

Abstract
How the ATPase activity of Heat shock protein 90 (Hsp90) is coupled to client protein activation remains obscure. Using truncation and missense mutants of Hsp90, we analysed the structural implications of its ATPase cycle. C-terminal truncation mutants lacking inherent dimerization displayed reduced ATPase activity, but dimerized in the presence of 5′-adenylamido-diphosphate (AMP-PNP), and AMP-PNP- promoted association of N-termini in intact Hsp90 dimers was demonstrated. Recruitment of p23/Sba1 to C-terminal truncation mutants also required AMP-PNP-dependent dimerization. The temperature- sensitive (ts) mutant T101I had normal ATP affinity but reduced ATPase activity and AMP-PNP-dependent N-terminal association, whereas the ts mutant T22I displayed enhanced ATPase activity and AMP-PNP-dependent N-terminal dimerization, indicating a close correlation between these properties. The locations of these residues suggest that the conformation of the ‘lid’ segment (residues 100–121) couples ATP binding to N-terminal association. Consistent with this, a mutation designed to favour ‘lid’ closure (A107N) substantially enhanced ATPase activity and N-terminal dimerization. These data show that Hsp90 has a molecular ‘clamp’ mechanism, similar to DNA gyrase and MutL, whose opening and closing by transient N-terminal dimerization are directly coupled to the ATPase cycle.
Keywords: chaperone/conformational switch/cross-linking/mutational analysis
Introduction
Heat shock protein 90 (Hsp90) is an essential molecular chaperone in eukaryotes, involved in activation of an important set of proteins involved in signal transduction and cell-cycle regulation. Unlike the Hsp70 and Hsp60/GroEL families, its biochemical mechanism has remained obscure (Bukau and Horwich, 1998). Structural and biochemical analyses identified an ATP/ADP-binding site in the N-terminal domain of Hsp90s (Grenert et al., 1997; Prodromou et al., 1997a), and ATP binding and hydrolysis have been shown to be essential for Hsp90 function in vivo (Obermann et al., 1998; Panaretou et al., 1998) and for activation of authentic client proteins, such as glucocorticoid receptor, in vitro (Grenert et al., 1999). The ATPase activity of Hsp90s is regulated by interaction with co-chaperones, and the ATP-dependent step in client protein activation has been localized to ‘mature’ complexes (Prodromou et al., 1999). The role of this inherent ATPase activity in the biochemistry of Hsp90 is yet to be defined.
We have now expressed and characterized Hsp90 mutants with C-terminal truncations of varying degree, and/or with missense mutations conferring temperature-sensitive (ts) phenotypes in yeast (Nathan and Lindquist, 1995). The behaviour of these mutants shows a clear association between nucleotide binding and association of the N-terminal domains, coupled by conformational changes in a segment of the N-terminus that provides a ‘lid’ for the nucleotide-binding pocket. These studies suggest a mechanism involving nucleotide-regulated association of the N-terminal domain of Hsp90, which with the enforced dimerization provided by the C-terminus drives an ATPase-coupled ‘clamp’ mechanism essentially identical to those proposed for DNA gyrase B (GyrB) and MutL.
Results and discussion
C-terminal truncation reduces ATPase activity
Previous studies identified the N-terminal domain as the nucleotide-binding site in Hsp90s (Grenert et al., 1997; Prodromou et al., 1997a), while C-terminal regions, particularly the last 13 kDa, were implicated in binding to tetratricopeptide repeat (TPR)-domain co-chaperones (Chen et al., 1998; Young et al., 1998; Carrello et al., 1999). However, the ATPase cycle of Hsp90 is clearly involved in client protein activation (Obermann et al., 1998; Panaretou et al., 1998; Grenert et al., 1999) and is regulated by TPR-domain co-chaperones (Prodromou et al., 1999) so that the N- and C-terminal functions are not simply separable. Furthermore, the isolated N-terminal domain, although fully able to bind ATP, has negligible ATPase activity, suggesting involvement of other regions of the molecule in the ATPase activity of Hsp90.
To identify other regions involved in the ATPase mechanism, we expressed a series of Hsp90 mutants with progressive truncation of the C-terminus, and determined their ATPase activities (Figure 1). Surprisingly, even the shortest truncation mutant we investigated (construct NC599), which lacks only the C-terminal 13 kDa, showed a significantly reduced ATPase activity, ∼22% that of wild type at 37°C. Loss of a further 47 C-terminal residues (NΔ39C551) caused a further reduction to ∼11% that of wild-type ATPase activity, while truncation of a further 100 residues (NC450) and all truncations beyond that, including the N-terminal domain itself, displayed ATPase activities <1% that of wild type (Figure 1).

Fig. 1. ATPase activities of C-terminal truncation mutants. (A) Schematics of yeast Hsp90 C-terminal truncation mutants. (B) ATPase activities of wild-type Hsp90 and C-terminal truncation mutants measured at 37°C.
Hsp90 N-terminal dimerization is nucleotide dependent
As well as providing TPR-domain-binding sites, the C-terminal region of Hsp90 mediates its dimerization (Chen et al., 1998; Carrello et al., 1999). The marked decrease in ATPase activity when this region was deleted (see above) suggested that the dimeric state of Hsp90 might be coupled to the ATPase activity or vice versa. To explore this, we analysed the oligomeric state of wild-type Hsp90 and the various C-terminal truncation mutants in the presence and absence of nucleotides, by cross-linking with dimethylsuberimidate (DMS). With wild-type Hsp90 containing an intact C-terminal dimerization region, cross-linked dimers were obtained in all conditions, as expected. However, there was a clear enhancement of dimer yield in the presence of the non-hydrolysable ATP analogue 5′-adenylamido-diphosphate (AMP-PNP) compared with ADP, suggesting that the nucleotide triphosphate either stabilized dimer formation or altered the extent of the interactions within the dimer (Figure 2A). With the C-terminal truncation mutant NC599, which lacks the C-terminal dimerization domain, no cross-linked dimers were obtained in the absence of nucleotide or in the presence of ADP. However, significant cross-linked dimer formation was observed in the presence of AMP-PNP (Figure 2B). Shorter truncation mutants (NΔ39C551 and NΔ0C531, which also contain linker modifications) also produced cross-linked dimers but only in the presence of AMP-PNP (Figure 2C and D), whereas the shorter construct NC450 and the isolated N-terminal domain (NC220) did not (Figure 2E and F). Geldanamycin and radicicol inhibited AMP-PNP-dependent formation of cross-linked dimers (Figure 2G), confirming that dimerization resulted from AMP-PNP binding to the characterized drug-sensitive nucleotide-binding site in the N-terminal domain of Hsp90 (Prodromou et al., 1997a; Stebbins et al., 1997; Roe et al., 1999). The ability of C-terminal deletion constructs to dimerize in the presence of AMP-PNP correlated with their ATPase activity. Thus, NC599, NΔ39C551 and NΔ0C531, which have measurable ATPase activity, also displayed AMP-PNP-dependent dimerization, whereas NC450 and NC220 do not dimerize in the presence of AMP-PNP and lack ATPase activity.

