results in the chemoselective attachment of a “clickable” alkyne group onto the affibody (HER2-AFP). Subsequent Cu(I)-catalyzed terminal alkyne-azide cycloaddition (CuAAC) between azide modified SPIO-NPs and HER2-AFP results in the site-specific attachment of the HER2-Affibody onto the SPIO NPs.
2. Results
2.1. HER2-Affibody Expression and Expressed Protein Ligation
Expression of the recombinant HER2-Affibody and the efficiency of the Her2-Affibody/AFP ligation were assessed by 16% Tricine SDS PAGE. Figure 2 shows the migration of the HER2-Affibody to an expected molecular weight of 6.6 kDa in lane 1. The HER2-AFP ligand, which was run in the adjacent lane, settled at a molecular weight of 8 kDa. Fluorescent images of the gel revealed significant fluorescence emitting from the HER2-AFP ligand in lane 2, but not from the unreacted HER2-Affibody in lane 1. The absence of an unligated HER2-Affibody band in lane 2 indicates that the HER2-Affibody was efficiently consumed by the AFP.
Figure 2.

Tricine SDS-PAGE analysis of HER2-Affibodies before and after expressed protein ligation with the alkynated-fluorescent peptide (AFP). A white light image of the gel reveals that the AFP was successfully conjugated to HER2-affibodies, as evidenced by the higher molecular weight of the conjugate. Formation of the conjugate was further confirmed by fluorescent imaging of the gel, which showed that only the HER2-AFP was fluorescent.
To ensure that the HER2-Affibody retained its ability to bind the HER2/neu receptor following ligation to AFP, HER2-AFP conjugates were incubated with HER2/neu-positive T6-17 cells and the cells were subsequently analyzed via flow cytometry (Figure S1A). A clear shift in the flow cytometry histogram was observed compared with unlabeled cells, confirming cell binding. Specificity was confirmed by performing competitive inhibition studies with excess free affibody (i.e. without the AFP). The free affibody prevented binding of HER2-AFP (Figure S1B), suggesting that the interactions were indeed specific. Further, no cell labeling was seen when HER2-AFP were incubated with HER2/neu-negative NIH/3T3 cells (Figure S1C). Analogous findings were obtained when HER2-AFP was incubated with HER2/neu-positive SK-BR-3 cells and HER2/neu-negative HCC38 cells (See Figure S2).
2.2. CuAAC Conjugation
To confirm that HER2-AFP could be efficiently attached to the azide-labeled SPIO nanoparticles via the CuAAC reaction in controllable fashion, a fixed concentration of SPIO (2 mg Fe/mL) was incubated with increasing concentrations of the HER2-AFP ligand. Since the unconjugated SPIO NPs do not elicit a fluorescent signal, the fluorescence intensity of the “clicked” SPIO following magnetic purification was used to determine the extent of labeling. As can be seen in Figure 3, the fluorescence intensity of HER2-SPIO increased with increasing HER2-AFP concentrations up to a saturating concentration of 30 μM HER2-AFP ligand.
Figure 3.

Controlled labeling of azide-modified SPIO with HER2-AFP. The saturation curve presented was generated by conjugating increasing concentrations of HER2-AFP to a fixed concentration of SPIO NPs (2 mg Fe/mL) using the EPL-Click conjugation strategy.
The functionality of HER2-SPIO was subsequently assessed by conducting cell-binding assays with HER2/neu-positive (T6-17) and -negative (NIH/3T3) cells. Flow cytometric analysis revealed strong labeling of T6-17 cells (Figure 4A). Competitive inhibition studies, performed with an excess of free affibody, confirmed that HER2-SPIO binding was specific (Figure 4B). Further, cell labeling was undetectable when HER2-SPIO was incubated with HER2/neu negative NIH/3T3 cells (Figure 4C). Analogous findings were obtained when HER2-SPIO was incubated with HER2/neu positive SK-BR-3 cells and HER2/neu negative HCC38 cells (See Figure S3).
Figure 4.

Flow cytometric analysis of cells incubated with HER2-SPIO. (A) Flow cytometry histogram of HER2/neu-positive T6-17 cells incubated in the presence of HER2-SPIO (grey line). (B) Flow cytometry histogram of HER2/neu-positive T6-17 cells incubated in the presence of HER2-SPIO and an excess of free HER2-affibody (grey line). (C) Flow cytometry histogram of HER2/neu-negative NIH/3T3 cells incubated in the presence of HER2-SPIO (grey line). Histograms of unlabeled cells are also shown (black line).
2.3. Internalization of HER2-Affibody SPIO
Cell-labeling studies were carried out at 4°C and 37°C to determine whether HER2-AFP and/or HER2-SPIO were internalized, following binding to the HER2/neu receptor on SK-BR-3 cells. At 4°C, fluorescence for both the HER2-AFP ligand and HER2-SPIO was constrained to the cell membrane (Figure S4), which was expected since receptor-mediated internalization is stalled at this temperature.[16] At 37°C, fluorescence from the HER2-AFP ligand remained on the outer cell surface; however, the HER2-SPIO appeared as punctate fluorescent spots. Live-cell staining with the lysosomal labeling dye, Lysotracker Red, indicated that the majority of SPIO were likely within lysosomes.
