

Abstract
Pseudomonas aeruginosa is naturally resistant to many antibiotics, and infections caused by this organism are a serious threat, especially to hospitalized patients. The intrinsic low permeability of P. aeruginosa to antibiotics results from the coordinated action of several mechanisms, such as the presence of restrictive porins and the expression of multidrug efflux pump systems. Our goal was to develop antimicrobial peptides with an improved bacterial membrane-permeabilizing ability, so that they enhance the antibacterial activity of antibiotics. We carried out a structure activity relationship analysis to investigate the parameters that govern the permeabilizing activity of short (8- to 12-amino-acid) lactoferricin-derived peptides. We used a new class of constitutional and sequence-dependent descriptors called PEDES (peptide descriptors from sequence) that allowed us to predict (Spearman's ρ = 0.74; P < 0.001) the permeabilizing activity of a new peptide generation. To study if peptide-mediated permeabilization could neutralize antibiotic resistance mechanisms, the most potent peptides were combined with antibiotics, and the antimicrobial activities of the combinations were determined on P. aeruginosa strains whose mechanisms of resistance to those antibiotics had been previously characterized. A subinhibitory concentration of compound P2-15 or P2-27 sensitized P. aeruginosa to most classes of antibiotics tested and counteracted several mechanisms of antibiotic resistance, including loss of the OprD porin and overexpression of several multidrug efflux pump systems. Using a mouse model of lethal infection, we demonstrated that whereas P2-15 and erythromycin were unable to protect mice when administered separately, concomitant administration of the compounds afforded long-lasting protection to one-third of the animals.
The increasing emergence of antibiotic-resistant bacteria is a major public health problem. Whereas therapeutic options for the treatment of infections due to Gram-positive bacteria (e.g., methicillin-resistant Staphylococcus aureus [MRSA]) are in expansion, there is an urgent need for the development of new drugs effective against Gram-negative pathogens. The latter organisms, in particular, Pseudomonas aeruginosa, Acinetobacter baumannii, Stenotrophomonas maltophilia, and several members of the Enterobacteriaceae, are rapidly evolving resistance to most of the currently available antibiotic treatments (37).
The resistance of Gram-negative bacteria to antibiotics is highly dependent on the integrity of the outer membrane (OM), since this structure acts as a permeability barrier and efficiently prevents many antimicrobial compounds from reaching their targets inside the cell (9). In addition, key mechanisms of resistance, such as antibiotic removal via efflux pump systems, rely on the ability of the OM to maintain concentrations of drugs in the periplasm at levels nontoxic for the cell (42). In turn, the OM owes many of its properties, including its low intrinsic permeability, to an external layer composed primarily of a negatively charged molecule called lipopolysaccharide (LPS). Stability of the OM depends to a large extent on the cross-bridging of neighboring LPS molecules by divalent cations (41).
Due to the essential role of LPS in OM stability and the structural conservation of this molecule in different species of Gram-negative bacteria, virtually all cellular organisms produce antibacterial agents targeting LPS. The most prominent example of these agents belongs to a family called host defense peptides or antimicrobial peptides (AMPs) (27). The cationic nature and amphiphilic structure characteristic of the vast majority of AMPs enable them to bind specifically to LPS, destabilize the OM, and kill the target cell by mechanisms that are still a matter of debate (6, 38). However, it is clear (at least for some AMPs) that when added at concentrations lower than their MICs, they alter the bacterial cell permeability barrier, thus making bacteria sensitive to agents that would be excluded by an intact outer membrane (54, 60).
Our model organism is Pseudomonas aeruginosa, one of the leading causative agents of infections in hospital settings. The intrinsic low permeability of the P. aeruginosa OM (at least 10 times lower than that of E. coli [see reference 19]), combined with the activity of efflux pumps of the Mex family and beta-lactamase expression (AmpC), confers to this pathogen its characteristic broad spectrum of antibiotic resistance (39). Moreover, P. aeruginosa often mutates and gives rise to strains with enhanced antibiotic resistance due to overexpression of efflux pumps, acquisition of plasmid-borne metallo-beta-lactamases (MBLs), and repression of the OprD porin (the uptake pathway for hydrophilic carbapenems, such as imipenem) (5). Consequently, some isolates of P. aeruginosa remain sensitive only to polymyxins, although the systemic use of these AMPs is not widely accepted by clinicians due to their alleged nephro- and neurotoxicity (35).
We have previously demonstrated that subinhibitory concentrations of synthetic peptides derived from lactoferrin, a human protein with antibacterial and LPS binding activities, sensitize P. aeruginosa to both hydrophobic and hydrophilic antibiotics in vitro (53). Although other researchers have reported similar findings using either polymyxin B (PMB) nonapeptide (PMBN; an enzymatic derivative of PMB) (43, 59) or other polycations (3, 33, 36, 46, 52, 56, 61), no one has studied whether OM-permeabilizing peptides share common structural features not necessarily related to their antibacterial activity. In this respect, it is important to point out that good permeabilizers do not necessarily correspond to compounds with potent bactericidal activity (53). To the contrary, it has been reported that those activities are inversely correlated in some instances (56, 64), suggesting that at least for some classes of AMPs, OM permeabilization and bactericidal activity may have different structural bases.
In this work, we designed a novel type of antimicrobial peptide descriptors for lactoferrin-derived peptides and performed a quantitative structure activity relationship (QSAR) analysis to predict the permeabilizing activity of a new peptide generation. We demonstrated that the most potent peptide from this new library enhances antibiotic activity in vivo and, as a consequence, confers protection to an animal model of lethal P. aeruginosa infection. This work sets the foundation for the rational design of improved lactoferrin-derived permeabilizers that could act synergistically with antibiotics and neutralize resistance mechanisms.
MATERIALS AND METHODS
Bacterial strains.
All the strains were grown at 37°C in LB (Luria-Bertani broth; Pronadisa, Madrid, Spain) or TSB (tryptic soy broth; BioMérieux, Marcy l'Etoile, France) supplemented with agar (Pronadisa, Alcobendas-Madrid, Spain) when necessary. Strains used in the present study are listed in Table 1.
TABLE 1.
Characterization of some relevant mechanisms of antibiotic resistance in the P. aeruginosa strains used in this study
| Strain | Relevant feature(s) | Source/reference | Expression of efflux pump systema |
Other mechanisms |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MexAB-OprM | MexCD-OprJ | MexEF-OprN | MexXY-OprM | OprDb | QRDR modificationsc | Carbd | AmpCe | Colf | |||
| PAO1 | Wild type | CECTg | + | + | + | + | + | − | − | + | − |
| PAOLC1-6 | nalB derivative of PAO1 | 53 | +++ | + | + | + | + | − | − | + | − |
| TNP004 | OprD− derivative of PAO1 | 53a | + | + | + | + | − | − | − | + | − |
| Ps1 | Wild type, clinical isolate | CUNh | +++ | + | ++ | +++ | + | + | − | +++ | − |
| Ps2 | Wild type, clinical isolate | CUN | + | +++ | +++ | +++ | − | + | − | NDi | − |
| Ps4 | Wild type, clinical isolate | CUN | +++ | + | + | + | + | + | − | ND | − |
| Ps6 | Wild type, clinical isolate | CUN | + | +++ | +++ | +++ | + | + | − | ND | − |
| Ps21 | Wild type, clinical isolate | CUN | ++ | + | + | + | − | ND | − | +++ | − |
| Ps64 | Wild type, clinical isolate | CUN | +++ | +++ | +++ | +++ | − | ND | − | ND | − |
| Ps71 | Wild type, clinical isolate | CUN | ++ | +++ | +++ | +++ | + | ND | + | + | − |
| Ps74 | Wild type, clinical isolate | CUN | +++ | +++ | +++ | +++ | − | ND | − | + | + |
Measured by reverse-transcription PCR (RT-PCR) and Western blotting. “+,” “++,” and “+++” denote the presence of basal, intermediate, and high levels of expression of the indicated efflux pump systems, respectively.
The presence (+) or absence (−) of the OprD porin was assessed by RT-PCR and sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
The presence (+) or absence (−) of mutations in the QRDRs of genes gyrA and parC was detected by sequencing.
Expression of carbapenemases (Carb) (+) was quantified by both phenotypic and molecular methods, as described in Materials and Methods.
Inducible (+) or constitutive (+++) AmpC expression was assessed by measuring the hydrolysis of cephalothin.
Resistance to colistin (Col).
CECT, Colección Española de Cultivos Tipo (Spanish Culture Type Collection).
Isolated at the Clínica Universidad de Navarra (CUN; University Hospital of Navarra).
ND, not done.
Peptides.
Peptides, purchased from PolyPeptide Laboratories (San Diego, CA), were synthesized with an amidated C terminus using 9-fluorenylmethyloxycarbonyl (Fmoc) solid-phase chemistry and were purified by reverse-phase high-pressure liquid chromatography (RP-HPLC). Purity (>96%) and identity were verified by RP-HPLC and mass spectroscopy analyses. Peptides sequences are shown in the tables (Table 2; see Table 4).
TABLE 2.