Fig. 2. DMS cross-linking of Hsp90 and C-terminal truncation mutants. (A–F) SDS–PAGE gels showing cross-linking of (A) wild-type Hsp90 and C-terminal truncation mutants NC599 (B), NΔ39C551 (C), NΔ0C530 (D), NC450 (E) and NC220 (F). In all cases ‘90’ indicates the Hsp90 construct; L, DMS linker; D, ADP; N, AMP-PNP. Cross-linked species corresponding to dimers are only observed with wild-type Hsp90 and with the NC599, NΔ39C551 and NΔ0C530 C-terminal truncation mutants, only in the presence of AMP-PNP. (G) Inhibition of cross-linking of the C-terminal truncation mutant NΔ0C530 by the specific competitive inhibitors of nucleotide binding in the N-domain, geldanamycin (G) and radicicol (R). Reaction conditions are given in Materials and methods.
p23/Sba1 recruitment requires N-terminal dimerization
Binding of the co-chaperone p23 and its yeast homologue Sba1 to Hsp90 is ATP dependent (Johnson and Toft, 1994, 1995; Fang et al., 1998), and mutations in the N-terminal domain of Hsp90s that abolish ATP binding also prevent p23/Sba1 binding (Obermann et al., 1998; Grenert et al., 1999). However, the isolated N-terminal domain, although fully capable of binding ATP or AMP-PNP, is not sufficient for p23/Sba1 binding. The data presented above suggest that dimeric association of regions outside the C-terminus is a consequence of ATP binding to Hsp90. We have therefore attempted to discover whether this ATP-dependent dimerization plays a role in the recruitment of p23/Sba1.
Yeast p23/Sba1 cross-linked by DMS in the absence of nucleotide or in the presence of ADP or AMP-PNP was overwhelmingly monomeric (data not shown). At comparable protein concentration, wild-type Hsp90 gives a substantial dimer band, suggesting that p23/Sba1 is probably a monomer in solution. When Hsp90 was included in cross-linking experiments with p23/Sba1, clear bands for Hsp90 monomers and dimers, and for p23/Sba1 monomers, were observed in the absence of nucleotide or in the presence of ADP (Figure 3A). In the presence of AMP-PNP, additional bands were observed at higher molecular weight than the cross-linked Hsp90 dimer, consistent with one or more bound p23/Sba1 molecules becoming cross-linked to Hsp90 dimers. With longer C-terminal truncation mutants (NC599, NΔ39C551), bands higher than Hsp90 dimers were again only seen in the presence of AMP-PNP (Figure 3B and C). The presence of p23/Sba1 in these higher bands was confirmed by western blotting with an anti-p23/Sba1 polyclonal serum (Figure 3F). Thus, constructs displaying ATPase activity and able to dimerize in the presence of AMP-PNP were also able to recruit p23/Sba1. With the shorter truncation mutants (NC450, NC431, NC220), which bind AMP-PNP but do not dimerize in its presence and lack ATPase activity, no cross-linking to p23/Sba1 was observed (Figure 3D and E). Thus, binding of p23/Sba1 to Hsp90 constructs correlated not with their ability to bind AMP-PNP (and by inference ATP), but with their ability to dimerize on doing so. The ATP dependence of p23/Sba1 binding to Hsp90 is thus indirect, so that p23/Sba1 binds selectively to the dimerized conformation of Hsp90 that is induced by ATP binding. The actual location of the p23/Sba-binding site on the Hsp90 dimer is not known, nor is the stoichiometry of the interaction.

Fig. 3. Recruitment of p23/Sba1 to Hsp90 and C-terminal truncation mutants. (A–E) SDS–PAGE gels showing cross-linking of (A) wild-type Hsp90 and C-terminal truncation mutants NC599 (B), NΔ39C551 (C), NC450 (D) and NC220 (E). In all cases ‘90’ indicates the Hsp90 construct; 23, p23/Sba1; L, DMS linker; D, ADP; N, AMP-PNP; T, ATP. A cross-linked band higher than the Hsp90 dimer is observed with wild-type Hsp90, NC599 and NΔ39C551, only in the presence of p23/Sba1 and AMP-PNP. (F) Western blot of NC599 and p23/Sba1 cross-linking experiment. A high molecular weight band cross-reacting with anti-p23/Sba1 antisera is only observed in the presence of AMP-PNP.
ATP binding promotes N-domain association
The N-terminal domain of Hsp90 itself (NC220) lacks ATPase activity and does not dimerize in isolation, but nonetheless provides the binding site for nucleotide and must therefore play an essential role in the mechanism of AMP-PNP-dependent dimerization. To determine whether the N-terminal domains themselves become associated in the AMP-PNP-bound state, we took advantage of the absence of native cysteine residues in yeast Hsp90 (HSP82) to make single cysteine mutations (see Materials and methods) at surface-accessible positions in the N-terminal domain (Prodromou et al., 1997b). Mutants E7C, Q9C and E11C were expressed, purified and analysed on SDS–PAGE gels. At concentrations where wild-type Hsp90 showed no cross-linked oligomers higher than dimers, all three mutants showed significant dimerization in non-reducing conditions, indicating formation of intra-dimer disulfide bonds between the cysteines in the N-terminal domains (Figure 4A). Only ∼50% of the protein could be obtained in disulfide form, probably due to a refractory sub-population of Hsp90 dimers in which one or both cysteines had become terminally oxidized to cysteic acid. Formation of intra-domain disulfide bonds in the absence of nucleotide shows that the N-termini within an Hsp90 dimer are capable of coming into very close proximity. To observe N-terminal association in an equilibrium system, where differences between ADP- and ATP-bound states could be observed, we modified the E11C mutant by specific covalent attachment of a pyrene group to the thiol side chain of Cys11. Pyrene molecules covalently attached to proteins display a marked alteration in their fluorescence emission spectrum at ∼450–500 nm when they come into close contact, due to the formation of an excited-state dimer (excimer) (Sen and Chakrabarti, 1990). The fluorescence emission spectrum of the pyrene-modified E11C showed significant excimer formation compared with unmodified wild-type Hsp90, or wild-type Hsp90 subjected to the pyrene modification procedure, indicating site-specific attachment of the pyrene (Figure 4B). The excimer signal was substantially enhanced by the presence of AMP-PNP compared with ADP (Figure 4C), confirming that AMP-PNP binding stabilized a conformation in which the N-terminal domains are closely associated.
