

Abstract
Late-onset GM2 gangliosidosis is an autosomal recessive, neurodegenerative, lysosomal storage disease, caused by deficiency of β-hexosaminidase A (Hex A), resulting from mutations in the HEXA (Tay–Sachs variant) or the HEXB (Sandhoff variant) genes. The enzyme deficiency in many patients with juvenile or adult onset forms of the disease results from the production of an unstable protein, which becomes targeted for premature degradation by the quality control system of the smooth endoplasmic reticulum and is not transported to lysosomes. In vitro studies have shown that many mutations in either the α or β subunit of Hex A can be partially rescued, i.e. enhanced levels of both enzyme protein and activity in lysosomes, following the growth of patient cells in the presence of the drug, pyrimethamine. The objectives of the present clinical trial were to establish the tolerability and efficacy of the treatment of late-onset GM2 gangliosidosis patients with escalating doses of pyrimethamine, to a maximum of 100 mg per day, administered orally in a single daily dose, over a 16-week period. The primary objective, tolerability, was assessed by regular clinical examinations, along with a panel of hematologic and biochemical studies. Although clinical efficacy could not be assessed in this short trial, treatment efficacy was evaluated by repeated measurements of leukocyte Hex A activity, expressed relative to the activity of lysosomal β-glucuronidase. A total of 11 patients were enrolled, 8 males and 3 females, aged 23 to 50 years. One subject failed the initial screen, another was omitted from analysis because of the large number of protocol violations, and a third was withdrawn very early as a result of adverse events which were not drug-related. For the remaining 8 subjects, up to a 4-fold enhancement of Hex A activity at doses of 50 mg per day or less was observed. Additionally marked individual variations in the pharmacokinetics of the drug among the patients were noted. However, the study also found that significant side effects were experienced by most patients at or above 75 mg pyrimethamine per day. We concluded that pyrimethamine treatment enhances leukocyte Hex A activity in patients with late-onset GM2 gangliosidosis at doses lower than those associated with unacceptable side effects. Further plans are underway to extend these trials and to develop methods to assess clinical efficacy.
Keywords: GM2 gangliosidosis, Pyrimethamine, Pharmacologic chaperone
Introduction
GM2 gangliosidosis (GM2), (MIM 230700), is an inherited neurodegenerative disorder characterized by progressive deterioration of motor, cerebral and spinocerebellar function caused by deficiency of lysosomal β-hexosaminidase A (Hex A). Normal human tissues contain two major β-hexosaminidase (Hex) isozymes, Hex A and Hex B. Hex A is a heterodimer made up of non-identical α and β subunits, encoded by two evolutionarily related genes, HEXA and HEXB, respectively. Hex B is a homodimer made up of two identical β-subunits. A third minor, unstable Hex isozyme, Hex S, is a dimer of α-subunits and is only detected unequivocally in tissues from patients with the Sandhoff disease variant (SD) (MIM 268800) of GM2. SD results from HEXB mutations producing a deficiency of functional β-subunits and both Hex A (αβ) and Hex B (ββ) activities. On the other hand, Tay–Sachs disease (TSD; MIM 272800) is caused by HEXA mutations resulting in a deficiency of α-subunits, and Hex A (αβ) activity, but normal levels of Hex B (reviewed in [1]). Deficiency of the non-catalytic GM2 activator protein, a substrate-specific cofactor for Hex A, results in the third very rare AB-variant form of GM2.
GM2 is characterized by a marked clinical heterogeneity. The most severe and devastating forms are the infantile or acute variants of TSD and SD, associated with <0.5% of normal Hex A activity. It is characterized by rapid neurodegeneration, culminating in death in infancy. By contrast, the late-onset variants, which are subdivided into juvenile or sub-acute and adult or chronic forms, are associated with residual Hex A activities 2–4% of normal. Asymptomatic individuals have been described with as low as 10% of normal Hex A activity, i.e. pseudo-deficiencies. Patients with juvenile GM2 usually present with evidence of neurodeterioration starting after one year of age, and experience a slower rate of progression than patients with the infantile forms. Patients with adult-onset disease may present with spinocerebellar, psychiatric and/or peripheral neuropathies, which do not significantly affect longevity in some cases (reviewed in [1,2]). The rate of disease progression and severity has been found to correlate roughly with the level of residual Hex A activity: generally, clinical disease does not develop unless residual Hex A activity drops below a critical threshold of 5–10% of normal, as measured in patient fibroblasts. Thus, only a low level of residual Hex A activity is apparently needed to prevent or reverse substrate-storage in these conditions [3], i.e. 1.5- to 3-fold enhancement of Hex A levels in late onset patients.