Novobiocin-enhancing activity of first-generation peptides on P. aeruginosa Ps4
| Peptide | Sequence | MIC (μg/ml)a | Novobiocin MIC at indicated peptide concn (μg/ml) |
FICb | MIC ratioc | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 6.25 | 12.5 | 25 | 50 | ||||||||||||||||||||
| LF11 | F | Q | W | Q | R | N | I | R | K | V | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | >256 | >0.5 | 0 | ||||
| C12LF11 | lauryl | F | Q | W | Q | R | N | I | R | K | V | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | >256 | >0.5 | 0 | |||
| P1-1 | F | W | Q | R | N | I | R | K | V | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | >256 | >0.5 | 0 | |||||
| P1-2 | F | W | Q | R | I | R | K | V | R | -NH2 | 250 | 512 | >256 | 256 | 128 | 32 | 0.262 | 16 | ||||||
| P1-3 | F | W | R | N | I | R | K | V | R | -NH2 | >250 | 512 | >256 | >256 | >256 | 256 | >0.5 | 2 | ||||||
| P1-4 | F | W | Q | R | N | I | R | V | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | >256 | >0.5 | 0 | ||||||
| P1-5 | F | W | Q | R | N | I | K | V | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | >256 | >0.5 | 0 | ||||||
| P1-6 | F | W | R | I | R | K | V | R | -NH2 | 250 | 512 | >256 | 256 | 128 | 32 | 0.262 | 16 | |||||||
| P1-7 | F | W | Q | R | N | I | R | K | V | R | R | -NH2 | 250 | 512 | >256 | >256 | 256 | 64 | 0.325 | 8 | ||||
| P1-8 | F | W | Q | R | N | I | R | K | V | K | K | -NH2 | >250 | 512 | >256 | 256 | 128 | 64 | 0.225 | 8 | ||||
| P1-9 | F | W | Q | R | N | I | R | K | V | R | R | R | -NH2 | >250 | 512 | >256 | 256 | 128 | 64 | 0.225 | 8 | |||
| P1-10 | F | W | Q | R | N | I | R | K | V | K | K | K | -NH2 | >250 | 512 | 256 | 128 | 32 | 32 | 0.113 | 16 | |||
| P1-11 | F | W | Q | R | N | I | R | K | V | R | R | R | R | -NH2 | 250 | 512 | >256 | 256 | 128 | 64 | 0.325 | 8 | ||
| P1-12 | F | W | Q | R | N | I | R | K | V | R | R | R | I | -NH2 | 250 | 512 | >256 | 256 | 64 | 32 | 0.225 | 16 | ||
| P1-13 | F | W | Q | R | N | I | R | K | V | K | K | K | K | -NH2 | >250 | 512 | >256 | 256 | 128 | 64 | 0.225 | 8 | ||
| P1-14 | F | W | Q | R | N | I | R | K | V | K | K | K | I | -NH2 | >250 | 1,024 | 256 | 128 | 64 | 32 | 0.113 | 32 | ||
| P1-15 | F | W | Q | R | R | I | R | K | V | R | R | -NH2 | 250 | 512 | 256 | 128 | 32 | 8 | 0.163 | 64 | ||||
| P1-16 | F | W | Q | R | K | I | R | K | V | K | K | -NH2 | >250 | 512 | >256 | 256 | 128 | 64 | 0.225 | 8 | ||||
| P1-17 | R | F | W | Q | R | N | I | R | K | V | R | R | -NH2 | >250 | 512 | >256 | >256 | >256 | 256 | >0.5 | 2 | |||
| P1-18 | K | W | Q | R | N | I | R | K | V | R | R | -NH2 | >250 | 512 | >256 | 256 | 128 | 64 | 0.225 | 8 | ||||
| P1-19 | R | W | Q | R | N | I | R | K | V | R | R | -NH2 | 250 | 512 | >256 | 256 | 128 | 32 | 0.263 | 16 | ||||
| P1-20 | R | R | W | Q | R | N | I | R | K | V | R | R | -NH2 | >250 | 512 | >256 | 256 | 64 | 32 | 0.163 | 16 | |||
| P1-21 | R | F | W | Q | R | N | I | R | K | Y | R | -NH2 | >250 | 512 | >256 | >256 | 256 | 128 | 0.350 | 4 | ||||
| P1-22 | R | F | R | W | Q | R | N | I | R | K | Y | R | R | -NH2 | 31.25 | 1,024 | >256 | 32 | 1 | NC | 0.431 | >1,024 | ||
| P1-23 | R | W | Q | R | N | I | R | K | Y | R | R | -NH2 | 250 | 1,024 | >256 | >256 | 256 | 64 | 0.263 | 16 | ||||
| P1-24 | R | R | W | Q | R | N | I | R | K | Y | R | R | -NH2 | >250 | 1,024 | >256 | >256 | 128 | 32 | 0.131 | 32 | |||
| P1-25 | C | F | W | Q | R | N | I | R | K | V | R | C | -NH2 | >250 | 1,024 | >256 | >256 | >256 | >256 | >0.5 | 0 | |||
| P1-26 | C | F | W | Q | R | N | I | R | K | V | C | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | >256 | >0.5 | 0 | |||
| P1-27 | C | W | Q | R | N | I | R | K | C | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | >256 | >0.5 | 0 | |||||
| P1-28 | C | F | W | Q | R | N | I | R | K | V | R | C | -NH2 | >250 | 1,024 | >256 | >256 | >256 | >256 | >0.5 | 0 | |||
| P1-29 | F | W | Q | R | N | I | R | K | I | R | -NH2 | 250 | 1,024 | >256 | >256 | >256 | 256 | 0.450 | 4 | |||||
| P1-30 | F | W | Q | R | N | I | R | K | L | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | 256 | 0.350 | 4 | |||||
| P1-31 | F | W | Q | R | N | I | R | K | W | R | -NH2 | 125 | 512 | >256 | >256 | >256 | >256 | >0.5 | 0 | |||||
| P1-32 | F | W | Q | R | N | I | R | K | Y | R | -NH2 | >250 | 512 | >256 | >256 | >256 | 256 | >0.5 | 2 | |||||
| P1-33 | F | W | Q | R | N | I | R | K | F | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | 256 | 0.350 | 4 | |||||
| P1-34 | F | Y | Q | R | N | I | R | K | V | R | -NH2 | >250 | 512 | >256 | >256 | >256 | >256 | >0.5 | 0 | |||||
| P1-35 | F | F | Q | R | N | I | R | K | V | R | -NH2 | >250 | 512 | >256 | >256 | >256 | >256 | >0.5 | 0 | |||||
| P1-36 | F | W | Q | R | N | I | R | I | R | R | -NH2 | 250 | 512 | >256 | >256 | >256 | 128 | 0.450 | 4 | |||||
| P1-37 | F | W | Q | R | N | W | R | K | V | R | -NH2 | >250 | 512 | >256 | >256 | >256 | 64 | 0.225 | 8 | |||||
| P1-38 | F | W | Q | R | N | F | R | K | V | R | -NH2 | 250 | 512 | >256 | >256 | >256 | 128 | 0.450 | 4 | |||||
| P1-39 | F | W | Q | R | N | Y | R | K | V | R | -NH2 | 250 | 512 | >256 | >256 | >256 | 256 | >0.5 | 2 | |||||
| P1-40 | F | W | Q | R | N | I | F | K | V | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | >256 | >0.5 | 0 | |||||
| P1-41 | F | W | Q | R | N | I | Y | K | V | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | >256 | >0.5 | 0 | |||||
| P1-42 | F | A | W | Q | R | N | I | R | K | V | R | -NH2 | >250 | 1,024 | >256 | >256 | 256 | 256 | 0.300 | 4 | ||||
| P1-43 | F | I | W | Q | R | N | I | R | K | V | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | 256 | 0.350 | 4 | ||||
| P1-44 | F | L | W | Q | R | N | I | R | K | V | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | 128 | 0.225 | 8 | ||||
| P1-45 | F | V | W | A | R | N | I | R | K | V | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | 256 | 0.350 | 4 | ||||
| P1-46 | F | W | A | R | N | I | R | K | V | R | -NH2 | >250 | 1,024 | >256 | >256 | 256 | 128 | 0.225 | 8 | |||||
| P1-47 | F | W | I | R | N | I | R | K | V | R | -NH2 | >250 | 1,024 | >256 | >256 | 128 | 32 | 0.131 | 32 | |||||
| P1-48 | F | W | L | R | N | I | R | K | V | R | -NH2 | 250 | 1,024 | >256 | 128 | 32 | 4 | 0.131 | 256 | |||||
| P1-49 | F | W | V | R | N | I | R | K | V | R | -NH2 | >250 | 1,024 | >256 | >256 | 256 | 128 | 0.225 | 8 | |||||
| P1-50 | F | W | P | R | N | I | R | K | V | R | -NH2 | >250 | 1,024 | >256 | >256 | 256 | 32 | 0.131 | 32 | |||||
| P1-51 | F | W | Q | R | P | I | R | K | V | R | -NH2 | >250 | 1,024 | >256 | >256 | 128 | 64 | 0.163 | 16 | |||||
| P1-52 | F | W | Q | R | G | I | R | K | V | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | 128 | 0.225 | 8 | |||||
| P1-53 | F | W | Q | S | N | I | R | K | V | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | >256 | >0.5 | 0 | |||||
| P1-54 | F | W | Q | R | N | I | S | K | V | R | -NH2 | >250 | 512 | >256 | >256 | >256 | >256 | >0.5 | 0 | |||||
| P1-55 | F | W | Q | R | N | I | R | S | V | R | -NH2 | >250 | 512 | >256 | >256 | >256 | >256 | >0.5 | 0 | |||||
| P1-56 | F | W | Q | R | N | I | R | K | V | S | -NH2 | >250 | 512 | >256 | >256 | >256 | >256 | >0.5 | 0 | |||||
MIC of the antibiotic determined by a microbroth-based assay in non-cation-adjusted Mueller-Hinton medium.
Fractional inhibitory concentration (FIC) index (see Materials and Methods for details). FIC indices indicative of synergy are shown in boldface.
Ratio of antibiotic MICs (determined as described in footnote a) in the absence and in the presence of the peptide. The ratio shown is the maximum value of those calculated for each peptide concentration. MIC ratios indicative of synergy are shown in boldface.
Antibiotics.
Antibiotic solutions were diluted and stored by following the manufacturer's recommendations. The antibiotics used were ampicillin, cefotaxime, chloramphenicol, erythromycin, fusidic acid, nalidixic acid, novobiocin, polymyxin B (PMB), polymyxin B nonapeptide (PMBN), tetracycline, and ticarcillin (Sigma-Aldrich, Madrid, Spain); amoxicillin and ciprofloxacin (Fluka, Madrid, Spain); meropenem (Meronem; AstraZeneca, Bilbao, Spain); and imipenem (Tienam; MSD, Madrid, Spain).
