

Abstract
Pathologic hyperplasia of various pancreatic endocrine cells is rare but has been long known. β cell hyperplasia contributes to persistent hyperinsulinemic hypoglycemia of infancy, which is commonly caused by mutations in the islet ATP-sensitive potassium channel, and to non-insulinoma pancreatogenous hypoglycemia in adults, which may or may not be associated with bariatric surgery. α cell hyperplasia may cause glucagonoma syndrome or induce pancreatic neuroendocrine tumors. An inactivating mutation of the glucagon receptor causes α cell hyperplasia and asymptomatic hyperglucagonemia. Pancreatic polypeptide cell hyperplasia has been described without a clearly-characterized clinical syndrome and hyperplasia of other endocrine cells inside the pancreas has not been reported to our knowledge. Based on morphological evidence, the main pathogenetic mechanism for pancreatic endocrine cell hyperplasia is increased endocrine cell neogenesis from exocrine ductal epithelium. Pancreatic endocrine cell hyperplasia should be considered in the diagnosis and management of hypoglycemia, elevated islet hormone levels, and pancreatic neuroendocrine tumors. Further studies of pathologic pancreatic endocrine cell hyperplasia will likely yield insights into the pathogenesis and treatment of diabetes and pancreatic neuroendocrine tumors.
Keywords: Glucagon receptor, Hyperplasia, Nesidioblastosis, Islet, Pancreatic endocrine cell, Neuroendocrine tumor
INTRODUCTION
The pancreas, a key regulator of nutrient digestion, absorption, and utilization, can be divided into two major components, the endocrine and exocrine pancreas. The endocrine pancreas consists of five distinct cell types, α, β, δ, ε, and pancreatic polypeptide (PP) cells, that produce glucagon, insulin, somatostatin, ghrelin, and PP, respectively[1-3]. The pancreatic endocrine cells may give rise to distinct neuroendocrine tumors such as insulinoma, gastrinoma, glucagonoma, VIPoma, and non-functioning tumors[4-6]. In contrast, pancreatic endocrine cell hyperplasia as a group of diseases is a relatively unexplored area. From the 1960s to the present day, there have been various reports regarding pancreatic endocrine cell hyperplasia. Much of the literature has focused on β cell hyperplasia in particular, but hyperplasia of other pancreatic endocrine cells has also been described, some in great detail. In this review we summarize the body of literature on pathologic pancreatic endocrine cell hyperplasia.
Hyperplasia refers to an increased number of a certain type of cells in a given organ or tissue than is ordinarily observed. Mechanisms regulating pancreatic endocrine cell number include proliferation (division of existing cells), apoptosis (controlled cell death), and neogenesis (differentiation of endocrine cells from the exocrine epithelium), and abnormalities in each could result in hyperplasia[7-9]. The diagnostic criteria of pancreatic endocrine cell hyperplasia are not universally agreed upon. Rindi et al[10] defines pancreatic endocrine cell hyperplasia as an expansion of the endocrine cell mass to more than 2% (in adults) or 10% (in infants) of the total pancreas mass. As it is impractical to do detailed pancreatic morphometry in a clinical specimen, the diagnosis of pancreatic endocrine cell hyperplasia is often subjective. Most would regard an islet size large than 250 μm in diameter and an increase in islet numbers as evidence of pancreatic endocrine cell hyperplasia[11-14].
Pancreatic endocrine cell hyperplasia can be non-specific and involve most or all types of islet cells or specific and involve predominantly one cell type. Non-specific, focal endocrine hyperplasia and microadenoma are not uncommon incidental pathological findings in the pancreas; if carefully screened, up to 10% of adults harbor these lesions at autopsy[15]. In those patients, all types of pancreatic endocrine cells could be focally hyperplastic. Most of those lesions probably do not indicate clinical significance. Diffuse pancreatic endocrine cell hyperplasia and microadenoma are a feature of multiple endocrine neoplasia type 1 (MEN1), and to a less extent, von Hippel-Lindau (VHL) disease[16-19]. All types of endocrine cells can be hyperplastic, but β and α cells are more often so, probably because these cells are normally more numerous than other types. In this article, we will focus on diffuse and specific pancreatic endocrine cell hyperplasia as a group of diseases. We define it pathologically as an overwhelming increase in islet size and/or number in all the pancreatic sections examined so that it is reasonable to assume that the remaining pancreas or unexamined pancreas blocks should exhibit similar changes. Moreover, the hyperplastic endocrine cells should be mainly limited to one type of islet cells which have apparently similar cell lineage supported by consistent hormone production profile and other cellular markers. Finally we will only discuss the literature on pathologic pancreatic endocrine cell hyperplasia in humans.
β CELL HYPERPLASIA
The pancreatic β cells are the only source of insulin, the hormone that decreases blood glucose levels by increasing glucose uptake and decreasing hepatic glucose output. Physiological hyperplasia of β cells is commonly seen in patients with insulin resistance and early-stage type 2 diabetes, and is intensely studied for diabetes treatment[20,21]. Except for postprandial hypoglycemia, we are not aware of any reports that physiological β cell hyperplasia causes clinical syndromes[22,23]. The physiological β cell hyperplasia appears to be tightly regulated and has not been reported to give rise to pancreatic neuroendocrine tumors. We thus consider it part of insulin resistance syndromes and will not address it further in this article.
Admittedly, pathologic β cell hyperplasia is a controversial term in both infants and adults in the absence of endocrine tumor syndromes. Although β cell hyperplasia has been recorded in non-insulinoma hyperinsulinemic hypoglycemia, it is rather moderate in most cases and may be even non-existent in some cases[24]. “Nesidioblastosis” has been used by some authors to denote the same pathologic changes[25-28]. The term nesidioblastosis was coined in the first half of the 20th century initially to describe islet neogenesis from pancreatic ductal epithelium in neonates with hyperinsulinemic hypoglycemia[29]. β cell hyperplasia and hypertrophy often accompany nesidioblastosis[24,30-32]. Although islet neogenesis is also observed in normal infants, by the 1970s, nesidioblastosis was used to describe all forms of persistent congenital hyperinsulinism in infants, whether the hyperinsulinemic states were associated with hyperplasia or not[25,26]. In recent years, nesidioblastosis has also been used to describe acquired hyperinsulinism with β cell hyperplasia in adults[27,28]. As the use of this term is not consistent, we use nesidioblastosis strictly as a morphological term in describing any endocrine cells (not limited to β cells) budding from the ductal epithelium and use “persistent hyperinsulinemic hypoglycemia of infancy (PHHI)” to describe the various forms of similar such diseases in neonates or infants and “non-insulinoma pancreatogenous hypoglycemia (NIPH)” to describe hypoglycemia syndromes without evidence of insulinoma in adults[24,30-41]. Both PHHI and NIPH are associated with pathologic β cell hyperplasia in most cases.