2.4. In vivo Targeting of HER2-Affibody SPIO
To examine whether HER2-SPIO could be used to effectively bind HER2/neu-positive tumors in living subjects, axial T2*-weighted magnetic resonance (MR) images of mice with T6-17 cell xenografts were acquired precontrast and 24 hours after retro-orbital injection of HER2-SPIO (10 mg Fe/kg) (Figure 5). In the pre-contrast images, there was little discernible intrinsic contrast between the tumor xenograft and surrounding muscle. At 24 hours, significant negative contrast was apparent at the site of the tumor relative to the surrounding muscle. When HER2-SPIO were injected into mice bearing HER2/neu negative tumor xenografts (NIH/3T3 cells), very little negative contrast was noted between the pre- and post-contrast images. Quantitative analysis of the MR images revealed that HER2-SPIO generated a statistically significant improvement (p < 0.05) in image contrast in HER2/neu-positive tumors compared with pre-contrast images and HER2/neu-negative tumors (Figure 5E).
Figure 5.

MR images of SCID mice pre-injection and 24 hours post- injection of HER2-SPIO NPs and corresponding image analysis. Mice with HER2/neu-positive T6-17 tumor xenografts were imaged (A) pre-injection and (B) 24 hours post retro-orbital injection of 10 mg Fe/kg HER2-SPIO NPs. Tumors are indicated by white arrows. Mice with HER2/neu-negative NIH/3T3 tumor xenografts were imaged (C) pre- and (D) post-injection of HER2-SPIO NPs. (E) Quantitative analysis of MR images. Signal intensity for each tumor was normalized to surrounding muscle tissue and the relative signal intensity, rSI, was calculated as the quotient of the 24-hour post-injection image and the pre-injection image. A t-test (two-tailed, unequal variance) was used to compare the rSI for each group of animals. A p<0.05 was considered statistically significant and is indicated by an asterisk.
2.5. Comparison of EPL-Click Bioconjugations to Popular Conjugation Methods
The benefits of attaching HER2-affibodies to SPIO NPs in a site-specific manner was highlighted by comparing the functionality of HER2-SPIO, prepared using the EPL-click approach, to HER2-SPIO prepared using two popularly employed conjugation methods, namely CuAAC and carbodiimide chemistries. The CuAAC approach involved the indiscriminate labeling of available affibody amines with propargyl-dPEG4-NHS ester. CuAAC was then used to attach the alkynated affibody to azide-labeled SPIO. The carbodiimide approach required the conversion of amines on the SPIO to carboxyl groups using succinic anhydride. These carboxyls were then directly coupled to available amines on the affibody using EDC/Sulfo-NHS chemistry. Both the CuAAC and carbodiimide method were optimized as previously described.[7] Cell binding assays were carried out by incubating HER2-SPIO conjugates with T6-17 cells for 1 hour at a final concentration of 150 μg/mL Fe. Since HER2-SPIO prepared using CuAAC and carbodiimide-based chemistries lack fluorescent tags, comparisons in cell binding were based on T2-relaxation times of cell pellets. All three conjugates showed a marked decrease in T2-relaxation times when compared to blank cells, with the EPL-click derived SPIO showing the greatest decrease (Figure 6). Quantitative analysis revealed that the EPL-click derived SPIO produced a statistically significant decrease in T2-relaxation time when compared to blank cells and cells labeled with HER2-SPIO prepared using CuAAC and carbodiimide chemistries (p < 0.05). Additionally, T2*-weighted MR images were collected for each cell pellet to highlight the impact of achieving improved cell targeting (Figure 6, right panel).
Figure 6.
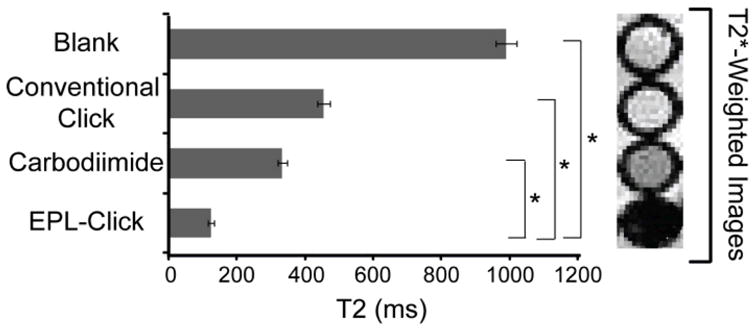
Comparison of HER-SPIO prepared via EPL-click, Carbodiimide, and CuAAC bioconjugation techniques. From the same stock of SPIO NPs, HER2-SPIO were generated by means of carbodiimide chemistry, conventional click chemistry or EPL-Click chemistry. Potency of each HER2-SPIO conjugate was tested by incubating 150 μg Fe/mL with 4 × 106 T6-17 cells. T2 relaxation times were collected for each cell sample as well as T2*-weighted MR images of the respective cell pellets. A t-test (two-tailed, unequal variance) was used to compare the relative signal intensity for each group of samples. A p<0.05 was considered statistically significant and is indicated by an asterisk.
