

Polymerization of MIP-1 chemokine (CCL3 and CCL4) and clearance of MIP-1 by insulin-degrading enzyme
The two closely related MIP chemokines, CCL3 and CCL4, assemble into high-order polymers that are protected from degradation. MIP-1 polymers can dissociate into active MIP-1 monomers that in turn are selectively degraded by the insulin-degrading enzyme.
Keywords: chemokine, chemotactic gradient, insulin-degrading enzyme, MIP-1 polymerization, X-ray crystallography
Abstract
Macrophage inflammatory protein-1 (MIP-1), MIP-1α (CCL3) and MIP-1β (CCL4) are chemokines crucial for immune responses towards infection and inflammation. Both MIP-1α and MIP-1β form high-molecular-weight aggregates. Our crystal structures reveal that MIP-1 aggregation is a polymerization process and human MIP-1α and MIP-1β form rod-shaped, double-helical polymers. Biophysical analyses and mathematical modelling show that MIP-1 reversibly forms a polydisperse distribution of rod-shaped polymers in solution. Polymerization buries receptor-binding sites of MIP-1α, thus depolymerization mutations enhance MIP-1α to arrest monocytes onto activated human endothelium. However, same depolymerization mutations render MIP-1α ineffective in mouse peritoneal cell recruitment. Mathematical modelling reveals that, for a long-range chemotaxis of MIP-1, polymerization could protect MIP-1 from proteases that selectively degrade monomeric MIP-1. Insulin-degrading enzyme (IDE) is identified as such a protease and decreased expression of IDE leads to elevated MIP-1 levels in microglial cells. Our structural and proteomic studies offer a molecular basis for selective degradation of MIP-1. The regulated MIP-1 polymerization and selective inactivation of MIP-1 monomers by IDE could aid in controlling the MIP-1 chemotactic gradient for immune surveillance.
Introduction
Chemokines (chemotactic cytokine) are 8–14 kDa peptides required for the recruitment of immune cells to the site of infection or inflammation and the development of secondary lymphoid organs (Allen et al, 2007). In humans, ∼50 chemokines and 20 G-protein-coupled chemokine receptors form a complex signalling network (Wells et al, 2006). Macrophage inflammatory protein-1 (MIP-1), MIP-1α and MIP-1β (also known as CC motif chemokine Iigand 3 (CCL3) and CCL4, respectively) were first found based on their proinflammatory properties (Wolpe et al, 1988). MIP-1 proteins are inducible in most immune cells in response to various proinflammatory stimuli and are potent chemoattractants to cells vital for innate and adaptive immunity (Menten et al, 2002; Maurer and von Stebut, 2004). MIP-1 can activate several chemokine receptors (CCR1 and CCR5), which initiate diverse cellular responses that regulate both acute and chronic inflammation. Consequently, the deregulation of MIP-1 proteins is associated with altered susceptibility to infection and human diseases (Kobayashi et al, 2002; Menten et al, 2002; Maurer and von Stebut, 2004). In addition, MIP-1α knockout mice show altered inflammation reactions to the infection of various viral, bacterial and parasitic pathogens (Cook et al, 1995; Sato et al, 1999; Lindell et al, 2001). On the basis of the protective activity of haematopoietic stem cells and the inhibition of HIV-1 infections by their binding with the HIV-1 coreceptor, CCR5, MIP-1 proteins were also explored as possible therapeutics for cancer and AIDS (Graham et al, 1990; Dunlop et al, 1992; Cocchi et al, 1995; Horuk, 2009).
The monomer of CC chemokines is presumed to be the form that binds and activates their cognate receptors. However, several CC chemokines can form oligomers, and mutations that alter assembly lead to selective reduction of biological activities. For example, the ability of MIP-1β, CCL5 (RANTES) and CCL2 (MCP-1) to oligomerize is crucial for their in vivo cellular recruitment into mouse peritoneal cavity (Proudfoot et al, 2003). The oligomerization of CCL5 is crucial for CCR1-mediated leukocyte arrest on inflamed endothelium (Baltus et al, 2003). Interestingly, oligomerization-defective mutations of CCL2 can suppress the normal and pathological responses to chemokines (Handel et al, 2008).
Of 28 CC chemokines, only MIP-1α and MIP-1β are highly acidic and can form high-molecular-weight (MW) aggregates (600 kDa or higher) (Graham et al, 1994). Such MIP-1 aggregates can form under physiologically relevant concentrations given the high levels of MIP-1 proteins that immune cells can secrete upon stimulation: up to 1 pg MIP-1β per hour per dendritic cell (Sallusto et al, 1999). Consistent with this notion, MIP-1 and RANTES are secreted as high-MW aggregates from HIV-1-specific cytotoxic T lymphocyte (Wagner et al, 1998). The aggregation of MIP-1 proteins is dynamic and reversible, allowing the intricate regulation of the level of MIP-1 monomer (Graham et al, 1994). NMR structures of MIP-1 dimer reveal a common fold for CC chemokines, in which two anti-parallel β-strands formed by the N-terminal loop of MIP-1 constitute the primary contact of the MIP-1 dimer (Lodi et al, 1994). However, these structures neither offer a molecular basis for reversible MIP-1 aggregation nor explain how point mutations located outside the dimer interface (such as D27A and E67A in MIP-1α) can effectively reduce MIP-1 aggregation (Graham et al, 1994; Czaplewski et al, 1999).
As in the case of other chemokines, MIP-1-mediated chemotaxis is highly regulated. Glycosaminoglycans (GAGs), a family of sulphated or acetylated polysaccharides composed of alternating glucosamine units that are the key components of proteoglycan, can fine-tune diverse mammalian physiological functions, including chemotaxis (Handel et al, 2005; Bishop et al, 2007). Proteoglycan-bound MIP-1β on endothelium is used to effectively activate and induce the adhesion of circulating leukocytes for their extravasation (Tanaka et al, 1993). Furthermore, the binding of chemokines to GAGs is postulated to facilitate the formation of chemokine gradients and the presentation of chemokines to leukocytes in tissues (Kuschert et al, 1998; Lortat-Jacob et al, 2002). Consequently, GAG binding defective MIP-1β mutant is inactive in vivo (Proudfoot et al, 2003). The activity of MIP-1 is also regulated by proteolysis (Wolf et al, 2008). Protease CD26/DPP IV can efficiently process MIP-1 proteins to more active forms, whereas cathepsin D can degrade and inactivate MIP-1 proteins (Proost et al, 2000; Wolf et al, 2003). The physiological relevance of such proteolytic modifications and their relationship with MIP-1 aggregation is currently unknown. Therefore, the protease(s) responsible for MIP-1 clearance and the mechanism of MIP-1 turnover in vivo remains to be elucidated.
In this report, we have determined polymer structures for MIP-1α, MIP-1β and an MIP-1α deaggregation mutant, D27A, to reveal the molecular basis for the rod-shaped, double-helical polymer formation of MIP-1. Using small angle X-ray scattering (SAXS) and size exclusion chromatography (SEC), we show that MIP-1 proteins form polydisperse, rod-shaped polymers in solution and that heparin binding can profoundly affect MIP-1 polymerization. Our structures reveal that polymerization buries the receptor-binding sites of MIP-1. Consistent with this notion, we show that depolymerization mutations enhance the ability of MIP-1α to arrest monocytes onto human endothelial monolayer under flow conditions. However, we also show that the same depolymerization mutations profoundly reduce the ability of MIP-1α to recruit cells into mouse peritoneum.