Clinically characterized by hyperinsulinemic hypoglycemia in infants and neonates, PHHI is not a single disease entity but a group of related diseases[33-35]. In the majority of patients, insulinoma is not identified but the pancreas exhibits focal or diffuse β cell abnormalities commonly associated with genetic mutations affecting β cells[35,42]. In about a third of cases, focal β cell hypertrophy and hyperplasia is observed[33,34]. The endocrine cells are arranged in huge islet-like structures separated by acinar cells or connective tissue, and some harbor large nuclei. The endocrine cell proliferation rate is generally increased. All types of endocrine cells are represented in the islet-like structures with normal spatial distribution. The percentage of β cells is higher (70%-90%) than normal (50%). The diffuse form is found in about two-thirds of cases. Although the β cell mass is only mildly increased compared with normal control, islet size varies and some islets are very large while others are poorly defined and irregularly shaped small endocrine cell clusters. As in the focal form, all types of endocrine cells are represented in the islet-like structures with normal spatial distribution, and some endocrine cells have large hyperchromatic nuclei. The endocrine cell proliferation rate, however, is not increased in the diffuse form[43]. Clinically it is important to differentiate the focal from the diffuse form as partial pancreatectomy is sufficient for the former while near-total or total pancreatectomy is required for the latter. There is a correlation between the underlying genetic abnormalities and the pancreas pathology. The focal hypertrophy and hyperplasia is associated with a paternally inherited ATP-sensitive potassium channel defect with loss of maternal heterozygosity on the 11p chromosome, and is thus sporadic[35,42]. The diffuse form is most commonly associated with traditional mutations leading to defects in the same potassium channel, and can be either sporadic or familial. It is still unclear how the genetic abnormalities lead to the unique islet structure and β cell morphology.
In adults, NIPH is a rare cause of hyperinsulinemic hypoglycemia[36-41]. NIPH is characterized by mostly postprandial hypoglycemia rather than fasting hypoglycemia which is usually seen in patients with insulinoma[38-40]. This syndrome is distinguished from reactive hypoglycemia in that in some patients, the development of severe neuroglycopenic symptoms including diplopia, dysarthria, confusion, disorientation, and even convulsions and coma, may occur in addition to adrenergic symptoms that predominate in reactive hypoglycemia[22,23,38-40]. This syndrome is also distinguished from PHHI in that these patients do not have mutations of the KIR6.2 (KCNJ11) and SUR1 (ABCC8) genes, which encode the subunits of the pancreatic ATP-sensitive potassium channel[38]. Evidence of β cell hyperplasia is present in every patient studied[36-41]. The islets exhibit normal structure but diffusely are more numerous and larger than those in normal control, and nesidioblastosis is pervasive. All types of islet cells are normally distributed throughout the islets but the predominant cell type is the β cell. Partial pancreatectomy often resolves hypoglycemia[36-41]. The association of NIPH and bariatric surgery is controversial. Hypoglycemia after bariatric surgery is common and is mostly caused by dumping syndrome[44,45]. In a minority of patients, hypoglycemia is associated with hyperinsulinemia and can only be controlled by partial pancreatectomy[44,46]. It was initially reported that the endocrine pancreas essentially exhibits the same changes as described above for patients without a history of obesity and bariatric surgery[44,46]; later studies, however, failed to demonstrate β cell hyperplasia when obese patients without bariatric surgery were used as controls[47]. Thus bariatric surgery may not by itself cause β cell hyperplasia.
In summary, pathologic β cell hyperplasia causes hypoglycemia and requires pancreatectomy in most patients. Pancreatic neuroendocrine tumor pathogenesis is a concern but has not been observed clinically. Increased neogenesis (morphologically as nesidioblastosis) appears to be the main mechanism responsible for β cell hyperplasia. The etiology of β cell hyperplasia commonly is mutations in the ATP-sensitive potassium channel in infants; in adults, it is still unknown but is probably related to unknown genetic changes. Further study of β cell hyperplasia will undoubtedly provide insights into fundamental β cell biology and suggest novel therapies for diabetes.
α CELL HYPERPLASIA
Although much research has focused on β cell hyperplasia, α cell hyperplasia also occurs and recent studies have shed light on its mechanism. The α cells are the second most common cells in an islet[1-3]. The first case of diffuse α cell hyperplasia and hyperglucagonemia was published more than 40 years ago in a 69-year-old man with hyperparathyroidism and calcific pancreatitis[48]. The cause of α cell hyperplasia and hyperglucagonemia in that case was not clear but the pancreatitis may have contributed to α cell hyperplasia or the patient may have had multiple endocrine neoplasia type 1 which encompasses primary hyperparathyroidism and pancreatic endocrine cell hyperplasia[16,17]. To our knowledge, nine more cases of α cell hyperplasia have been reported afterwards (Table 1)[49-54]. Glucagon levels were elevated in all cases where they were measured. The ethnicity of patients included Asian, Iranian, or presumably Caucasians. Both female and male patients were affected and all patients were adults aged from 25 to 74; none had a family history of endocrine tumors. Only one patient presented with typical glucagonoma syndrome in the form of necrolytic migratory erythema, deep vein thrombosis, and weight loss, and the other eight patients presented with non-specific symptoms in whom hyperglucagonemia was identified during the work-up for possible pancreatic neuroendocrine tumors.
Table 1.