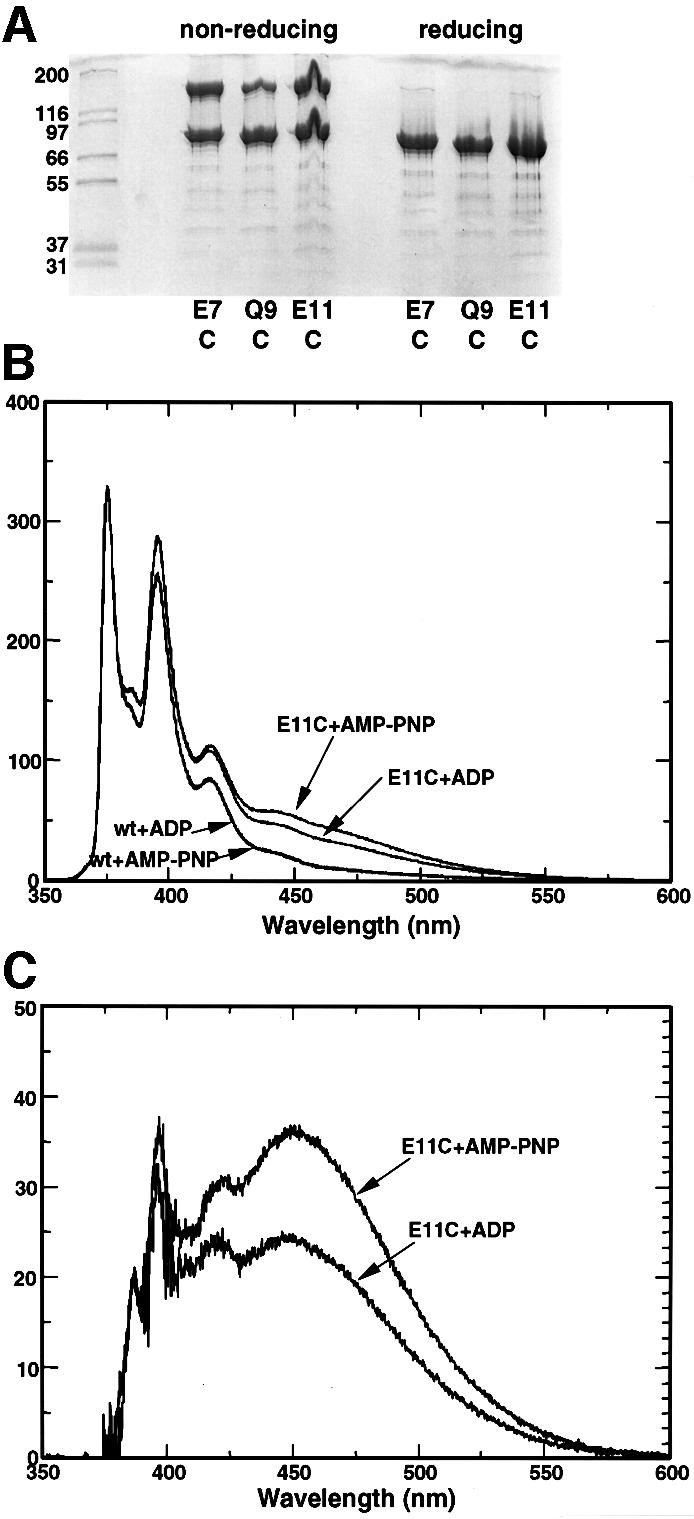
Fig. 4. N-terminal interactions in single-cysteine mutants. (A) Polyacrylamide gel of purified single-cysteine mutants E7C, Q9C and E11C without added nucleotide, under non-reducing conditions and in the presence of the non-thiol reducing agent TCEP. In all three mutants, intra-dimer disulfide bonds are formed in the absence of reducing agent. (B) Excimer formation in pyrene-modified single-cysteine mutants. E11C mutant and wild-type Hsp90s were treated with pyrene (see Materials and methods), and their fluorescence emission spectra recorded. Only the cysteine mutants displayed a significant excimer signal in the region 450–500 nm, and this was substantially enhanced in the presence of AMP-PNP compared with ADP. (C) Specific excimer fluorescence signals for the E11C mutant, corrected for non-specific signal due to adsorption of the hydrophobic pyrene onto the protein surface by subtraction of the corresponding emission spectrum for the wild-type Hsp90.
An ATPase-coupled molecular ‘clamp’
The C-terminus provides the major dimerization interface of Hsp90, so that C-terminal deletion constructs are inherently monomeric. Surprisingly, given that the N-terminal domain provides the nucleotide-binding site, loss of the C-terminal 13 kDa decreased the inherent ATPase to ∼20% of its normal activity. Although ordinarily monomeric, this mutant dimerized in the presence of AMP-PNP, whereas more severely truncated mutants, lacking ATPase activity, did not. The correlation between dimerization and ATPase activity is consistent with positive cooperativity between the two ATPase active sites in an Hsp90 dimer, so that N-terminal dimerization is required for efficient ATP hydrolysis. The possibility of direct association between the N-termini is demonstrated by the ability of cysteine mutants to form intra-dimer disulfide bonds. When the N-terminal domains were site-specifically modified by pyrene, excimer formation, indicative of close proximity, was promoted by AMP-PNP compared with ADP. Thus, bound ATP (but not ADP) promotes association of the N-terminal domains within the Hsp90 dimer, and subsequent ATP hydrolysis is highly cooperative and dependent on that dimerization. Together with the inherent dimerization provided by the C-terminus, these properties describe a molecular clamp whose opening and closing by transient dimerization of the N-termini are directly coupled to the ATPase cycle (Figure 5).

Fig. 5. Opening and closing of Hsp90 molecular clamp coupled to the ATPase cycle.
Dimerization of the N-terminus of Hsp90 has been controversial. In the crystal structure of an N-terminal domain of yeast Hsp90, we observed a dimer interface formed by association of C-terminal strands in an antiparallel β-sheet (Prodromou et al., 1997b). However, the crystal structure of the N-terminal domain of human Hsp90, in which this C-terminal strand was not included, was monomeric. The dimer in the yeast Hsp90 N-domain structure defined a channel whose dimensions suggested a possible peptide-binding function. However, in subsequent studies we have failed to establish such a function, and cannot dismiss the possibility that the observed strand-swap and consequent ‘channel’ are artefacts of crystallization of an isolated fragment. Nonetheless, other groups have observed peptide binding to isolated N-terminal fragments (Young et al., 1997; Scheibel et al., 1998, 1999), but the site of these interactions within the N-terminal domain is unknown, and the stoichiometry and affinity of the complexes have not been determined. The possibility of N-terminal dimerization was further challenged by electron microscopy studies, which showed a divergent structure for the Hsp90 dimer (Koyasu et al., 1986; Wearsch and Nicchitta, 1996) in which only the C-termini are associated. Recently, toroidal structures consistent with association of both N- and C-termini, as we describe above, have been observed in electron microscopy studies of Hsp90 following heat-shock, and to a lesser extent, in the presence of ATP (Maruya et al., 1999).