In search of the physiologically relevant protease that could render the depolymerized MIP-1 mutants less effective for their in vivo function, we found that insulin-degrading enzyme (IDE) can selectively inactivate only monomeric MIP-1, and reduced expression of IDE leads to elevated MIP-1 levels in cultured cells. IDE is a 110 kDa ubiquitously expressed, zinc metalloprotease that has a key role in the clearance of insulin and amyloid β (Aβ), peptides vital for the development of diabetes mellitus and Alzheimer's disease (AD), respectively (Farris et al, 2003; Leissring et al, 2003; Sladek et al, 2007; Malito et al, 2008). Our recent structural studies revealed that two roughly equally sized, 55 kDa domains of IDE (IDE-N and IDE-C) form an enclosed catalytic chamber (Shen et al, 2006). The unique properties of this catalytic chamber allow IDE to selectively bind substrates with a defined size, charge distribution, N-terminal position and tendency to form amyloid fibril (Shen et al, 2006; Malito et al, 2008; Manolopoulou et al, 2009; Guo et al, 2010). We also applied proteomics and structural analyses to decipher the molecular mechanism of MIP-1 inactivation by IDE.
Results
Polymer structures of MIP-1α and MIP-1β
To address the structural basis of MIP-1 aggregation, we purified and crystallized recombinant human MIP-1α and MIP-1β from E. coli in the absence of harsh conditions (such as low pH, high salt and organic solvent, which were used in the previous NMR analyses) (Table I). The chemotaxis activities of these proteins were comparable with commercial peptides (Supplementary Figure S1). MIP-1 proteins were readily crystallized in <3 days. The MIP-1α and MIP-1β structures were refined to 2.65 and 2.6 Å, respectively (Table I, Supplementary Figure S2).
Table 1. Crystallographic statistics of MIP-1 and MIP-1α-bound IDE.
| MIP-1α | MIP-1β | MIP-1α (D27A) | IDE/MIP-1α | |
|---|---|---|---|---|
| Crystallization conditiona | Na/KPO4 (1.6 M, ph 7) | 8% PEG8000 | 0.2 M NH4Ac | 11.5% PEGMME 5000 |
| 0.1 M Tris pH 7 | 0.1 M HEPES (pH 7.8) | 0.1 M HEPES pH 7 | ||
| 0.2 M MgCl2 | 26% PEG3350 | 10% Tacsimate | ||
| 10% Dioxane | ||||
| Data collection | ||||
| Beam line | APS-19ID | APS-19ID | APS-19ID | APS 14-BMC |
| Wavelength (Å) | 0.9792 | 0.9792 | 0.9792 | 0.9762 |
| Space group | P6222 | C2221 | P212121 | P65 |
| Cell dimension (Å) | ||||
| a | 182 | 60 | 57 | 263 |
| b | 182 | 88 | 114 | 263 |
| c | 77 | 187 | 174 | 91 |
| α | 90 | 90 | 90 | 90 |
| β | 90 | 90 | 90 | 90 |
| γ | 120 | 90 | 90 | 120 |
| Resolution (Å) | 50–2.65 | 50–2.6 | 50–2.18 | 50–3.0 |
| Rsym (%)b | 8.3 (46.0)c | 12.1 (49.9) | 7.6 (46.4) | 14.2 (53.0) |
| I/sigma | 40.7 (5.2)c | 17.6 (3.8) | 31.6 (4.8) | 16.9 (3.0) |
| Redundancyd | 10.6 (11.0)c | 7.2 (7.2) | 4.7 (4.9) | 5.0 (5.0) |
| Completeness (%) | 99.9 (100)c | 100 (100) | 99.8 (100.0) | 99.6 (100.0) |
| Unique reflections | 22 235 | 15 529 | 61 457 | 67 744 |
| Refinement | ||||
| Rworke | 22.3 | 18.6 | 21.1 | 18.2 |
| Rfreef | 26.5 | 26.0 | 28.5 | 23.7 |
| No. of atoms | ||||
| Protein | 2605 | 2649 | 9142 | 15678 |
| Water | 13 | 145 | 448 | 133 |
| B-factors | ||||
| Protein | 52.4 | 33.3 | 39.3 | 50.5 |
| Substrate | 73.2 | |||
| Water | 51.8 | 29.6 | 39.1 | 45.5 |
| r.m.s.d. | ||||
| Bond lengths (Å) | 0.023 | 0.008 | 0.008 | 0.014 |
| Bond angles (deg) | 1.946 | 1.045 | 1.112 | 1.588 |
| Ramachandran plot (%) | ||||
| Favourable | 93.1 | 92.5 | 97.3 | 88.3 |
| Allowed | 6.9 | 7.2 | 2.7 | 11.7 |
| Generously allowed | 0 | 0.3 | 0 | 0 |
| Disallowed | 0 | 0 | 0 | 0 |
| PDE code | 2X69 | 2X6 l | 2X6 g | 3H44 |
| aMIP-1 proteins and IDE/MIP-1α are crystallized at 2.5 and 5 mg/ml, respectively. | ||||
| bRmerge=Σ (I−〈I〉)/Σ 〈I〉. | ||||
| cThe outer resolution shell. Values in parentheses indicate the highest resolution shell. | ||||
| dNobs/Nunique. | ||||
| eRwork=Σhkl∥Fobs∣−∣Fcalc∥/Σhkl∣Fobs∣. | ||||
| fRfree, calculated the same as for Rwork, but on 5% data excluded from the refinement calculation. | ||||
MIP-1α and MIP-1β share 67% sequence identity and our MIP-1 polymer structures reveal that MIP-1α and MIP-1β form nearly identical rod-shaped, double-helical polymers (RMSD=0.766 Å for MIP-1α and MIP-1β tetramers) (Figure 1). MIP-1β polymer is slightly longer than MIP-1α polymer (∼0.3 Å per MIP-1 dimer). These structures can be viewed as polymerized MIP-1 dimers, a basic unit revealed by the NMR structures of MIP-1 proteins (Lodi et al, 1994; Czaplewski et al, 1999). In polymeric MIP-1, each dimer rotates 36° to bind to the adjacent dimer such that a decamer completes an 180° turn (Figure 1B and C). Each decamer of MIP-1 dimers is ∼44 Å wide and 110 Å long (Figure 1C). Both MIP-1 proteins exhibit highly negatively charged convex surfaces; the concave surface of MIP-1α is mostly neutral, whereas that of MIP-1β is weakly positively charged (Figure 1D).
Figure 1.

Structure of MIP-1α and MIP-1β polymer. (A) Sequence alignment of MIP-1α and MIP-1β with different residues highlighted in blue. (B) Crystal structure of MIP-1α polymer. A 40mer is depicted to show the periodicity of two 360° turns, and 20 dimers within the 40mer are labelled. The C-terminal helix of each dimer pair is coloured in red and green, respectively. (C) Secondary structure and (D) surface charge comparison of the MIP-1α and MIP-1β decamer. The molecular surface is coloured from negative (red, −6kT) to positve (blue, −6kT) as calculated by APBS (38). (E) Structural comparison of MIP-1 X-ray structure (solid colour) from this study with NMR structures of MIP-1α and MIP-1β (transparent colour). Structure comparison of MIP-1 monomer is on the left and that of dimer is on the right. The PDB code for MIP-1α and MIP-1β NMR structures are 1B50 and 1HUN, respectively. (F) Charge complementarity between MIP-1 dimers. The molecular surface is coloured as calculated by APBS (<−6kT in red, 0kT in white, and >+6kT in blue). (G) Detailed interaction of MIP-1α at the dimer–dimer interface formed by the interaction of monomers diagonal to each other (top right panel) and between the side-by-side monomers (bottom right panel).