Summary of 9 cases of α cell hyperplasia
| Study | Toda et al[49] | Brown et al[50] | Martignoni et al[51] | Chen et al[52] | Yu et al[53] | Henopp et al[54] |
| Location | Japan | USA | Germany | Taiwan | USA | Germany |
| Ethnicity | Japanese | ND | ND | Chinese | Persian | ND |
| Age (yr) | 74 | 48 | 54 | 45 | 60 | 25-44 |
| Sex | F | M | M | M | F | 2F/2M |
| Clinical | Diabetes | Diabetes | Mild diabetes | Mild diabetes | Nonspecific | Various |
| Glucagon (pg/mL) Pre-op | ND | 4200 | Elevated | ND | 59 284 | 4-25-fold |
| Glucagon (pg/mL) Post-op | ND | 5700 | ND | ND | Elevated | ND |
| Imaging | Negative | Mass in body | Negative | Diffusely enlarged | Mass on uncinate | ND |
| Octreotide scan | ND | ND | Negative | ND | Negative | ND |
| Pathology | Numerous micro-glucagonoma | Glucagonoma, α cell hyperplasia | Glucagonoma, α cell hyperplasia | α cell hyperplasia | NF-PNET, α cell hyperplasia | α cell hyperplasia |
ND: Not described; NF-PNET: Non-functioning pancreatic neuroendocrine tumor.
Regardless whether the α cell hyperplasia is associated with glucagonoma syndrome, its morphology is remarkably similar in all cases[49-54]. We have performed detailed histological studies on the α cell hyperplasia of our patient (Figure 1)[53]. In this patient’s pancreas, hyperplastic islets were innumerable and classic nesidioblastosis was commonly seen. Most of these hyperplastic islets (60%-80%) contain endocrine cells positive for glucagon but negative for insulin, but smaller, normal-looking islets exhibit normal insulin and glucagon hormonal expression. Accompanying the α cell hyperplasia, there are a 4-cm nonfunctioning pancreatic neuroendocrine tumor and multiple microadenomas. Henopp et al[54] further note that it is difficult to distinguish microglucagonomas from hyperplastic islets in α cell hyperplasia, and in some very large islets (> 300-500 μm), there appears to be an imperceptible transition from α cell hyperplasia to microglucagonoma. Thus the morphological studies suggest that α cell hyperplasia gives rise to glucagonoma and other pancreatic endocrine tumors.
Figure 1.
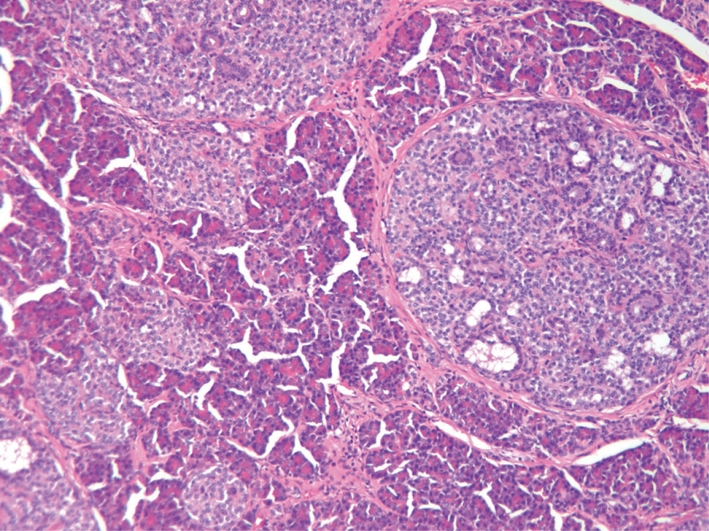
α cell hyperplasia of a patient with homozygous inactivating mutation of the glucagon receptor. Note the large islets with nesidioblastosis. Most of the islets are positive for glucagon but negative for insulin. 100 ×.
The pathogenesis of α cell hyperplasia has been elucidated in our patient[55]. As the patient has extremely elevated glucagon levels but without glucagonoma syndrome, which resembles the phenotype of mice without a glucagon receptor[56,57], we sequenced the patient’s glucagon receptor gene and identified a novel homozygous inactivating P86S mutation[55]. When tested in vitro, the P86S mutant glucagon receptor exhibits partial cytoplasmic localization and decreased glucagon binding. Compared with the wild-type glucagon receptor, the P86S mutant produces less cAMP under physiological concentrations of glucagon. The hyperplastic α cells in our patient also produce glucagon-like peptide 1 and PP, suggesting immature, more embryonic traits. We believe that our patient has a novel disease which we term “Mahvash disease” because it has a distinct etiology (inactivating glucagon receptor mutation), pathology (α cell hyperplasia), and clinical syndrome (hyperglucagonemia and pancreatic neuroendocrine tumors).
Thus clinically, there appear to be at least two types of α cell hyperplasia, functional and reactive. Functional α cell hyperplasia is analogous to adult β cell hyperplasia (which produces non-insulinoma pancreatogenous hyperinsulinemic hypoglycemia, NIPH) and produces non-glucagonoma hyperglucagonemic glucagonoma syndrome. Partial or total pancreatectomy may be a logical treatment. Currently only one case of functional α cell hyperplasia is known[54]. Our case and possibly a few others represent reactive α cell hyperplasia (equivalent to Mahvash disease) which produces hyperglucagonemia as a result of inactivated glucagon signaling and consequently does not cause glucagonoma syndrome[49-54]. The clinical significance of reactive α cell hyperplasia is pancreatic neuroendocrine tumors so that clinical, laboratory, and imaging surveillance are required to identify those tumors early. Once identified, these tumors should be treated as a regular pancreatic neuroendocrine tumor.