2.6. Applicability of EPL-click to other Nanoparticle Platforms
To underscore the versatility of the EPL-click conjugation strategy, HER2-AFP ligands were “clicked” to other popular nanoparticle platforms, i.e. fifth-generation PAMAM dendrimers and liposomes. The functionality of the HER2/neu-targeted dendrimers and liposomes was subsequently assessed by conducting cell-binding assays and evaluating the extent of cell labeling by flow cytometry. Both the targeted dendrimer and liposome conjugates efficiently labeled T6-17 cells (Figure 7). Specificity of each nanoparticle platform was confirmed through competitive inhibition assays using an excess of free HER2 Affibodies and by performing cell binding assays on HCC38 cells. Fluorescent microscopy was also performed on T6-17 cells labeled with HER2/neu targeted liposomes. Rhod-PE was incorporated into the liposomal membrane so that the liposome could be directly imaged. Further, HER2-AFP could be imaged through the attached fluorescein dye. Merging the two images confirmed the co-localization of the two fluorescent signals (Figure S5).
Figure 7.

Flow cytometric analysis of cells incubated with HER2-dendrimers or HER2-liposomes. (A) Flow cytometry histogram of HER2/neu-positive T6-17 cells incubated in the presence of HER2-dendrimers (grey line). (B) Flow cytometry histogram of Her2/neu-positive T6-17 cells incubated in the presence of HER2-dendrimer and an excess of free HER2-affibody (grey line). (C) Flow cytometry histogram of HER2/neu-negative HCC38 cells incubated in the presence of HER2-dendrimer (grey line). (D) Flow cytometry histogram of HER2/neu-positive T6-17 cells incubated in the presence of HER2-liposomes (grey line). (E) Flow cytometry histogram of Her2/neu-positive T6-17 cells incubated in the presence of HER2-liposomes and an excess of free HER2-affibody (grey line). (F) Flow cytometry histogram of HER2/neu-negative HCC38 cells incubated in the presence of HER2-liposomes (grey line). Histograms of unlabeled cells are also shown (black line).
3. Discussion
Prior to combining EPL with click chemistry, we used EPL to directly couple HER2-Affibodies to cysteine-labeled SPIO nanoparticles; however, the EPL proved to be extremely inefficient between the small protein and the large nanoparticle. These findings were recently corroborated by several reports where proteins were attached to liposomes and dendrimers via EPL, also with low efficiency.[12, 13]
The investigated EPL reaction between expressed Affibodies and the AFP has several advantages over direct ligation to nanoparticles. Foremost, the EPL reaction involving the AFP is highly efficient. PAGE gel analysis revealed a complete consumption of the HER2-Affibody by the AFP during EPL, as is evident by the absence of an unligated HER2-Affibody band in lane 2 (Figure 2). Subsequent click conjugation to various nanoparticles was also highly efficient, allowing for potentially 100% of the expressed protein to be conjugated to the nanoparticle’s surface. Utilization of the AFP also maintains the site-specificity and chemoselectivity of the conjugation, ensuring proper orientation of the targeting ligands following click conjugation with SPIO NPs. While the proposed conjugation strategy does introduce an extra step to the conjugation process, the ligation with an intermediate peptide also allows for the introduction of additional functionality. For example, in this study a fluorophore was readily introduced onto the Affibody, which allowed us to confirm HER2-AFP coupling to SPIO. It also allowed us confirm binding of HER2-SPIO to cells via fluorescence microscopy and flow cytometry. Of course, aside from fluorophores other functional groups could also be incorporated onto the alkynated peptide and ultimately HER2-SPIO, including haptens, metal chelates, chromophores, etc.[17–19] As the size of these agents are relatively small, they are not expected to impede the efficiency of the EPL reaction.
The EPL-Click reaction is also highly controllable. Figure 3 demonstrates the degree of control provided by the EPL-Click strategy. Several iterations of HER2-SPIO nanoparticles were created with increasing densities of HER2-Affibodies on the SPIO surface up to a saturating concentration. This degree of control is often unachievable with popular crosslinking chemistries (e.g. carbodiimide chemistries), which require saturating levels of ligand to achieve a successful conjugation. For this study a saturating density of HER2-Affibodies on the SPIO NPs was utilized for targeting studies; however, several studies have shown saturating ligand densities are not ideal for producing the highest degree of cell labeling and biomolecule binding.[20–22] Therefore, it is envisioned that the control afforded by the EPL-click strategy could have a significant impact on the optimization of targeted nanoparticles in both diagnostic and therapeutic applications.
The potency of EPL-Click derived HER2-SPIO NPs was evident in both in vitro and in vivo cell targeting studies. Flow cytometric analysis of cell binding experiments revealed a high degree of cell labeling on HER2/neu-positive T6-17 and SK-BR-3 cells. Moreover, HER2-SPIO was shown to possess a high specificity for the HER2/neu receptor, as was evidenced by both competitive inhibition assays and a lack of binding to HER2/neu-negative HCC38 and NIH/3T3 cells. Internalization studies further revealed that conjugating HER2-Affibodies to SPIO NPs augmented their cell labeling abilities by facilitating their internalization, most likely through receptor mediated endocytosis.