Within the MIP-1 polymer, the MIP-1 monomer displays the common structural fold of CC chemokines: a 10 amino acid long N-terminal loop followed by a short 310 helix, a β-sheet composed of three anti-parallel β-strands, and a C-terminal α-helix that packs against the β-sheet (Figure 1E, Supplementary Figure S3). The crystal structures of MIP-1 monomer overlap well with the available NMR structures of wild-type MIP-1β and MIP-1α D27A mutant (Figure 1E) (Lodi et al, 1994; Czaplewski et al, 1999). RMSD values range from 1.6 to 2.0 and 1.0 to 1.1 Å for MIP-1α and MIP-1β, respectively, whereas the corresponding RMSD values within the NMR structures range from 1.1 to 1.5 and 0.1 to 0.3 Å, respectively. Similar to the NMR structures, the N-terminal loop of two MIP-1 monomers within MIP-1 polymers also comes together to form an antiparallel β-sheet to form an MIP-1 dimer (Figure 1E). However, comparison of MIP-1 dimers in the NMR and crystal structures reveals a noticeable rigid body movement between MIP-1 monomers (Figure 1E). The centre of mass of MIP-1β monomer shifts 7 Å and undergoes a 27° rotation (relative to the Cα atom of threonine 9), whereas MIP-1α has a smaller translation (5 Å) and rotation (7°). This movement renders the dimer of MIP-1α and MIP-1β inside the MIP-1 polymers to be more compact.
Our MIP-1 polymer structures reveal for the first time the novel interaction between MIP-1 dimers that lead to polymerization. The dimer–dimer interface of MIP-1 proteins has ∼1100 Å2 buried surface (Supplementary Figure S4). As a monomer, MIP-1 proteins have a dipolar charge distribution (Figure 1F). By formation of the MIP-1 polymer, the positively charged patch of MIP-1 crucial for receptor binding is buried (Figure 1F). The salt bridges between D27/E67 and R46/R48 and the hydrogen bond between D6 and S33 are key contacts between MIP-1 dimers (Figure 1G, Supplementary Figure S4). Consistent with this, D27 and E67 are highly conserved among the chemokines that tend to form high-MW aggregates (MIP-1 and RANTES), and mutation at either of these sites to alanine destabilizes MIP-1α and MIP-1β aggregates (Supplementary Figure S5) (Czaplewski et al, 1999). Our structures show that F24, F29 and Y28, which are conserved among MIP-1α, MIP-1β and RANTES, also contribute to MIP-1 polymerization by hydrophobic interactions (Figure 1G, Supplementary Figure S5).
Structure of depolymerization mutant MIP-1α D27A
MIP-1α has been explored as a potential therapeutic agent (BB-10010) to protect haematopoietic progenitors (Hunter et al, 1995; Bernstein et al, 1997; Owen-Lynch et al, 1998; Hough et al, 2003). To avoid the complication of MIP-1α polymerization and increase its solubility, a mutation of aspartate 27 to alanine is introduced. The MIP-1 D27A mutation has the most dramatic effect in reducing the aggregation (Czaplewski et al, 1999). However, the mechanism by which the D27A mutation hinders MIP-1α polymerization was unclear. To understand this mechanism, we purified MIP-1α D27A and, as expected, it is active in an in vitro chemotaxis assay (Supplementary Figure S1). We subsequently solved the polymer structure of this mutant at 2.1 Å resolution.
Our MIP-1α D27A structure reveals two polymer states (Figure 2A). The first state is a MIP-1α D27A polymer identical to those formed by wild-type MIP-1α, revealing its full potential to form rod-shaped polymers. Interestingly, the second state is a rod-shaped helical octamer decorating the rod-shaped polymer (Figure 2A). These two states make minimal contact and thus are unlikely to be biologically significant. However, the structure of the MIP-1α D27A octamer is noticeably different from the MIP-1α D27A polymer (Figure 2A). Instead of the 36° rotation found in the MIP-1α D27A polymer, the adjacent dimer within the MIP-1α D27A octamer has only rotated ∼31°, leading to a total of 15° under-rotation of this octamer (Figure 2B and C). The octamer is less ordered, on the basis of the fact that its overall thermal B factor is significantly higher than that of the MIP-1α D27A polymer, and its terminal monomers have high individual B factors (Figure 2D). In addition, one of the four dimers within the MIP-1α D27A octamer has a noticeable conformational change in the C-terminal α-helix (Figure 2E). Together, our data suggest that the MIP-1α D27A octamer represents a less-stable intermediate state of MIP-1 polymer and offers the molecular basis for depolymerization caused by D27A mutation.
Figure 2.

Characterization and structure analysis of MIP-1α (D27A) polymer. (A) Overall structure of MIP-1α D27A polymer, which consists of the MIP-1α D27A polymer (coloured in red and green on their C-terminal α-helices) and MIP-1α D27A octamer. Comparison of MIP-1α D27A polymer and octamer either as tetramer (B) or octamer (C), revealing the difference in their degree of rotation. (D) Comparison of MIP-1α D27A polymer and octamer based on their thermal B factor. (E) Comparison of two dimers within the MIP-1α D27A octamer, revealing the conformational change in the C-terminal α-helices in one of the four dimers within the MIP-1α D27A octamer.
MIP-1 proteins form polydisperse, rod-shaped polymers in solution
We then characterized MIP-1 polymers in solution biophysically, particularly, with respect to their size and shape. The SEC data confirmed that both MIP-1α and MIP-1β behaved as high-MW polymers as previously reported (Figure 3A). The MW of MIP-1α and MIP-1β at 1 mg/ml are approximately 400–600 and 600–3000 kDa, respectively. This indicates that MIP-1 can form polymers that contain more than 50 monomers in solution (Supplementary Table S2). We then used SAXS to assess the shape of MIP-1 in solution. If MIP-1 polymers were rod shaped, we would expect to observe linearity of the cross-section, Guinier rod plot over a wide but intermediate range of scattering vectors, q. This is indeed the case (Figure 3B). This is further supported by the asymmetric shape of the distance distribution function (P(r)) with a linear slope on the right side (Supplementary Figure S6). Thus, MIP-1α and MIP-1β form rod-shaped polymers in the physiological buffer. The radii of the rod-shape particles (49 and 47 Å for MIP-1α and MIP-1β, respectively) are close to the width of the MIP-1 polymer structures (44 Å plus a layer of water). Furthermore, the values of the cross-section radius of gyration (Rc) for MIP-1α and MIP-1β are nearly identical. Thus, the SAXS data agree with the MIP-1 polymer structures. Consistent with our SEC data, the SAXS data also indicate that MIP-1β forms polymers with an averaged length greater than MIP-1α at a given concentration (Supplementary Table S2).
Figure 3.