Pharmacological chaperones (PC) are small molecules which are often also competitive inhibitors of their target enzyme. Thus PCs bind to and stabilize the “native” folded conformation of the protein. This results in a functional, active enzyme, unless the mutation affects a functional residue in the enzyme; e.g. αR178H [4] associated with the B1-variant of TSD [5]. Arg178 in the α-subunit is directly involved in first binding the substrate and then in stabilizing the reaction intermediate [6]. Once formed into its native structure, the enzyme ceases to be a substrate for the endoplasmic reticulum (ER)-associated degradation pathway (ERAD) and is transported to lysosomes [7]. In lysosomes it is believed that the stored substrate will displace the PC and continue to stabilize the enzyme. However, some PCs, like pyrimethamine (PYR), have the added advantage of inhibiting their target enzyme better at the neutral pH of the ER than at the acidic pH of lysosomes. Additional stability is gained once a lysosomal enzyme is folded correctly in the ER, because disulfide bonds are then formed [8], and the mutant subunit assembles with a wild-type subunit to form the Hex A heterodimer. Dimerization involves an extensive subunit-subunit interface, ~2700 Å2, which occurs between the catalytic domains of the two subunits, with several residues from one subunit structurally completing and stabilizing active site residues of the other [6]. Stability is further increased upon lysosomal compartmentalization, because Hex A, like most lysosomal enzymes; e.g., glucocerebrosidase [9], is more stable at acidic pH than at the neutral pH of the ER, i.e. the melting temperature of wild-type Hex A increases from 52 °C at pH 7.0 to 59 °C at pH 4.3 (Tropak et al., unpublished data). The PC approach has been shown to enhance the enzyme levels of five different mutant lysosomal enzymes causing chronic forms of the lysosomal storage diseases, GM2 gangliosidosis [4,10], Fabry [11], Gaucher [9,12–14], and GM1 gangliosidosis [15].
By screening a 1040-compound library of FDA approved drugs obtained from the National Institute of Neurological Diseases and Stroke (NINDS) for Hex inhibitors, we identified PYR as a μM competitive inhibitor of Hex and thus a potential PC [4]. Pyrimethamine is a selective inhibitor of parasitic dihydrofolate reductase (folate antagonist) [http://www.rxlist.com/daraprim-drug.htm]. It is approved for the treatment of chloroquine-resistant malaria and malaria prophylaxis and toxoplasmosis treatment. It is not generally recommended alone for the treatment of acute malaria, but is used for malaria prophylaxis in situations where the parasite is not resistant to the drug. It is used as a first-line treatment of toxoplasmosis, along with sulfonamides, particularly in immunocompromised individuals. The drug is used in dosages up to 75 mg per day in adults. The toxic side effects relate primarily to hypersensitivity reactions and the folate antagonist properties of the drug and are preventable by concomitant treatment with folinic acid.
The characterization of the relevant PC properties of the drug was performed using fibroblast cell-lines from TSD- or SD-variant patients with subacute (juvenile) and late-onset (adult) GM2. Each of the mutant cell lines from our patients’ responded differently to PYR [4,16]. Recently an increased turn-over rate, comparable to the enhancement levels of Hex A, of a fluorescent derivative of GM2 ganglioside loaded into the lysosomes of adult Tay–Sachs fibroblasts treated with 12 μM (~3 mg/L) PYR has been demonstrated [17]. However one potential problem is that PYR contains two primary amines that could, if high intra-lysosomal concentrations were reached, decrease lysosomal pH leading to increased secretion of lysosomal enzymes through inhibition of mannose-6-phosphate receptor recycling, i.e. a lysosomotropic effect. In tissue culture the level of PYR needed to produce such an effect was ~100 μM (~25 mg/L) [4].
The present study was undertaken to examine the potential clinical benefit of the treatment of late-onset forms of both TSD- and SD-variants of GM2 gangliosidosis with doses of PYR similar to those used for the treatment of parasitic diseases, such as malaria. The protocol was designed as a Phase I/II clinical trial with a primary focus on the establishment of the tolerability of the treatment and indications of efficacy based on measurements of leukocyte Hex A activity, and the levels of PYR in patients’ plasma. Clinical assessments were done frequently, primarily to identify early evidence of adverse drug-related reactions. The clinical assessments were not sufficiently sensitive to identify any subtle clinical improvements in the patients during this short-term study.
Subjects
All the subjects, both males and females, were 18 years of age or older. All had biochemically and genetically confirmed GM2 gangliosidosis caused by known HEXA or HEXB mutations, with clinical characteristics consistent with juvenile- or adult-onset disease. Each provided formal, signed consent for participation in the study, with the consent forms and protocol approved by the Institutional Review Boards of the Hospital for Sick Children (Toronto) and the New York University Medical Center (New York).
Inclusion criteria
biochemically and genetically confirmed diagnosis of GM2 gangliosidosis caused by β-hexosaminidase deficiency resulting from mutations in the HEXA or HEXB genes;
having HEXA or HEXB mutations shown to be responsive to PYR in vitro;
18 years of age or over at the time of study initiation;
able to understand and cooperate with the requirements of the study protocol;
mentally competent, have the ability to understand and willingness to sign the informed consent form;
able to travel to one of the two participating study sites;
women of child-bearing potential must use accepted contraceptive methods and must have a negative serum or urine pregnancy test within one week prior to treatment initiation;
fertile men must practice effective contraceptive methods during the study period, unless documentation of infertility exists;
laboratory values≤2 weeks prior to beginning the trial must show adequate hematologic, hepatic, renal, and coagulation function; and body weight >40 kg.
Exclusion criteria
serious medical illness, significant cardiac disease or severe debilitating pulmonary disease;
any hematologic abnormality, especially megaloblastic anemia, leukopenia, thrombocytopenia, pancytopenia;
any active uncontrolled bleeding or any bleeding diathesis (e.g., active peptic ulcer disease);
possible folate deficiency, and those receiving therapy (such as phenytoin) affecting folate levels;
any complex disease that may confound treatment assessment;
pregnant women or women of child-bearing potential not using reliable means of contraception;
lactating females;
fertile men unwilling to practice contraceptive methods during the study period;
unwilling or unable to follow protocol requirements;
known hypersensitivity reactions, intolerance or adverse reactions to PYR;
evidence of active infection, or serious infection within the past month;
HIV infection;
a history of cancer of any type;
receiving any other standard or investigational treatment for any indication within the past 4 weeks prior to initiation of PYR treatment;
receiving immunotherapy of any type within the past 4 weeks prior to initiation of PYR treatment; or any condition or abnormality which may, in the opinion of the investigator, compromise the safety of patients.