Specific targeting of the HER2/neu receptor with HER2-SPIO was also demonstrated in vivo in a murine tumor model. Visual analysis of tumor images underscores the striking difference in negative contrast between HER2/neu positive and HER2/neu negative tumor xenografts. Quantitative analysis of MR images revealed that HER2-SPIO targeted to HER2/neu positive cells showed a statistically significant improvement in image contrast compared to HER2-SPIO targeted to HER2/neu negative cells (p < 0.05). It is surprising that the negative control tumors did not experience a larger signal decrease, since a small extent of HER2-SPIO were expected to accumulate in the tumor due to enhanced permeability and retention (EPR) effects. It is hypothesized that although HER2-SPIO likely did accumulate in the tumor, the concentration was too low to generate negative contrast, a product of the low sensitivity of MR.
To fully underscore the versatility of EPL-click conjugations, the strategy was applied to fifth-generation PAMAM dendrimers and liposomes. Similar to the SPIO NPs, EPL-Click derived HER2/neu targeting dendrimers and liposomes were able to effectively and specifically target T6-17 cells. Further, both HER2-dendrimers and HER2-liposomes did not associate with HER2/neu negative HCC38 cells. Because the fluorescence detected in the flow cytometry studies is inherent to the HER2-AFP ligand and not the nanoparticles, it was necessary to ensure that the HER2-AFP ligand was in fact bound to the nanoparticle surface. Since the liposome was doped with Rhodamine PE and the HER2-AFP ligand was labeled with a FITC fluorophore, fluorescent microscopy confirmed the colocalization of the two fluorescent signals on labeled cells.
Perhaps the best demonstration of the advantages derived from the EPL-Click conjugation strategy can be seen from the comparison study performed utilizing carbodiimide chemistries, conventional click chemistry, and EPL-Click chemistry. HER2-Affibodies were conjugated to SPIO nanoparticles using the aforementioned chemistries, creating three distinct stocks of HER2-SPIO. As the ultimate application of these HER2-SPIO conjugates is for diagnostic evaluation of tumor malignancies, determining each conjugates ability to lower a cells spin-spin relaxation time (T2) was used as the most critical assessment of each conjugates applicability in vivo. Additionally, as each HER2-SPIO conjugate was derived from the same stock of SPIO NPs, there exist no expected discrepancies in their magnetic characteristics.
As befits their widespread application in the literature, all three HER2-SPIO conjugates were successful in significantly lowering the T2 times of HER2/neu positive cells; however, HER2-SPIO conjugates from the EPL-Click strategy showed the most dramatic reduction in T2 relaxation times. It is hypothesized that the site-specific attachment of HER2-AFP to SPIO via the EPL-Click reaction can account for the differences in T2-relaxation times. Both conventional click and carbodiimide chemistries couple the primary amines of the HER2-Affibody to SPIO indiscriminately. The HER2-Affibody is a lysine rich protein (>10%), providing multiple amines through which a conjugation can be made. Therefore, conjugation through many of these amines can produce a poorly orientated HER2-Affibody on the SPIO surface, which we believe will lower its effectiveness as a targeting agent.
While the benefits derived from a site-specific click conjugation were expected, it was surprising that the carbodiimide derived HER2-SPIO bound cells better than the conventional click derived HER2-SPIO. It has been well established that carbodiimide chemistries are plagued by low reaction efficiencies while click chemistry reactions have been shown to be up to 100% efficient with small molecules and proteins. We hypothesize that the binding of click-reactive ligands to lysine residues on the HER2-Affibody interfered with normal protein function. It is common practice to functionalize targeting ligands by incubating them with an excess of click-reactive ligands, and accordingly it is expected that many of the lysines on the affibody will be coupled with these click ligands.[7, 23] Affinity maturation studies have shown that at least one lysine residue in the HER2-Affibody directly influences the protein’s affinity,[14] thus it is reasonable to assume that coupling through this amino acid could diminish the affibody’s targeting abilities.
The effect that improved cell binding has on improving image contrast is evident from the T2*-weighted images of labeled cell pellets (Figure 6). While all three HER2-conjugates decreased the T2-relaxation times of targeted cells, only HER2/neu positive cells from the carbodiimide and EPL-click targeted studies could be visualized in the MR images. The discrepancies between decreases in T2-relaxation times and negative contrast can likely be attributed to the specific pulse sequence that was applied and the intrinsic sensitivities of the systems.
4. Conclusions
The evolution of bioconjugation techniques towards more efficient, chemoselective reactions serves to greatly expand the role of NP carriers in diagnostic imaging and drug delivery applications. Herein, we have shown that the benefits afforded by expressed protein ligation and click chemistry conjugations can be combined to produce a highly efficient, site-specific conjugation strategy. With the ever growing library of peptide and protein-based targeting ligands being developed from phage display and homology studies, it is expected that the utility provided by the EPL-Click conjugation strategy will lead to its expanded application in the functionalization of nanoparticle carriers.