PP CELL HYPERPLASIA
PP-producing cells represent about 10% of endocrine cells in an islet[1-3]. The PP cells often take up a peripheral position, mixed with α and δ cells. The physiologic effects of PP are not very clear but include inhibiting gallbladder contraction and pancreatic enzyme secretion and decreasing appetite and food intake[58,59]. PP cell hyperplasia was first described in 1980 and a total of eight cases of diffuse PP cell hyperplasia have been reported to our knowledge (Table 2)[60-65]. As in α cell hyperplasia, both sexes were affected and patients were aged from 37 to 76 years; all patients were without a family history of endocrine tumors. Watery diarrhea was a common symptom and four of the eight patients had simultaneous or a history of gastrinoma and Zollinger-Ellison syndrome. The clinical significance of PP cell hyperplasia and elevated PP levels remain relatively unknown. It is not clear if PP cell hyperplasia indeed causes elevated PP levels. First of all, there does not appear to be a direct relationship between the number of PP cells in the pancreas and the serum levels of circulating PP[66]. In addition, PP levels were unknown in most patients and were measured in only two of the eight patients; in one of these two patients, the PP levels were comparable to those in asymptomatic patients with pancreatic neuroendocrine tumors. Lastly, the association of PP cell hyperplasia and gastrinoma may confound the clinical presentations of PP cell hyperplasia as diarrhea is a common symptom of gastrinoma. Thus far, a clinical syndrome of elevated PP levels cannot be established and it is not clear whether PP cell hyperplasia causes watery diarrhea. PP cell hyperplasia has also been found to be positive on somatostatin receptor scintigraphy, an imaging modality used to visualize neuroendocrine tumors[65].
Table 2.
Summary of 8 cases of pancreatic polypeptide cell hyperplasia
| Study | Tomita et al[60] | Farley et al[61] | Martella et al[62] | Pasieka et al[63] | Albazaz et al[64] | Bunning et al[65] |
| Location | USA | USA | Italy | Canada | UK | USA |
| Ethnicity | ND | ND | ND | ND | ND | ND |
| Age (yr) | 70 | 66 | 50-70 | 37 | 76 | 71 |
| Sex | F | M | 3F | F | M | M |
| Clinical | Diarrhea | Diarrhea | ZES | Diarrhea | Bowel obstruction | Nausea ZES |
| PP (pg/mL) Pre-op | ND | Highly elevated | About 3 fold | ND | ND | ND |
| PP (pg/mL) Post-op | ND | ND | ND | ND | ND | ND |
| Imaging | Mass in pancreas head | Mass in pancreas head | Nonspecific | Normal | Mass in pancreas head | Normal |
| Octreotide scan | ND | ND | ND | ND | ND | Uptake in pancreas head |
| Pathology | PP cell hyperplasia | PP cell hyperplasia | PP cell hyperplasia | PP cell hyperplasia | PP cell hyperplasia | PP cell hyperplasia |
ND: Not described; ZES: Zollinger-Ellison syndrome; PP: Pancreatic polypeptide.
The histology of PP cell hyperplasia is very similar to that of α or β cell hyperplasia but the hyperplastic cells are PP cells[60-65]. There are numerous PP cell clusters, some of which are very large and dysplastic. The PP cells are the predominant cells found inside or outside the islets. Extensive PP cell neogenesis is inferred from the evident nesidioblastosis. While the association with gastrinoma suggests that it may be secondary to gastrinoma, the etiology of PP cell hyperplasia remains elusive.
HYPERPLASIA OF OTHER PANCREATIC ENDOCRINE CELLS
Somatostatin is secreted by the δ cells of the islets. Islet somatostatin probably plays a paracrine role in inhibiting insulin and glucagon secretion from the neighboring β and α cells[67]. Somatostatin cell hyperplasia inside the pancreas appears to be extremely rare and only one case of focal δ cell hyperplasia has been reported which is in association with pancreatic cancer[68]. Hyperplasia of gastrin cells inside the pancreas has not been reported. We could not identify any literature on hyperplasia of ghrelin cells or vasoactive intestinal peptide cells either inside the pancreas or in other organs.
PANCREATIC ENDOCRINE CELL HYPERPLASIA AND PATHOGENESIS OF NEUROENDOCRINE TUMORS
It is not clear if pancreatic endocrine cell hyperplasia represents precursor lesions for pancreatic neuroendocrine tumors[69,70]. Diffuse endocrine cell hyperplasia, dysplasia, and microadenoma are present in the pancreas of patients with MEN1 and VHL, and are indeed considered as precursor lesions[16-19]. The hyperplastic pancreatic endocrine cells in patients with MEN1 and in mice with a heterozygous menin mutation are polyclonal and retain the normal menin allele, indicating that deletion of one copy of menin causes pancreatic endocrine cell to proliferate without tumorigenesis[71,72]. Loss of heterogeneity (LOH) of the menin locus is present in adenomas as small as 0.3 mm in diameter, demonstrating that these microadenomas are true tumors according to Knudson’s two-hit hypothesis of tumor development[73]. Interestingly, the exact pattern of LOH is different between microadenomas, suggesting that these microadenomas arise independently from the hyperplastic background[71,72]. As only a select number of clinical adenomas eventually develop while there are numerous microadenomas, additional mutations have to accrue to form larger and clinically significant pancreatic neuroendocrine tumors (PNETs). There have been no reports of similar precursor lesions for sporadic PNETs. Endocrine hyperplasia, dysplasia, and microadenoma, however, are not uncommon findings in the pancreas[27]. It is not known if these lesions are monoclonal. Although most of these lesions probably do not indicate clinical significance, they could represent precursor lesions giving rise to sporadic PNETs, since all clinical PNETs have to pass through a microadenoma stage during their growth[73]. It is thus plausible that PNETs develop from precursor (pre-malignant) lesions such as hyperplasia and microadenoma in familial PNET syndromes and at least partly in sporadic cases such as the patient we describe with α cell hyperplasia and pancreatic neuroendocrine tumors[53]. The key question of what additional genetic changes are needed to transform a microadenoma to a clinical PNET remains unanswered.
CONCLUSION
Pathologic pancreatic endocrine cell hyperplasia is a distinct group of diseases with various clinical, histological, and etiological features. β cell hyperplasia in adults causes non-insulinoma pancreatogenous hypoglycemia and probably contributes to persistent hyperinsulinemic hypoglycemia in infants. α cell hyperplasia causes glucagonoma syndrome or pancreatic neuroendocrine tumors. The main pathogenetic mechanism for pancreatic endocrine cell hyperplasia is increased endocrine cell neogenesis rather than proliferation of existing cells. The etiology has only been elucidated in some patients with demonstration of mutations of relevant genes regulating pancreatic endocrine cell phenotype. Although rare, this group of diseases does affect a significant number of patients and should be considered in the diagnosis and management hypoglycemia, elevated islet hormone levels, and pancreatic neuroendocrine tumors. Moreover, as the pancreatic endocrine cells are critical in regulating glucose metabolism and their hyperplasia may result in tumorigenesis, further studies of pathologic pancreatic endocrine cell hyperplasia will likely yield insights into the pathogenesis and treatment of diabetes and pancreatic neuroendocrine tumors.