Study design
The study was a two-center, open-label, study of 10 patients with late-onset forms of GM2 gangliosidosis, examining the effect of escalating doses of PYR to a maximum of 100 mg per day administered orally. All subjects received folinic acid, 5 mg per day, throughout the study. Tolerability was assessed by regular clinical assessments, as well as frequent measurements of a wide range of hematologic and biochemical parameters. Efficacy was evaluated by weekly measurements of leukocyte Hex A activity normalized to β-glucuronidase (Glcr) activity to compensate for variations in the quality of the leukocyte pellet preparation and to monitor for any non-specific lysosomotropic effects of the treatment. Additionally, levels of PYR and Glcr were measured in plasma samples in order to roughly assess the drug’s pharmacokinetics and as a secondary method of detecting any lysosomotropic effects of the treatment, respectively.
Protocol
The clinical trial protocol is summarized in Table 1.
Table 1.
Outline of clinical trial protocol.
| Parameters | Treatment period (weeks) |
|||||||
|---|---|---|---|---|---|---|---|---|
| Pre |
PYR treatment 25 mg/day |
50 mg/day |
75 mg/day |
100 mg/day |
Post |
|||
| −2 to 0 | 1 | 2–3 | 4 | 5–8 | 9–12 | 13–16 | 17–18 | |
| General | ||||||||
| Medical history | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |
| Pregnancy test for woman of child-bearing potential | ✓ | ✓ | ✓ | ✓ | ||||
| Height | ✓ | ✓ | ✓ | ✓ | ||||
| Body weight | ✓ | ✓ | ✓ | ✓ | ||||
| Concomitant meds querya |
✓ | ✓ | Weekly | ✓ | Weekly | ✓ | Weekly | Weekly |
| Safety assessment | ||||||||
| Physical exam including vital signsb | ✓ | ✓ | ✓ | 8th wk | 12th wk | 16th wk | 18th wk | |
| Adverse event querya |
✓ | ✓ | Weekly | ✓ | Weekly | Weekly | Weekly | Weekly |
| Clinical pathologyc | ✓ | 4th wk | 12th wk | 16th wk | 18th wk | |||
| Urinalysis | ✓ | 4th wk | 8th wk | 12th wk | 16th wk | 18th wk | ||
| Efficacy assessment | ||||||||
| Plasma and leukocyte Hex A and B, β-glucuronidase and acid phosphatased | ✓ | ✓ | Weekly | ✓ | Weekly | Weekly | Weekly | Weekly |
| Reserve bloods for measurement of GM2e | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |
| Reserve bloods for measurement of PYRe | ✓ | ✓ | Weekly | ✓ | Weekly | Weekly | Weekly | Weekly |
Concomitant meds query and adverse event query may be done by telephone.
Pulse rate, respiratory rate, blood pressure, and temperature.
Clinical pathology can be done at a local lab, includes chemistry, hematology and coagulation. Renal function will be assessed utilizing the Cockcroft–Gault formula and the serum creatinine.
These measurements must be done at least 3 times at intervals of not less than 1 week BEFORE starting PYR. The last measurement can be done on the same day as PYR treatment is started, before the patient receives the first dose of the drug.
The total volume of blood needed is estimated on the basis of the needs for lysosomal enzyme assays, PYR assays, and the safety labs: Enzyme assays, 10 mL; PYR assays, 5 mL; Safety labs, 15 mL.
Materials and methods
Isolation of peripheral blood leukocyte pellets
Two samples of venous blood (8–12 mL each) collected into EDTA containing tubes were divided into equal aliquots for centrifugation (1550g for 5 min at 10 °C). The two upper phases corresponding to plasma were combined and stored at −80 °C. To the lower phase containing pelleted red cells and nucleated blood cells, 0.9% saline was added to a final volume of ~8 mL. Then 2–3 mL of freshly prepared 5% Dextran (MW150,000–200,000) in 0.7% saline was added and the tubes mixed by repeated inversion. The samples were allowed to stand at room temperature for approximately 30–45 min until the red cells had settled out. The supernatants containing leukocytes were transferred to new 10 mL tubes for centrifugation (2 min as above).
To each leukocyte pellet 4 mL of 0.2% saline were added and re-suspended by gentle up and down pipetting with a wide bore pasteur pipette for about 1 min to lyse any remaining red cells. The leukocyte suspension was brought to isotonicity by addition of 3.2 mL 1.8% NaCl and centrifuged for 2 min (as above). These steps were repeated once. The final leukocyte pellets were stored at −80 °C. When required samples were shipped frozen on dry ice to the Hospital for Sick Children for enzyme assays.
Preparation of leukocyte pellet lysates for lysosomal enzyme assays
Frozen leukocyte pellets resuspended in 0.9% saline were sonicated by using a sonic dismembrator (Systems Corporation) with a micro probe at the maximum power setting of 60. Each sample, immersed in ice water, was sonicated three times for 6 second bursts with a 6 seconds rest period between each burst. The sonicates were transferred to microcentrifuge tubes and spun at maximum speed for 10 min in a benchtop centrifuge. The supernatant was transferred and used for enzyme analyses.