5. Experimental Section
5.1. Materials
Azido-dPEG4-NHS ester and propargyl-dPEG-NHS ester were purchased from Quanta BioDesign Ltd. (Powell, OH). The SPIO coating material, dextran T10, was purchased from Pharmacosmos (Denmark). Human breast carcinoma (HCC38), human breast adenocarcinoma (SK-BR-3) and mouse fibroblast (NIH/3T3) cell lines were obtained from the American Type Culture Collection (Manassas, VA). NIH/3T3 cells that were engineered to stably express the Her2/neu receptor (T6-17) were kindly provided by Mark Greene, MD/PhD (University of Pennsylvania). Bathocuproinedisulfonic acid (BCS) was acquired from Acros Organics (Geel, Belgium). Hydrogenated soy phosphatidylcholine (HSPC), cholesterol, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (Rhod-PE), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000] (NH2-PEG(2000)-DSPE) were obtained from Avanti Polar Lipids (Alabaster, AL). PAMAM dendrimers (ethylenediamine core, generation 5) were purchased as methanol solutions from Dendritech Inc. (Midland, MI). The 70 mm volume coil used for radiofrequency transmission and reception was purchased from Insight Neuroimaging Systems, LLC (Worcester, MA). All other reagents were purchased from Thermo Fisher Scientific (Waltham, MA) unless otherwise noted.
5.2. SPIO NPs Synthesis and Amination
SPIO NPs were prepared by chemical coprecipitation, as previously described.[24] Briefly, 0.7313 g FeCl2 and 1.97 g FeCl3 were each dissolved in 12.5 mL diH2O and added to 25g dextran T10 in 50 mL diH2O at 4°C. Ammonium hydroxide (15 mL) was slowly added to this mixture, turning the light yellow–colored solution black. This NP slurry was then heated to 90°C for 1 hour and cooled overnight. Purification of SPIO NPs was accomplished by ultracentrifugation of the mixture at 20,000 relative centrifugal force (RCF) for 30 minutes. Pellets were discarded, and the supernatant was subjected to diafiltration against greater than 20 volumes of 0.02 M citrate, 0.15 M sodium chloride buffer using a 100 kDa cutoff membrane filter (GE Healthcare). The purified particles were then cross-linked by reacting the particles (10 mg Fe/mL) with 25% (v/v) 10 M NaOH and 33% epichlorohydrin. After mixing for 24 hours, the particles were briefly dialyzed and then functionalized with amines by adding 25% ammonium hydroxide. This reaction was allowed to continue for another 24 hours followed by diafiltration as described above.
5.3. SPIO Characterization
The hydrodynamic diameter of SPIO NPs was measured to be 30 nm by dynamic light scattering using a Zetasizer Nano-z (Malvern Instruments, Malvern, UK). SPIO NPs were diluted in phosphate-buffered saline (PBS) to a concentration of approximately 0.5 mg Fe/mL and read in triplicate. The values reported are the intensity peak values. The transverse relaxivity (R2) of the SPIO was determined to be 89 mM−1s−1 using a Bruker mq60 MR relaxometer. T2 measurements were made using τ = 1.5 milliseconds and two dummy echoes and fitted assuming monoexponential decay.
5.4. Liposome Preparation
The desired lipid components, HSPC/cholesterol/NH2-PEG(2000)-DSPE, were dissolved in chloroform at a at a molar ratios of 55:40:5. A small molar percentage (0.1%) of Rhod-PE was also doped during film preparation. Lipids were dried of organic solvent using a stream of N2, and then further dried under vacuum for at least 4 hours to ensure removal of all the chloroform. The sample was then hydrated with aqueous buffer. Following incubation of the sample in a 55 °C water bath for 30 minutes, the mixture was subjected to ten freeze-thaw-vortex cycles in liquid nitrogen and warm water (55°C), respectively. Finally, the solution was extruded (21 times) through a 100-nm Nuclepore polycarbonate filter using a stainless steel extruder (Avanti Polar Lipids, Inc.).
5.5. Azide Modification of Liposomes, Dendrimers and SPIO NPs
Surface amines on liposomes, dendrimers and SPIO NPs were reacted with the amine-reactive azido-dPEG4-NHS, diluted 10 times from stock in dimethyl sulfoxide (DMSO), in 0.1 M sodium phosphate buffer, pH 9. The linker was added at 100 times molar excess to the nanoparticles. All nanoparticles solutions were mixed for 8 hours at room temperature. Liposomes and SPIO NPs were purified via superdex 200 chromatography columns (GE Healthcare, Piscataway, NJ). Dendrimer nanoparticles were purified by several rounds of washing on Ultracel 3,000 MWCO filters (Millipore, Billerica, MA).
5.6. Cloning of HER2-Affibody recombinant protein into pTXB1 vector
Two complimentary oligonucleotides encoding the HER2-Affibody amino acid sequence[14] and flanked at both ends by 15 base sequences homologous to the desired restriction sites of the destination vector were ordered from Integrated DNA Technologies (Coralville, IA). To improve subsequent affinity column cleavage, an additional 9 base pairs encoding a “MRM” amino acid sequence were included in the oligonucleotides at the C-terminal end of the HER2-Affibody sequence. The full nucleotide and amino acid sequence for the HER2-Affibody can be found in Supporting Information (Figure S6). Oligonucleotides were incubated together at a final concentration of 5 μM and hybridized at room temperature for 30 minutes. The resulting HER2-Affibody sequence was gel purified and directly ligated with gel-purified NdeI-XhoI double digested pTXB1 vector (New England Biolabs, Inc) via the CloneEZ kit (Genscript). Insertion of the HER2-Affibody sequence was verified by DNA sequencing using the T7 promoter as the sequencing primer.