Susceptibility assays.
MICs of the peptides were determined in Mueller-Hinton (MH) medium (Difco Laboratories, Detroit, Michigan) by the broth microdilution assay by following the recommendations of the Clinical Laboratory Standards Institute (CLSI; formerly NCCLS) (40), as described elsewhere (53).
Synergy studies.
The existence of synergistic interactions between peptides and antibiotics was assessed by the checkerboard assay (13, 53), using Mueller-Hinton medium (Difco Laboratories). The fractional inhibitory concentration (FIC) index of each combination was calculated according to the following formula: [(A)/MICA] + [(P)/MICP] = FICA + FICP = FIC index, where MICA and MICP are the MICs of the antibiotic and peptide, respectively, determined separately and (A) and (P) are the MICs of the antibiotic and peptide, respectively, determined in combination.
Combinations were classified as synergistic (FIC ≤ 0.5), indifferent (0.5 < FIC < 4), and antagonist (FIC > 4). For those organisms resistant to the highest concentration tested, the MIC was arbitrarily assigned a value double of that concentration.
Combined therapy in vivo.
Female ICR (CD-1) mice weighing 20 to 23 g were purchased from Harlan Spain (Harlan Interfauna Iberica S.A., Barcelona, Spain) and randomly distributed in experimental groups (n = 16). Animals were immunosuppressed by the subcutaneous administration of two doses of cyclophosphamide 4 days before (3 mg/animal) and 1 day before (2 mg/animal) bacterial inoculation. In preliminary experiments, this treatment was shown to cause severe neutropenia (PMN < 1,000) in the animals 24 h after the second administration of the immunosuppressant. Inocula for experimental infections consisted of 105 CFU of Pseudomonas aeruginosa PAO1 administered intraperitoneally. In pilot experiments, this dose was found to be lethal for more than 90% of the animals at day 2 postinoculation. One hour after bacterial inoculation, a group of animals was intraperitoneally injected with a combination of 500 μg of the test peptide (25 mg/kg of body weight) and 500 μg of erythromycin resuspended in 200 μl of saline. Animal mortality was monitored at daily intervals for 7 days. In parallel experiments, a group of animals received a second dose of the combined treatment by the same route 5 h after the first dose administration. As controls for these experiments, a group of PAO1-inoculated animals was treated either with 500 μg of the peptide or with 500 μg of the antibiotic alone. Finally, one experimental group inoculated with bacteria was left untreated. The results of animal mortality at all experimental time points were analyzed using the Kaplan-Meier survival analysis. When the survival plots were parallel, data were compared by the log rank test, whereas for those plots that intersected, the Breslow-Gehan-Wilcoxon test was applied. P values were always obtained by comparing data from the same experiment (mortality in treated versus untreated groups). All the mouse experiments were approved by the Ethical Committee for Animal Experimentation of the University of Navarra (permission number 069/09).
Calculation of peptide descriptors.
Peptide descriptors were calculated using in-house PEDES (peptide descriptors from sequence) computer software (24) (see the supplemental material). The PEDES algorithm calculates composition and sequence-dependent physicochemical descriptors based entirely on the peptide amino acid sequence. Descriptors are based on the recapitulation of the mechanism of the antimicrobial peptide mode of action. The PEDES output consists of a file in which 32 descriptors are listed for each individual peptide (Table 3) and a file which represents the input matrix for QSAR calculation. Definitions of constitutional and sequence-dependent descriptors are provided in Table 3. LogP values were calculated using HyperChem 6.0 software.
TABLE 3.
Definitions of peptide descriptors used in the QSAR study
| Type of descriptor | Abbreviation | Definition |
|---|---|---|
| Constitutionala | length | No. of amino acid residues in the peptide |
| MW(peptide-NH2): | Mol wt of the peptide | |
| no_basic | No. of basic residues | |
| no_aromatic | No. of aromatic residues | |
| no_hydrophobic | No. of hydrophobic residues | |
| no_W | No. of Trp residues | |
| no_basic/length | No. of basic residues divided by the length of the peptide (linear density of basic residues in the sequence) | |
| % basic_res | Percentage of basic residues | |
| % hydrophobic+basic_res | Percentage of sum of hydrophobic and basic residues | |
| sum_aromatic+basic | Sum of aromatic and basic residues | |
| He | Hydrophobicity according to the Eisenberg scale (11) | |
| Hk | Hydrophobicity according to the Kyte-Doolittle scale (34) | |
| Sequence dependentb | max_dis_W | Maximum distance between Trp residues in the sequence |
| max_dis_basic | Maximum distance between basic residues in the sequence | |
| max_dis_ aromatic | Maximum distance between aromatic residues in the sequence | |
| max_dis_ hydrophobic | Maximum distance between hydrophobic residues in the sequence | |
| hmom(100deg) | Hydrophobic moment (μ) at 100° angle (5/9 π rad). 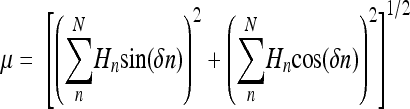 where Hn is the hydrophobicity of nth residue and δ is the angle between two sequential reinsured measured in radians where Hn is the hydrophobicity of nth residue and δ is the angle between two sequential reinsured measured in radians |
|
| hmom(160deg) | Hydrophobic moment (μ) at 160° angle | |
| hmom(180deg) | Hydrophobic moment (μ) at 180° angle | |
| max_avg_seq_chg(frame = 2) | Maximum avg charge of 2 sequential residues | |
| max_avg_seq_chg(frame = 4) | Maximum avg charge of 3 sequential residues | |
| max_avg_seq_chg(frame = 3) | Maximum avg charge of 4 sequential residues | |
| no_2-aromatic-res-clust | No. of 2 aromatic residue clusters | |
| no_2-hydrophobic-res-clust | No. of 2 hydrophobic residue clusters | |
| no_2-polar-res-clust | No. of 2 polar residue clusters | |
| no_2-basic-res-clust: | No. of 2 basic residue clusters | |
| max_basic_clust_chg | Maximum charge of basic residue cluster | |
| avg_dist_between_chg | Avg distance between basic residues | |
| avg_dist_hydrophobic_chg | Avg distance between hydrophobic residues | |
| (cen_pos_chg_res)-(cen_hydrophobic_res) | Distance between the centers of basic and hydrophobic residues | |
| (c.o.m._pos_chg_res)-(c.o.m._hydrophobic_res) | Distance between the centers of gravity of basic and hydrophobic residues | |
| linear_amphiphatic_moment |
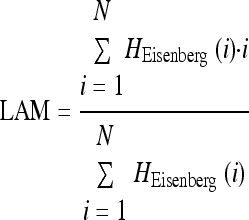 where i is the sequential residue no. in the peptide from the N-terminal part and HEisenberg (i) is the hydrophobicity, according to the Eisenberg scale where i is the sequential residue no. in the peptide from the N-terminal part and HEisenberg (i) is the hydrophobicity, according to the Eisenberg scale |
Constitutional descriptors depend fundamentally on the amino acid composition of the peptides.
Sequence-dependent descriptors depend more on the geometry and topology of the peptides.
QSAR models.
MIC activities against Pseudomonas aeruginosa were measured for a set of 93 peptides, including LF11, lauryl-LF11, 56 peptides from the first generation, and 35 peptides from the second generation. Experimental data were filtered by including only exact values and excluding all nondetermined values (e.g., MIC > 250 μg/ml). This condition reduced the original set of 93 peptides to 30 peptides for MIC activities, 53 for novobiocin MIC ratios, and 51 for FIC values.
For series of peptides with the same mode of action, it can be shown that the biological activity of a peptide, i, can be written as a linear combination of physicochemical descriptors, as follows:
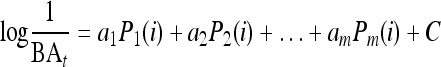 |
where m is the number of independent physicochemical parameters, Pj(i) is the jth parameter of the peptide i, BAi is the measurement of the biological activity that is expressed as the 50% inhibitory concentration (IC50) or MIC, and aj is the coefficient in the multiple linear equation corresponding to the parameter Pj(i) (49). Biological activity was modeled with PLS (partial least-squares projection to latent structures) analysis using CODESSA software (32). Peptide descriptors were centered and normalized prior to analysis to ensure that they had equal influence in the model.
RESULTS
Characterization of mechanisms of antibiotic resistance.
To study to what extent the mechanisms of antibiotic resistance expressed by Pseudomonas aeruginosa can be neutralized by peptide-mediated permeabilization, we first characterized the most relevant mechanisms of antibiotic resistance in a collection of clinical strains with distinct antibiotic susceptibility profiles. Specifically, we studied the presence of representatives of the major classes of antibiotic resistance mechanisms such as (i) systems involved in the active efflux of antibiotics (i.e., Mex family), (ii) repression of porins mediating antibiotic entry (i.e., OprD for imipenem), (iii) modifications in the intracellular antibiotic target (i.e., quinolone resistance-determining region [QRDR] of gyrase for quinolones), and (iv) production of hydrolytic enzymes (AmpC and carbapenemase production for beta-lactam antibiotics). As controls for these studies, we included nalB and oprD mutant derivatives of the wild-type P. aeruginosa PAO1 strain.
The strains chosen for this study expressed different antibiotic resistance mechanisms (Table 1), and each pattern was associated with a distinct profile of antibiotic susceptibility (data not shown). This characterization provided us with a means to infer the sensitivity of each particular combination of mechanisms to peptide-mediated permeabilization.
Quantification of the permeabilizing activity of the peptides.