Lysosomal enzyme assays
The following synthetic fluorogenic substrates, 4-methylumbelliferyl-β-D-glucuronide (MUGlcr) purchased from Sigma-Aldrich (Canada), and 4-methylumbelliferyl-2-acetamido-2-deoxy-β-D-glucopyranoside-6-sulfate (MUGS) from Toronto Research Chemicals (Canada), were used to assay Glcr, and Hex A, respectively. The levels of Hex A activity were measured using 50 μL of white cell lysate (0.9% sodium chloride), to which 50 μL of 0.5% (w/v) human serum albumin (Hex-free) and 100 μL MUGS (3.2 mM), dissolved in McIlvaine buffer (0.2 M Sodium Phosphate; 0.1 M citric acid) pH 4.5, were added. After incubation at 37 °C for 2 h, the reaction was stopped by the addition of 1 mL of 0.1 M 2-amino 2-methyl 1-propanol (MAP), pH 10.5. Fluorescence was measured using an excitation wavelength of 365 nm and emission wavelength of 450 nm as previously described [18]. Glcr was measured in a similar manner except that only 20 μL of cell lysate or plasma was used. For lysate, 80 μL of human serum albumin (0.5% w/v) and 100 μL of MUGlcr (10 mM), dissolved in 0.2 M sodium acetate buffer pH 5.0, were added and the mixture incubated for 1 h at 37 °C. For plasma, the human serum albumin was omitted and 80 μL of the acetate buffer was added along with the 100 μL of substrate. All enzyme assays were done in triplicate. Protein was determined by the method of Lowry [19].
Immunoselection assay to determine Hex A levels in leukocyte lysates from Sandhoff patients
Because leukocytes from Sandhoff patients contain significant amounts of Hex S that are also enhanced upon treatment with a PC [10], Hex A was first separated from Hex S using an immobilized sheep anti-β-subunit IgG as previously described [4]. The immobilized Hex A was then directly assayed using MUGS as described above.
Mass spectrometric (MS) quantitation of plasma pyrimethamine levels
Patient plasma (50 μL) was precipitated with 4 volumes of acetonitrile. The precipitate was pelleted using a microcentrifuge at maximum speed for 15 min at 4 °C. The resulting supernatant (200 μL) was dried down in a speedvac. PYR levels were determined in the AIMS Laboratory at the Dept. of Chemistry, University of Toronto, Canada. Each dried sample was resuspended in methanol (10 μL) and applied via an Autosampler (Leap Technologies HTS PAL) onto a Gemini-NX C18 50 mm×4.6 mm, 3 μ column backed with a C18 guard column connected to an HPLC (Agilent) equipped with a binary pump (G1312A) and Degasser (G132A). Between each sample the injection syringe and valves were washed three times with methanol followed by three washes with water. Mobile phases consisted of A) 0.1% formic acid Milli-Q Water, and B; Methanol. Using this gradient PYR had a retention time of 4.24 min. Material from the column (flow rate 500 μL/min) was injected onto an Applied Biosystems (MDS Sciex, Toronto, Canada) API4000 triple-quadrapole mass spectrometer operating in positive MRM mode. PYR levels were monitored using parent to product ion transitions of 248.9–> 233.1 with DF and CE set to 53, 27 and 35 and 19, respectively. Dwell time for each compound was set to 100 ms. For each batch of unknown samples, a standard curve for PYR (known concentration spiked into control plasma) spanning the range 1 nM to 1000 nM were generated with an LOQ of 3 nM (average accuracy 97.1%, range 90.1–102%).
Results
Patient characteristics
The baseline characteristics of the initial 11subjects participating in the study are shown in Table 2. One subject, 1001, undergoing screening was rejected from participation because of mild thrombocytopenia, which was considered probably related to prior treatment with a different drug. A large number of protocol violations were encountered with subject 1005, making interpretation of the results of analysis impossible. The data from this subject were not included in subsequent analyses of the results. Subject 1011 experienced a severe psychotic episode, unrelated to PYR treatment, at the outset of the trial and she was withdrawn before any data other than base-line Hex A values, were obtained relevant to the study. Thus the data presented in this report are from 8 patients (1002–1004 from Toronto and 1006–1011 from New York, Table 2).
Table 2.
Characteristics of subjects enrolled in the clinical trial.
| Subject | Age | Sex | Hex A, % control | Mutationsa | Baselineb | Stagec | Comments |
|---|---|---|---|---|---|---|---|
| 1001 | 36 | M | NDd | αW474C/Nulle | 3 | Screen failure | |
| 1002 | 33 | M | 6.3±0.8f | αW474C/Null | 3 | I | Withdrawal, seizures |
| 1003 | 25 | M | 2.9±0.6 | βR505Q/IVS11 +5 g>a | 3 | III | Withdrawal, dizziness, incoordination |
| 1004 | 23 | M | 3.3±0.4 | βR505Q/IVS11 +5 g>a | 4 | III | Withdrawal, altered consciousness |
| 1005 | 36 | F | 3.3±0.7 | αG269S/Null | 3 | III | Amenorrhea, drug-related |
| 1006 | 23 | F | 3.1±0.2 | αG269S/Null | 2 | III | Nausea, blurred vision, drug-related |
| 1007 | 50 | M | 4.0 | αG269S/Null | 3 | III | Increased ataxia, drug-related |
| 1008 | 25 | M | 3.6±1.5 | αG269S/Null | 2 | III | Increased tremors, mobility worse |
| 1009 | 30 | M | 3.6 | αG269S/Null | 4–5 | III | Weakness, malaise, drug-related |
| 1010 | 44 | M | 4.7 | αG269S/Null | 2 | III | Metallic taste, drug-related |
| 1011 | 38 | F | 3.9 | αG269S/Null | 4–5 | I | Withdrawal, psychotic episode |
β—SD and α—TSD patient.
