

Abstract
The structurally intriguing bicyclic ketal moiety of tirandamycin is common to several acyl-tetramic acid antibiotics, and is a key determinant of biological activity. We have identified the tirandamycin biosynthetic gene cluster from the environmental marine isolate Streptomyces sp. 307-9, thus providing the first genetic insight into the biosynthesis of this natural product scaffold. Sequence analysis revealed a hybrid-polyketide synthase-nonribosomal peptide synthetase gene cluster with a colinear domain organization entirely consistent with the core structure of the tirandamycins. We also identified genes within the cluster that encode candidate tailoring enzymes for elaboration and modification of the bicyclic ketal system. Disruption of tamI, which encodes a presumed cytochrome P450, led to a mutant strain deficient in production of late stage tirandamycins that instead accumulated tirandamycin C, an intermediate devoid of any post-assembly line oxidative modifications.
Keywords: biosynthesis, natural products, polyketide
Introduction
The antibiotic tirandamycin is included in the tetramic acid class of natural products defined by a 2,4-pyrrolidinedione ring system that is biosynthetically derived from condensation of an amino acid to a polyketide-derived acyl chain to yield a 3-acyl tetramate moiety that confers metal chelating activity.[1] Beyond this common feature, this class encompasses a substantial breadth of structural diversity and biological activities represented by the HIV-1 integrase inhibitor equisetin,[2] the antimycotic dihydromaltophilin,[3] and the first discovered tetramate antibiotic streptolydigin (Str, 1),[4] which targets bacterial RNA polymerase (RNAP).[5]
Recently, Str has generated considerable interest as a chemical tool for inhibition of RNAP,[6] and as a subject of biosynthetic study in isotope feeding experiments.[7, 8] The structural feature of predominant biosynthetic interest in Str is an intriguing bicyclic ketal skeleton (Figure 1) that is common to the tirandamycins, tirandalydigin (2), BU-2313B (3), and nocamycin II (4), all of which possess antimicrobial activity.[9] Tirandamycin A (TirA, 5) and tirandamycin B (TirB, 6) were originally discovered in the 1970s from the fermentation broths of Streptomyces species as antimicrobial agents,[10, 11] and we have recently described the identification of tirandamycin C (TirC, 7) and tirandamycin D (TirD, 8) from the environmental isolate Streptomyces sp. 307-9.[12] These four tirandamycins differ in the oxidized groups present on the bicyclic ketal moiety, and the resulting structural variation was found to be a key determinant of potency against vancomycin-resistant Enterococcus faecalis.[13]
Figure 1.

Tetramic acid natural products bearing a bicyclic ketal moiety (red) with varying degrees of oxidative modification.
Additionally, the co-crystal structures of Str complexed with either Escherichia coli or Thermus thermophilus RNAP display extensive contacts with the bicyclic ketal.[13, 14] Together, these observations indicate that formation and modification of this unusual ring system is critical for biological activity, but there has been a lack of genetic or biochemical characterization relating to the assembly and tailoring of this class of molecules. Herein, we report the elucidation of the tirandamycin gene cluster (tam) and the identification by genetic disruption of the presumed cytochrome P450 (CYP450) gene product of tamI as an enzyme required for oxidative modification of the bicyclic ketal moiety.
Results
Cloning and sequencing of tam cluster
Our search for genes encoding tirandamycin biosynthetic proteins was informed by isotope feeding studies of Str production in Streptomyces lydicus,[7, 15] and the organization of previously elucidated gene clusters for natural products bearing a tetramic acid moiety,[3, 16–18] all of which are derived from hybrid-polyketide synthase (PKS) nonribosomal peptide synthetase (NRPS) assembly line systems. Type I PKSs are large biosynthetic enzymes organized as a linear series of modules containing conserved catalytic domains that direct the selection and incorporation of a single coenzyme A (CoA)-activated short chain acyl group into a nascent polyketide.[19] To initiate synthesis, the acyltransferase domain (AT) of a loading module (module 0) recognizes an acyl-CoA substrate and transfers the acyl group to the phosphopantetheine arm of the adjacent acyl carrier protein (ACP) domain. In subsequent extension cycles, a ketosynthase domain (KS) catalyzes the decarboxylative condensation of a downstream ACP-bound α-carboxy subunit (typically from malonyl-CoA or methylmalonyl-CoA) with the acyl chain from the previous module, thus extending the polyketide chain by two carbon atoms and transferring it to the next module. Modules can also contain auxiliary catalytic domains that introduce structural diversity through the β-keto reduction of units incorporated in the previous extension cycle to yield β-hydroxy (formed by ketoreductase domain, KR), α-β olefin (formed by KR and dehydratase domain, DH), or β-methylene groups (formed by KR, DH, and enoyl reductase domain, ER).