The structure of the N-terminal ATP-binding domain of Hsp90s does not conform to the ‘Walker’ consensus (Walker et al., 1982). Instead, it resembles the ATP-binding sites in GyrB (Wigley et al., 1991), the MutL mismatch repair protein (Ban et al., 1999) and histidine kinases (Tanaka et al., 1998; Bilwes et al., 1999). The ATP-binding domain in GyrB regulates opening and closing of a molecular clamp and consequent binding and release of a DNA duplex, by promoting association of the N-termini on ATP binding, and dissociation on hydrolysis to ADP (Kampranis et al., 1999). The inherent ATPase of MutL (Ban et al., 1999) functions in much the same way, although its role in mismatch repair is unclear. In these systems the C-terminus provides a dimerization interface independent of the nucleotide-loaded state of the N-terminal domain, and converts the ATP-dependent dimerization of the N-terminus into the opening and closing of a molecular clamp, hinged at the C-terminus. The data we present here describe an essentially identical mechanism for an ATPase-coupled molecular clamp in Hsp90, and unite Hsp90 structurally and mechanistically with MutL and GyrB.
ATPase activity of ts missense mutants
Genetic studies (Nathan and Lindquist, 1995) identified missense mutations in the yeast Hsp90 (HSP82) that confer ts viability phenotypes. Four of these (T22I, A41V, T101I, G170D) map in the N-terminal ATP-binding domain (Figure 6A), suggesting that their phenotypes might result from defects in ATPase activity. G170D has attracted particular interest, as unlike T22I, A41V and T101I, which display growth defects at all temperatures, it displays a true ts phenotype with near-normal growth at permissive temperatures and a catastrophic loss of viability at non-permissive temperatures. In contrast to the other mutants used in this study, G170D proved impossible to over-express and purify from Escherichia coli, displaying a strong tendency to aggregate and precipitate during purification, even when handled entirely at ‘permissive’ temperatures, suggesting a considerably destabilized structure. Gly170 occupies a conformationally restricted position in the Hsp90 N-domain structure (Prodromou et al., 1997b), requiring the left-handed helical conformation uniquely available to glycine. Replacement with aspartic acid would generate a structural defect that destabilizes the protein, causing loss of function at non-permissive temperatures. While it may be mitigated by interaction with co-chaperones and by general molecular crowding in vivo at permissive temperatures, this instability is manifest when the protein is isolated. We were thus unable to determine the effect of this mutation on the ATPase of the protein.

Fig. 6. ATPase activities and dimerization efficiency of ts missense mutants. (A) Schematic of yeast Hsp90 primary structure with positions of missense mutations conferring ts phenotypes indicated. (B) ATPase activities of wild-type Hsp90 and ts mutants at 22, 30, 37 and 45°C. (C) Polyacrylamide gel of Hsp90 ATPase mutants T22I and T101I (6 µM) cross-linked with DMS in the presence of 10 mM AMP-PNP at 37°C for 1 h (see Materials and methods). Numbers below the lanes indicate the molar ratio of cross-linker to protein primary amine content in that reaction. At all linker concentrations the ATPase hyperactive mutant T22I shows a much higher degree of cross-linked dimers than the hypoactive T101I mutant.
The A41V mutation maps to the bottom of the nucleotide-binding pocket and was predicted to interfere with ATP binding (Panaretou et al., 1998). Consistent with this, A41V bound AMP-PNP much less tightly than wild-type Hsp90 and displayed a greatly reduced ATPase activity at all temperatures (Table I and Figure 6B). A41V did not affect the structural stability of the protein relative to wild type, as judged by its melting temperature (see Materials and methods). With the identification of an ATP-binding site in the N-domain similar to that in GyrB (Prodromou et al., 1997a), Thr101 was revealed as part of a ‘lid’ segment that in GyrB closes over the bound ATP (Wigley et al., 1991). The T101I mutation did not affect the stability of the protein or its affinity for AMP-PNP (Table I). Nonetheless, the ATPase activity of T101I is <5% that of wild type over the temperature range 22–45°C (Figure 6B), suggesting that the mutation affects the ATPase at a step subsequent to ATP binding. The temperature sensitivity and general growth defects displayed by T101I and A41V in vivo are most likely the result of their respective defects in ATPase activity and in nucleotide binding, attributes essential for Hsp90 function in vivo (Obermann et al., 1998; Panaretou et al., 1998).
Table I. AMP-PNP affinity, stability and relative ATPase activity of ts mutants.
| Construct | Kd AMP-PNP-Mg2+ | Tm | ATPase at 37°C |
|---|---|---|---|
| (µM) | (°C) | (% of wild type) | |
| Wild type | 33 | 65.2 | 100 |
| T22I | 110 | 61.4 | 456 |
| A41V | 154 | 65.1 | 1.3 |
| T101I | 37 | 63.6 | 9.4 |
| A587T | 37 | 63.6 | 94.3 |
Thr22 is some way from the mouth of the nucleotide-binding pocket and is unable to make direct contact with bound nucleotide. Surprisingly, the T22I mutation has a dramatic effect on the ATPase activity, increasing by 6-fold that of wild type at 30°C, although its affinity for AMP-PNP is somewhat decreased. As the T22I mutant is fully capable of binding and hydrolysing ATP, it is not immediately obvious why this ‘hyperactive’ mutant is defective in vivo. We had previously shown (Prodromou et al., 1999) that the lifetime of the ATP-bound state of Hsp90 corresponds to that of the ‘mature’ Hsp90–client protein complex (Smith, 1993; Smith et al., 1995): ∼4–5 min at 30°C. In T22I, which is barely viable at 30°C, the lifetime of the ATP-bound state is reduced to ∼30–40 s. As the ATP-bound mature Hsp90 complexes are the stage in which client protein activation takes place, a substantial decrease in the lifetime of that state would inevitably impair activation of essential client proteins even under normal growth conditions. Significantly, p60/Hop/Sti1, which inhibits Hsp90 ATPase activity (Prodromou et al., 1999), partially rescues T22I when expressed at high levels (Chang et al., 1997). One further ts mutant mapping outside the N-terminal domain was also characterized. A587T displayed no decrease in stability and possessed ‘wild-type’ AMP-PNP binding and ATPase activity at all temperatures (Figure 6B). Its loss of function at non-permissive temperatures presumably results from defects in some other aspect of Hsp90 biochemistry.
ATPase mutants show altered ATP-dependent N-terminal dimerization
The defect in the ATPase activity of A41V can be understood in terms of its significantly reduced nucleotide affinity, whereas the aberrant activities of T22I and T101I result from changes in the behaviour of Hsp90 in stages of the ATPase cycle subsequent to ATP binding. To gain further insight into these changes, we examined the dimerization behaviour of T22I and T101I in the presence of AMP-PNP, by DMS cross-linking. Cross-linking by DMS requires the primary amines of lysine residues on the interacting protein molecules to be close enough together for a sufficient time to allow the reaction to take place. Assuming a more or less random distribution of lysines on the surface of the interacting molecules, then for a given concentration of cross-linking reagent the amount of cross-linked products obtained will depend on the size of the molecular surfaces that interact and the stability of that interaction. Cross-linking is therefore a sensitive probe of the conformation of the Hsp90 dimer, as well as of its stoichiometry. Thus, with intact Hsp90 (Figure 2A), more cross-linked product is obtained in the ATP-bound state when both N- and C-terminal regions are in proximity (see above), than in the ADP-bound or unliganded state when only the C-termini interact strongly. With the ts mutants that display altered ATPase activities, the degree of cross-linking obtained when saturated with AMP-PNP at all linker concentrations is significantly lower for the hypoactive T101I mutant than for the hyperactive T22I under the same conditions (Figure 6C). Thus, the T101I mutant, which binds AMP-PNP with wild-type affinity, has a diminished ability to dimerize its N-terminal domains in the ATP-bound state. Conversely, T22I, whose apparent ATP affinity is lower than wild type, displays enhanced N-terminal dimerization. The respective enhancement and reduction relative to wild type in the ATPase activities of T22I and T101I, respectively, reflects their correspondingly enhanced and reduced ATP-dependent N-terminal dimerization, and provides strong support for a mechanism in which ATP binding promotes N-terminal association, which is in turn required for ATP hydrolysis.