5.7. Expression and Purification of HER2-Affibody recombinant protein
The pTXB1-HER2-Affibody vector was transformed in Rosetta 2(DE3)pLysS Competent Cells (Novagen). Bacterial cell cultures were initially grown overnight in an air shaker (225 rpm) at 37 °C in 3 mL of LB medium. Cultures were scaled up to fifty mL of LB medium and grown overnight under the same conditions, and then inoculated into 1 L LB containing 50 mg/L of ampicillin. At OD600 nm = 0.6, IPTG was added at a final concentration of 0.5 mM to induce T7 RNA polymerase-based expression. Cultures were allowed to express for 2 hours at 37 °C. Bacterial cultures were centrifugally pelleted at 10,000 x g for 5 minutes, resuspended in 5 mL of column buffer (20 mM Na-HEPES, 0.5 M NaCl, 1 mM EDTA, pH 8.5) containing 0.75 g/L lysozyme and 50 mM phenylmethylsulfonyl fluoride. Cells were lysed by pulse sonication on ice. Cells were centrifuged at 15,000 g for 30 minutes at 4 °C. Supernatant was collected and stored at −20 °C. For the following purification steps, all procedures were run at 4 °C. One mL of the supernatant was incubated for 10 minutes in a 10 mL Poly-Prep chromatography column (Bio-Rad, Hercules, CA) packed with 1 mL of chitin beads (New England Biolabs, Inc). Supernatant was allowed to pass through the column and chitin beads were washed with 50 mL of column buffer at a flow rate of approximately 2 mL/min. Three mL of 50 mM MESNA was quickly passed through the column in order to evenly distribute the MESNA throughout the chitin beads, and flow was stopped. The column was incubated for 16 hours at 4 °C. HER2-Affibody proteins, now containing a C-terminal thioester, were eluted from the column in a total 4 mL buffer (0.1 M Tris-HCl, pH 8.5) and concentrated to a volume of 500 μL using an Ultracell 3,000 (Millipore, Billerica, MA). Tricine-SDS-PAGE was used to identify the presence of the HER2-Affibody protein.
5.8. Expressed Protein Ligation
Expressed protein ligation was carried about between the thioester containing HER2-Affibody and an alkynated fluorescent peptide (AFP) with an N-terminal cysteine. The sequence of the AFP was NH2-CDPEK(5-FAM)DSG-D(Pra)-CONH2. The K(5-FAM) represents a lysine with a fluorescein covalently attached to its ε-amino group and the D(Pra) represents a glycine with a propargyl group (i.e. alkyne) attached to its side-arm. The AFP (0.1 mM) was incubated with approximately 0.01 mM HER2-Affibody. The EPL reaction was mixed overnight at room temperature. The EPL product and excess AFPs were separated on a Superdex 30 chromatography column. 16% Tricine-SDS-PAGE gels were used to visualize the separation between reacted and unreacted HER2-Affibodies. Gels were run in accordance with previously published methods for the visualization of small proteins.[25]
5.9. Alkynation of HER2-Affibody
For conventional CuAAC reactions, alkyne functionalization of HER2-Affibodies was accomplished by the addition of 10% v/v propargyl-dPEG12-NHS in DMSO to approximately 5 mg/mL HER2-Affibody in 0.1 M sodium phosphate buffer, pH 9. HER2-Affibodies were purified on Superdex 30 column equilibrated with PBS and then reconcentrated using Ultracel 3,000 filters. HER2-Affibody concentrations were assessed spectrophotometrically at 280 nm using a molar extinction coefficient of 1280 M−1cm−1.
5.10. CuAAC Conjugation
Azido-SPIO NPs (2 mg/mL) were mixed with increasing concentrations of HER2-AFP ligand, 5 mM BCS, 1 mM CuSO4 and 5 mM sodium ascorbate. Reactions were mixed overnight at room temperature and then purified on MACS MS columns ((Miltenyi Biotec, Bergisch Gladbach, Germany) equilibrated with PBS. For conventional click reactions, 30 μM of the alkynated-HER2 Affibody was click conjugated to azido-SPIO NPs according to the conditions above.
5.11. Carboxylation of SPIO NPs
Carboxylated NPs were prepared by reacting the amine-functionalized NPs (described above) with an excess of succinic anhydride in basic solution. Specifically, to 450 μL of NH2-SPIO NPs (5 mg Fe/mL) in 0.02 M citrate buffer, pH 8, 40 μL of 1 M NaOH was added followed by 40 μL of 4 M succinic anhydride in dimethylformamide. The reaction was allowed to mix overnight. Carboxylated SPIO NPs were subsequently precipitated three times in four volumes of isopropanol to remove excess reactants.