In an analogous synthesis strategy, an NRPS module is composed of an adenylation domain (A-domain) that activates and transfers amino acids to the phosphopantetheine arm of an adjacent peptidyl carrier protein domain (PCP), after which a condensation domain (C-domain) mediates amide bond formation between a downstream PCP-bound amino acid and the peptide chain from the previous module.[19] In PKSs and NRPSs, the linear precursor that is formed on the thiolation domain of the terminal module is typically released by cyclization or hydrolysis catalyzed by a thioesterase domain. Notably, the chemistry of both pathways involves condensation between acyl groups tethered to phosphopantetheinylated thiolation domains (ACP or PCP), and this common biosynthetic scheme enables the exchange of nascent chains between PKS and NRPS modules. Accordingly, hybrid PKS-NRPS enzymes incorporate both acyl-CoA monomers and amino acids into a single product, as was anticipated for tirandamycin biosynthesis. We also expected that the gene cluster would contain oxidative tailoring enzymes to modify the bicyclic ketal group, and in particular we noted that epoxide and hydroxyl groups are often installed by CYP450 monooxygenases.[20]
To interrogate the genome of Streptomyces sp. 307-9 for putative tirandamycin biosynthetic genes, we designed degenerate PCR primers within conserved regions of KS domains and CYP450 enzymes (Supporting Information Figure S1), and used these primers to amplify gene probes from genomic DNA. Amplification using the KS and CYP450 primers generated products of the expected size with high sequence homology to known corresponding genes. These probes were radiolabelled and hybridized to a ~1200 clone fosmid library of Streptomyces sp. 307-9 genomic DNA immobilized on nitrocellulose membranes. Five KS and 23 CYP450 hybridizing clones were identified, with fosmid 6G9 positive for both gene probes. Beginning with 6G9, a total of three overlapping fosmids containing KS hybridizing DNA fragments were subcloned, sequenced, and assembled. This provided ~92 kb of contiguous sequence of which ~56 kb was assigned as the tirandamycin biosynthetic gene cluster.
We conducted an extensive bioinformatic analysis of the entire sequenced region to initiate examination of tirandamycin biosynthesis. First, we were particularly interested in characterizing the enzyme(s) responsible for an oxidative modification cascade, anticipated to mediate a multi-step conversion of TirC to TirB that likely proceeds through the intermediates TirD and TirA (Figure 1). Second, we sought to elucidate the origin of the C-11/C-12 double bond of TirC, which is unusual for a polyketide in part due to its cis configuration, and additionally because it spans an intra-subunit bond as opposed to the inter-subunit bonds typically installed by KR-DH domains of PKSs. Finally, we were interested in analyzing the tirandamycin polyketide synthase to predict its yet undiscovered post-assembly line product and identify candidate enzymes for the maturation of this linear intermediate.
To this end, we searched the ~92 kb of sequence for potential coding regions and identified 46 open reading frames (ORF) with translated sequences bearing homology to proteins of known function or conserved hypothetical proteins. Fifteen of these ORFs constitute a contiguous 56 kb assigned as the tirandamycin biosynthetic cluster (Figure 2 and Supporting Information Figure S2) that is flanked by putative transposable elements. On either side of this region are 31 ORFs that comprise the remaining 36 kb and encode presumed primary metabolic, regulatory, and ribosomal proteins. The tirandamycin cluster includes ORFs that encode three PKS and one NRPS proteins, and eleven proposed to mediate post-assembly line tailoring steps, self-resistance, and regulation of tam gene expression.
Figure 2.

Organization of the tirandamycin biosynthetic gene cluster (tam). Genes are classified by color for encoding PKS or NRPS modules (blue), tailoring enzymes (red), proteins involved in regulation and resistance (yellow), and proteins with accessory or unknown functions (green).
aLength of predicted gene product, in amino acids; bPercent identity to closest homologue; cPercent similarity to closest homologue
Analysis of genes encoding assembly-line enzymes
The PKS genes tamAI, tamAII, and tamAIII encode proteins containing four, two, and two PKS modules, respectively, and tamD encodes a NRPS monomodule. To predict the assembly line-derived linear precursor, we analyzed the domain organization of these modules using online resources from Pfam[21] and the PKS-NRPS Analysis Website (http://www.tigr.org/jravel/nrps) and by in-house alignments with modules from the rapamycin,[22] erythromycin,[23] lipomycin,[17] and nystatin clusters.[24] We also used previously described sequence motifs to predict the substrate specificity of incorporation and stereochemistry of modifications where applicable. Because the AT domain of each module is responsible for selecting and loading a specific short-chain acyl group onto its ACP, it is possible to predict the subunit incorporation at a given step by examining specificity determining sequence motifs within this domain. We performed an alignment of this sequence motif from the eight tirandamycin AT domains with the consensus sequences that specify malonate and methylmalonate incorporation according to Haydock et al.[25] This clearly indicated that module 0 and extension modules 2, 6, and 7 are specific for malonate, while the other modules are specific for methylmalonate incorporation (Figure 3A). Module 0 contains an apparent KSQ domain hallmarked by the presence of a glutamine residue in place of an otherwise conserved active site cysteine (Figure 3B).[26] This KSQ domain has been shown in other pathways to catalyze the decarboxylation of malonyl-ACP of a loading module to furnish an acetate starter unit. The presence of this KSQ domain, and the predicted specificities of the ATs are entirely consistent with the carbon skeleton of tirandamycin, and also with labeled acetate/propionate feeding studies of Str.[7] We were also able to predict the amino acid incorporated by TamD through analysis of its A-domain specificity-conferring code using the NRPS Predictive Blast Server.[27] The top three matches represent glycine activating A-domains (Figure 3F), as is expected for the glycine unit present within the tetramate moiety of tirandamycin.
Figure 3.