Using P. aeruginosa Ps4 as a test strain, we studied the ability of the compounds from a peptide library derived from human lactoferricin (53) to sensitize P. aeruginosa to novobiocin, a hydrophobic antibiotic that cannot reach its intracellular target (gyrase) due to its inability to cross the intact outer membrane. To quantify the existence of synergy, we calculated the ratio of novobiocin MICs in the absence (MIC ≥ 512 μg/ml) and in the presence of subinhibitory concentrations of the peptides (MIC ratio). To correct for the differences in peptides' MICs, we also calculated the FIC indices for all the peptide-novobiocin combinations (Table 2). The majority of authors consider a combination synergistic when its FIC index is ≤0.5 or when the MIC ratio is at least 4 (13).
Based on those criteria, most of the peptides (36 of 56) acted in synergy with novobiocin, and in all cases, the two parameters used to detect synergy were in agreement. However, the information provided by FIC indices and MIC ratios was not quantitatively comparable (Table 2). Based on these results and other comparative physical data (not shown), a second set of peptides was generated (called second-generation peptides).
QSAR model of the antibiotic-enhancing activity of peptides.
We hypothesized that we could predict the permeabilizing activity of the new generation of peptides based on the novobiocin-enhancing activity of the first generation of peptides (henceforth second and first generation, respectively). To correlate physicochemical properties to biological activity, we performed a QSAR analysis using experimentally determined biological data, including antimicrobial activity, affinity for LPS, and structural data based on determined tertiary structures, and combined it with partial least-squares regression (also known as partial least-squares projection to latent structures [PLS]). Since peptides are highly difficult to treat using a conventional QSAR approach due to their complex structure soft-modeling PLS, we devised a new set of 32 structural and constitutional parameters called PEDES. These parameters describe physicochemical properties of the peptides mechanistically related to their antimicrobial and permeabilizing activities (Table 3), as detailed in Materials and Methods.
Peptide descriptors contain both compositional and sequence-related information, such as the content of basic residues or the number of hydrophobic residue clusters, respectively. Descriptors were arranged in the input x matrix, where each peptide constituted a row and each descriptor constituted a column. Similarly, MIC values (factor 1), novobiocin MIC ratios at peptide concentrations of 50 μg/ml (factor 2), and FIC values (factor 3) were used to construct a 93-by-3 matrix.
A principal component analysis of log(1/MIC of peptide) showed that peptides distribute into approximately three clusters according to their number of basic residues (see Fig. S1b in the supplemental material). A first cluster consisting of P1-7, P1-15, P1-11, P1-12, and P1-19 contained peptides with five or more basic residues. Most of the peptides which clustered around the origin contained four basic residues, whereas P2-23, P2-29, P2-30, and P2-31 each possessed three basic residues. In addition, each cluster could be subdivided according to the number of hydrophobic residues. Variation in the descriptors could be related to variation in MIC values in the loading plots (see Fig. S1a in the supplemental material). Most of the variation could be assigned to the first two principle components, which explained 64% of the total variation. Since peptides with lower MIC values have larger log(1/factor 1) values, descriptors which are oriented in the same direction as that of the log(1/factor 1) vector in the factor 1-factor 2 plane will have an increase in antimicrobial activity, and the opposite (mirrored through the origin) will occur to those descriptors that are negatively correlated. The PLS-loading plot in Fig. S1a in the supplemental material shows that the most important descriptors governing antibacterial activity are the number of Trp residues, hydrophobicity, the number of aromatic residues, and the percentage of hydrophobic plus basic residues. Negative correlation was observed for the following descriptors: the number of basic residues, maximum basic cluster charge, number of 2-polar residue clusters, and length. The success of QSAR analysis was confirmed by the agreement found between experimental and calculated MIC values (R2 = 0.94 for the 5-parameter model), as shown in Fig. 1A. The model was validated using leave-one-out cross-validation (CV), which rendered an R2(CV) value of 0.90 (58).
FIG. 1.

Partial least-squares projection to latent structures (PLS) analysis of the correlation between descriptors and antimicrobial activities, expressed as MIC values (A), novobiocin MIC ratio values at a 50 μg/ml peptide concentration (B), and FIC values (C) against P. aeruginosa. Models illustrating the correlation between experimental and calculated values are shown.
A similar approach was used to analyze novobiocin MIC ratios at a 50 μg/ml peptide concentration and FIC data. For novobiocin MIC ratios at a 50 μg/ml peptide concentration, PLS scores showed that peptides clustered according to their numbers of basic residues (see Fig. S1d in the supplemental material), in which we found a clear distinction between peptides of the first and second generations, except for peptides with the same number of cationic residues. The PLS-loading plot in Fig. S1c in the supplemental material revealed a positive correlation between log(1/factor 2) and the following descriptors: the number of Trp, the hydrophobic moment, the maximum distance between aromatic residues, and hydrophobicity. A negative correlation was found between log(1/factor 2) and length, molecular weight (MW), maximum basic cluster charge, maximum average sequential charge, and the number of two basic residue clusters. The model which contained five significant components (factors 1, 3, 4, 5, and 23) explained 85% of the variation and showed good agreement with experimental data (R2 = 0.838) (Fig. 1B). The most important parameters that positively correlated with factor 3 were the hydrophobic moment, basic residue density (the number of basic residues/length), and the number of basic residues (see Fig. S1e in the supplemental material), whereas negative correlation was observed in terms of the number of hydrophobic residues and hydrophobicity, suggesting that charge plays an important role in FIC activity. The PLS regression model is shown in Fig. 1C (R2 = 0.844).
To test the validity of our QSAR analysis, we used the PLS models to predict the permeabilizing activity of the second generation of peptides. In parallel, we quantified the novobiocin-enhancing activity of the new peptide generation, as expressed by their MIC ratios and FIC indices. The results from these assays are shown in Table 4, along with the relative position that each compound occupies in the ranking of permeability-enhancing activity, according to our predictions. Almost all the compounds with significant permeabilizing activity were correctly predicted to rank among the first two groups, whereas those peptides lacking novobiocin-potentiating activity fell into the third and fourth groups, with a few exceptions. More importantly, the predicted MIC ratios were found to correlate with the corresponding experimental values to a large extent (Spearman's ρ = 0.74; P < 0.001). These experiments led to the selection of two of the most potent permeabilizing compounds, P2-15 and P2-27, for the following assays.
TABLE 4.
Novobiocin-enhancing activity of second-generation peptides on P. aeruginosa Ps4f
| Peptide | Sequence | MIC (μg/ml)a | Novobiocin MIC at indicated peptide concn (μg/ml)e |
FICb | MIC ratioc | Predicted MIC ratio (group)d | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 6.25 | 12.5 | 25 | 50 | ||||||||||||||||||
| P2-1 | F | W | Q | R | N | I | R | I | R | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | >256 | >0.5 | 0 | 3 | ||
| P2-2 | L | W | Q | R | N | I | R | I | R | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | >256 | >0.5 | 0 | 3 | ||
| P2-3 | I | W | Q | R | N | I | R | I | R | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | >256 | >0.5 | 0 | 3 | ||
| P2-4 | F | F | W | Q | R | N | I | R | I | R | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | >256 | >0.5 | 0 | 3 | |
| P2-5 | F | W | Q | R | N | W | R | I | R | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | >256 | >0.5 | 0 | 4 | ||
| P2-6 | F | W | Q | R | N | I | R | W | R | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | >256 | >0.5 | 0 | 4 | ||
| P2-7 | F | W | Q | R | N | I | R | F | R | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | >256 | >0.5 | 0 | 4 | ||
| P2-8 | F | W | Q | R | N | W | R | W | R | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | >256 | >0.5 | 0 | 3 | ||
| P2-9 | F | W | Q | R | N | W | R | F | R | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | >256 | >0.5 | 0 | 4 | ||
| P2-10 | F | W | R | N | I | R | I | R | R | -NH2 | >250 | 1,024 | >256 | >256 | 256 | 128 | 0.23 | 8 | 2 | |||
| P2-11 | F | W | R | N | I | R | I | R | R | -NH2 | 125 | 1,024 | >256 | >256 | 256 | 8 | 0.408 | 128 | 2 | |||
| P2-12 | F | W | G | R | N | I | R | I | R | R | -NH2 | >250 | 1,024 | >256 | >256 | 256 | 128 | 0.225 | 8 | 2 | ||
| P2-13 | F | W | Q | R | I | R | I | R | R | -NH2 | >250 | 1,024 | >256 | >256 | 256 | 128 | 0.225 | 8 | 2 | |||
| P2-14 | F | W | Q | R | N | L | R | L | R | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | >256 | >0.5 | 0 | 4 | ||
| P2-15 | F | W | R | I | R | I | R | R | -NH2 | 62,5 | 1,024 | 256 | 16 | 2 | NG | 0.216 | >4,096 | 1 | ||||
| P2-16 | F | W | R | N | I | R | I | W | R | R | -NH2 | 250 | 1,024 | >256 | >256 | 16 | 2 | 0.116 | 512 | 3 | ||
| P2-17 | F | W | Q | R | N | W | I | R | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | >256 | >0.5 | 0 | 4 | |||
| P2-18 | R | F | W | Q | R | N | I | R | I | R | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | >256 | >0.5 | 0 | 2 | |
| P2-19 | R | W | Q | R | N | I | R | I | R | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | >256 | >0.5 | 0 | 2 | ||
| P2-20 | F | W | Q | R | N | I | R | F | V | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | >256 | >0.5 | 0 | 4 | ||
| P2-21 | F | W | Q | R | N | I | W | R | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | >256 | >0.5 | 0 | 4 | |||
| P2-22 | F | W | R | R | N | F | W | R | R | -NH2 | 250 | 1,024 | >256 | >256 | 64 | 16 | 0.163 | 64 | 3 | |||
| P2-23 | F | W | R | W | R | R | -NH2 | 125 | 1,024 | >256 | >256 | >256 | 128 | 0.525 | 8 | 1 | ||||||
| P2-24 | F | W | R | R | W | R | R | -NH2 | 125 | 1,024 | >256 | 256 | 64 | 4 | 0.263 | 256 | 1 | |||||
| P2-25 | F | W | R | R | W | I | R | R | -NH2 | 125 | 1,024 | >256 | 32 | 4 | 0,5 | 0.131 | 2,048 | 1 | ||||
| P2-26 | F | W | R | G | W | R | I | R | R | -NH2 | 125 | 1,024 | >256 | 128 | 32 | 4 | 0.225 | 256 | 1 | |||
| P2-27 | F | W | R | R | F | W | R | R | -NH2 | 62.5 | 1,024 | 128 | 8 | NG | NG | 0.208 | >4,096 | 1 | ||||
| P2-28 | F | W | R | W | R | W | R | -NH2 | 62.5 | 1,024 | >256 | 128 | 8 | 1 | 0.325 | 1,024 | 1 | |||||
| P2-29 | F | W | R | I | W | R | W | R | -NH2 | 31.25 | 1,024 | >256 | 256 | 1 | NG | >0.5 | >4,096 | 2 | ||||
| P2-30 | F | W | R | W | R | I | W | R | -NH2 | 31.25 | 1,024 | >256 | 256 | 2 | 0,25 | >0.5 | 4,096 | 2 | ||||
| P2-31 | F | W | R | I | W | R | I | W | R | -NH2 | 62.5 | 1,024 | >256 | >256 | >256 | NG | >0.5 | 0 | 3 | |||
| P2-32 | F | I | W | R | W | R | W | R | -NH2 | 62.5 | 1,024 | >256 | >256 | >256 | NG | >0.5 | 0 | 2 | ||||
| P2-33 | P | F | W | R | I | R | I | R | R | -NH2 | 62.5 | 1,024 | 128 | 32 | 8 | 1 | 0.225 | 1,024 | 1 | |||
| P2-34 | F | W | Q | R | R | I | R | I | R | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | 128 | 0.225 | 8 | 1 | ||
| P2-35 | F | Q | W | Q | R | N | I | R | I | R | R | -NH2 | >250 | 1,024 | >256 | >256 | >256 | >256 | >0.5 | 0 | 4 | |
MIC of the antibiotic determined by a microbroth-based assay in non-cation-adjusted Mueller-Hinton medium.