Characterization of MIP-1α and MIP-1β polymers in solution. (A) SEC of 1 mg/ml MIP-1α, MIP-1β and MIP-1α D27A with or without 0.1 mg/ml heparin. SEC was performed using phosphate-buffered saline (PBS). (B) Experimental scattering curves for 1 mg/ml MIP-1α, MIP-1β and MIP-1α D27A with or without 0.1 mg/ml heparin in PBS. Insets: cross-section, Guinier rod plots. (C) Schematic representation of our model for the polymerization process. The detailed mathematical modelling is appended in the Supplementary Material. (D) Fitting of the experimental data (solid line) with the calculated scattering from either the best polymer distribution given by our model (dashed line) or the best single polymer (dotted line) with difference between the experimental and fitted theoretical curves shown on bottom. Inset: size distribution of the polymers length distribution. The fitting errors I0 × Itheoa(q)−Iexp(q)+Ib are represented in the deviation curve. Parameter values were chosen to minimize the fit function 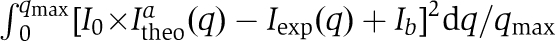 , as described in the Supplementary Material.
, as described in the Supplementary Material.
Our data also support the idea that the D27A mutation hinders MIP-1α polymerization. The SEC data showed that the D27 mutation reduces MIP-1α aggregation from 400–600 kDa to 50–150 kDa (Figure 3A). Consistent with this, SAXS data indicated that the MIP-1α D27A mutant yields shorter polymers than the wild-type MIP-1α (Figure 3B, Supplementary Table S2). In agreement with our MIP-1α D27A structure, the scattering data indicated that this mutant also forms rod-shaped polymers in solution (Figure 3B, Supplementary Figure S6).
Heparin, a highly sulphated GAG, is frequently used to examine the effect of GAG on the aggregation of chemokines. Our SEC data showed that the addition of heparin increased the elution volume of the MIP-1 peak and broadened the elution profile (Figure 3A). Thus, heparin can trigger a reduction in MIP-1α and MIP-1β polymer size and increase their polydispersity. Our SAXS analysis also showed that heparin significantly altered the scattering pattern of MIP-1α in such a way that MIP-1α maintained its rod shape but reduced its size from a 40–50mer to a 20–30mer (Figure 3B, Supplementary Table S2). Together, our data indicate that heparin binding leads to MIP-1 depolymerization. Our SEC data showed that heparin increased the averaged size of MIP-1α D27A larger, which may be due to the molecular mass of heparin and/or the altered polymerization of the MIP-1α mutant (Figure 3A). However, the addition of heparin did not alter the SAXS pattern for MIP-1α D27A, suggesting that heparin does not significantly affect the polymerization of MIP-1α D27A (Figure 3B, Supplementary Table S2).
To better understand the polymer length distribution, we used a simple equilibrium model for the polymerization process (see Supplementary Mathematical Modeling for details; van Dongen and Ernst, 1984). We propose that the basic unit in the model is the MIP-1 dimer. This is based on the fact that MIP-1α and MIP-1β stay dimeric under relatively harsh conditions to reduce the aggregation of MIP-1 in previous NMR studies (Lodi et al, 1994; Czaplewski et al, 1999). Furthermore, the binding energy calculation using our MIP-1 polymer structures and PISA web-server shows that the dimerization energy of MIP-1 monomers is much higher than the polymerization energy of MIP-1 dimers. In our polymerization model, dimers come together to form polymers, and two polymers of arbitrary sizes can coalesce to form a longer one (Figure 3C). This process is reversible, and therefore a long polymer can break into two smaller polymers. Based on this, we derived analytical expressions for the concentrations of polymers of all sizes as a function of only two parameters: the binding affinity between MIP-1 dimers (which we assume to be independent of the dimer position within the polymer) and MIP-1 concentration (Supplementary Mathematical Modeling). This model implies that the average MIP-1 polymer size is concentration dependent and polymers of different sizes coexist. Consistent with these hypotheses, our SEC data show that the average MIP-1 polymer size is concentration dependent and the elution profile of MIP-1α and MIP-1β is asymmetric and skewed towards low-MW fractions (Figure 3A, Supplementary Figure S7).
To further examine the polydispersity of MIP-1 proteins in solution, we then fit our experimental scattering data with the theoretical scattering intensity expected for either a monodisperse or polydisperse polymer solution. We found that polydisperse polymer populations expected from our reversible polymerization model fit our experimental data for MIP-1α D27A significantly better than any unique polymer size (Figure 3D). In addition, our polymer model offered a fit of comparable quality with that obtained for a single polymer size for the SAXS data of wild-type MIP-1α and MIP-1β (Supplementary Figure S8). Thus, our data together support the formation of reversible rod-shaped MIP-1 polymers in solution.
Effect of depolymerization mutations on the biological functions of MIP-1α
Our structures reveal that polymerization would bury the CCR binding site of MIP-1. Thus, MIP-1 polymers should have a reduced potency in MIP-1-mediated responses and the depolymerization mutations could render MIP-1 more effective. To test this hypothesis, we examined the ability of MIP-1 to arrest human peripheral blood mononuclear cells (PBMCs) onto the human umbilical vein endothelial cell (HUVEC) monolayers that were activated by tumour necrosis factor-α (TNF-α) under flow conditions. We show that while the moderate ability of wild-type MIP-1α (WT) to recruit PBMC is almost indistinguishable from the buffer control, the treatment with MIP-1α D27A induces a three-fold increase in PBMC arrest onto the activated HUVEC monolayers (Figure 4A).
Figure 4.

Effects of MIP-1 depolymerization mutations on biological functions of MIP-1. (A) Arrest of PBMCs onto TNFα-activated HUVECs under flow conditions after preincubation without or with MIP-1α WT or D27A or P8A (1 μg/ml). The P-values for MIP-1α WT versus D27A or P8A are <0.02. (B) Theoretical model for the MIP-1 monomer profile away from its secretion site in the presence of diffusion and degradation by IDE. Three curves are plotted, corresponding to three different set of chemical reactions: degradation only (solid line; M (monomer only)); degradation and dimerization (dashed line; M+D); degradation, dimerization and polymerization of the dimer (dotted line; M+D+P). The arrow marks the cutoff point, where the concentration drop across the cell is larger than 1% on the M+D+P gradient so that effective chemotaxis occurs far away from the source of MIP-1 (where it is steep), but not in its vicinity (where it plateaus). The parameters of the model are chosen as follows: diffusion constant: 1 μm2/s; degradation rate: 10−3/s; MIP-1 secretion rate at the secretion site: 2 × 10−11 mol/m2/s; dimerization rate: 106/M/s; dimer disassembly rate: 10−2/s; dimer–dimer binding rate: 105/M/s; dimer–dimer unbinding rate: 10−2/s. The details in this mathematical modelling are appended in the Supplementary Material. (C) Peritoneal cell accumulations in response to wild-type MIP-1α, MIP-1α D27A or MIP-1α P8A. Male C57BL/6 mice were intraperitoneally injected with 10 μg of either MIP-1α WT or D27A or P8A mutant for 20 h. Cells accumulated in the peritoneal cavity were collected and counted. Values shown are means±s.e.m. of measurements using five mice per group. The P-values for the comparison of MIP-1α WT with PBS and MIP-1α D27A mutant are <0.0001 and that with MIP-1α WT and P8A mutant is 0.0012.