Sequence motifs from the tirandamycin PKS-NRPS assembly line enzymes. A) AT sequence motifs correlating with incorporation of malonate (red) or methylmalonate (blue); residues in gray are conserved in all active ATs; asterisk denotes active site serine. B) KS sequence motifs correlating with KSQ loading module (gold) and canonical extender modules (gray); residues in red are conserved in all active KS domains; asterisk denotes active site cysteine. C) KR sequence motifs correlating with reduction to A-type (gold) and B-type (gray) hydroxyl group stereochemistry; residues in red are conserved in all active KRs; asterisk denotes active site tyrosine that is mutated in the TamAI module 1 KR. D) Intact Rossmann fold motif of all KRs; residues in green denote the canonical G/AxG/AxxG/AxxxG/A motif; residues in red are also conserved in active KRs. E) Critical active site and conserved motifs within DH domains; residues in green denote HxxxGxxxxP motif; asterisk denotes active site histidine; red residues are conserved in functional DH domains. F) A-domain sequences correlating with substrate specificity; residues in green comprise the eight residues of the specificity conferring code; residues in red are conserved in functional A-domains.
Next, we analyzed the reductive processing domains within PKS genes to predict the functional groups present at the β-keto position following each extension step. Modules 1–6 each contain a KR with an intact Rossmann fold motif (Figure 3D), but in TamAI module 1 the conserved tyrosine present in all functional KRs is replaced by a phenylalanine residue (Figure 3C). Reid et al. demonstrated that this same Y→F mutation leads to inactivation of the DEBS3 module 6 KR in vivo and in vitro, implicating this tyrosine as the critical proton donor to the β-keto group.[28] The presence of this mutation in TamAI module 1 KR indicates that this domain is inactive, resulting in a keto group at the C-13 position of the tirandamycin linear precursor. The stereochemistry of each KR domain was also predicted using sequence motifs described by Caffrey,[29] which indicated that the TamAI module 4 KR specifies formation of a hydroxyl with A-type stereochemistry, while all other Tam KRs result in hydroxyl groups with B-type stereochemistry (Figure 3C). In addition to KRs, DH domains were observed in modules 3, 5, and 6 that contain all conserved and active site motifs (Figure 3E). Apart from the inactive KR of TamAI module 1, we conclude that all other PKS and NRPS domains are functional based on conserved motifs and active site residues that have been previously observed in other functional natural product gene clusters (Supporting Information Figure S3).[22, 23, 30]
This sequence analysis results in a prediction for assembly line biosynthesis (Figure 4) that generates the final chain elongation intermediate 9 (Scheme 1) tethered to the terminal NRPS module. Note that the stereochemistry of this intermediate cannot be predicted at position C-12 because any configuration present in 9 would be lost upon conversion to the sp2 center of TirC, and established anew with epoxide formation toward TirA.
Figure 4.
Domain organization and deduced assembly line biosynthesis of tirandamycin. Modules and subunits in gold denote incorporation of the starter unit, those in blue denote incorporation from methylmalonyl-CoA, those in red denote incorporation from malonyl-CoA, and the one in silver denotes incorporation of glycine. Bonds in black denote inter-subunit linkages and oxidative modifications. The presumed inactive KR of TamAI module 1 is shown in black.
The terminal module (TamD) lacks a C-terminal domain typical for termination in assembly line enzymes (i.e. a thioesterase or reductase domain), thus leaving the mechanism of tirandamycin chain release unresolved. In the biosynthesis of the fungal tetramic acid equisetin, the terminal NRPS module contains a domain that catalyzes chain release coincident with tetramic acid ring formation in a Dieckmann condensation.[31] This releasing domain is common for fungal tetramic acid pathways, but bacterial tetramic acid pathways contain either a terminal thioesterase domain, as for heat-stable antifungal factor,[3] or no apparent C-terminal release domain, as for lipomycin[17] and tirandamycin. Although the streptolydigin gene cluster has not been identified, it was recently reported that an ACP from the fatty acid biosynthesis [32] operon of S. lydicus may be involved in tetramic acid formation for streptolydigin.[33] Genetic disruption of fabC abolished streptolydigin production and led to the accumulation of the streptolol moiety in culture, suggesting that a trans-ACP may be involved in bridging the PKS and NRPS steps of tetramic acid biosynthesis. We find no evidence of a trans-ACP present within or adjacent to the tam cluster, and note that each Tam module contains an embedded thiolation domain as expected for canonical assembly line biosynthesis. We propose that tirandamycin tetramic acid formation proceeds non-enzymatically by attack of the methylene Cα-3′ on the TamD PCP thioester (Scheme 1B), as proposed for biosynthesis of lipomycin and the tetronic acid tetronomycin.[34] An analogous non-enzymatic route to tetramic acids has recently been described by Janda and coworkers (Scheme 1A) in which a homoserine lactone undergoes rearrangement by intramolecular attack on the lactone ring,[35] thereby providing a natural precedent for the predicted tirandamycin pathway termination chemistry.