Fractional inhibitory concentration (FIC) index (see Materials and Methods for details).
Ratio of antibiotic MICs (determined as described in footnote a) in the absence and in the presence of the peptide.
Classification of the peptide according to its predicted FIC index into 4 groups. Group 1, 25% of highest values; group 2, 50 to 75% of highest values; group 3, 50 to 25% of lowest values; group 4, 25% of lowest values.
NG, no growth in a control well treated with the peptide alone, thus indicating inhibitory activity of the peptide by itself at that concentration.
Activity observed versus activity predicted by QSAR analysis of the previous peptide generation.
Neutralization of P. aeruginosa resistance mechanisms.
To study to what extent lactoferricin-derived peptides could neutralize antibiotic resistance mechanisms expressed by P. aeruginosa, we combined either P2-15 or P2-27 with representatives of antibiotic classes differing in their mechanism of action. For these experiments, synergy was assessed by the checkerboard assay, using the P. aeruginosa strains whose antibiotic resistance mechanisms had been previously characterized as test organisms (Table 1). As shown in Table 5, subinhibitory concentrations of P2-15 or P2-27 sensitized wild-type strain PAO1 to several antibiotics, including a macrolide (erythromycin), a quinolone (nalidixic acid), chloramphenicol, and fusidic acid. Peptides also acted in synergy (FIC indices < 0.5) with beta-lactamic antibiotics, but sensitization was more modest than that measured for the previous antibiotics (examples shown in Table 5). P2-15 and P2-27 were able to counteract efflux pump overexpression when tested with strain PAOLC1-6, since they formed synergistic combinations with antibiotics that are known substrates of the MexAB-OprM system, with the exception of cefotaxime. In addition, both peptides efficiently sensitized strain TNP004, an isogenic PAO1 mutant lacking the imipenem-specific OprD porin, to imipenem, demonstrating that this mechanism is susceptible to peptide-mediated permeabilization. Similar results were obtained with the P. aeruginosa Ps21 and Ps64 clinical strains that not only lack OprD but also express other mechanisms contributing to imipenem resistance, such as AmpC constitutive derepression in the former strain and global efflux pump overexpression in the latter strain. In contrast, even the most potent peptide P2-15 was barely able to neutralize a mechanism of carbapenem resistance based, at least partially, on carbapenemase expression (see results from strain Ps71 in the presence of imipenem or meropenem). This phenomenon resembles the one mentioned above for AmpC, another antibiotic-hydrolyzing enzyme. Nevertheless, the clinical strain opposing the highest resistance to peptide-mediated permeabilization was, not unexpectedly, the one displaying peptide insensitivity, as observed with the colistin-resistant strain Ps74.
TABLE 5.
Antibiotic-enhancing activity of peptides added at subinhibitory concentrations to P. aeruginosa strainse
| Strain | Antibiotic | MIC (μg/ml)a | P2-15 |
P2-27 |
PMBNc |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MIC atb: |
FICd | MIC at: |
FIC | MIC at: |
FIC | |||||||||
| 6.25 | 12.5 | 25 | 6.25 | 12.5 | 25 | 6.25 | 12.5 | 25 | ||||||
| PAO1 | Erythromycin | 128 | 16 | 8 | 1 | 0.325 | 16 | 2 | NG | 0.325 | 0.25 | 0.25 | NG | 0.052 |
| Fusidic acid | 1,024 | 64 | 8 | 0.5 | 0.263 | 32 | 8 | ≤1 | 0.231 | 0.5 | 0.5 | 0.5 | 0.050 | |
| Nalidixic acid | 512 | 32 | 8 | 0.5 | 0.263 | 32 | 16 | 1 | 0.263 | 1 | 0.5 | 0.5 | 0.058 | |
| Chloramphenicol | 256 | 16 | 8 | 1 | 0.225 | 8 | 1 | 0.25 | 0.231 | 1 | 1 | 1 | 0.066 | |
| Amoxicillin | 2,048 | 256 | 128 | 32 | 0.325 | 512 | 128 | 32 | 0.350 | 2 | 1 | 1 | 0.052 | |
| Ampicillin | 1,024 | 256 | 32 | 8 | 0.431 | 512 | 32 | NG | 0.431 | 16 | 8 | 4 | 0.066 | |
| PAOLC1-6 (nalB) | Tetracycline | 32 | 4 | 4 | 1 | 0.325 | 8 | 4 | NG | 0.350 | 2 | 2 | 2 | 0.081 |
| Fusidic acid | 4,096 | >1,024 | 128 | 16 | 0.431 | >1,024 | 32 | 4 | 0.408 | 1 | 1 | 1 | 0.050 | |
| Nalidixic acid | 1,024 | 256 | 32 | 4 | 0.431 | 16 | 8 | 4 | 0.216 | 4 | 2 | 2 | 0.066 | |
| Chloramphenicol | 256 | 8 | 8 | 0.25 | 0.163 | 4 | 2 | 2 | 0.163 | 4 | 4 | 4 | 0.066 | |
| Cefotaxime | 64 | >16 | 16 | 8 | 0.650 | 32 | 16 | NG | 0.550 | 2 | 2 | 1 | 0.081 | |
| Ticarcillin | 64 | 32 | 8 | 4 | 0.525 | 16 | 16 | 4 | 0.350 | 1 | <0.12 | <0.12 | 0.081 | |
| TNP004 (oprD) | Imipenem | 8 | 2 | 0.5 | <0.12 | 0.450 | 2 | 2 | 0.25 | 0.350 | <0.125 | <0.12 | <0.12 | 0.066 |
| Ps2 | Imipenem | 32 | 32 | 32 | 8 | 0.350 | 32 | 16 | 16 | 0.550 | 2 | 2 | 1 | 0.131 |
| Meropenem | 8 | 4 | 2 | 2 | 0.350 | 16 | 8 | 4 | 0.6 | 1 | 1 | 0.5 | 0.07 | |
| Ps21 | Imipenem | 64 | 32 | 16 | 2 | 0.231 | 64 | 16 | 8 | 0.325 | 0.125 | 0.063 | <0.06 | 0.116 |
| Meropenem | 32 | 16 | 8 | 4 | 0.325 | 16 | 8 | 8 | 0.350 | 1 | 0.125 | 0.125 | 0.163 | |
| Ps64 | Imipenem | 16 | 4 | 4 | 2 | 0.225 | 8 | 8 | 8 | 0.525 | <0.06 | <0.06 | <0.06 | 0.011 |
| Meropenem | 32 | 16 | 8 | 4 | 0.175 | 16 | 8 | 4 | 0.175 | <0.06 | <0.06 | <0.06 | 0.005 | |
| Ps71 | Imipenem | 16 | 16 | 16 | 0.5 | 0.431 | 16 | 16 | 16 | 1.003 | 16 | 8 | 8 | 0.600 |
| Meropenem | 32 | 16 | 8 | 2 | 0.450 | 32 | 32 | 32 | 1.003 | 8 | 2 | 2 | 0.163 | |
| Ps74 | Imipenem | 32 | 32 | 32 | 32 | 1.003 | 32 | 32 | 32 | 1.003 | 8 | 8 | 2 | 0.350 |
| Meropenem | 8 | 8 | 4 | 4 | 0.525 | 8 | 8 | 4 | 0.550 | 1 | 1 | 0.25 | 0.250 | |
| Ps1 | Nalidixic acid | 2,048 | 64 | 32 | 16 | 0.16 | 128 | 64 | 32 | 0.23 | 64 | 32 | 8 | 0.06 |
| Ciprofloxacin | 128 | 32 | 32 | 16 | 0.65 | 32 | 32 | 32 | 0.65 | 16 | 16 | 8 | 0.28 | |
| Ps6 | Nalidixic acid | 2,048 | 512 | 64 | 8 | 0.43 | 512 | 128 | 32 | 0.45 | 32 | 32 | ND | 0.21 |
| Ciprofloxacin | 128 | 32 | 32 | 16 | 0.65 | >32 | 32 | 16 | 0.65 | 16 | 16 | 8 | 0.28 | |
MIC of the antibiotic determined by a microbroth-based assay in non-cation-adjusted Mueller-Hinton medium.