Footnotes
Peer reviewer: Antonio Basoli, Professor, General Surgery “Paride Stefanini”, Università di Roma - Sapienza, Viale del Policlinico 155, Rome 00161, Italy
S- Editor Tian L L- Editor Cant MR E- Editor Zheng XM
References
- 1.Kim SK, MacDonald RJ. Signaling and transcriptional control of pancreatic organogenesis. Curr Opin Genet Dev. 2002;12:540–547. doi: 10.1016/s0959-437x(02)00338-6. [DOI] [PubMed] [Google Scholar]
- 2.Murtaugh LC, Melton DA. Genes, signals, and lineages in pancreas development. Annu Rev Cell Dev Biol. 2003;19:71–89. doi: 10.1146/annurev.cellbio.19.111301.144752. [DOI] [PubMed] [Google Scholar]
- 3.Piper K, Brickwood S, Turnpenny LW, Cameron IT, Ball SG, Wilson DI, Hanley NA. Beta cell differentiation during early human pancreas development. J Endocrinol. 2004;181:11–23. doi: 10.1677/joe.0.1810011. [DOI] [PubMed] [Google Scholar]
- 4.Rindi G, Bordi C. Highlights of the biology of endocrine tumours of the gut and pancreas. Endocr Relat Cancer. 2003;10:427–436. doi: 10.1677/erc.0.0100427. [DOI] [PubMed] [Google Scholar]
- 5.Massironi S, Sciola V, Peracchi M, Ciafardini C, Spampatti MP, Conte D. Neuroendocrine tumors of the gastro-entero-pancreatic system. World J Gastroenterol. 2008;14:5377–5384. doi: 10.3748/wjg.14.5377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ehehalt F, Saeger HD, Schmidt CM, Grützmann R. Neuroendocrine tumors of the pancreas. Oncologist. 2009;14:456–467. doi: 10.1634/theoncologist.2008-0259. [DOI] [PubMed] [Google Scholar]
- 7.Bonner-Weir S. Perspective: Postnatal pancreatic beta cell growth. Endocrinology. 2000;141:1926–1929. doi: 10.1210/endo.141.6.7567. [DOI] [PubMed] [Google Scholar]
- 8.Heit JJ, Karnik SK, Kim SK. Intrinsic regulators of pancreatic beta-cell proliferation. Annu Rev Cell Dev Biol. 2006;22:311–338. doi: 10.1146/annurev.cellbio.22.010305.104425. [DOI] [PubMed] [Google Scholar]
- 9.Ackermann AM, Gannon M. Molecular regulation of pancreatic beta-cell mass development, maintenance, and expansion. J Mol Endocrinol. 2007;38:193–206. doi: 10.1677/JME-06-0053. [DOI] [PubMed] [Google Scholar]
- 10.Rindi G, Solcia E. Endocrine hyperplasia and dysplasia in the pathogenesis of gastrointestinal and pancreatic endocrine tumors. Gastroenterol Clin North Am. 2007;36:851–865, vi. doi: 10.1016/j.gtc.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 11.Weidenheim KM, Hinchey WW, Campbell WG Jr. Hyperinsulinemic hypoglycemia in adults with islet-cell hyperplasia and degranulation of exocrine cells of the pancreas. Am J Clin Pathol. 1983;79:14–24. doi: 10.1093/ajcp/79.1.14. [DOI] [PubMed] [Google Scholar]
- 12.Ueda Y, Kurihara K, Kondoh T, Okanoue T, Chiba T. Islet-cell hyperplasia causing hyperinsulinemic hypoglycemia in an adult. J Gastroenterol. 1998;33:125–128. doi: 10.1007/s005350050057. [DOI] [PubMed] [Google Scholar]
- 13.Kim YW, Park YK, Park JH, Lee SM, Lee J, Ko SW, Yang MH. Islet cell hyperplasia of the pancreas presenting as hyperinsulinemic hypoglycemia in an adult. Yonsei Med J. 2000;41:426–429. doi: 10.3349/ymj.2000.41.3.426. [DOI] [PubMed] [Google Scholar]
- 14.Starke A, Saddig C, Kirch B, Tschahargane C, Goretzki P. Islet hyperplasia in adults: challenge to preoperatively diagnose non-insulinoma pancreatogenic hypoglycemia syndrome. World J Surg. 2006;30:670–679. doi: 10.1007/s00268-005-0543-6. [DOI] [PubMed] [Google Scholar]
- 15.Kimura W, Kuroda A, Morioka Y. Clinical pathology of endocrine tumors of the pancreas. Analysis of autopsy cases. Dig Dis Sci. 1991;36:933–942. doi: 10.1007/BF01297144. [DOI] [PubMed] [Google Scholar]
- 16.Thompson NW, Lloyd RV, Nishiyama RH, Vinik AI, Strodel WE, Allo MD, Eckhauser FE, Talpos G, Mervak T. MEN I pancreas: a histological and immunohistochemical study. World J Surg. 1984;8:561–574. doi: 10.1007/BF01654938. [DOI] [PubMed] [Google Scholar]
- 17.Anlauf M, Schlenger R, Perren A, Bauersfeld J, Koch CA, Dralle H, Raffel A, Knoefel WT, Weihe E, Ruszniewski P, et al. Microadenomatosis of the endocrine pancreas in patients with and without the multiple endocrine neoplasia type 1 syndrome. Am J Surg Pathol. 2006;30:560–574. doi: 10.1097/01.pas.0000194044.01104.25. [DOI] [PubMed] [Google Scholar]
- 18.Lubensky IA, Pack S, Ault D, Vortmeyer AO, Libutti SK, Choyke PL, Walther MM, Linehan WM, Zhuang Z. Multiple neuroendocrine tumors of the pancreas in von Hippel-Lindau disease patients: histopathological and molecular genetic analysis. Am J Pathol. 1998;153:223–231. doi: 10.1016/S0002-9440(10)65563-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Périgny M, Hammel P, Corcos O, Larochelle O, Giraud S, Richard S, Sauvanet A, Belghiti J, Ruszniewski P, Bedossa P, et al. Pancreatic endocrine microadenomatosis in patients with von Hippel-Lindau disease: characterization by VHL/HIF pathway proteins expression. Am J Surg Pathol. 2009;33:739–748. doi: 10.1097/PAS.0b013e3181967992. [DOI] [PubMed] [Google Scholar]