Analysis of genes encoding oxidative enzymes
TirC is the earliest intermediate that has been identified from culture broths.[12] Transformation into TirB, the most highly modified and final product, requires several oxidative modifications. We had originally anticipated that one or more cytochrome P450 enzymes would catalyze epoxidation and hydroxylation steps, and the presence of the P450 homolog TamI was consistent with this hypothesis. Sequence analysis of TamI revealed that it contains all of the highly conserved motifs of this enzyme class, including the Helix I, ExxR, and heme binding motifs (Supporting Information Figure S4).[36]
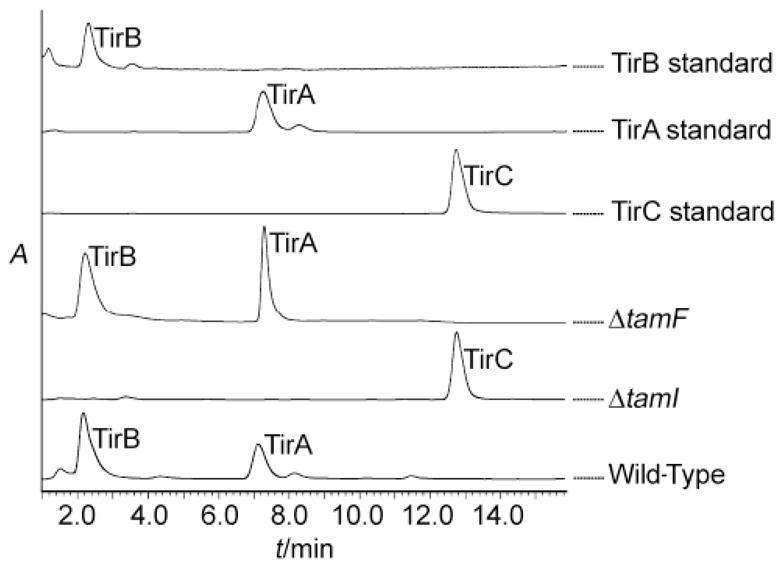
We confirmed the role of TamI in vivo by genetic disruption of the relevant ORF, tamI. A DNA transfer protocol for Streptomyces sp. 307-9 was developed using conjugation from an E. coli S-17 donor strain[39] in conjunction with the REDIRECT recombination system.[40] Double-crossover allelic exchange mutants that contained an in-frame replacement of tamI by an apramycin resistance gene were selected and verified by PCR screening of genomic DNA. Verified mutants were grown in fermentation media containing apramycin, the broths were extracted, and tirandamycin metabolite profiles were analyzed. Wild-type cultures accumulated TirA and TirB whereas the ΔtamI P450 mutant strain accumulated exclusively TirC (Figure 5), indicating that TamI is critical for at least the first step in the oxidative tailoring process. Within the gene cluster we also identified tamL, which encodes a predicted flavin-dependent oxidoreductase bearing conserved Rossmann fold nucleotide binding motifs[37] and a berberine bridge enzyme sequence motif that contains the site of covalent co-factor attachment in a sub-class of FAD-dependent enzymes (Supporting Information Figure S4).[38] Because flavin-dependent enzymes can catalyze a variety of oxidative chemistries, TamL is a likely candidate for tailoring of the bicyclic ketal and its role is the subject of ongoing investigation.
Figure 5.

LC-MS metabolite profile of gene disruption mutants. A TamI P450 mutant shows accumulation of TirC, but a TamF mutant still accumulates TirA and TirB. Peaks were identified by MS and comparison to authentic standards. Traces indicate absorbance at 354 nm.
Pathway for bicyclic ketal formation
Our predicted intermediate 10, which follows from tetramic acid cyclization and chain release, must undergo bicyclic ketal formation to generate TirC.[12] The connectivity of 10 suggests that formation of bridging ether bonds must occur between C-7/C-13 and C-13/C-9, and double bond formation between C-11/C-12. Hemiketalization between the C-7 hydroxyl and the C-13 carbonyl would afford the first of these bridging ethers, but the origin of the second bridging ether remains unclear.
On the 3′ flank of the assembly line genes we identified the terpene synthase homolog tamF, which led us to hypothesize that tirandamycin cyclization might be effected by a reaction similar to terpene cyclization. In these cascading cyclizations, a carbocation is generated through olefin protonation or diphosphate ionization and quenched by an apposed double bond, water (solvolysis), or an elimination reaction. For 10, we noted that protonation at C-8 would form a carbocation that might be quenched by the hemiketal C-13 hydroxyl for subsequent C-O bond formation in an intramolecular solvolysis-like reaction (Scheme 2). We thus anticipated that a tamF disruption strain would not produce TirC (or any downstream products) but would instead accumulate 10 or another intermediate. Contrary to our hypothesis, the ΔtamF mutant strain still accumulated TirA and TirB, with a metabolite profile similar to the wild type strain (Figure 5). This result leaves no apparent role for TamF in tirandamycin biosynthesis, suggesting that the proposed cyclization mechanism is not employed, or is achieved by a functional (and possibly redundant) terpene cyclase homolog located elsewhere on the genome of Streptomyces sp. 307-9.