MICs are given for antibiotics in the presence of the indicated subinhibitory concentrations (μg/ml) of peptides P2-15, P2-27, and PMBN. The MICs of all the peptides on most of the strains were 31.25 to 64.5 μg/ml. Strains Ps21, Ps2, Ps64, and Ps74, in that order, were the most resistant (MICs of 125 to 250 mg/ml).
Polymyxin B nonapeptide.
Fractional inhibitory concentration (FIC) index (see Materials and Methods for details).
ND, not determined; NG, no growth in a control well treated with the peptide alone, thus indicating inhibitory activity of the peptide by itself at that concentration.
Peptides had varied success with sensitizing quinolone-resistant clinical strains, such as Ps1 and Ps6, to quinolones, and this appeared to depend on the profile of resistance mechanisms displayed by the organism. Thus, at low peptide concentrations, strain Ps6 was harder to sensitize to nalidixic acid than Ps1. This may be due to the interplay of generalized global efflux pump overexpression and the presence of modifications at the QRDR site in the former strain (Table 1). In contrast, none of the peptides could neutralize the mechanisms implicated in resistance to ciprofloxacin.
Our control permeability-increasing agent, PMBN, clearly outperformed our peptides in almost all the cases and strongly sensitized P. aeruginosa strains even to beta-lactamic antibiotics and ciprofloxacin, although it failed to counteract imipenem resistance in the carbapenemase-expressing strain Ps71.
Peptide cytotoxicity.
Before performing in vivo experiments, we measured the cytotoxicity of the peptides on a human culture cell line (HeLa cells) by using two complementary assays, the neutral red and the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) tests. As shown in Table 6, neither P2-15 nor P2-27 significantly reduced cell viability when tested by the MTT assay at 100 μg/ml, a concentration almost double their MICs and at least 10 times higher than their minimum permeabilizing activity levels. In contrast, PMB was notably more cytotoxic under the same conditions and reduced cell viability to 54%. Unlike the MTT test, which measures the cell respiratory rate, the neutral red assay detects alteration of the cell membrane. As judged by the latter assay, P2-15 was less cytotoxic than P2-27 and PMB.
TABLE 6.
Cytotoxicity of selected peptides on human cellsa
| Compound | Cytotoxicity results |
|
|---|---|---|
| Neutral red test | MTT test | |
| P2-15 | 93.7 ± 18.3 | 97.5 ± 11.2 |
| P2-27 | 80.9 ± 5.9 | 99.1 ± 9.8 |
| P2-29 | 87.3 ± 0.1 | 53.7 ± 8.0 |
| DMSOb | 4.2 ± 2.1 | 1.3 ± 3.5 |
| Melittin | 12.8 ± 11.9 | 9.9 ± 0.6 |
| Polymyxin B | 75.5 ± 10.2 | 54.2 ± 12.0 |
Cells were exposed to 100 μg/ml of the indicated compound for 60 min. Results are expressed as the percentages of cell viability ± standard deviations, with respect to an untreated control (100%).
Treatment with dimethyl sulfoxide (DMSO; 100 μg/ml) was used as a positive control of toxicity.
Peptide-mediated antibiotic-enhancing activity in vivo.
Based on its low toxicity and high permeabilizing activity in vitro, we selected compound P2-15 to evaluate whether this peptide could potentiate antibiotic activity in vivo. As an infection model, we used mice that had been rendered neutropenic by treatment with cyclophosphamide. We chose this model to try to reproduce the clinical scenario, where immunocompromised patients are precisely the most common P. aeruginosa hosts. We combined P2-15 with erythromycin, an antibiotic with very low activity against P. aeruginosa (MIC = 128 μg/ml) but which was efficiently potentiated in vitro (MIC = 1 μg/ml) in the presence of subinhibitory amounts (25 μg/ml) of P2-15. As shown in Fig. 2A, administration of either the peptide (0.5 mg/animal intraperitoneally [i.p.]) or the antibiotic (0.5 mg/animal i.p.) at 45 to 60 min postinoculation conferred no protection to the P. aeruginosa PAO1-infected mice. In addition, animals receiving a second dose of the same compound 5 h after the first dose were equally unprotected. In contrast, when P2-15 and erythromycin were given to the animals (0.5 mg of each per animal i.p.) as a combined treatment (Fig. 2B), 50% of the animals were significantly (P = 0.024) protected, although such protection was short lasting (24 h). Interestingly, administration of a second dose of the combined treatment 5 h after the first dose afforded long-lasting (7 days, at least) protection to 33% of the mice (P = 0.027).
FIG. 2.

Therapeutic efficacy of a peptide-antibiotic combination against an intraperitoneal infection of P. aeruginosa PAO1. Prior to infection, groups (n = 16) of mice were rendered neutropenic (immunocompromised) by the subcutaneous administration of two doses of cyclophosphamide. On day 0, mice were i.p. infected with 105 CFU per animal, and 60 min later, they i.p. received the following treatment: either a single dose of the peptide P2-15 or erythromycin (0.5 mg/animal in both cases) (A) or a combination of the peptide and the antibiotic administered at the same concentration as that used in the single-dose treatment (B). Groups of animals received second doses of the corresponding treatment 5 h after the first doses. Statistical differences were analyzed by the Kaplan-Meier survival test (*, P < 0.05).
DISCUSSION
This study identified peptides that efficiently permeabilize the P. aeruginosa OM and sensitize this organism not only to hydrophobic antibiotics (i.e., erythromycin and fusidic acid) but also to much more hydrophilic drugs, such as the beta-lactams amoxicillin, ampicillin, imipenem, and meropenem. Whereas the capacity of cationic peptides of diverse natures to enhance the activity of hydrophobic antibiotics has been repeatedly shown (23, 44, 61, 62), references of peptides endowed with the latter ability are more scarce (18, 59). In fact, polycations acting in synergy with hydrophobic antibiotics often fail to enhance beta-lactams on P. aeruginosa (17, 47, 56).
Our results obtained from using the wild-type strain indicate that mechanisms of antibiotic resistance relying on the basal activity of the efflux system in conjunction with the OM intrinsic permeability barrier are susceptible to peptide permeabilization. Thus, when peptides were combined with either nalidixic acid or chloramphenicol, two known substrates of the MexAB-OprM efflux system, MICs dropped down to levels as low as those obtained for these antibiotics with an isogenic mutant of PAO1 lacking the MexAB-OprM system (strain K1119) (data not shown). In addition, peptides enhanced to even a further extent the activity of erythromycin, novobiocin, and fusidic acid, which are also substrates of the MexAB-OprM system, resulting in MIC values 8 times lower than those obtained for the same antibiotics with strain K1119. This observation implies that peptides counteract not only efflux pump activity but also the resistance mechanism based on intrinsic low OM permeability. Nevertheless, peptides potentiated the beta-lactams amoxicillin and ampicillin to a lesser extent than the other antibiotics tested, suggesting that the mechanisms providing P. aeruginosa PAO1 with intrinsic antibiotic resistance (involved in resistance to the latter classes of antibiotics) are more vulnerable to permeabilization than those relying on the beta-lactamase activity of AmpC (Table 1, profile of strain PAO1).
Using strain PAOLC1-6, we also showed that mechanisms of resistance based on overexpression of the MexAB-OprM efflux system are vulnerable to peptides, at least partially. Thus, peptides potentiated the antibiotics tested—all of them substrates of the MexAB-OprM system, except the most hydrophilic ones, the beta-lactams cefotaxime and ticarcillin. It has been hypothesized that the ability of lactoferricin to counteract efflux pump activity is due not only to its membrane-disorganizing nature but also to dissipation of the proton-motive force, resulting in decreased activity of ATP-dependent multidrug efflux pumps (63). The latter activity has also been reported in peptides unrelated to lactoferricin (51). Interestingly, unlike P2-15 and P2-27, the first-generation peptide P1-22 was unable to sensitize strain PAOLC1-6 to antibiotics (53).
In contrast to the susceptibility to the previous mechanisms, strains carrying mutations in the binding sites of quinolones were not vulnerable to permeabilization. In these strains, it is likely that the antibiotic does not reach an intracellular concentration high enough to compensate for the loss of affinity of its target molecule, gyrase, and topoisomerase, as previously suggested (7).
While considerable effort to study how to rationally improve the antibacterial activity and selectivity of AMPs has been made (2, 8, 14, 15, 16, 26, 28, 29, 30, 31, 45, 48, 50), to our knowledge, this is the first report investigating the structural basis of peptide-mediated OM permeabilization as a function of the peptide's ability to enhance antibiotic activity. In this regard, our results confirm (53, 56, 64) that antibacterial activity and antibiotic-enhancing ability are not necessarily associated, since peptides with poor antimicrobial activity levels showed significant novobiocin-enhancing potential (e.g., compounds P1-14 and P1-48). This behavior resembles that of compounds such as PMBN (59) and CEMA (cecropin A-melittin hybrid peptide) (46).
Conversely, some peptides with high antimicrobial activity levels were barely able to act in synergy with novobiocin (e.g., P1-22, P2-29 and P2-30), a result in agreement with that of previous authors (36, 55), who reported that the lethal (bactericidal) activity of a compound may be so great that it is not possible to observe nonlethal (permeabilization) activity. A similar explanation of this phenomenon was given by Zhang and collaborators, who proposed that the highly bactericidal peptides cannot exhibit synergy because they cause cell death at the same concentration at which they permeabilize the OM (64). Finally, we showed that it is possible to design potent permeabilizers that rank among the best antibacterial agents, as exemplified by our lead compounds P2-15 and P2-27.