N-domain dimer interface and ‘lid’ conformation
With the identification of an ATP-binding function in the N-terminal domain of Hsp90, and in light of the mechanistic and structural similarity between Hsp90, GyrB and MutL that has subsequently emerged, we have re-examined the dimerization interface of the Hsp90 N-terminal domain as originally proposed in ignorance of those data (Prodromou et al., 1997b). Yeast Hsp90 N-terminal domain crystallized in space group P43212 with a monomer in the asymmetric unit, so that two different dimers are generated by the 2-fold symmetry axes of the crystal lattice. When compared with the crystal structure of the dimeric AMP-PNP-bound N-terminal 40 kDa fragment of GyrB (not publicly available at the time of the original study) it became apparent that the alternative dimer, in which the ATP-binding pockets of the N-domains face each other, constitutes a more biologically probable dimer than that originally proposed (Figure 7A). In GyrB and MutL, a short loop segment folds over the nucleotide bound in the N-terminal domain, acting as a ‘lid’ on the nucleotide-binding pocket. The exposed surface of the lids in the dimers forms a major part of the N-domain dimer interface, and it is the stabilization of the interface by the ATP-bound conformation of the lids that is believed to promote ATP-dependent dimerization in these systems (Wigley et al., 1991; Ban et al., 1999). In yeast Hsp90 crystals, the more structured lid segments are ‘open’ and folded away from the nucleotide-binding pocket onto hydrophobic patches on the protein surface. The interface in this dimer is not as extensive as in the GyrB and MutL structures due to the open lid conformation. Nonetheless, the mechanistic similarities we have observed between Hsp90, GyrB and MutL suggest that ATP binding will also promote closure of the Hsp90 lid with formation of a more intimate dimer interface, although this has yet to be observed directly.

Fig. 7. Hsp90 N-terminal dimer interface and lid closure. (A) Comparison of GyrB 40 kDa N-terminal fragment dimer (top) with alternative dimers in the yeast Hsp90 N-domain crystal lattice (bottom). The relative orientation of the leftmost pair (red–yellow) of Hsp90 N-domains more closely resembles that of the N-domains in GyrB than does the rightmost pair (yellow–green), originally suggested. The conformations of the ‘lid’ segments and the N-terminal strands differ between GyrB and Hsp90. AMP-PNP bound to GyrB and ADP bound to Hsp90 are shown as blue CPK models. (B) Comparison of the open conformation of the ‘lid’ segment observed in the crystal structure of yeast Hsp90 N-domain (left) with the modelled structure of the closed conformation (right). Ala107, which is exposed in the open conformation, comes close to a hydrophilic patch provided by residues Ser39, Asp40 and Asp43 in the closed conformation. Its mutation to asparagine, which is expected to stabilize this conformation, produces a dramatic increase in ATPase activity (see text). The positions of Thr22 and Thr101 are also indicated. (C) Polyacrylamide gel showing cross-linking as a function of incubation time for the C-terminal truncation mutant NΔ0C551 (wt), the A107N-NΔ0C551 double mutant (107) and the T101I-NΔ0C551 double mutant (101) cross-linked with a 15-fold molar excess of DMS, in the presence of AMP-PNP.
ATP-dependent lid-closure promoting dimerization explains the behaviour of the T22I and T101I mutants. In the re-interpreted Hsp90 N-domain dimer, Thr22 residues are exposed on the surface forming the dimer interface and the side chains would become buried in interactions with the opposite monomer on dimer formation. Mutation to the more hydrophobic isoleucine in T22I would render the interacting surface ‘stickier’, favouring its burial in the dimer interface and stabilizing N-domain dimerization. If, as in GyrB, N-terminal dimerization is necessary for ATP hydrolysis, then a mutation favouring the dimeric state would promote ATP hydrolysis and produce the hyperactive ATPase observed in the T22I mutant. Thr101 lies within the lid segment in Hsp90 that would close on ATP binding. Its mutation to isoleucine has no significant effect on AMP-PNP binding, but markedly decreases ATPase activity. In the open lid conformation observed in Hsp90 structures, the side chain of Thr101 packs into a hydrophobic hole formed by the side chains of Ile12 and Leu15, and the Cα and Cβ of Glu11. Lid closure necessitates disruption of this interaction and exposes the side chain of Thr101 on the top surface of the lid. Mutation to isoleucine would stabilize the hydrophobic interaction in the open lid conformation and destabilize the exposure of the side chain in the closed lid. If, as in GyrB, lid closure is necessary for dimerization and subsequent ATP hydrolysis, a mutation favouring the open lid conformation would disfavour dimerization and consequent ATP hydrolysis, producing the lower ATPase activity observed in the T101 mutation.
As no experimental structures have so far been reported for an Hsp90 N-terminal construct with the lid in a closed conformation, we have constructed a model based on the known structures of the open lid conformation of yeast Hsp90, and of the GyrB dimer, with lid closure achieved by hinging at Gly100 and Gly121 (Figure 7B). In the open lid structure, Ala107 near the tip of the lid is fully exposed and makes no contact with other parts of the structure. In the hypothetical closed lid model, Ala107 undergoes a substantial change of environment, coming close to a hydrophilic patch formed by Ser39, Asp40 and Asp43. Mutation of Ala107 to asparagine should have little effect on the stability or mobility of the lid, but should provide additional favourable interactions with the polar side chains of Ser39, Asp40 and Asp43 in the closed lid conformation.
To test the reasonableness of the model experimentally, we constructed an A107N mutation in yeast Hsp90 and expressed and purified the mutant protein (see Materials and methods). As expected, the A107N mutant protein produced no change in AMP-PNP affinity relative to wild type (Table II). However, the ATPase activity of this mutant was substantially activated over wild-type ATPase activity to an even greater degree than with the ts T22I mutant. To determine whether this enhanced ATPase activity of A107N was accompanied by any change in the conformational response to ATP binding, the A107N missense mutation was combined with a truncation mutation, NΔ0C551, which lacks the C-terminal region required for inherent dimerization. Compared with the equivalent truncation mutant with a wild-type N-terminal domain, this A107N-NΔ0C551 double mutant showed a substantially higher yield of cross-linked dimer in the presence of AMP-PNP (Figure 7C). In contrast, a double mutant in which truncation was combined with the ATPase-deactivating mutation T101I showed little cross-linked dimer formation under identical conditions. The increased ATPase activity of A107N, accompanied by its enhanced dimerization in the presence of nucleotide triphosphate, is absolutely consistent with improved interactions stabilizing lid closure, which in turn promotes the N-terminal domain association and closure of the ‘clamp’ required for efficient ATPase activity.