Analysis of genes with accessory or unknown functions
Apart from the biosynthetic enzymes described above, there are several other accessory proteins that we have assigned to the gene cluster (Figure 2). tamM encodes a predicted phosphopantetheinyl transferase enzyme responsible for generating the holo forms of thiolation domains (ACPs and PCPs),[41] and the proximity of this gene to the tirandamycin cluster suggests that it may be dedicated for this biosynthetic pathway. tamB encodes a predicted type II thioesterase, an enzyme that has been shown to have proofreading activity in other PKS-NRPS pathways.[42] Though located immediately adjacent to the tamD NRPS gene, tamB is a discrete ORF outside of tamD and is not thought to be involved in chain release. Indeed, gene disruption of this homolog in the streptolydigin producer S. lydicus resulted in a decrease, but not complete abrogation of streptolydigin production, which is consistent with a proofreading role.[43] tamC encodes a conserved hypothetical protein of unknown function for which no significant domain motifs were found by Pfam analysis, but a homolog of TamC exists in the gene cluster for the tetramic acid lipomycin.[17] tamE encodes a predicted O-glycosyl hydrolase of the laminarinase family and contains a conserved catalytic motif consistent with this function (Supporting Information Figure S4), but there are no predicted glycosyltransferases within the cluster that might append an O-linked sugar. TamE does not contain an N-terminal secretion signal as determined by SignalP 3.0 analysis,[44] therefore making it unlikely to serve a role similar to that of DesR,[45] the extracellular glucosidase in the pikromycin pathway that activates a glycosylated methymycin prodrug after export. It is perhaps noteworthy that the tirandamycins contain a ketal carbon at C-13, which is structurally analogous to the anomeric carbon of a glycoside linkage that is hydrolyzed by these enzymes. Hydrolysis at this intramolecular glycoside center would therefore be a ring opening reaction, suggesting that perhaps TamE inactivates excess intracellular tirandamycin as a mechanism of self-protection for the producing organism. tamG and tamH encode, respectively, a predicted ATP-dependent DNA helicase and a LuxR type regulatory protein that are proposed to be pathway specific positive regulators of tirandamycin biosynthesis.[46] tamJ and tamK encode a predicted ABC-type transporter and a divergently oriented TetR-like transcriptional regulator, respectively. A similar arrangement of genes is found in the tetracycline resistance determinant of E. coli, in which the TetR repressor allows expression of the efflux pump TetA only when bound to tetracycline.[47] It seems likely that the TamJ efflux pump affords a similarly regulated mechanism of self-resistance.
Discussion
Identification and analysis of the tirandamycin biosynthetic gene cluster has revealed a hybrid PKS-NRPS whose domain organization corresponds with the carbon skeleton of the tirandamycins. However, maturation of the predicted linear intermediate 9 into TirC, which requires tetramic acid ring closure and bicyclic ketal formation, is not fully understood. Beyond bicyclic ketal formation, installation of the C-11/C-12 cis double bond is also required to generate TirC. It is important to note that the position of this bond deviates from that expected by a canonical PKS embedded DH domain because it spans an intra-subunit bond derived from an intact methylmalonate precursor, whereas the conventional DH activity results in formation of a double bond between subunits. Based on sequence analysis, TamAI module 2 contains no discernible DH domain adjacent to the KR that reduces the C-11 keto group to a hydroxyl, and the only proximal DH activity occurs in the downstream TamAI module 3. In epothilone biosynthesis it has been demonstrated that cis double bond formation during the fourth extension cycle is actually the result of an unexpected DH activity of EpoC module 5, as no EpoC module 4 DH is present.[48] However, this dehydration still forms an α-β double bond, whereas a parallel strategy in tirandamycin biosynthesis would require formation of a β-γ olefin, making this route unlikely. It is more plausible that a post-PKS dehydration step yields the cis olefin, as is the case for the six-membered ring lactone olefin of phoslactomycin.[49] No evident enzyme candidate is encoded within the tirandamycin gene cluster, suggesting that the corresponding dehydratase gene might exist elsewhere in the Streptomyces sp. 307-9 genome.
It is also relevant to consider the timing of cis double bond formation relative to bicyclic ketal formation, since cyclization may be influenced by the conformational constrains imposed by the cis olefin. Str also contains a cis double bond within the bicyclic ketal, though its C-10/C-11 position is consistent with canonical PKS mediated olefin formation. Thus, in Str biosynthesis a post-assembly line cyclization step likely proceeds on a cis-olefin constrained substrate, but this is less clear in the case of tirandamycin biosynthesis. Indeed, caution must be taken in drawing parallels between the cyclizations of Str and tirandamycin. Str feeding studies with [13C, 18O]-labelled precursors indicate that the C-7/C-13 ether oxygen is incorporated with the C-7/C-8 propionate unit,[8] and the C-9/C-13 bridging ether oxygen originates from the C-9/C-10 acetate unit. This conflicts with our PKS domain analysis that prescribes elimination of the tirandamycin C-9 oxygen by module 3 DH to create the C-8/C-9 olefin. In the absence of a characterized Str biosynthetic gene cluster, no genetic comparison is possible, but these preliminary results suggest that multiple routes to bicyclic ketal formation may exist.
Formation of the bicyclic ketal skeleton alone is not sufficient to produce an effective bioactive molecule, as the potency of the early intermediate TirC (MIC 110 μM) is far less than that of TirA (MIC 2.25 μM), making the oxidative tailoring of the bicyclic ketal a crucial element of tirandamycin biosynthesis. Our gene disruption experiments demonstrated that a ΔtamI mutant accumulates TirC, an intermediate that lacks oxidative tailoring of the bicyclic ketal. This P450 homolog is thus critical for at least the first, and potentially several, of these modifications. Indeed, our ongoing work has shown that TamI is responsible for many of these oxidative tailoring steps, which will be reported in due course (J. C. Carlson, S. Li, Y. Anzai, D. A. Burr, D. H. Sherman, in preparation).
Conclusion
The tirandamycins represent a small class of acyl tetramic acid antibiotics bearing a bicyclic ketal moiety of great biosynthetic interest. In tirandamycin biosynthesis, this scaffold undergoes extensive oxidative modification that enhances bioactive potency. Identification and initial characterization of the tam biosynthetic pathway from Streptomyces sp. 307-9 has provided the first genetic insight into this class of compounds and revealed enzyme candidates for several intriguing biochemical transformations. These results, along with our previous isolation of novel tirandamycin intermediates,[12] provide the foundation for further investigation of the molecular mechanisms of tirandamycin biosynthesis.