- 20.Bouwens L, Rooman I. Regulation of pancreatic beta-cell mass. Physiol Rev. 2005;85:1255–1270. doi: 10.1152/physrev.00025.2004. [DOI] [PubMed] [Google Scholar]
- 21.Gleason CE, Gross DN, Birnbaum MJ. When the usual insulin is just not enough. Proc Natl Acad Sci USA. 2007;104:8681–8682. doi: 10.1073/pnas.0702844104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hofeldt FD. Reactive hypoglycemia. Endocrinol Metab Clin North Am. 1989;18:185–201. [PubMed] [Google Scholar]
- 23.Brun JF, Fedou C, Mercier J. Postprandial reactive hypoglycemia. Diabetes Metab. 2000;26:337–351. [PubMed] [Google Scholar]
- 24.Rahier J, Fält K, Müntefering H, Becker K, Gepts W, Falkmer S. The basic structural lesion of persistent neonatal hypoglycaemia with hyperinsulinism: deficiency of pancreatic D cells or hyperactivity of B cells? Diabetologia. 1984;26:282–289. doi: 10.1007/BF00283651. [DOI] [PubMed] [Google Scholar]
- 25.Heitz PU, Klöppel G, Häcki WH, Polak JM, Pearse AG. Nesidioblastosis: the pathologic basis of persistent hyperinsulinemic hypoglycemia in infants. Morphologic and quantitative analysis of seven cases based on specific immunostaining and electron microscopy. Diabetes. 1977;26:632–642. doi: 10.2337/diab.26.7.632. [DOI] [PubMed] [Google Scholar]
- 26.Goossens A, Gepts W, Saudubray JM, Bonnefont JP, Nihoul-Fekete , Heitz PU, Klöppel G. Diffuse and focal nesidioblastosis. A clinicopathological study of 24 patients with persistent neonatal hyperinsulinemic hypoglycemia. Am J Surg Pathol. 1989;13:766–775. [PubMed] [Google Scholar]
- 27.Cummings DE. Gastric bypass and nesidioblastosis--too much of a good thing for islets? N Engl J Med. 2005;353:300–302. doi: 10.1056/NEJMe058170. [DOI] [PubMed] [Google Scholar]
- 28.Cryer PE, Axelrod L, Grossman AB, Heller SR, Montori VM, Seaquist ER, Service FJ. Evaluation and management of adult hypoglycemic disorders: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2009;94:709–728. doi: 10.1210/jc.2008-1410. [DOI] [PubMed] [Google Scholar]
- 29.Laidlaw GF. Nesidioblastoma, the islet tumor of the pancreas. Am J Pathol. 1938;14:125–134.5. [PMC free article] [PubMed] [Google Scholar]
- 30.Jack MM, Walker RM, Thomsett MJ, Cotterill AM, Bell JR. Histologic findings in persistent hyperinsulinemic hypoglycemia of infancy: Australian experience. Pediatr Dev Pathol. 2000;3:532–547. doi: 10.1007/s100240010117. [DOI] [PubMed] [Google Scholar]
- 31.Suchi M, MacMullen C, Thornton PS, Ganguly A, Stanley CA, Ruchelli ED. Histopathology of congenital hyperinsulinism: retrospective study with genotype correlations. Pediatr Dev Pathol. 2003;6:322–333. doi: 10.1007/s10024-002-0026-9. [DOI] [PubMed] [Google Scholar]
- 32.Pronicki M, Grajkowska W, Iwanicka K, Szymañska-Dêbiñska T, Taybert J, Drewniak T. Pathology of endocrine pancreas in persistent hyperinsulinemic hypoglycemia in children - practical diagnostic considerations. Ann Diagn Pediatr Pathol. 2002;6:101–106. [Google Scholar]
- 33.Klöppel G, Reinecke-Lüthge A, Koschoreck F. Focal and Diffuse Beta Cell Changes in Persistent Hyperinsulinemic Hypoglycemia of Infancy. Endocr Pathol. 1999;10:299–304. doi: 10.1007/BF02739772. [DOI] [PubMed] [Google Scholar]
- 34.Rahier J, Guiot Y, Sempoux C. Persistent hyperinsulinaemic hypoglycaemia of infancy: a heterogeneous syndrome unrelated to nesidioblastosis. Arch Dis Child Fetal Neonatal Ed. 2000;82:F108–F112. doi: 10.1136/fn.82.2.F108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kapoor RR, James C, Hussain K. Hyperinsulinism in developmental syndromes. Endocr Dev. 2009;14:95–113. doi: 10.1159/000207480. [DOI] [PubMed] [Google Scholar]
- 36.Stefanini P, Carboni M, Patrassi N, Basoli A. Hypoglycemia and insular hyperplasia: review of 148 cases. Ann Surg. 1974;180:130–135. doi: 10.1097/00000658-197407000-00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harrison TS, Fajans SS, Floyd JC Jr, Thompson NW, Rasbach DA, Santen RJ, Cohen C. Prevalence of diffuse pancreatic beta islet cell disease with hyperinsulinism: problems in recognition and management. World J Surg. 1984;8:583–589. doi: 10.1007/BF01654942. [DOI] [PubMed] [Google Scholar]
- 38.Service FJ, Natt N, Thompson GB, Grant CS, van Heerden JA, Andrews JC, Lorenz E, Terzic A, Lloyd RV. Noninsulinoma pancreatogenous hypoglycemia: a novel syndrome of hyperinsulinemic hypoglycemia in adults independent of mutations in Kir6.2 and SUR1 genes. J Clin Endocrinol Metab. 1999;84:1582–1589. doi: 10.1210/jcem.84.5.5645. [DOI] [PubMed] [Google Scholar]