To further assess the function of MIP-1α polymerization, we sought to analyse an additional depolymerization mutant. The mutation of proline 8 to alanine restricts MIP-1β to its monomeric form and causes MIP-1β to be ineffective in peritoneal cell recruitment (Laurence et al, 2000; Proudfoot et al, 2003). On the basis of our MIP-1 structures, proline 8 is not involved in the dimerization or polymerization directly, but its impact on the structure could affect assembly indirectly. We thus predicted and confirmed that the P8A mutation decreases MIP-1α polymerization relative to wild-type MIP-1α at various protein concentrations (Supplementary Figure S7). Accordingly, we show that the treatment of MIP-1α P8A mutant also leads to an elevated arrest of PBMC onto the activated HUVEC monolayers (Figure 4A).
The reversible equilibrium of MIP-1 monomer with MIP-1 dimer and polymers would make MIP-1 a more effective chemoattractant over a longer range, if proteases that selectively degrade MIP-1 monomer but not MIP-1 dimer or polymer exist (Figure 4B; see Supplementary Mathematical Modeling for details). Our mathematical modelling predicts that the MIP-1 monomer concentration would decay exponentially if MIP-1 exists only as monomers. By allowing MIP-1 to dimerize, a fraction of the MIP-1 is protected from degradation, which results in a shallower gradient with a longer chemoattraction range. These effects would be more pronounced when taking into account both the dimerization and polymerization of MIP-1 (Figure 4B). Thus dimerization and polymerization would extend the effective range of the chemotactic gradient of MIP-1.
This model predicts that the depolymerization mutation would reduce the effective range for cell recruitment in vivo by MIP-1. To test this, we examined the ability of MIP-1α D27A to recruit cells into mouse peritoneum. Intra-peritoneal injection of wild-type MIP-1α significantly increased cell infiltration into mouse peritoneum by three- to four-fold over baseline (PBS control) (Figure 4C). However, MIP-1α D27A failed to elicit such a response despite its normal in vitro chemotactic activity (Figure 4C, Supplementary Figure S1). We then show that MIP-1α P8A mutant also has reduced potency for in vivo cell recruitment into mouse peritoneum (Figure 4C), despite its activity in an in vitro chemotaxis assay (Supplementary Figure S1). Together, our studies using two independent depolymerization mutations in MIP-1α demonstrate that polymerization is crucial for the biological activities of MIP-1α.
Identification of monomeric MIP-1α and MIP-1β as novel substrates of IDE
In searching for the candidate protease that selectively degrades MIP-1 monomer, we turned our attention to IDE. The size, charge distribution and the location of the N-termini of MIP-1α and MIP-1β monomer make them putative substrates for IDE (Malito et al, 2008; Manolopoulou et al, 2009; Guo et al, 2010). Our MIP1 structures reveal that MIP-1α and MIP-1β monomers have a volume of ∼14 000 Å3, which fits comfortably inside the catalytic chamber of IDE (Figure 5A). Both MIP-1 molecules possess dipolar charge distributions that complement well the charge distribution in the inner surface of the IDE catalytic chamber (Figure 5A). Upon proper alignment of electrostatic interactions between MIP-1 and the IDE catalytic chamber, the N-terminal end of MIP-1 can be readily inserted into the exosite of IDE, a key feature of high-affinity binding exhibited by IDE substrates (Malito et al, 2008; Guo et al, 2010). Whereas the closely related chemokine RANTES has a mostly positively charged surface, which would be repelled from the IDE-C catalytic chamber (Figure 5A). Thus, we predict that MIP-1α and MIP-1β monomers are high-affinity substrates for IDE, whereas RANTES monomer is not.
Figure 5.

Identification of MIP-1α as a high-affinity substrate for IDE. (A) MIP-1α/MIP-1β/RANTES are modelled into the catalytic chamber of IDE. IDE is depicted as light green and the chamber as dark green. Molecular surface of MIP-1α/MIP-1β/RANTES is coloured from negative (red, −6kT) to positive (blue, +6kT) as calculated by APBS (38). (B) Representative MS spectra of MIP-1α WT (top), D27A and MIP-1β (middle), RANTES (bottom) with and without IDE. MIP-1α WT and D27A proteins (8 μM) alone or digested by IDE (0.16 μM) with or without the presence of heparin at 37°C for 30 s were analysed by MALDI-TOF MS. MIP-1β and RANTES samples were treated similarly at 37°C for 15 min. (C) IDE expression of BV-2 stable lines encoding shRNA against mouse IDE. The total cell lysates of BV-2 cells that expressed the control shRNA or IDE shRNA were immunoblotted with anti-IDE (top panel) and anti-ERK1/2 (bottom panel). (D) The secretion of MIP-1 and RANTES by BV-2 cells that have reduced IDE expression. Secreted MIP-1 chemokine levels from IDE knockdown BV-2 cells with or without 10 ng/ml LPS stimulation for 3 h were determined by ELISA assays.
MALDI-TOF mass spectrometry (MS) analyses indicate that IDE indeed can cleave both MIP-1α and MIP-1β. MIP-1α fragments were readily observed after a 30-second incubation of MIP-1α with IDE in a 50:1 molar ratio. The cleavage rate is estimated to be ∼0.7 per second. IDE can degrade MIP-1α D27A faster (∼5 per second), comparable with the cleavage rate of insulin by IDE (Manolopoulou et al, 2009). This is consistent with the notion that IDE can only degrade MIP-1α monomers, as the D27A mutation induces MIP-1α depolymerization. We found that heparin modestly accelerated the degradation of MIP-1α by IDE to ∼1 per second, which correlates well with the effect of heparin in depolymerizing MIP-1α. IDE could also degrade MIP-1β, and the addition of heparin increased the cleavage rate (from 0.4 to 3 per minute) (Figure 5B). However, we did not observe any appreciable degradation of RANTES by IDE (Figure 5B).
We then examined whether IDE could also selectively degrade MIP-1 in mouse microglia BV-2 cells, which are known to secrete IDE and various chemokines (Ralat et al, 2009). We used short hairpin RNA (shRNA) delivered by lentivirus to selectively knockdown IDE expression (Figure 5C). We then examined the secretion of MIP-1 and RANTES from these cells by ELISA under basal- and lipopolysaccharide (LPS)-stimulated conditions (Figure 5D). We found elevated levels of MIP-1α and MIP-1β under both basal- and LPS-stimulated conditions in the IDE knockdown BV-2 cells as compared with BV-2 cells infected with control lentivirus. Interestingly, the RANTES level was decreased in IDE knockdown cells, suggesting a possible compensatory mechanism for RANTES clearance. These data suggest that IDE is involved in the clearance of MIP-1α and MIP-1β, but not in RANTES.
Analysis of the cleavage sites of MIP-1α by IDE and functional consequences
We then focused on the degradation of MIP-1α by IDE, as IDE exhibits much faster cleavage of MIP-1α than MIP-1β. We first assessed the ability of MIP-1α to compete with the degradation of the fluorogenic bradykinin-mimetic peptide, Substrate V by recombinant human IDE (Figure 6A). The IC50 value of MIP-1α is ∼170 nM, comparable with other high-affinity substrates such as insulin (Manolopoulou et al, 2009). We also examined whether cleavage by IDE could affect the biological activities of MIP-1. We first studied the in vitro chemotactic activity of intact and IDE-digested MIP-1α proteins on THP-1 cells, a human acute monocytic leukaemia cell line in a modified Boyden chamber assay. As expected, we observed a typical bell-shaped dose–response curve for intact MIP-1α with a peak effective concentration at 1 nM (Figure 6B). The treatment of MIP-1 by IDE led to a reduction or loss of chemotactic activity (Figure 6B).