Experimental Section
General Experimental Procedures
LC-MS analysis was performed on a Shimadzu 2010 EV ESI spectrometer using an XBridgeTM C18 3.5 μm 50 mm column with a MeCN and H2O solvent system supplemented with formic acid (0.1% v/v). DNA sequencing was performed at the University of Michigan DNA Sequencing Core Facility using the dideoxy chain terminator method. Sequence assembly and analysis was performed using the VectorNTI (Invitrogen) and Lasergene (DNAStar) packages.
Culture Maintenance and Fermentation
Streptomyces sp. 307-9 was maintained on ISP2 agar plates and as glycerol stocks. All culture incubations were at 30°C, and with shaking at 150 rpm for liquid cultures. Seed cultures were of 2xYT media (10 mL) inoculated with a loopful of vegetative cells from plate culture. Fermentation cultures were shake flasks of Md2 media (10 g dextrose, 2 g NZ-Amine A, 1 g yeast extract, 0.77 g meat extract, 30 g NaCl per 1 L H2O) seeded with a 1% inoculum of 2xYT culture and grown for 5 days.
Generation and Probing of Fosmid Library
Genomic DNA from Streptomyces sp. 307-9 was isolated using standard procedures[39] and fractionated on an agarose gel by pulsed field electrophoresis. Fragments of ~40 kb were excised, recovered from the gel, and cloned into the pCCFOS1 fosmid vector using the Epicentre Copy Control Fosmid Kit according to manufacturers directions. The library was packaged using MaxPlax Lambda phage extracts (Epicentre) and transduced into the appropriate E. coli host strain according to manufacturers directions. ~1200 resulting clones were outgrown in LB broth and transferred by 96-pin replicator to nitrocellulose membranes. These membranes were overlaid onto LB agar and grown overnight, after which the membrane bound clones were lysed and the DNA immobilized using standard methods.[50]
Probes for P450 and KS genes were generated by PCR amplification off of Streptomyces sp. 307-9 gDNA using degenerate primers. Resulting products of the expected size were sequenced and found to be mixed products representing homologues of the anticipated genes. These PCR products were labeled with 32P dCTP using a Radprime Labeling Kit (Invitrogen) according to manufacturers instructions. The labeled probes were hybridized to the nitrocellulose-immobilized library and the membranes were exposed to X-ray film overnight. Clones that generated positive spots were cultured from the parent library, validated by PCR screening and end-sequencing of inserts. Fosmids 2H2, 6G9, and 9H8 were chosen for shotgun sequencing. Briefly, preparations of fosmid DNA were randomly fragmented by nebulization (AeroMist Treatment reservoir, Inhalation Plastics Inc.), blunt-ended using the End-It™ DNA end repair kit (Epicentre), and size selected for fragments of 2–4 kb by gel electrophoresis. The recovered target fragments were cloned into the pSMART HC-Kan vector (Lucigen) and transformed into E. cloni electrocompetent cells (Lucigen), after which plasmid DNA was harvested from the resultant clones and sequenced.
Sequence Assembly
Sequence reads were scanned for vector and contamination derived sequences, yielding a set of approximately 1000 reads that assembled into several large contigs, after which primer walking was used to bridge the few remaining gaps, yielding 92,267 bp of contiguous sequence. Functional predictions were made for gene products of ORFs of at least 50 codons in length initiated by ATG or GTG start codons.
Generating Gene Disruption Mutants
Knockout vectors were constructed by PCR targeted mutagenesis of appropriate cosmids as described previously.[40] This REDIRECT system was used to completely replace tamI of cosmid 6G9 with a cassette bearing an apramycin resistance gene and an origin of transfer for conjugation. The AprR mutagenized cosmid was introduced into E. coli S17-1 by transformation and then transferred to Streptomyces sp. 307-9 by conjugation. Intergeneric conjugation between E. coli S17-1 and Streptomyces sp. 307-9 was performed as described previously, with minor modification.[39] An overnight culture of the E. coli donor strain was diluted into fresh medium and incubated for 3 hours. The cells were harvested, washed twice and concentrated 10-fold in TS broth. Cells of Streptomyces sp. 307-9 were grown in 2xYT broth for several days, harvested by centrifugation, washed and re-suspended in a half volume of TS broth. These recipient cells were mixed with a half volume of E. coli donor cells, and 150 μL were plated on MS agar. The plates were incubated at 30°C for 20 hours, and then covered with water (1 mL) containing nalidixic acid (500 μg) to inhibit further growth of E. coli and apramycin (1 mg) to select Streptomyces sp. 307-9 exconjugants. Incubation at 30°C was continued for 1 – 2 weeks to allow outgrowth of the exconjugants. AprR exconjugants were screened for chloramphenicol sensitivity to detect double-crossover allelic exchange. Exconjugants with the correct antibiotic resistance phenotype were confirmed for gene replacement by PCR and restriction digest of the PCR products. Metabolite profiles for mutants were determined by culturing and extraction as described earlier, but fermentation was in media supplemented with apramycin (50 μg/mL).
Supplementary Material
Scheme 1. Routes to tetramic acid ring formation. A) Observed rearrangement of a homoserine lactone into a tetramic acid moiety. B) Analogous proposed mechanism of tetramic acid ring formation in tirandamycin biosynthesis.
Scheme 2. Terpene synthase reactions. The proposed mechanism of tirandamycin bicyclic ketal formation by TamF proceeds through an analogous carbocation formation and “intramolecular solvolysis” to effect C-O bond formation.