Clinical status: 1, pre-symptomatic; 2, mildly impaired; 3, moderately impaired; 4, severely impaired; 5, very severely impaired.
Most advanced dosage completed: I, 25 mg/day; II, 50 mg/day; III, 75 mg/day.
Not determined.
Any mutation allele known to totally prevent the production of functional enzyme in patient cells, e.g. 1278insTATC in the HEXA gene.
Standard errors are given when the number of independent patient samples used to determine the baseline residual Hex A activity was ≥ 3.
Tolerability
The lower doses of PYR were well-tolerated. However, all but one of the subjects experienced significant side-effects of the treatment soon after the dose of the drug was raised from 50 to 75 mg per day (Table 2). There was no hematologic or clinical evidence of folate deficiency. In all cases, the adverse reactions were of a neurologic nature, such as increased ataxia or incoordination, and remitted on interruption of drug treatment. Only one serious adverse event (SAE) was experienced, in Subject 1002, who required hospitalization for treatment of status epilepticus shortly after beginning treatment with 50 mg/day of PYR. The patient had a history of seizures, but had been seizure-free for several years and was not on anti-convulsant medication at the time of this study. Seizures have been reported as an adverse side-effect of PYR treatment [http://www.rxlist.com/daraprim-drug.htm], though the mechanism is unclear. The relationship of the seizure disorder in Subject 1002 to treatment with PYR is unclear; however, it was treated as probably drug-related.
Additional measurements
In addition to total protein, Hex A and Glcr, we analyzed total Hex, β-galactosidase and glucocerebrosidase activities. Initial experiments concluded that the most consistent results were obtained when Hex A values were normalized using Glcr rather than total protein or any of the other lysosomal enzyme activities examined (data not shown). A single control value was used to calculate all “% Control” levels for all patient samples. This value was the average of those from three healthy adults; i.e.,74 nmol MUGS hydrolysed/((h) (mg total protein)), or a ratio of 0.88 MUGS units relative to MUGlrc units. Measurements of the Glcr levels in the plasma of the Toronto patients (1002–1004) produced no significant upwards trends with increasing plasma PYR levels, indicating no lysosomotropic effects resulting from the treatment (data not shown).
Effects on Hex A activities
The effects of PYR treatment on Hex A activities for the three Toronto subjects in the trial are shown in Fig. 1 and for the 5 New York subjects in Fig. 2. The combined data from all 8 subjects are shown in Fig. 3. All of these are expressed as fold increase over their baseline value (Table 2). The results from the Toronto patients indicate that a clear enhancement of Hex A levels over baseline values can be achieved when plasma levels of PYR reach 1–1.5 mg/L (~4–6 μM). It is particularly noteworthy that no inhibition of Hex A activity was observed at even the highest plasma levels of PYR reached by patients in this study (~2.5 mg/L).
Fig. 1.
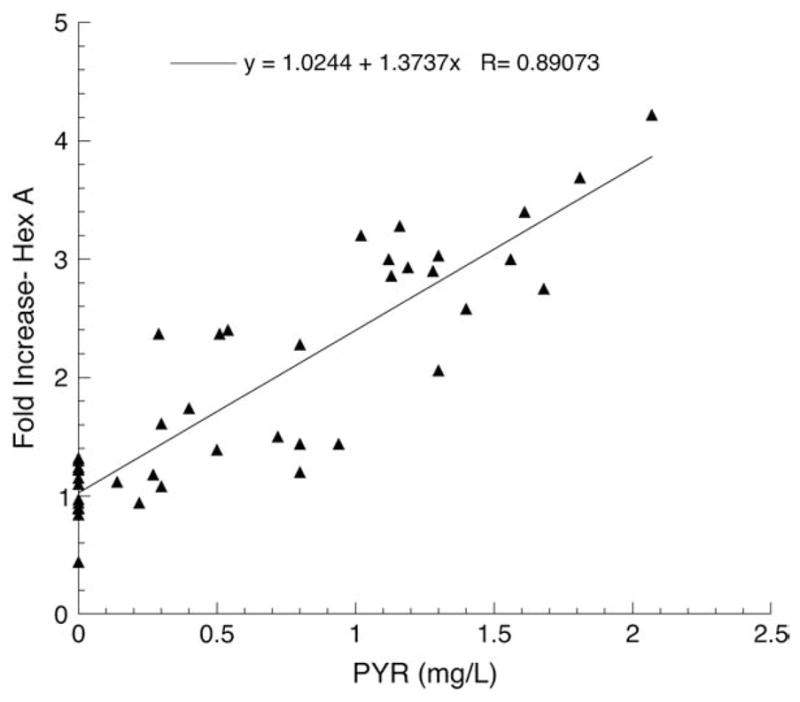
Plot of fold-increase over baseline in residual Hex A activity versus plasma PYR concentration for all three Toronto Subjects, 1002–1004.