Figure 6.

Functional analyses on the effect of MIP-1 degradation by IDE. (A) Inhibition of IDE-mediated degradation of substrate V by MIP-1α. IDE activity on substrate V was measured at 37°C in the presence of indicated concentrations of MIP-1α. (B, C) Effects of IDE on the chemotaxis and increase of [Ca2+]i of MIP-1α on THP-1 Cells. MIP-1α was preincubated with IDE at the indicated molar ratio at 37°C for 15 min. For the chemotactic activity (B), chemotactic response was expressed as mean chemotaxis index. For the MIP-1-mediated increase of [Ca2+]i (C), fura-2 loaded THP-1 cells were stimulated with either 1 or 10 nM MIP-1α as the positive control or the same concentrations of MIP-1α that were pretreated with IDE (MIP-1α+IDE). MIP-1α-dependent increases of [Ca2+]i were monitored by ratio of fluorescence at 340 over 380 nm. The change of [Ca2+]i is indicated by ΔF340/F380, which is the difference between the peak F340/F380 value after addition of MIP-1α and the basal level before stimulation. (D) Summary of IDE cleavage sites on the MIP-1α primary sequence by mass spectrometry analysis is illustrated. Initial cleavage sites and secondary cutting sites are shown as big and small arrows, respectively. (E) Representative ESI-FTICR-MS spectrum of MIP-1α after a 2-min IDE digestion.
We then investigated whether IDE also affects the MIP-1α-mediated increase of intracellular calcium in THP-1 cells. As expected, we observed a dose-dependent increase of [Ca2+]i by MIP-1α and that 1 and 10 nM MIP-1α could elicit ∼50 and 100% responses, respectively (Figure 5C, Supplementary Figure S9A). The treatment of MIP-1α with IDE at a molar ratio of 10:1 caused a modest decrease in [Ca2+]i change (Figure 6C). Thus, our data indicated that IDE cleavage could both inactivate MIP-1α-mediated chemotaxis and modulate MIP-1 signalling. It is worth noting that the treatment of MIP-1α by the lower level of IDE (50:1, MIP-1 to IDE ratio) resulted in noticeable cleavage of MIP-1α and reduced the chemotactic activity, but did not significantly reduce the potency of MIP-1 to raise [Ca2+]i (Figure 6B and C, Supplementary Figure S9B). Thus, the cleavage of MIP-1α by IDE is less effective at reducing the change of [Ca2+]i than chemotaxis (Figure 6B and C). It is conceivable that IDE-cleaved MIP-1α products that are held together by disulphide bonds are still able to partially activate CCR receptors for the transient increase of [Ca2+]i, but lose their potency in chemotaxis, which occurs on a much longer time scale.
To identify the cleavage sites of MIP-1α by IDE, we took advantage of the high mass accuracy and high resolution of Fourier-transform ion cyclotron resonance (FTICR) MS-MS analysis to characterize the IDE cleavage sites. MIP-1α contains two disulphide bonds (Figure 6D). IDE degrades MIP-1α without the reduction of its disulphide bonds. The addition of a reducing agent after the completion of IDE digestion revealed more than 10 major peaks in the MS spectrum (Figure 6E). A total of 20 fragments were identified, with excellent matches to the calculated mass (Supplementary Tables S3 and S4). From the IDE-digested MIP-1α fragments, eight could be generated from the stochastic, single cleavage of MIP-1α at residues between 18–19, 45–46, 46–47 and 48–49. They include the paired MIP-1α segments: 1–18 and 19–70, 1–45 and 46–70, 1–46 and 47–70, 1–48 and 49–70 (Figure 6E). These cleavage sites were further confirmed by the proteolytic 18O-labelling during IDE digestion (Supplementary Figure S10). Thus, our data indicate that these four sites are the preferred initial cleavage sites of MIP-1α by IDE.
Structure analysis of MIP-1α-bound IDE
To elucidate the molecular basis of MIP-1α recognition by IDE, the crystal structure of catalytically inactive IDE in complex with MIP-1α was solved at 3.0 Å resolution (Table I, Supplementary Figure S2). The overall fold of IDE is nearly identical to structures of IDE in complex with other known substrates (Figure 7A). Two discrete electron densities representing segments of MIP-1α are clearly visible inside the catalytic chamber of IDE; one is at the exosite and the other one is at the catalytic site (Figure 7A and B). At the catalytic site of IDE, residues 45–50 of MIP-1α and a zinc ion coordinated by H108, H112 and E189 of IDE are clearly visible (Figure 7B, Supplementary Figure S4). E111, a catalytic acid known to deprotonate the catalytic water, was mutated to glutamine for catalytic inactivation. This catalytic residue is close to the scissile bond between residues R48 and Q49 of MIP-1α, one of the four initial cleavage sites identified by our MS analysis. At the exosite, the continuous density could be modelled well with the four N-terminal residues of MIP-1α; which resembles the previously observed binding of exosite with the N terminus of IDE substrates (Shen et al, 2006; Manolopoulou et al, 2009; Guo et al, 2010). In this case, the N-terminal loop of MIP-1α forms a β-strand and inserts into the existing β-sheet of IDE, parallel to the β12 strand. Residues L359, G361 and Q363 in strand β12 form hydrogen bonds with N-terminal residues of MIP-1α. Residue E341 from IDE is also in close vicinity and can form a hydrogen bond with the amino terminus of MIP-1α. The remaining regions of MIP-1α are not visible and are likely disordered as we did find intact MIP-1α by MS in the MIP-1α-bound IDE crystal (data not shown).
Figure 7.

Structural analyses on MIP-1α-bound IDE. (A) Overall secondary structure of IDE-CF-E111Q in complex with MIP-1α. N- and C-terminal domains of IDE are coloured cyan and grey, respectively. MIP-1α is coloured orange. (B) The detailed interaction of MIP-1α at the exosite (left) and catalytic site (right). Composite omit map was coloured orange at 1σ level; the atoms oxygen, nitrogen and carbon of substrates are shown in red, blue and orange, respectively. (C) Comparison of MIP-1α in the free form (1B50, green) with IDE-bound form (3H44, red). The corresponding regions of MIP-1α found in IDE-bound form are coloured as red. (D) The three-dimensional structure of MIP-1α (2X69). The residues for IDE initial cleavage sites and secondary sites are coloured red and light green, respectively. Residues involved in receptor CCR5 binding are highlighted yellow. (E) Structural comparison of IDE in complex with Aβ (2G47), insulin (2WBY), bradykinin (3CWW) and MIP-1α (3H44). IDE and its substrates are coloured green and red, respectively.
To bind the catalytic chamber of IDE, both segments of MIP-1α in the IDE-bound MIP-1α structure must undergo a substantial conformational change upon their interaction with IDE (Figure 7C). The N-terminal segment of MIP-1α alters its orientation in order to bind the exosite. The loop region of residues 45–50 also forms a β-strand that fits into the catalytic cleft of IDE. Compared with other substrate-bound IDE structures, we propose that IDE also uses the open-closed conformational switch to entrap and unfold MIP-1α for its degradation (Supplementary Figure S11) (Shen et al, 2006; Manolopoulou et al, 2009; Guo et al, 2010).