Table II. AMP-PNP affinity and ATPase activity of the A107N mutant.
| Construct | Kd AMP-PNP-Mg2+ (µM) | ATPase activity (mol/min/mol) |
|||
|---|---|---|---|---|---|
| 22°C | 30°C | 37°C | 45°C | ||
| Wild type | 33 | 0.06 | 0.23 | 0.75 | 2.16 |
| T22I | 110 | 0.35 | 1.48 | 3.41 | 6.42 |
| A107N | 37 | 0.33 | 1.34 | 3.81 | 9.22 |
Conclusion
We have shown here that Hsp90 is related to GyrB and MutL, sharing a common structure in the N-terminal domain, a strong C-terminal dimerization interface and an essentially identical ATPase-coupled clamp mechanism. However, their biological roles and the other components of the molecular systems in which they participate are markedly different. In GyrB the molecular clamp controls binding and release of a DNA duplex during the topoisomerase strand-passage reaction. The surfaces of the ‘jaws’ brought into opposition in the ‘closed’ ATP-bound state are lined by positively charged residues able to interact with the phosphate backbone of the DNA (Wigley et al., 1991; Kampranis et al., 1999). By analogy, the ‘jaws’ in the Hsp90 molecular clamp might provide surfaces for interaction with proteins, with their ability to bind client proteins altering with the changes in conformational state that accompany the ATPase cycle. The hydrophobicity of Hsp90 decreases on ATP binding (Sullivan et al., 1997), which could represent a decreased affinity for unfolded proteins in the ‘closed’ ATP-bound state of the clamp, comparable to the occlusion of hydrophobic binding surfaces that occurs in the ATP-bound state of the GroEL molecular chaperone (Xu et al., 1997). However, there is no direct evidence that Hsp90 interacts with unfolded proteins in vivo, nor that it is involved in de novo folding or refolding following heat shock (Nathan et al., 1997), unlike GroEL. Rather, in its authenticated in vivo functions, Hsp90 appears to bind and stabilize client proteins in substantially folded states that are maintained in a receptive conformation during the ATP-bound stage of the cycle for interaction with other proteins and/or ligands (Meyer and Bukau, 1999). The rate of ATP hydrolysis by Hsp90 will define the lifetime of these receptive Hsp90-based complexes (Smith et al., 1995; Prodromou et al., 1999) and must be finely tuned, as under- and over-activity (as in the T22I ts mutant) both impair in vivo function. It remains to be determined whether the closure of the ‘clamp’ achieved by ATP-dependent dimerization of the N-terminal domains actually entraps the client protein, or rather acts as a ‘tense’ state stabilizing a transient receptive conformation in the client, which is then released as the Hsp90 ‘relaxes’ and the N-termini dissociate, following ATP hydrolysis.
Materials and methods
Mutagenesis and plasmid construction
Plasmid p82-2b (Cheng et al., 1992) was used as the PCR template for construction of C-terminal truncation mutants. Deletion mutants were subsequently cloned between the NheI and HindIII, BamHI or EcoRI sites of pRSETA for expression of His6-tagged proteins. Wild type (pRSETA-p90), all linker and C-terminal truncation mutants were PCR amplified using the same 5′ N-terminal primer (N1), CGGACTGGATCCATATGGCTAGCGAAACTTTTGAATTTCAAGCTG. The 3′ primers were CGGAGGAAGCTTGCATGCTTAATCTACCTCTTCCATTTCGG (C1) for pRSETA-p90, CGGACGGATCCTTATGGAATTGGAACTTCCTTTTC for NC220, CGGACTGAATTCTTACTCGAGGGACTTGGTAGAGTTGTAAC for NC450 and GGAGGACATAAGCTTTCATCTCAAGGCTTGAGCCTTCATG for NC599. NΔ0C530 was PCR amplified with N1 and the 3′ primer CGGAGCCTGCAGTGGCTTCTTTTCCTCTTCTT and ligated to a C-terminal construct PCR amplified using the 5′ primer CGGAGG CTGCAGCATATGGCTAGCGTTAAAGAAGAAGTTCAAGAGATA (C2) and the 3′ primer CGGACAAGCTTACAATTCGAAATC TTTAGTAATGTC. NΔ0C530 has the amino acid linker sequence LysThrLysLys (residues 256–259) replaced by LeuGlnHisMetAlaSer, and the native C-terminal sequence begins again at residue Val260 and ends at residue Leu530. NΔ39C551 was PCR amplified using N1 and the 3′ primer CGGAGGCTGCAGTGGAATTGGAACTTCCTTTTC to produce N220LeuGln, which was ligated to a C-terminal PCR construct amplified using C2 and a 3′ primer CGGACAAGCTTAGGTCAATGGTTCATATTCTTTGA. NΔ39C551 has 39 amino acid residues deleted from the linker (221–259) and replaced by LeuGlnHisMetAlaSer. The native C-terminal sequence begins again at Val260 and ends at Thr551.
Single amino acid changes were generated in pRSETA-p90 using the QuickChange mutagenesis system (Stratagene). The following mutations were introduced: E7C, Q9C, E11C, T22I, A41V, T101I, A107N, G170D and A587T (GAA to TGT, CAA to TGT, GAA to TGT, ACC to ATC, GCG to GTG, ACC to ATC, GCT to AAC, GGT to GAT and GCT to ACT, respectively). Mutations were confirmed by dye terminator cycle sequencing (ABI).
Expression and purification
Expression and purification of all His6-tagged constructs were as described in Panaretou et al. (1998). Buffers for the purification of the Cys mutants contained 1 mM dithiothreitol (DTT) (ion-exchange and gel-filtration buffers) and 1 mM β-mercaptoethanol (Talon buffers).
Determination of Tm by circular dichroism
Melting temperatures were determined by measuring loss of ellipticity in circular dichroism (CD) signals attributable to polypeptide secondary structure. Measurements were made in 0.1 cm path length quartz cells using an Aviv 202SF spectropolarimeter. Protein concentrations were 3.7 µM, based on an extinction coefficient of 54 100 M–1cm–1, in 25 mM Tris buffer at pH 7.5. Proteins were heated from 25 to 95°C in 2°C steps, with 1 min equilibration at each temperature. The change in ellipticity at 222 nm was recorded using a 2 s averaging time. Melting profiles were fitted to a sigmoidal (Boltzman) curve and normalized to percentage change in ellipticity. The reported Tms are the midpoints of these fits.