5.12. Carbodiimide Conjugation
HER2-Affibodies were conjugated to SPIO-NPs using carbodiimide chemistries as previously described.[7] Briefly, carboxyl groups on the SPIO-NPs were reacted with 50 mM EDC and 200 mM sulfo-NHS to produce reactive NHS esters on the SPIO surface. SPIO were precipitated in four volumes of isopropanol and excess reactants were removed. SPIO-NPs were then resuspended in 100 μL of HER2-Affibodies (5 mg/mL) for 24 hours. Subsequent reaction products were purified via MACS MS columns equilibrated with PBS.
5.13. Cell Culture
NIH/3T3 and T6-17 cells were cultured and maintained in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin at 37°C and 5% CO2. SK-BR-3 cells were cultured and maintained in McCoy’s 5A medium supplemented with 10% FBS and 1 penicillin/streptomycin at 37°C and 5% CO2. HCC-38 cells were cultured and maintained in RPMI 1640 medium supplemented with 10% FBS and 1 penicillin/streptomycin at 37°C and 5% CO2.
5.14. Flow Cytometric Analysis
Cells were dissociated from culture flasks using PBS-based enzyme free dissociation buffer and transferred to sterile 96-well plates at a final concentration of 50,000 cells per well. HER2-AFP ligand or HER2-SPIO were added to the wells for 30 minutes at 37°C at a final concentration of 1 μM and 100 μg Fe/mL, respectively. Cells were transferred to 1.5 mL centrifuge tubes and washed in triplicate by pelleting cells at 1000 RCF for 3 minutes and then resuspending in PBS. Cells were resuspended in 250 μL of PBS and seeded in a 96-well plate (50,000 cells per well) and analyzed using a Guava Easycyte Plus system (Guava Technologies, Hayward, CA). Flow cytometry data were analyzed using FlowJo software (TreeStar Inc., San Francisco, CA).
5.15. Internalization study
SK-BR-3 cells were seeded in chambered coverglass slides at a concentration of 10 × 103 cell per chamber. HER2-AFP ligands and HER2-SPIO were incubated with cells for 1 hour at final concentrations of 1 μM and 125 μg Fe/mL respectively. After labeling, cells were washed in triplicate and incubated with 5 nM Lyostracker Red (Invitrogen) for 5 minutes at 37°C. Images were acquired with an Olympus IX 81 inverted fluorescence microscope using a LUC PLAN 40× objective (numerical aperture 0.6; Olympus) and an X-cite 120 excitation source (EXFO, Quebec, QC). Micrographs were acquired using a back-illuminated electron multiplying charge-coupled device camera (Andor Technology PLC, Belfast, Northern Ireland).
5.16. Cell Relaxation Studies
T6-17 cells were dissociated using PBS-based enzyme free dissociation buffer and transferred to sterile 48-well plates at a concentration of 4 × 106 cells per well. HER2-SPIO conjugates were incubated with these cells in the 48-well plate at a final concentration of 150 μg Fe/mL for 1 hour at 37°C (n=3 for each targeting agent). Cells were transferred to 1.5 mL centrifuge tubes and washed in triplicate by pelleting cells at 1 RCF for 3 minutes and then resuspending in PBS. Cells were suspended in a final volume of 300 μL PBS and T2 measurements were taken using the benchtop relaxometer.
5.17. Cell Pellet MR Imaging
Following relaxation measurements, T6-17 cells were combined and centrifugally pelleted. Cells were then transferred to a 96-well plate that was cut into a smaller piece (62 mm × 80 mm, 12 × 106 cells per well). The 96-well plate was centrifugally spun at 2,000 RCF for 2 minutes and the supernatant was carefully removed from each cell pellet. The cells were then imaged on 9.4-T magnet interfaced to a Varian INOVA console using a 70 mm inner diameter volume coil for radiofrequency transmission and reception. T2*-weighted gradient echo (GRE) MR images were collected using parameters as follows: repetition time (TR) = 200 ms, echo time (TE) = 5 ms, flip angle = 20°, slice thickness = 0.5 mm, number of acquisitions = 8.
5.18. In vivo MR Imaging
Approximately 6-week old female Fox Chase SCID mice (Charles River Laboratory, Charles River, MS) were maintained in accordance with the Institutional Animal Care and Use Committee of the University of Pennsylvania. Mice were anesthetized via isoflurane and T6-17 or NIH/3T3 cells were injected subcutaneously into the back right flank (2 × 106 cells in 0.2 mL PBS). Tumors were grown to an approximate size of 100 mm3 and pre-contrast tumor images were acquired using a 9.4-T magnet interfaced to a Varian INOVA console. T2*-weighted GRE images were collected using parameters as follows: TR = 200 ms, TE = 5 ms, flip angle = 20°, number of acquisitions = 8, slice thickness = 1 mm. Immediately following the MR scan, HER2-SPIO nanoparticles were injected retro-orbitally ( 10 mg/kg Fe in 0.2 mL). Post-contrast images were collected 24 hours post-injection under the same imaging parameters used for pre-contrast images.