Our MIP-1α-bound IDE structure offers a molecular basis for the selective cleavage of MIP-1α by IDE at the defined sites, leading to MIP-1 inactivation. The four IDE cleavage sites (between residues 18–19, 45–46, 46–47 and 48–49) reside at the two structurally adjacent loops and they overlap with residues that are indispensable for the biological functions of MIP-1α (Figure 7D). When residues R18, Q19, K45, R46 and R48 were mutated into alanines, MIP-1α-binding affinity to its cognate receptor CCR5 was greatly attenuated (Teng et al, 2008). Interestingly, residues of MIP-1α crucial for receptor binding all map to the same side of the molecule, where four IDE initial cleavage sites reside (Figure 7D). Thus, IDE can specifically target functional residues of MIP-1α and cleavage of MIP-1α at any one of these four sites by IDE would likely perturb the local structure to prevent MIP-1α from activating its receptor.
Structural comparison of MIP-1α- and insulin-bound IDE structures revealed common and unique features for the interaction of IDE with these two structurally distinct substrates (Figure 7E). Both insulin and MIP-1α have a dipolar charge distribution. Similar to the interaction of insulin with IDE, the mostly negatively charged surface of MIP-1α is facing the positively charged IDE-C domain. The interaction with both exosite and IDE-C domain is likely to greatly enhance the binding affinity of MIP-1α and insulin to IDE. In contrast of insulin, where the initial cleavages are ∼15 Å away from the catalytic site, our MIP-1α-bound IDE model places the four initial cleavages of MIP-1α close to the catalytic site. Thus, instead of the major conformational change required for insulin to be cut by IDE, a relatively small repositioning of two adjacent loops that contain the four cleavage sites would be needed for IDE to cut any of these four sites in a stochastic manner. Furthermore, based on the NMR structures, MIP-1α molecules are highly flexible, especially at the N-terminal end (Figure 7C). This flexibility is also likely to contribute to the unfolding and cleavage of MIP-1α by IDE.
Discussion
Protein polymerization governs diverse biological and pathological events (Frieden, 2007). Our studies simplify the polymerization of MIP-1 from a potentially complex process, such as amyloid fibre formation to a simple dimer–dimer interaction by charge and shape complementarity. Furthermore, our structural and biophysical analyses reveal that MIP-1 forms ordered, rod-shaped helical polymers and explain how the reversible polymerization of these chemokines occurs in solution. Our structural and biophysical analyses also offer the molecular basis of MIP-1 polymerization and how the D27A mutation affects MIP-1α polymerization. Using two depolymerization mutants (D27A and P8A), we also demonstrate vital roles of polymerization for two MIP-1α-mediated functions. We show that while depolymerization mutations allow MIP-1α to enhance the arrest of monocytes onto activated human endothelial monolayer, the same mutations render MIP-1α ineffective in recruiting cells into mouse peritoneum. Various chemokines have different degrees of oligomerization (Handel et al, 2005). Together with previous studies of other CC chemokines, our data clearly show that the ordered and reversible self-assembly of chemokines has key roles in their biological functions (Baltus et al, 2003; Proudfoot et al, 2003; Handel et al, 2008).
GAG is known to bind and regulate the functions of various chemokines (Handel et al, 2005; Bishop et al, 2007). We found that the addition of heparin (an exemplary GAG) leads to depolymerization of MIP-1. The positively charged residues (R18, K45 and R46) residing at the dimer–dimer interface of MIP-1 polymers are critical for GAG binding (Koopmann et al, 1999; Proudfoot et al, 2003; Handel et al, 2005). Thus, the binding of heparin could reduce the degree of MIP-1 polymerization by capping the end of MIP-1 polymer or affecting the equilibrium of MIP-1 polymerization. However, the effect of GAG binding in MIP-1 polymerization in vivo is likely far more complicated. In different tissues, the heparan sulphate GAG chains are highly diverse in structure and their linked protein partners also vary. In addition to depolymerization, GAG could facilitate MIP-1 polymer formation by anchoring and clustering MIP-1 polymers together on the cell surface or extracellular matrix as well. The effects of GAGs on reversible MIP-1 polymerization in vivo and MIP-1-mediated biological functions await future investigation.
MIP-1 polymerization shares interesting similarities with the formation of actin filaments. Actin polymerization also requires minimal conformational change and results in rod-shaped, double-helical filaments (Dominguez, 2009; Kueh and Mitchison, 2009). In addition, diverse cellular components are involved in regulating the polymerization of both proteins (Disanza et al, 2005). GAGs and proteoglycans on cell surfaces or extracellular matrix could cap the end of MIP-1 polymers to regulate MIP-1 polymerization. The reversible binding of GAGs and proteoglycans to the end of MIP-1 polymers would immobilize MIP-1 and render cell recruitment from a chemotactic response to haptotaxis. Alternatively, GAGs could also sever MIP-1 polymers to facilitate the distribution of MIP-1 polymers for their biological functions. Similarly, a plethora of actin-binding proteins modulate the formation and distribution of actin filaments (Disanza et al, 2005). Capping proteins can block the elongation of actin polymerization. Through its severing activity, cofilin could increase the free barbed end of actin polymer for actin-based protrusion. Furthermore, multiple structural states exist in actin filament (Kueh and Mitchison, 2009). The less stable polymer state found in the MIP-1α D27A octamer structure highlights the potential complexity in conformational states of chemokine polymers.
Our structural analyses of the wild-type and mutant forms of MIP-1 reveal that CC chemokine polymerization may be more common than previously anticipated. For example, RANTES shares 44% sequence identity with MIP-1α and is also known to form large aggregates (Supplementary Figure S5). Although the isoelectric point (pI) for RANTES is 9.2, whereas that of MIP-1α is 4.8, RANTES preserves the charged residues (D27, E67, R46 and R48) vital for polymer formation (Supplementary Figure S5). As expected, our preliminary SAXS analysis suggests that RANTES indeed forms rod-shaped polymers (data not shown). The sequence alignment of 27 CC chemokines reveals that those charged residues crucial for the polymerization of MIP-1α and MIP-1β are not absolutely conserved. We found that D27A mutation reduces but does not abolish MIP-1α polymerization. Thus, a chemokine missing one of these key contact sites, such as CCL8, may still be capable of forming polymers. The corresponding residues for D27 and E67 in CCL14 are serine and lysine, respectively (Supplementary Figure S5). This may explain why the packing of the CCL14 tetramer structure is distinct from that in the MIP-1 polymers (Supplementary Figure S3).