Determination of Kd values by CD
Disassociation constants (Kd) for binding of AMP-PNP to Hsp90 constructs were determined by measuring the difference CD signal at 259 nm as a function of ligand concentration and fitting to a single-site binding model by non-linear regression, as previously described (Freeman et al., 1998; Prodromou et al., 1999). For each titration, the difference CD was calculated by subtracting the spectrum of the unbound ligand at n molar ratio from that of the protein + ligand mixture at 1 to n molar ratio (dilution corrected). CD spectra were recorded on a nitrogen-flushed JASCO J720 spectropolarimeter, with multiple scanning for maximum data precision. CD titrations were carried out in 0.1 cm cells, with protein concentrations in the range 0.145–0.158 mM.
ATPase activities
ATPase activities were measured spectrophotometrically using a regenerating enzyme-coupled assay as previously described (Panaretou et al., 1998; Prodromou et al., 1999; Roe et al., 1999). As Hsp90 notoriously co-purifies with amounts of non-specific ATPase activity, the specific Hsp90 ATPase was determined by its sensitivity to inhibition by the Hsp90-specific inhibitors geldanamycin or radicicol, which bind to the nucleotide-binding site and are potent competitive inhibitors of the Hsp90 ATPase (Roe et al., 1999). All activities are averages of three or more separate measurements, typically with Hsp90 at 2 µM. Mutants with low activity were assayed at concentrations up to 20 µM.
Cross-linking
Proteins for cross-linking were diluted to a final concentration of 0.25–0.5 mg/ml in 100 mM HEPES pH 8.5, 150 mM KCl and 5 mM MgCl2. Either no additions, 10 mM ADP-Mg2+, 10 mM ATP-Mg2+, 10 mM AMP-PNP-Mg2+, 1.5 µM radicicol or 1.5 µM geldanamycin was added to the diluted proteins. In experiments shown in Figures 2, 3 and 4, DMS (15-fold molar excess over the protein’s primary amines) was added and allowed to cross-link for 2 h following a 2 h incubation at room temperature. In Figure 6, cross-linker was added at varying molar ratio and reacted for 2 h. Very little enhancement in cross-linking was observed between molar ratios of 7.5 and 30, confirming that the reactions were effectively saturated at a ratio of 7.5. For Figure 7, AMP-PNP (10 mM) and DMS (15-fold molar excess over primary amines) were added simultaneously and incubated for the times shown. All reactions were stopped by the addition of 25 mM Tris pH 6.8 and SDS loading buffer and analysed on 7.5 or 10% SDS–PAGE gels.
Fluorescent labelling
Proteins for fluorescent labelling were desalted on Sephadex G50 (Nick Spin Columns; Pharmacia) equilibrated in 100 mM HEPES pH 8 containing 150 mM KCl. Samples were diluted to 10 mg/ml and the non-thiol reducing agent tris(2-carboxyethyl)-phosphine hydrochloride (TCEP) added to a final concentration of 10 mM. Following incubation at room temperature for 30 min, N-(1-pyrene)-maleimide (in 100% dimethylformamide) was added to a final concentration of 10 mM and incubated for a further 60 min. Unreacted label was removed by desalting twice with Nick Spin columns equilibrated in 10 mM Tris pH 7.4 containing 1 mM EDTA. Fluorescence measurements were carried out at a final labelled protein concentration of 4.8 µM in 100 mM Tris pH 7.4 containing 1 mM EDTA, 10 mM MgCl2, 150 mM KCl and either 10 mM ADP or AMP-PNP. Labelled proteins were excited at 341 nm and their emission spectra analysed over the range of 220–600 nm on a Shimadzu RF-5301PC spectrofluorophotometer.
Acknowledgments
Acknowledgements
We are grateful to Keith Willison and David Lilley for useful discussion and technical advice. We are grateful to Dale Wigley for access to the coordinates of the GyrB dimer. This work was supported by a project grant from the Wellcome Trust to P.W.P. and L.H.P.
References
- Ban C., Junop,M. and Yang,W. (1999) Transformation of MutL by ATP binding and hydrolysis: a switch in DNA mismatch repair. Cell, 97, 85–97. [DOI] [PubMed] [Google Scholar]
- Bilwes A.M., Alex,L.A., Crane,B.R. and Simon,M.I. (1999) Structure of CheA, a signal-transducing histidine kinase. Cell, 96, 131–141. [DOI] [PubMed] [Google Scholar]
- Bukau B. and Horwich,A.L. (1998) The Hsp70 and Hsp60 chaperone machines. Cell, 92, 351–366. [DOI] [PubMed] [Google Scholar]
- Carrello A., Ingley,E., Minchin,R.F., Tsai,S. and Ratajczak,T. (1999) The common tetratricopeptide repeat acceptor site for steroid receptor associated immunophilins and hop is located in the dimerization domain of Hsp90. J. Biol. Chem., 274, 2682–2689. [DOI] [PubMed] [Google Scholar]
- Chang H.C.J., Nathan,D.F. and Lindquist,S. (1997) In vivo analysis of the Hsp90 cochaperone Sti1 (p60). Mol. Cell. Biol., 17, 318–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.Y., Sullivan,W.P., Toft,D.O. and Smith,D.F. (1998) Differential interactions of p23 and the TPR-containing proteins Hop, Cyp40, FKBP52 and FKBP51 with Hsp90 mutants. Cell Stress Chaperones, 3, 118–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L., Hirst,K. and Piper,P.W. (1992) Authentic temperature regulation of a heat-shock gene in yeast on a high copy number vector. Influence of overexpression of Hsp90 protein on high-temperature growth and thermotolerance. Biochim. Biophys. Acta, 1132, 26–34. [DOI] [PubMed] [Google Scholar]
- Fang Y., Fliss,A.E., Rao,J. and Caplan,A.J. (1998) SBA1 encodes a yeast Hsp90 cochaperone that is homologous to vertebrate p23 proteins. Mol. Cell. Biol., 18, 3727–3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman D.J., Pattenden,G., Drake,A.F. and Siligardi,G. (1998) Marine metabolites and metal ion chelation. Circular dichroism studies of metal binding to Lissoclinum cyclopeptides. J. Chem. Soc. Perkin Trans. 2, 1, 129–136. [Google Scholar]
- Grenert J.P. et al. (1997) The amino-terminal domain of heat shock protein 90 (hsp90) that binds geldanamycin is an ATP/ADP switch domain that regulates hsp90 conformation. J. Biol. Chem., 272, 23843–23850. [DOI] [PubMed] [Google Scholar]
- Grenert J.P., Johnson,B.D. and Toft,D.O. (1999) The importance of ATP binding and hydrolysis by Hsp90 in formation and function of protein heterocomplexes. J. Biol. Chem., 274, 17525–17533. [DOI] [PubMed] [Google Scholar]