Supplementary Material
Footnotes
This work was supported in part by the National Institute of Health (NCI) R21 CA-132658, (NCI) R21 CA-140695, and the Department of Defense Breast Cancer Research Program of the Office of the Congressionally Directed Medical Research Programs (W81XWH-07-1-0457). We would also like to thank Hilary Harris, Mia Maamuri, Mark Mitchell and Mustafa Tumen for their contributions to this work
Supporting Information is available on the WWW under http://www.small-journal.com or from the author.
References
- 1.Thorek DL, Chen AK, Czupryna J, Tsourkas A. Ann Biomed Eng. 2006;34:23. doi: 10.1007/s10439-005-9002-7. [DOI] [PubMed] [Google Scholar]
- 2.Hwu JR, Lin YS, Josephrajan T, Hsu MH, Cheng FY, Yeh CS, Su WC, Shieh DB. J Am Chem Soc. 2009;131:66. doi: 10.1021/ja804947u. [DOI] [PubMed] [Google Scholar]
- 3.Zhao M, Kircher MF, Josephson L, Weissleder R. Bioconjug Chem. 2002;13:840. doi: 10.1021/bc0255236. [DOI] [PubMed] [Google Scholar]
- 4.Kocbek P, Obermajer N, Cegnar M, Kos J, Kristl J. J Control Release. 2007;120:18. doi: 10.1016/j.jconrel.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 5.Tsourkas A, Shinde-Patil VR, Kelly KA, Patel P, Wolley A, Allport JR, Weissleder R. Bioconjug Chem. 2005;16:576. doi: 10.1021/bc050002e. [DOI] [PubMed] [Google Scholar]
- 6.Kolb HC, Finn MG, Sharpless KB. Angew Chem Int Ed Engl. 2001;40:2004. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 7.Thorek DL, Elias DR, Tsourkas A. Mol Imaging. 2009;8:221. [PubMed] [Google Scholar]
- 8.von Maltzahn G, Ren Y, Park JH, Min DH, Kotamraju VR, Jayakumar J, Fogal V, Sailor MJ, Ruoslahti E, Bhatia SN. Bioconjug Chem. 2008;19:1570. doi: 10.1021/bc800077y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berrade L, Camarero JA. Cell Mol Life Sci. 2009;66:3909. doi: 10.1007/s00018-009-0122-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flavell RR, Muir TW. Acc Chem Res. 2009;42:107. doi: 10.1021/ar800129c. [DOI] [PubMed] [Google Scholar]
- 11.Muralidharan V, Muir TW. Nat Methods. 2006;3:429. doi: 10.1038/nmeth886. [DOI] [PubMed] [Google Scholar]
- 12.van Baal I, Malda H, Synowsky SA, van Dongen JL, Hackeng TM, Merkx M, Meijer EW. Angew Chem Int Ed Engl. 2005;44:5052. doi: 10.1002/anie.200500635. [DOI] [PubMed] [Google Scholar]
- 13.Reulen SW, Brusselaars WW, Langereis S, Mulder WJ, Breurken M, Merkx M. Bioconjug Chem. 2007;18:590. doi: 10.1021/bc0602782. [DOI] [PubMed] [Google Scholar]
- 14.Orlova A, Magnusson M, Eriksson TL, Nilsson M, Larsson B, Hoiden-Guthenberg I, Widstrom C, Carlsson J, Tolmachev V, Stahl S, Nilsson FY. Cancer Res. 2006;66:4339. doi: 10.1158/0008-5472.CAN-05-3521. [DOI] [PubMed] [Google Scholar]
- 15.Nygren PA. FEBS J. 2008;275:2668. doi: 10.1111/j.1742-4658.2008.06438.x. [DOI] [PubMed] [Google Scholar]
- 16.Nielsen UB, Kirpotin DB, Pickering EM, Drummond DC, Marks JD. BMC Immunol. 2006;7:24. doi: 10.1186/1471-2172-7-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garanger E, Weissleder R, Josephson L. Bioconjug Chem. 2009;20:170. doi: 10.1021/bc800392t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garanger E, Blois J, Hilderbrand SA, Shao F, Josephson L. J Comb Chem. 2010;12:57. doi: 10.1021/cc900141b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Helmick L, Antunez de Mayolo A, Zhang Y, Cheng CM, Watkins SC, Wu C, LeDuc PR. Nano Lett. 2008;8:1303. doi: 10.1021/nl073144l. [DOI] [PubMed] [Google Scholar]
- 20.Hellman P, Andersson L, Eriksson H. Cell Immunol. 2009;258:123. doi: 10.1016/j.cellimm.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 21.Heldt CL, Gurgel PV, Jaykus LA, Carbonell RG. Biotechnol Prog. 2009;25:1411. doi: 10.1002/btpr.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuan H, Zhang S. Applied Physics Letters. 2010:96. doi: 10.1063/1.3309593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun EY, Josephson L, Weissleder R. Mol Imaging. 2006;5:122. [PubMed] [Google Scholar]
- 24.Thorek DL, Tsourkas A. Biomaterials. 2008;29:3583. doi: 10.1016/j.biomaterials.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schagger H. Nat Protoc. 2006;1:16. doi: 10.1038/nprot.2006.4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.