- 39.Thompson GB, Service FJ, Andrews JC, Lloyd RV, Natt N, van Heerden JA, Grant CS. Noninsulinoma pancreatogenous hypoglycemia syndrome: an update in 10 surgically treated patients. Surgery. 2000;128:937–944;discussion 944-945. doi: 10.1067/msy.2000.110243. [DOI] [PubMed] [Google Scholar]
- 40.Anlauf M, Wieben D, Perren A, Sipos B, Komminoth P, Raffel A, Kruse ML, Fottner C, Knoefel WT, Mönig H, et al. Persistent hyperinsulinemic hypoglycemia in 15 adults with diffuse nesidioblastosis: diagnostic criteria, incidence, and characterization of beta-cell changes. Am J Surg Pathol. 2005;29:524–533. doi: 10.1097/01.pas.0000151617.14598.ae. [DOI] [PubMed] [Google Scholar]
- 41.Won JG, Tseng HS, Yang AH, Tang KT, Jap TS, Lee CH, Lin HD, Burcus N, Pittenger G, Vinik A. Clinical features and morphological characterization of 10 patients with noninsulinoma pancreatogenous hypoglycaemia syndrome (NIPHS) Clin Endocrinol (Oxf) 2006;65:566–578. doi: 10.1111/j.1365-2265.2006.02629.x. [DOI] [PubMed] [Google Scholar]
- 42.Shah JH, Maguire DJ, Brown D, Cotterill A. The role of ATP sensitive channels in insulin secretion and the implications in persistent hyperinsulinemic hypoglycaemia of infancy (PHHI) Adv Exp Med Biol. 2007;599:133–138. doi: 10.1007/978-0-387-71764-7_18. [DOI] [PubMed] [Google Scholar]
- 43.Sempoux C, Guiot Y, Dubois D, Nollevaux MC, Saudubray JM, Nihoul-Fekete C, Rahier J. Pancreatic B-cell proliferation in persistent hyperinsulinemic hypoglycemia of infancy: an immunohistochemical study of 18 cases. Mod Pathol. 1998;11:444–449. [PubMed] [Google Scholar]
- 44.Service GJ, Thompson GB, Service FJ, Andrews JC, Collazo-Clavell ML, Lloyd RV. Hyperinsulinemic hypoglycemia with nesidioblastosis after gastric-bypass surgery. N Engl J Med. 2005;353:249–254. doi: 10.1056/NEJMoa043690. [DOI] [PubMed] [Google Scholar]
- 45.McLaughlin T, Peck M, Holst J, Deacon C. Reversible hyperinsulinemic hypoglycemia after gastric bypass: a consequence of altered nutrient delivery. J Clin Endocrinol Metab. 2010;95:1851–1855. doi: 10.1210/jc.2009-1628. [DOI] [PubMed] [Google Scholar]
- 46.Patti ME, McMahon G, Mun EC, Bitton A, Holst JJ, Goldsmith J, Hanto DW, Callery M, Arky R, Nose V, et al. Severe hypoglycaemia post-gastric bypass requiring partial pancreatectomy: evidence for inappropriate insulin secretion and pancreatic islet hyperplasia. Diabetologia. 2005;48:2236–2240. doi: 10.1007/s00125-005-1933-x. [DOI] [PubMed] [Google Scholar]
- 47.Meier JJ, Butler AE, Galasso R, Butler PC. Hyperinsulinemic hypoglycemia after gastric bypass surgery is not accompanied by islet hyperplasia or increased beta-cell turnover. Diabetes Care. 2006;29:1554–1559. doi: 10.2337/dc06-0392. [DOI] [PubMed] [Google Scholar]
- 48.Paloyan E, Lawrence AM, Straus FH 2nd, Paloyan D, Harper PV, Cummings D. Alpha cell hyperplasia in calcific pancreatitis associated with hyperparathyroidism. JAMA. 1967;200:757–761. [PubMed] [Google Scholar]
- 49.Toda K, Souda S, Sueki H, Momiyama T, Kuratani T, Yamabe K. A case report of asymptomatic malignant microglucagonoma. Jpn J Gastroenterol Surg. 1991;24:3012–3016. [Google Scholar]
- 50.Brown K, Kristopaitis T, Yong S, Chejfec G, Pickleman J. Cystic glucagonoma: A rare variant of an uncommon neuroendocrine pancreas tumor. J Gastrointest Surg. 1998;2:533–536. doi: 10.1016/s1091-255x(98)80053-x. [DOI] [PubMed] [Google Scholar]
- 51.Martignoni ME, Kated H, Stiegler M, Büchler MW, Friess H, Zimmermann A, Schirp U, Nitzsche EU. Nesidioblastosis with glucagon-reactive islet cell hyperplasia: a case report. Pancreas. 2003;26:402–405. doi: 10.1097/00006676-200305000-00016. [DOI] [PubMed] [Google Scholar]
- 52.Chen HW, Chen HW, Su DH, Shun CT, Liu KL. Rare presentation of endocrine pancreatic tumor: a case of diffuse glucagonoma without metastasis and necrolytic migratory erythema. J Formos Med Assoc. 2005;104:363–366. [PubMed] [Google Scholar]
- 53.Yu R, Nissen NN, Dhall D, Heaney AP. Nesidioblastosis and hyperplasia of alpha cells, microglucagonoma, and nonfunctioning islet cell tumor of the pancreas: review of the literature. Pancreas. 2008;36:428–431. doi: 10.1097/MPA.0b013e31815ceb23. [DOI] [PubMed] [Google Scholar]
- 54.Henopp T, Anlauf M, Schmitt A, Schlenger R, Zalatnai A, Couvelard A, Ruszniewski P, Schaps KP, Jonkers YM, Speel EJ, et al. Glucagon cell adenomatosis: a newly recognized disease of the endocrine pancreas. J Clin Endocrinol Metab. 2009;94:213–217. doi: 10.1210/jc.2008-1300. [DOI] [PubMed] [Google Scholar]