Fig. 2.

Plot of fold-increase over baseline in residual Hex A activity versus plasma PYR concentration for New York Subjects1006–1010.
Fig. 3.

Plot of fold-increase over baseline line in residual Hex A activity versus plasma PYR concentration for Subjects 1002–1004 and 1006–1010 combin.
Subject 1005 is a young woman with TSD variant GM2 gangliosidosis who was the first to be enrolled in the study and lives the furthest from Toronto and New York. No clear correlation between her levels of plasma PYR (mg/L) versus enhancement of enzyme activity (% of control) was observed; i.e., linear regression produced a best fit line; % control=3.2 +0.21 X PYR (mg/L), R=0.21, for n=16, data not shown). However problems with the preparation and then shipping of the leukocyte pellets to Toronto, along with a large number of protocol violations, make confident interpretation of the data impossible. The subject had been treated in the past for a pituitary tumor unrelated to her GM2 gangliosidosis. On higher doses of PYR she developed amenorrhea, which resolved with cessation of the treatment, but then developed again on re-initiation of the drug. The problem only occurred at high doses of PYR; lower doses of the drug were tolerated without adverse reactions.
Subject 1002 is a young man with TSD variant GM2 gangliosidosis who showed robust enhancement of Hex A activities in leukocytes (best fit line; % control=6.2 +9.2 X PYR (mg/L), R=0.94 for n=8, data not shown). Plasma concentrations of PYR were also higher than those observed in other subjects at comparable doses of the drug reaching 1.6 mg/L after a single week on the 50 mg/d dosage (Fig. 4). At this time the subject developed status epilepticus and was withdrawn from the study. He had a history of occasional seizures in the past, but he was not on anticonvulsant therapy at the time of his enrolment into this clinical trial.
Fig. 4.

Relationship between oral doses of PYR and the resulting plasma levels of the drug. The daily doses of PYR, followed by the week(s) on that dose, are plotted against the resulting plasma levels (mg/L) for; (A) the Toronto patients, and (B) the New York patients.
Subjects1003 and 1004 are young brothers with SD variant GM2 gangliosidosis. They also showed robust enhancement of Hex A activities in leukocytes, with a close relationship between their plasma PYR concentrations and the degree of enzyme enhancement (best fit line; % control=3.3 +3.9 X PYR (mg/L), R=0.89 for n=35, data not shown). They both developed anorexia, nausea, and abdominal discomfort on high doses of PYR and were withdrawn from the study prior to advancement to the highest planned dose of the drug (100 mg/day).
Discussion
The results of this small initial clinical trial show clearly that PYR, administered orally to patients with late-onset GM2 gangliosidosis, in doses lower than those generally used to treat the parasitic diseases for which it is normally prescribed, produced enhancement of Hex A activity in peripheral blood leukocytes. Moreover, the enhancement occurred equally well in late onset patients with either the Sandhoff or Tay–Sachs variant of GM2 gangliosidosis, and the degree of enhancement was proportional to the plasma PYR concentration (Figs. 1–3). The observation that levels of Glcr activity in neither plasma (nmole MU/h/mL) or leukocytes (nmole MU/h/mg protein) and β-galactosidase activity in leukocytes (nmole MU/h/mg protein) were not enhanced by PYR treatment, indicates that the drug effect was specific to mutant Hex A and not the result of a non-specific lysosomotropic effect. The increased scatter and decreased slope of the best-fit lines generated for the New York subjects, versus those from Toronto, can be partially explained by numerous problems encountered in the preparation, storage and shipment to Toronto of the leukocyte pellets for analyses. These problems were likely confounded by the known decrease in heat-stability of Hex A containing the G269S substituted α-subunit [20]. Additionally genotype differences may have played a role in the variations in response to PYR. However, when tested in cultured fibroblasts, cells from the Toronto TSD patient (1002), as well as other fibroblast with the same genotype as all the New York patients (1006–1011), were enhanced ~2-fold by PYR treatment (3 μg/mL of medium over 5 days) (data not shown). On the other hand, fibroblasts from one of the Toronto SD patients (1004) were enhanced ~4-fold by the same PYR treatment (data not shown).
The relationship between drug dosage and plasma PYR concentrations proved to be highly variable between different subjects in the study (Fig. 4), a phenomenon reported informally by an Israeli group undertaking a similar evaluation of PYR treatment of GM2 gangliosidosis (Naftali Stern, personal communication). In a study of plasma PYR concentrations during long-term treatment for cerebral toxoplasmosis in patients with AIDS, Klinker and colleagues showed that on weekly doses of the drug, steady-state concentrations of PYR were achieved in 12 to 20 days [21]. There was, however, marked variability between patients in the relationship between drug dosage and plasma levels. Concomitant treatment with metabolism-inducing medications, such as anticonvulsants, tended to decrease plasma PYR concentrations.