- Johnson J.L. and Toft,D.O. (1994) A novel chaperone complex for steroid-receptors involving heat-shock proteins, immunophilins and p23. J. Biol. Chem., 269, 24989–24993. [PubMed] [Google Scholar]
- Johnson J.L. and Toft,D.O. (1995) Binding of p23 and Hsp90 during assembly with the progesterone-receptor. Mol. Endocrinol., 9, 670–678. [DOI] [PubMed] [Google Scholar]
- Kampranis S.C., Bates,A.D. and Maxwell,A. (1999) A model for the mechanism of strand passage by DNA gyrase. Proc. Natl Acad. Sci. USA, 96, 8414–8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyasu S., Nishida,E., Kadowaki,T., Matsuzaki,F., Iida,K., Harada,F., Kasuga,M., Sakai,H. and Yahara,I. (1986) Two mamalian heat shock proteins, HSP90 and HSP100 are actin binding proteins. Proc. Natl Acad. Sci. USA, 83, 8054–8058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruya M., Sameshima,M., Nemoto,T. and Yahara,I. (1999) Monomer arrangement in HSP90 dimer as determined by decoration with N and C-terminal region specific antibodies. J. Mol. Biol., 285, 903–907. [DOI] [PubMed] [Google Scholar]
- Meyer M.P. and Bukau,B. (1999) Molecular chaperones: the busy life of Hsp90. Curr. Biol., 9, R322–R325. [DOI] [PubMed] [Google Scholar]
- Nathan D.F. and Lindquist,S. (1995) Mutational analysis of Hsp90 function: interactions with a steroid receptor and a protein kinase. Mol. Cell. Biol., 15, 3917–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan D.F., Vos,M.H. and Lindquist,S. (1997) In vivo functions of the Saccharomyces cerevisiae Hsp90 chaperone. Proc. Natl Acad. Sci. USA, 94, 12949–12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermann W.M.J., Sondermann,H., Russo,A.A., Pavletich,N.P. and Hartl,F.U. (1998) In vivo function of Hsp90 is dependent on ATP binding and ATP hydrolysis. J. Cell Biol., 143, 901–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panaretou B., Prodromou,C., Roe,S.M., O’Brien,R., Ladbury,J.E., Piper,P.W. and Pearl,L.H. (1998) ATP binding and hydrolysis are essential to the function of the Hsp90 molecular chaperone in vivo. EMBO J., 17, 4829–4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prodromou C., Roe,S.M., O’Brien,R., Ladbury,J.E., Piper,P.W. and Pearl,L.H. (1997a) Identification and structural characterisation of the ATP/ADP binding site in the Hsp90 molecular chaperone. Cell, 90, 65–75. [DOI] [PubMed] [Google Scholar]
- Prodromou C., Roe,S.M., Piper,P.W. and Pearl,L.H. (1997b) A molecular clamp in the crystal structure of the N-terminal domain of the yeast Hsp90 chaperone. Nature Struct. Biol., 4, 477–482. [DOI] [PubMed] [Google Scholar]
- Prodromou C., Siligardi,G., O’Brien,R., Woolfson,D.N., Regan,L., Panaretou,B., Ladbury,J.E., Piper,P.W. and Pearl,L.H. (1999) Regulation of Hsp90 ATPase activity by tetratricopeptide repeat (TPR)-domain co-chaperones. EMBO J., 18, 754–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe S.M., Prodromou,C., O’Brien,R., Ladbury,J.E., Piper,P.W. and Pearl,L.H. (1999) The structural basis for inhibition of the Hsp90 molecular chaperone, by the anti-tumour antibiotics radicicol and geldanamycin. J. Med. Chem., 42, 260–266. [DOI] [PubMed] [Google Scholar]
- Scheibel T., Weikl,T. and Buchner,J. (1998) Two chaperone sites in Hsp90 differing in substrate specificity and ATP dependence. Proc. Natl Acad. Sci. USA, 95, 1495–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheibel T., Siegmund,H.I., Jaenicke,R., Ganz,P., Lilie,H. and Buchner,J. (1999) The charged region of Hsp90 modulates the function of the N-terminal domain. Proc. Natl Acad. Sci. USA, 96, 1297–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen A.C. and Chakrabarti,B. (1990) Proximity of sulfhydryl groups in lens proteins. Excimer fluorescence of pyrene-labeled crystallins. J. Biol. Chem., 265, 14277–14284. [PubMed] [Google Scholar]
- Smith D.F. (1993) Dynamics of heat-shock protein 90–progesterone receptor binding and the disactivation loop model for steroid receptor complexes. Mol. Endocrinol., 7, 1418–1429. [DOI] [PubMed] [Google Scholar]
- Smith D.F., Whitesell,L., Nair,S.C., Chen,S., Prapapnich,V. and Rimerman,R.A. (1995) Progesterone receptor structure and function altered by geldanamycin, an Hsp90 binding agent. Mol. Cell. Biol., 15, 6804–6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stebbins C.E., Russo,A.A., Schneider,C., Rosen,N., Hartl,F.U. and Pavletich,N.P. (1997) Crystal structure of an Hsp90–geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell, 89, 239–250. [DOI] [PubMed] [Google Scholar]
- Sullivan W., Stensgard,B., Caucutt,G., Bartha,B., McMahon,N., Alnemri,E.S., Litwack,G. and Toft,D.O. (1997) Nucleotides and two functional states of Hsp90. J. Biol. Chem., 272, 8007–8012. [DOI] [PubMed] [Google Scholar]
- Tanaka T. et al. (1998) NMR structure of the histidine kinase domain of the E.coli osmosensor EnvZ. Nature, 396, 88–92. [DOI] [PubMed] [Google Scholar]
- Walker J.E., Saraste,M., Runswick,M.J. and Gay,N.J. (1982) Distantly related sequences in the α-subunits and β-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J., 1, 945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wearsch P.A. and Nicchitta,C.V. (1996) Endoplasmic reticulum chaperone GRP94 subunit assembly is regulated through a defined oligomerization domain. Biochemistry, 35, 16760–16769. [DOI] [PubMed] [Google Scholar]
- Wigley D.B., Davies,G.J., Dodson,E.J., Maxwell,A. and Dodson,G. (1991) Crystal structure of an N-terminal fragment of the DNA gyrase B protein. Nature, 351, 624–629. [DOI] [PubMed] [Google Scholar]
- Xu Z.H., Horwich,A.L. and Sigler,P.B. (1997) The crystal structure of the asymmetric GroEL–GroES–(ADP)(7) chaperonin complex. Nature, 388, 741–750. [DOI] [PubMed] [Google Scholar]
- Young J.C., Schneider,C. and Hartl,F.U. (1997) In vitro evidence that hsp90 contains two independent chaperone sites. FEBS Lett., 418, 139–143. [DOI] [PubMed] [Google Scholar]
- Young J.C., Obermann,W.M.J. and Hartl,F.U. (1998) Specific binding of tetratricopeptide repeat proteins to the C-terminal 12-kDa domain of hsp90. J. Biol. Chem., 273, 18007–18010. [DOI] [PubMed] [Google Scholar]