Acknowledgments
This work was supported by NIH grant U01 TW007404 as part of the International Cooperative Biodiversity Group initiative at the Fogarty International Center, and the Hans W. Vahlteich Professorship (to D.H.S.).
References
- 1.Schobert R, Schlenk A. Bioorg Med Chem. 2008;16:4203–4221. doi: 10.1016/j.bmc.2008.02.069. [DOI] [PubMed] [Google Scholar]
- 2.Hazuda D, et al. Antiviral Chem Chemother. 1999;10:63–70. doi: 10.1177/095632029901000202. [DOI] [PubMed] [Google Scholar]
- 3.Yu F, Zaleta-Rivera K, Zhu X, Huffman J, Millet JC, Harris SD, Yuen G, Li XC, Du L. Antimicrob Agents Chemother. 2007;51:64–72. doi: 10.1128/AAC.00931-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rinehart KL, Beck JR, Borders DB, Epstein WW, Kinstle TH, Spicer LD, Krauss D, Button AC. Antimicrob Agents Chemother. 1963;161:346–348. [PubMed] [Google Scholar]
- 5.von der Helm K, Krakow JS. Nature New Biol. 1972;235:82–83. doi: 10.1038/newbio235082a0. [DOI] [PubMed] [Google Scholar]
- 6.Vassylyev DG, Vassylyeva MN, Zhang J, Palangat M, Artsimovitch I, Landick R. Nature. 2007;448:163–168. doi: 10.1038/nature05931. [DOI] [PubMed] [Google Scholar]
- 7.Chen H, Harrison PH. Org Lett. 2004;6:4033–4036. doi: 10.1021/ol048317h. [DOI] [PubMed] [Google Scholar]
- 8.Chen H, Olesen SG, Harrison PH. Org Lett. 2006;8:5329–5332. doi: 10.1021/ol0621304. [DOI] [PubMed] [Google Scholar]
- 9.Royles BJL. Chem Rev. 1995;95:1981–2001. [Google Scholar]
- 10.Meyer CE. J Antibiot. 1971;24:558–560. doi: 10.7164/antibiotics.24.558. [DOI] [PubMed] [Google Scholar]
- 11.Hagenmaier H, Jaschke KH, Santo L, Scheer M, Zähner H. Arch Microbiol. 1976;109:65–74. doi: 10.1007/BF00425114. [DOI] [PubMed] [Google Scholar]
- 12.Carlson JC, Li S, Burr DA, Sherman DH. J Nat Prod. 2009 doi: 10.1021/np9005597. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Temiakov D, Zenkin N, Vassylyeva M, Perederina A, Tahirov T, Kashkina E, Savkina M, Zorov S, Nikiforov V, Igarashi N. Mol Cell. 2005;19:655–666. doi: 10.1016/j.molcel.2005.07.020. [DOI] [PubMed] [Google Scholar]
- 14.Tuske S, et al. Cell. 2005;122:541–552. doi: 10.1016/j.cell.2005.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schobert R. Naturwissenschaften. 2007;94:1–11. doi: 10.1007/s00114-006-0152-8. [DOI] [PubMed] [Google Scholar]
- 16.Sims JW, Fillmore JP, Warner DD, Schmidt EW. Chem Commun. Cambridge, U. K: 2005. pp. 186–188. [DOI] [PubMed] [Google Scholar]
- 17.Bihlmaier C, Welle E, Hofmann C, Welzel K, Vente A, Breitling E, Müller M, Glaser S, Bechthold A. Antimicrob Agents Chemother. 2006;50:2113–2121. doi: 10.1128/AAC.00007-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song Z, Cox RJ, Lazarus CM, Simpson TJ. Chembiochem. 2004;5:1196–1203. doi: 10.1002/cbic.200400138. [DOI] [PubMed] [Google Scholar]
- 19.Cane DE, Walsh CT, Khosla C. Science. 1998;282:63–68. doi: 10.1126/science.282.5386.63. [DOI] [PubMed] [Google Scholar]
- 20.Urlacher VB, Lutz-Wahl S, Schmid RD. Appl Microbiol Biotechnol. 2004;64:317–325. doi: 10.1007/s00253-003-1514-1. [DOI] [PubMed] [Google Scholar]
- 21.Finn RD, et al. Nucleic Acids Res. 2006;34:D247–251. doi: 10.1093/nar/gkj149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aparicio JF, Molnár I, Schwecke T, König A, Haydock SF, Khaw LE, Staunton J, Leadlay PF. Gene. 1996;169:9–16. doi: 10.1016/0378-1119(95)00800-4. [DOI] [PubMed] [Google Scholar]
- 23.Donadio S, Katz L. Gene. 1992;111:51–60. doi: 10.1016/0378-1119(92)90602-l. [DOI] [PubMed] [Google Scholar]
- 24.Brautaset T, Sekurova ON, Sletta H, Ellingsen TE, StrLm AR, Valla S, Zotchev SB. Chem Biol. 2000;7:395–403. doi: 10.1016/s1074-5521(00)00120-4. [DOI] [PubMed] [Google Scholar]
- 25.Haydock SF, Aparicio JF, Molnár I, Schwecke T, Khaw LE, König A, Marsden AF, Galloway IS, Staunton J, Leadlay PF. FEBS Lett. 1995;374:246–248. doi: 10.1016/0014-5793(95)01119-y. [DOI] [PubMed] [Google Scholar]
- 26.Bisang C, Long PF, Cortés J, Westcott J, Crosby J, Matharu AL, Cox RJ, Simpson TJ, Staunton J, Leadlay PF. Nature. 1999;401:502–505. doi: 10.1038/46829. [DOI] [PubMed] [Google Scholar]