The prevalence and nature of side effects experienced by almost all the subjects on higher doses of PYR was unexpected. However, treatment with lower doses of the drug was well-tolerated and was associated with significant enhancement of Hex A enzyme activities in leukocytes. Because of the side effects, further enrolment of subjects into the current clinical trial protocol has been suspended. Instead, we have developed a modified drug treatment regimen to assess more rigorously the tolerability and efficacy of the treatment with lower doses of PYR. Considering the narrow therapeutic index of PYR in late onset GM2, these observations underscore the importance of monitoring plasma PYR levels closely in all patients with the disease undergoing treatment with the drug. The observation that leukocyte Hex A activity was apparently not inhibited even at the highest plasma PYR concentrations achieved indicates that dissociation of the drug from the mutant enzyme in the acidic environment of lysosomes, where it is 4-fold less inhibitory, prevents significant loss of enzyme activity, suggesting that there is no need for employing an interrupted dosage regimen such as has been proposed for PC treatment of other lysosomal disorders. This conclusion is consistent with recent in celullo substrate hydrolysis data from adult Tay–Sachs fibroblasts first treated with PYR and then loaded with a fluorescent derivative of GM2 ganglioside [17].
On the basis of what is known about the relationship between residual enzyme activity and clinical severity of the disease, the degree of enhancement (up to 4 fold) would, over time, be sufficient to produce a clinically significant improvement in the course of the disease—if the same enhancement occurred in important target tissues, especially the brain. Studies on human subjects have shown that the concentration of PYR in cerebrospinal fluid (CSF) is 13–27% of the concentration in plasma [22] and in studies on human subjects undergoing neurosurgical procedures, Weiss et al showed that the ratio of brain parenchymal-to-serum PYR concentrations varied from 2.5 to 5.2 after a single dose of 50 mg or 100 mg of the drug [22]. Assuming that the same relationship between the concentration of PYR in brain and plasma occurs in patients with GM2 gangliosidosis, plasma concentrations of 1–1.5 mg/L, that were generally well tolerated and in the Canadian patients produced an unequivocal enhancement in Hex A activity, might be expected to be associated with brain concentrations of 2.5 to 7.8 mg/L (10 to 31 μM). Our pre-clinical studies showed that optimum enhancement of Hex A activity in fibroblasts from late-onset GM2 patients occurred at comparable concentrations [4]. On the basis of these findings, therefore, doses of PYR sufficient to maintain plasma concentrations of the drug in the range of 1 to 1.5 mg/L might be expected to be sufficient to produce clinically significant enhancement of Hex A levels in brain without undesirable side-effects. It would also appear that plasma concentrations between 2.5 and 3 mg/L represent levels that are too high and likely to produce negative side-effects.
The unexpectedly robust response observed to relatively low doses of PYR underscores the need to determine the extent to which the drug becomes concentrated in neurons of the CNS, which is not ethically practical in humans. However, further studies to determine the concentration of the drug in other human cells, such as peripheral blood leukocytes, will be undertaken. Further studies are also needed to evaluate the effect of PYR-treatment over an extended period of time and using a dosage regime optimized for each patient, on clinical outcomes, such as mobility, speech, coordination, and cognition.
Conclusions
The results of this study showing enhancement of Hex A activity with increasing concentrations of PYR in plasma are consistent with the results of the pre-clinical studies done in cultured fibroblasts, confirming that the enhancement observed in vitro can be reproduced in vivo. Secondly, we discovered that significant enhancement was achievable at relatively low plasma PYR concentrations. We found that subjects varied considerably in the pharmacokinetics of the drug: plasma concentrations in some subjects were significantly higher than in other subjects on the same dosages of the drug. This observation, along with the experience of adverse drug reactions at high drug doses, indicates that optimum results with regard to benefits, in terms of enhanced enzyme activity, versus undesirable side effects requires careful monitoring of plasma PYR levels and individualization of dosage regimens. Further studies are necessary to evaluate the effect of the treatment on the clinical evolution of the disease.
Acknowledgments
We are particularly grateful to Sophia Pesotchinsky and Jack Keimel, whose steadfast belief in the importance of the project and their unstinting personal and material support were critically important to its success. We are grateful for the dedicated technical assistance of Marie-Anne Skomorowski and Paola Torres and the organizational skills of Mohammed Hussein and Michele Ford, study coordinators. This study was supported in part by grants from the ExSAR Incorporated, New Hope Research Foundation, the National Tay–Sachs and Allied Diseases Foundation, the Uger Estate, and the Hospital for Sick Children Foundation.