The correlation found between the predicted parameters and the corresponding experimental values confirm the utility of the QSAR-PLS analysis. In this regard, our results confirm that to maximize antimicrobial activity against Gram-negative bacteria, it is favorable to have larger amounts of hydrophobic residues, which cause distortions in the membrane (12, 21, 22). On the other hand, activity is decreased by an excessive concentration of basic residues in the molecule and by large peptide size, with the latter factor probably reflecting the steric hindrance that high-molecular weight peptides will encounter when crossing the hydrophilic carbohydrate chains of LPS (1).
Regarding permeabilizing activity, positive and negative correlations observed between FIC/MIC ratios and several parameters indicate that a high localization of charge (two or more sequential basic residues in the sequence) reduces synergistic activity. According to this model, the peptides should have larger amounts of aromatic residues, amphiphilic structure, and lower localization of positively charged residues to achieve optimal activity. Recent studies support the importance of some of these parameters, such as aromatic residues, in permeabilizing activity (10). These conclusions are consistent with the notion that the dual nature (cationic and hydrophobic) of a peptide is the driving force for its initial interaction with the OM (1).
Several QSAR studies correlating the antimicrobial activities of AMPs to their structural properties were recently published (8, 14, 15, 16, 26, 28, 29, 31, 45). The strategies used in those reports to build mathematical models include artificial neural networks, PLS-based methods, k-nearest neighbor analysis, and multiple linear regression. In these studies, structural properties of the peptides are presented as physicochemical descriptors. Ideally, these descriptors are calculated from three-dimensional models, but since a very limited number of structures is available (e.g., from nuclear magnetic resonance studies), in most cases, homology modeling is used to infer the required structures. The main disadvantage of this approach is that small changes in peptide sequence can drastically affect structure (20, 27, 28). Moreover, peptide conformation can be strongly influenced by binding to different structures of the bacterial membrane (lipopolysaccharide and phospholipids) unless stabilized by covalent bonds (4, 25). The main advantage of the PEDES (peptide descriptors from sequence) methodology is—as the name suggests—that it avoids structure-related descriptors by using a set of constitutional and sequence-dependent descriptors based on amino acid sequence. These descriptors were deduced from previous QSAR studies of lactoferricin-derived AMPs (21, 22, 57). As illustrated in this study, PEDES methodology offers a rapid and robust alternative for the calculation of descriptors which can then be analyzed using the PLS method to successfully predict activities of related AMPs.
Finally, although the ability of our leading peptide to enhance erythromycin activity in vivo and to protect animals from a lethal infection is far from optimal, it demonstrates the therapeutic potential of our compounds. Further studies are in progress to improve the efficiency of the experimental treatment and to rationally design new generations of peptides with enhanced permeabilizing activity by applying the conclusions of our QSAR analysis.
Supplementary Material
Acknowledgments
We are grateful to M. Carmen Conejo, Luis Martínez-Martínez, and Alvaro Pascual (from the Department of Microbiology, University of Seville, Spain) for providing us with strain PAOLC1-6.
G.M.d.T. was funded by grants from Ministerio de Sanidad y Consumo (FIS-PI050768), Spain, and from Proyectos de Investigación Universidad de Navarra (PIUNA-2008-11), Spain. The study has been carried out with financial support from the Commission of the European Communities, specific RTD program “Quality of Life and Management of Living Resources,” QLCK2-CT-2002-01001, “Antimicrobial Endotoxin Neutralizing Peptides To Combat Infectious Diseases.”
Footnotes
Published ahead of print on 18 October 2010.
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1.Andrä, J., K. Lohner, S. E. Blondelle, R. Jerala, I. Moriyon, M. H. Koch, P. Garidel, and K. Brandenburg. 2005. Enhancement of endotoxin neutralization by coupling of a C12-alkyl chain to a lactoferricin-derived peptide. Biochem. J. 385:135-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andrä, J., D. Monreal, G. Martinez de Tejada, C. Olak, G. Brezesinski, S. Sanchez-Gomez, T. Goldmann, R. Bartels, K. Brandenburg, and I. Moriyon. 2007. Rationale for the design of shortened derivatives of the NK-lysin-derived antimicrobial peptide NK-2 with improved activity against gram-negative pathogens. J. Biol. Chem. 282:14719-14728. [DOI] [PubMed] [Google Scholar]
- 3.Balakrishna, R., S. J. Wood, T. B. Nguyen, K. A. Miller, E. V. Suresh Kumar, A. Datta, and S. A. David. 2006. Structural correlates of antibacterial and membrane-permeabilizing activities in acylpolyamines. Antimicrob. Agents Chemother. 50:852-861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berman, H. M., J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov, and P. E. Bourne. 2000. The Protein Data Bank. Nucleic Acids Res. 28:235-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonomo, R. A., and D. Szabo. 2006. Mechanisms of multidrug resistance in Acinetobacter species and Pseudomonas aeruginosa. Clin. Infect. Dis. 43(Suppl. 2):S49-S56. [DOI] [PubMed] [Google Scholar]
- 6.Brogden, K. A. 2005. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 3:238-250. [DOI] [PubMed] [Google Scholar]
- 7.Campos, M. A., P. Morey, and J. A. Bengoechea. 2006. Quinolones sensitize gram-negative bacteria to antimicrobial peptides. Antimicrob. Agents Chemother. 50:2361-2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cherkasov, A., K. Hilpert, H. Jenssen, C. D. Fjell, M. Waldbrook, S. C. Mullaly, R. Volkmer, and R. E. W. Hancock. 2009. Use of artificial intelligence in the design of small peptide antibiotics effective against a broad spectrum of highly antibiotic-resistant superbugs. ACS Chem. Biol. 4:65-74. [DOI] [PubMed] [Google Scholar]
- 9.Delcour, A. H. 2009. Outer membrane permeability and antibiotic resistance. Biochim. Biophys. Acta 1794:808-816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dewan, P. C., A. Anantharaman, V. S. Chauhan, and D. Sahal. 2009. Antimicrobial action of prototypic amphipathic cationic decapeptides and their branched dimers. Biochemistry 48:5642-5657. [DOI] [PubMed] [Google Scholar]
- 11.Eisenberg, D., R. M. Weiss, and T. C. Terwilliger. 1984. The hydrophobic moment detects periodicity in protein hydrophobicity. Proc. Natl. Acad. Sci. U. S. A. 81:140-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eliassen, L. T., B. E. Haug, G. Berge, and O. Rekdal. 2003. Enhanced antitumor activity of 15-residue bovine lactoferricin derivatives containing bulky aromatic amino acids and lipophilic N-terminal modifications. J. Pept. Sci. 9:510-517. [DOI] [PubMed] [Google Scholar]
- 13.Eliopoulos, G. M., and R. C. Moellering, Jr. 1996. Antimicrobial combinations, p. 330-393. In V. Lorian (ed.), Antimicrobial in laboratory medicine, 4th ed. Williams and Wilkins, Baltimore, MD.
- 14.Fjell, C. D., H. Jenssen, K. Hilpert, W. A. Cheung, N. Panté, R. E. W. Hancock, and A. Cherkasov. 2009. Identification of novel antibacterial peptides by chemoinformatics and machine learning. J. Med. Chem. 52:2006-2015. [DOI] [PubMed] [Google Scholar]
- 15.Frecer, V., B. Ho, and J. L. Ding. 2004. De novo design of potent antimicrobial peptides. Antimicrob. Agents Chemother. 48:3349-3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frecer, V. 2006. QSAR analysis of antimicrobial and haemolytic effects of cyclic cationic antimicrobial peptides derived from protegrin-1. Bioorg. Med. Chem. 14:6065-6074. [DOI] [PubMed] [Google Scholar]
- 17.Giacometti, A., O. Cirioni, F. Barchiesi, M. Fortuna, and G. Scalise. 1999. In-vitro activity of cationic peptides alone and in combination with clinically used antimicrobial agents against Pseudomonas aeruginosa. J. Antimicrob. Chemother. 44:641-645. [DOI] [PubMed] [Google Scholar]
- 18.Giacometti, A., O. Cirioni, W. Kamysz, G. D'Amato, C. Silvestri, A. Licci, P. Nadolski, A. Riva, J. Lukasiak, and G. Scalise. 2005. In vitro activity of MSI-78 alone and in combination with antibiotics against bacteria responsible for bloodstream infections in neutropenic patients. Int. J. Antimicrob. Agents 26:235-240. [DOI] [PubMed] [Google Scholar]
- 19.Hancock, R. E., and D. P. Speert. 2000. Antibiotic resistance in Pseudomonas aeruginosa: mechanisms and impact on treatment. Drug Resist. Updat. 3:247-255. [DOI] [PubMed] [Google Scholar]
- 20.Hancock, R. E. W., and H. G. Sahl. 2006. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 24:1551-1557. [DOI] [PubMed] [Google Scholar]
- 21.Haug, B. E., and J. S. Svendsen. 2001. The role of tryptophan in the antibacterial activity of a 15-residue bovine lactoferricin peptide. J. Pept. Sci. 7:190-196. [DOI] [PubMed] [Google Scholar]
- 22.Haug, B. E., M. L. Skar, and J. S. Svendsen. 2001. Bulky aromatic amino acids increase the antibacterial activity of 15-residue bovine lactoferricin derivatives. J. Pept. Sci. 7:425-432. [DOI] [PubMed] [Google Scholar]
- 23.Hogg, G. M., J. G. Barr, and C. H. Webb. 1998. In-vitro activity of the combination of colistin and rifampicin against multidrug-resistant strains of Acinetobacter baumannii. J. Antimicrob. Chemother. 41:494-495. [DOI] [PubMed] [Google Scholar]
- 24.Japelj, B. 2005. PEDES reference manual. National Institute of Chemistry, Ljubljana, Slovenia.