- 27.Challis GL, Ravel J, Townsend CA. Chem Biol. 2000;7:211–224. doi: 10.1016/s1074-5521(00)00091-0. [DOI] [PubMed] [Google Scholar]
- 28.Reid R, Piagentini M, Rodriguez E, Ashley G, Viswanathan N, Carney J, Santi DV, Hutchinson CR, McDaniel R. Biochemistry. 2003;42:72–79. doi: 10.1021/bi0268706. [DOI] [PubMed] [Google Scholar]
- 29.Caffrey P. Chembiochem. 2003;4:654–657. doi: 10.1002/cbic.200300581. [DOI] [PubMed] [Google Scholar]
- 30.Marahiel MA, Stachelhaus T, Mootz HD. Chem Rev. 1997;97:2651–2674. doi: 10.1021/cr960029e. [DOI] [PubMed] [Google Scholar]
- 31.Sims JW, Schmidt EW. J Am Chem Soc. 2008;130:11149–11155. doi: 10.1021/ja803078z. [DOI] [PubMed] [Google Scholar]
- 32.Henrissat B, Callebaut I, Fabrega S, Lehn P, Mornon JP, Davies G. Proc Natl Acad Sci U S A. 1995;92:7090–7094. doi: 10.1073/pnas.92.15.7090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao GR, Luo T, Zhou YJ, Jiang X, Qiao B, Yu FM, Yuan YJ. Appl Microbiol Biotechnol. 2009;82 doi: 10.1007/s00253-009-1872-4. Online Edition. [DOI] [PubMed] [Google Scholar]
- 34.Demydchuk Y, Sun Y, Hong H, Staunton J, Spencer JB, Leadlay PF. Chembiochem. 2008;9:1136–1145. doi: 10.1002/cbic.200700715. [DOI] [PubMed] [Google Scholar]
- 35.Kaufmann GF, Sartorio R, Lee SH, Rogers CJ, Meijler MM, Moss JA, Clapham B, Brogan AP, Dickerson TJ, Janda KD. Proc Natl Acad Sci U S A. 2005;102:309–314. doi: 10.1073/pnas.0408639102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Werck-Reichhart D, Feyereisen R. Genome Biol. 2000;1:3003. doi: 10.1186/gb-2000-1-6-reviews3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kleiger G, Eisenberg D. J Mol Biol. 2002;323:69–76. doi: 10.1016/s0022-2836(02)00885-9. [DOI] [PubMed] [Google Scholar]
- 38.Kutchan TM, Dittrich H. J Biol Chem. 1995;270:24475–24481. doi: 10.1074/jbc.270.41.24475. [DOI] [PubMed] [Google Scholar]
- 39.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. Practical Streptomyces Genetics. John Innes Foundation; Norwich, U.K: 2000. [Google Scholar]
- 40.Gust B, Challis GL, Fowler K, Kieser T, Chater KF. Proc Natl Acad Sci U S A. 2003;100:1541–1546. doi: 10.1073/pnas.0337542100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lambalot RH, Gehring AM, Flugel RS, Zuber P, LaCelle M, Marahiel MA, Reid R, Khosla C, Walsh CT. Chem Biol. 1996;3:923–936. doi: 10.1016/s1074-5521(96)90181-7. [DOI] [PubMed] [Google Scholar]
- 42.Heathcote ML, Staunton J, Leadlay PF. Chem Biol. 2001;8:207–220. doi: 10.1016/s1074-5521(01)00002-3. [DOI] [PubMed] [Google Scholar]
- 43.Yu FM, Qiao B, Zhu F, Wu JC, Yuan YJ. Appl Biochem Biotechnol. 2006;135:145–158. doi: 10.1385/abab:135:2:145. [DOI] [PubMed] [Google Scholar]
- 44.Bendtsen JD, Nielsen H, von Heijne G, Brunak S. J Mol Biol. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 45.Zhao L, Sherman DH, Liu HW. J Am Chem Soc. 1998;120:9374–9375. [Google Scholar]
- 46.He W, Lei J, Liu Y, Wang Y. Arch Microbiol. 2008;189:501–510. doi: 10.1007/s00203-007-0346-2. [DOI] [PubMed] [Google Scholar]
- 47.Hillen W, Berens C. Annu Rev Microbiol. 1994;48:345–369. doi: 10.1146/annurev.mi.48.100194.002021. [DOI] [PubMed] [Google Scholar]
- 48.Tang L, Ward S, Chung L, Carney JR, Li Y, Reid R, Katz L. J Am Chem Soc. 2004;126:46–47. doi: 10.1021/ja030503f. [DOI] [PubMed] [Google Scholar]
- 49.Alhamadsheh MM, Palaniappan N, Daschouduri S, Reynolds KA. J Am Chem Soc. 2007;129:1910–1911. doi: 10.1021/ja068818t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sambrook J, Russell DW. Molecular Cloning - A Laboratory Manual. 3. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 2001. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Scheme 1. Routes to tetramic acid ring formation. A) Observed rearrangement of a homoserine lactone into a tetramic acid moiety. B) Analogous proposed mechanism of tetramic acid ring formation in tirandamycin biosynthesis.
Scheme 2. Terpene synthase reactions. The proposed mechanism of tirandamycin bicyclic ketal formation by TamF proceeds through an analogous carbocation formation and “intramolecular solvolysis” to effect C-O bond formation.