References
- 1.Mahuran DJ. Biochemical consequences of mutations causing the GM2 gang-liosidoses. Biochim Biophys Acta. 1999;1455:105–138. doi: 10.1016/s0925-4439(99)00074-5. [DOI] [PubMed] [Google Scholar]
- 2.Gravel RA, Clarke JTR, Kaback MM, Mahuran D, Sandhoff K, Suzuki K. The GM2 gangliosidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. Vol. 2. McGraw-Hill; New York: 1995. pp. 2839–2879. [Google Scholar]
- 3.Leinekugel P, Michel S, Conzelmann E, Sandhoff K. Quantitative correlation between the residual activity of beta-hexosaminidase A and arylsulfatase A and the severity of the resulting lysosomal storage disease. Hum Genet. 1992;88:513–523. doi: 10.1007/BF00219337. [DOI] [PubMed] [Google Scholar]
- 4.Maegawa GHB, Tropak M, Butner J, Stockley T, Kok F, Clarke JTR, Mahuran DJ. Pyrimethamine as a potential pharmacological chaperone for late-onset forms of GM2 gangliosidosis. J Biol Chem. 2007;282:9150–9161. doi: 10.1074/jbc.M609304200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ohno K, Suzuki K. Mutation in GM2-Gangliosidosis B1 variant. J Neurochem. 1988;50:316–318. doi: 10.1111/j.1471-4159.1988.tb13266.x. [DOI] [PubMed] [Google Scholar]
- 6.Mark BL, Mahuran DJ, Cherney MM, Zhao D, Knapp S, James MN. Crystal structure of human beta-hexosaminidase B: understanding the molecular basis of Sandhoff and Tay–Sachs disease. J Mol Biol. 2003;327:1093–1109. doi: 10.1016/s0022-2836(03)00216-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tropak MB, Mahuran D. Lending a helping hand, screening chemical libraries for compounds that enhance β-hexosaminidase A activity in GM2 Gangliosidosis cells. FEBS J. 2007;274:4951–4961. doi: 10.1111/j.1742-4658.2007.06040.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernales S, Papa FR, Walter P. Intracellular signaling by the unfolded protein response. Annu Rev Cell Dev Biol. 2006;22:487–508. doi: 10.1146/annurev.cellbio.21.122303.120200. [DOI] [PubMed] [Google Scholar]
- 9.Rigat B, Mahuran D. Diltiazem, a L-type Ca(2+) channel blocker, also acts as a pharmacological chaperone in Gaucher patient cells. Mol Genet Metab. 2009;96:225–232. doi: 10.1016/j.ymgme.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tropak MB, Reid S, Guiral M, Withers SG, Mahuran DJ. Pharmacological enhancement of β-hexosaminidase activity in fibroblasts from adult Tay–Sach and Sandhoff patients. J Biol Chem. 2004;279:13478–13487. doi: 10.1074/jbc.M308523200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benjamin ER, Flanagan JJ, Schilling A, Chang HH, Agarwal L, Katz E, Wu X, Pine C, Wustman B, Desnick RJ, Lockhart DJ, Valenzano KJ. The pharmacological chaperone 1-deoxygalactonojirimycin increases alpha-galactosidase A levels in Fabry patient cell lines. J Inherit Metab Dis. 2009;32:424–440. doi: 10.1007/s10545-009-1077-0. [DOI] [PubMed] [Google Scholar]
- 12.Khanna R, Benjamin ER, Pellegrino L, Schilling A, Rigat BA, Soska R, Nafar H, Ranes BE, Feng J, Lun Y, Powe AC, Palling DJ, Wustman BA, Schiffmann R, Mahuran DJ, Lockhart DJ, Valenzano KJ. The pharmacological chaperone isofagomine increases the activity of the Gaucher disease L444P mutant form of beta-glucosidase. FEBS J. 2010;277:1618–1638. doi: 10.1111/j.1742-4658.2010.07588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maegawa GHB, Tropak MB, Buttner JD, Rigat BA, Fuller M, Pandit D, Tang L, Kornhaber GJ, Hamuro Y, Clarke JTR, Mahuran DJ. Identification and characterization of ambroxol as an enzyme-enhancement agent for Gaucher disease. J Biol Chem. 2009;284:23502–23516. doi: 10.1074/jbc.M109.012393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sawkar AR, D’Haeze W, Kelly JW. Therapeutic strategies to ameliorate lysosomal storage disorders–a focus on Gaucher disease. Cell Mol Life Sci. 2006;63:1179–1192. doi: 10.1007/s00018-005-5437-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suzuki Y. Beta-galactosidase deficiency: an approach to chaperone therapy. J Inherit Metab Dis. 2006;29:471–476. doi: 10.1007/s10545-006-0287-y. [DOI] [PubMed] [Google Scholar]
- 16.Maegawa GH, Stockley T, Tropak M, Banwell B, Blaser S, Kok F, Giugliani R, Mahuran D, Clarke JT. The natural history of juvenile or subacute GM2 gang-liosidosis: 21 new cases and literature review of 134 previously reported. Pediatrics. 2006;118:e1550–e1562. doi: 10.1542/peds.2006-0588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tropak MB, Bukovac SW, Rigat BA, Yonekawa S, Wakarchuk W, Mahuran DJ. A sensitive fluorescence-based assay for monitoring GM2 ganglioside hydrolysis in live patient cells and their lysates. Glycobiology. 2010;20:356–365. doi: 10.1093/glycob/cwp183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hou Y, McInnes B, Hinek A, Karpati G, Mahuran D. A Pro504Ser substitution in the β-subunit of β-hexosaminidase A inhibits α-subunit hydrolysis of GM2 ganglioside, resulting in chronic Sandhoff disease. J Biol Chem. 1998;273:21386–21392. doi: 10.1074/jbc.273.33.21386. [DOI] [PubMed] [Google Scholar]
- 19.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 20.Brown CA, Mahuran DJ. β-hexosaminidase isozymes from cells co-transfected with α and β cDNA constructs: Analysis of α subunit missense mutation associated with the adult form of Tay–Sachs disease. Am J Hum Genet. 1993;53:497–508. [PMC free article] [PubMed] [Google Scholar]
- 21.Klinker H, Langmann P, Richter E. Plasma pyrimethamine concentrations during long-term treatment for cerebral toxoplasmosis in patients with AIDS. Anti-microb Agents Chemother. 1996;40:1623–1627. doi: 10.1128/aac.40.7.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weiss LM, Harris C, Berger M, Tanowitz HB, Wittner M. Pyrimethamine concentrations in serum and cerebrospinal fluid during treatment of acute Toxoplasma encephalitis in patients with AIDS. J Infect Dis. 1988;157:580–583. doi: 10.1093/infdis/157.3.580. [DOI] [PubMed] [Google Scholar]