- 25.Japelj, B., P. Pristovsek, A. Majerle, and R. Jerala. 2005. Structural origin of endotoxin neutralization and antimicrobial activity of a lactoferrin-based peptide. J. Biol. Chem. 280:16955-16961. [DOI] [PubMed] [Google Scholar]
- 26.Jenssen, H., T. J. Gutteberg, and T. Lejon. 2005. Modelling of anti-HSV activity of lactoferricin analogues using amino acid descriptors. J. Pept. Sci. 11:97-103. [DOI] [PubMed] [Google Scholar]
- 27.Jenssen, H., P. Hamill, and R. E. Hancock. 2006. Peptide antimicrobial agents. Clin. Microbiol. Rev. 19:491-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jenssen, H., T. Lejon, K. Hilpert, C. D. Fjell, A. Cherkasov, and R. E. W. Hancock. 2007. Evaluating different descriptors for model design of antimicrobial peptides with enhanced activity toward P. aeruginosa. Chem. Biol. Drug Des. 70:134-142. [DOI] [PubMed] [Google Scholar]
- 29.Jenssen, H., C. D. Fjell, A. Cherkasov, and R. E. W. Hancock. 2008. QSAR modeling and computer-aided design of antimicrobial peptides. J. Pept. Sci. 14:110-114. [DOI] [PubMed] [Google Scholar]
- 30.Juretić, D., D. Vukicević, N. Ilić, N. Antcheva, and A. Tossi. 2009. Computational design of highly selective antimicrobial peptides. J. Chem. Infect. Model. 49:2873-2882. [DOI] [PubMed] [Google Scholar]
- 31.Karakoc, E., S. C. Sahinalp, and A. Cherkasov. 2006. Comparative QSAR- and fragments distribution analysis of drugs, druglikes, metabolic substances, and antimicrobial compounds. J. Chem. Infect. Model. 46:2167-2182. [DOI] [PubMed] [Google Scholar]
- 32.Katritzky, A. R., V. Lobanov, and M. Karelson. 1996. CODESSA reference manual. University of Florida, Gainesville, FL.
- 33.Kwon, D. H., and C. D. Lu. 2007. Polyamine effects on antibiotic susceptibility in bacteria. Antimicrob. Agents Chemother. 51:2070-2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kyte, J., and R. F. Doolittle. 1982. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157:105-132. [DOI] [PubMed] [Google Scholar]
- 35.Landman, D., C. Georgescu, D. A. Martin, and J. Quale. 2008. Polymyxins revisited. Clin. Microbiol. Rev. 21:449-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li, C., M. R. Lewis, A. B. Gilbert, M. D. Noel, D. H. Scoville, G. W. Allman, and P. B. Savage. 1999. Antimicrobial activities of amine- and guanidine-functionalized cholic acid derivatives. Antimicrob. Agents Chemother. 43:1347-1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Livermore, D. M. 2009. Has the era of untreatable infections arrived? J. Antimicrob. Chemother. 64(Suppl. 1):i29-i36. [DOI] [PubMed] [Google Scholar]
- 38.Lohner, K., and S. E. Blondelle. 2005. Molecular mechanisms of membrane perturbation by antimicrobial peptides and the use of biophysical studies in the design of novel peptide antibiotics. Comb. Chem. High Throughput Screen. 8:241-256. [DOI] [PubMed] [Google Scholar]
- 39.Mesaros, N., P. Nordmann, P. Plésiat, M. Roussel-Delvallez, J. Van Eldere, Y. Glupczynski, Y. Van Laethem, F. Jacobs, P. Lebecque, A. Malfroot, P. M. Tulkens, and F. Van Bambeke, F. 2007. Pseudomonas aeruginosa: resistance and therapeutic options at the turn of the new millennium. Clin. Microbiol. Infect. 13:560-578. [DOI] [PubMed] [Google Scholar]
- 40.National Committee for Clinical Laboratory Standards (NCCLS). 2003. Methods for dilution antimicrobial susceptibility test for bacteria that grow aerobically, sixth edition. M7-A6. National Committee for Clinical Laboratory Standards, Wayne, PA.
- 41.Nikaido, H. 2003. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 67:593-656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nikaido, H. 2009. Multidrug resistance in bacteria. Annu. Rev. Biochem. 78:119-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ofek, I., S. Cohen, R. Rahmani, K. Kabha, D. Tamarkin, Y. Herzig, and E. Rubinstein. 1994. Antibacterial synergism of polymyxin B nonapeptide and hydrophobic antibiotics in experimental gram-negative infections in mice. Antimicrob. Agents Chemother. 38:374-377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ohta, M., H. Ito, K. Masuda, S. Tanaka, Y. Arakawa, R. Wacharotayankun, and N. Kato. 1992. Mechanisms of antibacterial action of tachyplesins and polyphemusins, a group of antimicrobial peptides isolated from horseshoe crab hemocytes. Antimicrob. Agents Chemother. 36:1460-1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ostberg, N., and Y. Kaznessis. 2005. Protegrin structure-activity relationships: using homology models of synthetic sequences to determine structural characteristics important for activity. Peptides 26:197-206. [DOI] [PubMed] [Google Scholar]
- 46.Piers, K. L., M. H. Brown, and R. E. Hancock. 1994. Improvement of outer membrane-permeabilizing and lipopolysaccharide-binding activities of an antimicrobial cationic peptide by C-terminal modification. Antimicrob. Agents Chemother. 38:2311-2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Plesiat, P., J. R. Aires, C. Godard, and T. Köhler. 1997. Use of steroids to monitor alterations in the outer membrane of Pseudomonas aeruginosa. J. Bacteriol. 179:7004-7010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Powers, J. P., and R. E. Hancock. 2003. The relationship between peptide structure and antibacterial activity. Peptides 24:1681-1691. [DOI] [PubMed] [Google Scholar]
- 49.Purcell, W. P., G. E. Bass, and J. M. Clayton. 1973. Strategy of drug design: a guide to biological activity. John Wiley & Sons, New York, NY.
- 50.Rathinakumar, R., W. F. Walkenhorst, and W. C. Wimley. 2009. Broad-spectrum antimicrobial peptides by rational combinatorial design and high-throughput screening: the importance of interfacial activity. J. Am. Chem. Soc. 131:7609-7617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rintoul, M. R., B. F. de Arcuri, R. A. Salomón, R. N. Farías, and R. D. Morero. 2001. The antibacterial action of microcin J25: evidence for disruption of cytoplasmic membrane energization in Salmonella newport. FEMS Microbiol. Lett. 204:265-270. [DOI] [PubMed] [Google Scholar]
- 52.Saha, S., P. B. Savage, and M. Bal. 2008. Enhancement of the efficacy of erythromycin in multiple antibiotic-resistant gram-negative bacterial pathogens. J. Appl. Microbiol. 105:822-828. [DOI] [PubMed] [Google Scholar]
- 53.Sánchez-Gómez, S., M. Lamata, J. Leiva, S. E. Blondelle, R. Jerala, J. Andrä, K. Brandenburg, K. Lohner, I. Moriyón, and G. Martínez-de-Tejada. 2008. Comparative analysis of selected methods for the assessment of antimicrobial and membrane-permeabilizing activity: a case study for lactoferricin derived peptides. BMC Microbiol. 8:196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53a.Satake, S., H. Yoneyama, and T. Nakae. 1991. Role of OmpD2 and chromosomal beta-lactamase in carbapenem resistance in clinical isolates of Pseudomonas aeruginosa. J. Antimicrob. Chemother. 28:199-207. [DOI] [PubMed] [Google Scholar]
- 54.Savage, P. B. 2001. Multidrug-resistant bacteria: overcoming antibiotic permeability barriers of gram-negative bacteria. Ann. Med. 33:167-171. [DOI] [PubMed] [Google Scholar]
- 55.Savage, P. B., C. Li, U. Taotafa, B. Ding, and Q. Guan. 2002. Antibacterial properties of cationic steroid antibiotics. FEMS Microbiol. Lett. 217:1-7. [DOI] [PubMed] [Google Scholar]
- 56.Scott, M. G., H. Yan, and R. E. Hancock. 1999. Biological properties of structurally related alpha-helical cationic antimicrobial peptides. Infect. Immun. 67:2005-2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Strom, M. B., B. E. Haug, O. Rekdal, M. L. Skar, W. Stensen, and J. S. Svendsen. 2002. Important structural featuresof 15-residue lactoferricin derivatives and methods for improvement of antimicrobial activity. Biochem. Cell Biol. 80:65-74. [DOI] [PubMed] [Google Scholar]
- 58.Tian, D., P. Zhou, F. Lv, R. Song, and Z. Li. 2007. Three-dimensional holograph vector of atomic interaction fields (3D-HoVAIF): a novel rotation-translation invariant 3D structure descriptor and its applications to peptides. J. Pept. Sci. 13:549-566. [DOI] [PubMed] [Google Scholar]
- 59.Vaara, M., and T. Vaara. 1983. Polycations sensitize enteric bacteria to antibiotics. Antimicrob. Agents Chemother. 24:107-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vaara, M. 1992. Agents that increase the permeability of the outer membrane. Microbiol. Rev. 56:395-411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vaara, M., and M. Porro. 1996. Group of peptides that act synergistically with hydrophobic antibiotics against gram-negative enteric bacteria. Antimicrob. Agents Chemother. 40:1801-1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vaara, M., J. Fox, G. Loidl, O. Siikanen, J. Apajalahti, F. Hansen, N. Frimodt-Møller, J. Nagai, M. Takano, and T. Vaara. 2008. Novel polymyxin derivatives carrying only three positive charges are effective antibacterial agents. Antimicrob. Agents Chemother. 52:3229-3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wakabayashi, H., S. Teraguchi, and Y. Tamura. 2002. Increased Staphylococcus-killing activity of an antimicrobial peptide, lactoferricin B, with minocycline and monoacylglycerol. Biosci. Biotechnol. Biochem. 66:2161-2167. [DOI] [PubMed] [Google Scholar]
- 64.Zhang, L., R. Benz, and R. E. Hancock. 1999. Influence of proline residues on the antibacterial and synergistic activities of alpha-helical peptides. Biochemistry 38:8102-8111. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.