- 55.Zhou C, Dhall D, Nissen NN, Chen CR, Yu R. Homozygous P86S mutation of the human glucagon receptor is associated with hyperglucagonemia, alpha cell hyperplasia, and islet cell tumor. Pancreas. 2009;38:941–946. doi: 10.1097/MPA.0b013e3181b2bb03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Parker JC, Andrews KM, Allen MR, Stock JL, McNeish JD. Glycemic control in mice with targeted disruption of the glucagon receptor gene. Biochem Biophys Res Commun. 2002;290:839–843. doi: 10.1006/bbrc.2001.6265. [DOI] [PubMed] [Google Scholar]
- 57.Gelling RW, Du XQ, Dichmann DS, Romer J, Huang H, Cui L, Obici S, Tang B, Holst JJ, Fledelius C, et al. Lower blood glucose, hyperglucagonemia, and pancreatic alpha cell hyperplasia in glucagon receptor knockout mice. Proc Natl Acad Sci USA. 2003;100:1438–1443. doi: 10.1073/pnas.0237106100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Greenberg GR, McCloy RF, Adrian TE, Chadwick VS, Baron JH, Bloom SR. Inhibition of pancreas and gallbladder by pancreatic polypeptide. Lancet. 1978;2:1280–1282. doi: 10.1016/s0140-6736(78)92042-1. [DOI] [PubMed] [Google Scholar]
- 59.Batterham RL, Le Roux CW, Cohen MA, Park AJ, Ellis SM, Patterson M, Frost GS, Ghatei MA, Bloom SR. Pancreatic polypeptide reduces appetite and food intake in humans. J Clin Endocrinol Metab. 2003;88:3989–3992. doi: 10.1210/jc.2003-030630. [DOI] [PubMed] [Google Scholar]
- 60.Tomita T, Kimmel JR, Friesen SR, Mantz FA Jr. Pancreatic polypeptide cell hyperplasia with and without watery diarrhea syndrome. J Surg Oncol. 1980;14:11–20. doi: 10.1002/jso.2930140104. [DOI] [PubMed] [Google Scholar]
- 61.Farley DR, van Heerden JA, Myers JL. Adult pancreatic nesidioblastosis. Unusual presentations of a rare entity. Arch Surg. 1994;129:329–332. doi: 10.1001/archsurg.1994.01420270107022. [DOI] [PubMed] [Google Scholar]
- 62.Martella EM, Ferraro G, Azzoni C, Marignani M, Bordi C. Pancreatic-polypeptide cell hyperplasia associated with pancreatic or duodenal gastrinomas. Hum Pathol. 1997;28:149–153. doi: 10.1016/s0046-8177(97)90098-8. [DOI] [PubMed] [Google Scholar]
- 63.Pasieka JL, Hershfield N. Pancreatic polypeptide hyperplasia causing watery diarrhea syndrome: a case report. Can J Surg. 1999;42:55–58. [PMC free article] [PubMed] [Google Scholar]
- 64.Albazaz R, Da Costa PE, Verbeke CS. Pancreatic polypeptide cell hyperplasia of the pancreas. J Clin Pathol. 2006;59:1087–1090. doi: 10.1136/jcp.2005.030478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bunning J, Merchant SH, Crooks LA, Hartshorne MF. Indium-111 pentetreotide uptake by pancreatic polypeptide cell hyperplasia: potential pitfall in somatostatin receptor scintigraphy. Pancreas. 2007;35:372–375. doi: 10.1097/mpa.0b013e31811ea2a2. [DOI] [PubMed] [Google Scholar]
- 66.O'Dorisio T, Vinik A. Pancreatic polypeptide and mixed peptide-producing tumors of the gastrointestinal tract. In: Cohen S, Soloway R, editors. Hormone-producing tumors of the gastrointestinal tract. New York: Churchill Livingstone; 1985. p. 117. [Google Scholar]
- 67.Hauge-Evans AC, King AJ, Carmignac D, Richardson CC, Robinson IC, Low MJ, Christie MR, Persaud SJ, Jones PM. Somatostatin secreted by islet delta-cells fulfills multiple roles as a paracrine regulator of islet function. Diabetes. 2009;58:403–411. doi: 10.2337/db08-0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Iki K, Pour PM. Somatostatin cell hyperplasia in the pancreas. Pancreas. 2006;33:471. [Google Scholar]
- 69.Anlauf M, Perren A, Klöppel G. Endocrine precursor lesions and microadenomas of the duodenum and pancreas with and without MEN1: criteria, molecular concepts and clinical significance. Pathobiology. 2007;74:279–284. doi: 10.1159/000105810. [DOI] [PubMed] [Google Scholar]
- 70.Klöppel G, Anlauf M, Perren A. Endocrine precursor lesions of gastroenteropancreatic neuroendocrine tumors. Endocr Pathol. 2007;18:150–155. doi: 10.1007/s12022-007-0025-5. [DOI] [PubMed] [Google Scholar]
- 71.Perren A, Anlauf M, Henopp T, Rudolph T, Schmitt A, Raffel A, Gimm O, Weihe E, Knoefel WT, Dralle H, et al. Multiple endocrine neoplasia type 1 (MEN1): loss of one MEN1 allele in tumors and monohormonal endocrine cell clusters but not in islet hyperplasia of the pancreas. J Clin Endocrinol Metab. 2007;92:1118–1128. doi: 10.1210/jc.2006-1944. [DOI] [PubMed] [Google Scholar]
- 72.Crabtree JS, Scacheri PC, Ward JM, Garrett-Beal L, Emmert-Buck MR, Edgemon KA, Lorang D, Libutti SK, Chandrasekharappa SC, Marx SJ, et al. A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Proc Natl Acad Sci USA. 2001;98:1118–1123. doi: 10.1073/pnas.98.3.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pritchard DM. Pathogenesis of gastrinomas associated with multiple endocrine neoplasia type 1. Gut. 2007;56:606–607. doi: 10.1136/gut.2006.113985. [DOI] [PMC free article] [PubMed] [Google Scholar]