In the presence of IDE or other monomer-selective proteases, the reversible oligomerization (dimerization and polymerization) of MIP-1 offers two unique features in their chemotactic gradient. First, the oligomerization protects MIP-1 from proteolytic degradation, so that they have longer effective range for recruitment of macrophages and neutrophils. Second, the oligomerization enables the MIP-1 gradient to reflect the severity of infection or inflammation. In a mild infection, when little MIP-1 is made and MIP-1 dimer rarely forms, an exponentially decaying MIP-1 spatial gradient is likely to form in the steady state (similar to the purely monomeric case shown in Figure 4B). Such a gradient is steep so that the cells are targeted to the centre of the infection. However, in a moderate-to-severe infection, MIP-1 production is high, which favours the formation of MIP-1 dimers and polymers. In addition to the benefit of having a longer effective range, our model predicts that the MIP-1 gradient near the centre of secretion is less effective for directional chemoattraction (Figure 4B). Thus, cells are expected to move mostly to the periphery of the infection site, rather than to the centre. This would help to prevent the spread of invading pathogens in a severe infection. This intricate regulation of chemotactic gradient by the interplay between chemokine oligomerization and extracellular proteases may be applicable to other chemokines as well. The validity of this hypothesis on the chemotactic gradient of MIP-1 and other chemokines that also form oligomers and are subject to proteolytic degradation awaits future investigation (Allen et al, 2007; Wolf et al, 2008).
MIP-1 D27A mutant (BB-10010) was tested in clinical trials for reducing chemotherapy-associated myelotoxicity (Bernstein et al, 1997; Owen-Lynch et al, 1998; Hough et al, 2003). The choice of MIP-1 D27A mutant was made to avoid issues related to MIP-1 aggregation (Hunter et al, 1995). However, the treatment revealed no significant improvement in protecting haematopoietic stem cells. Our finding of the defective in vivo chemotactic function of MIP-1α D27A may partially explain why BB-10010 did not exhibit the anticipated therapeutic benefit. The structural basis of MIP-1 polymerization and selective degradation of MIP-1 monomer by IDE may help bring better control of MIP-1 in the development of MIP-1-based therapy for several human diseases (Maurer and von Stebut, 2004).
In addition, our findings on how IDE can affect the clearance of MIP-1 in microglia add another layer of complexity to the role of IDE for the progression of AD. Studies in transgenic mice have revealed the role of IDE in Aβ degradation in vivo (Farris et al, 2003; Leissring et al, 2003). Furthermore, the IDE gene resides on chromosome 10q, and lymphoblast samples from chromosome 10-linked AD families have reduced IDE activity (Kim et al, 2007). Microglia, the resident macrophages of CNS, are recruited to the site of Aβ deposit and have a key role in Aβ clearance (El Khoury and Luster, 2008). We show here that reduced IDE levels can lead to an increase in MIP-1 accumulation. Thus, the reduced IDE activity observed in AD patients may affect Aβ accumulation through the ability of MIP-1 to compete with IDE and/or alter the recruitment of microglia to the Aβ deposit in the brain. Therefore, our studies open a new avenue of investigation to identify novel IDE substrates and their roles in the progression of AD, diabetes mellitus and other human diseases. Such studies will undoubtedly aid the development of IDE-related therapeutics.
Materials and methods
Cloning, expression and purification of recombinant chemokines and IDE
The synthetic genes that encode the mature forms of human MIP-1 and RANTES with optimization for E. coli codon usage were constructed with an enterokinase cutting site (DDDDK) in front of these chemokines. MIP-1α mutants were generated by site-directed mutagenesis. MIP-1 and RANTES were expressed as thioredoxin and His6-tag fusion proteins in E. coli BL21(DE3) cells. The fusion proteins were purified by Ni-NTA column (Qiagen) followed by a Source-Q column (GE). Recombinant MIP-1 and RANTES were obtained by removing fusion tags with enterokinase and further purified by heparin affinity chromatography. Final products of MIP-1 and RANTES were analysed by MALDI-TOF MS, and its biological activity was assayed on THP-1 cells by chemotaxis assay (Supplementary Figure S1). Human IDE proteins were expressed in E. coli as described (Manolopoulou et al, 2009).
Structural analysis
The diffracting MIP-1 and MIP-1α-bound IDE crystals were grown in the distinct crystallization conditions at 18°C by hanging drop, vapour diffusion (Table I). X-ray diffraction data were measured at 100 K on the ID-19 or 14-BMC at the SBC, Argonne National Laboratory and were processed using HKL3000 (Table I). The initial phase was solved by SAD using selenomethionyl MIP-1α (V3M/L63M) (Supplementary Table S1, Supplementary Figure S2). Native MIP-1α (WT and D27A) and MIP-1β structure determinations were performed by molecular replacement using the MIP-1α (V3M/L63M), as a search model with the program Phaser. MIP-1α-bound IDE structure was determined by molecular replacement using IDE-E111Q structure (2G47) as a search model. Structural building and refinement were performed by using REFMAC, Phoenix and Coot.
SEC and SAXS analysis of MIP-1
The size distribution of MIP-1 polymers was determined by using a S200 10/300 GL column. SAXS data were collected at the 18-ID (BioCAT) beamline using the Mar 165 CCD detector at room temperature (23°) using 1.033 Å as the incident X-ray wavelength. All data processing was carried out using Igor Pro (WaveMetrics Inc.) with macros written by the BioCAT staff following standard procedures and further analysed with GNOM (Svergun, 1991).
PBMC recruitment onto the activated HUVEC monolayer
Endothelial cell medium and HUVEC were obtained from Promocell (Heidelberg, Germany). HUVECs were grown to confluency on 35 mm dishes and activated with 10 ng/ml TNF-α for 3 h before the experiment. Human blood was obtained in citrate/dextran by venipuncture from healthy donors, who gave informed consent. PBMCs were isolated by density gradient centrifugation using Ficoll-Paque medium and washed with PBS before suspension in Hepes-buffered Hank's balanced salt solution containing 0.5% human serum albumin (HH buffer) at 500 000 cells/ml. Activated HUVECs were incubated with 1 μg/ml either MIP-1α WT or P8A or D27A or H2O at 37°C. After 60 min, HUVEC dishes were assembled in a parallel wall flow chamber and PBMCs were perfused at 1.5 dynes/cm2. Adherent cells were recorded by video microscopy at × 100 magnification and quantified in multiple high-power fields.
In vivo cell accumulation assay
Nine-week-old male C57BL/6 mice (Charles River laboratories; n=5) were intraperitoneally injected with 10 μg of either MIP-1α WT or D27A mutant or P8A mutant in 200 μl PBS. After 20 h, mice were killed by CO2 asphyxiation. Then the peritoneal cavity was washed 3–4 times with a total of 8 ml of ice-cold Ca2+/Mg2+-free HBSS containing 1 mM EDTA. The cavity lavage was collected and total cells in the lavage were counted. The experiments were conducted using protocols approved by the Institutional Animal Care and Use Committee at the University of Illinois at Chicago.
MIP-1 degradation by IDE
MIP-1α competition assay and MS analysis of IDE degradation were performed as previously described (Manolopoulou et al, 2009). For MALDI/TOF MS or LC-ESI-FTICR MS/MS analysis, enzyme reactions were performed at 37°C and a given chemokine/IDE molar ratio (usually 50:1) with and without 0.8 mg/ml heparin. Reactions were stopped by the addition of an equal volume of stop solution (200 mM EDTA, 0.2% TFA). Samples were further incubated with 10 mM TCEP at room temperature for 30 min to reduce the disulphide bonds before being applied to MS analysis.
shRNA knockdown of IDE in BV-2 cells
IDE expression in BV-2 cells is knockdown by lentivirus-mediated shRNA expression. Plasmids (a set of five) encoding shRNA that could knockdown mouse IDE expression were purchased from Open Biosystem (RMM4534-NM-031156), and lentivirus were produced by co-transfecting shRNA plasmids with pHR8.2ΔR packaging plasmid and pCMV VSVG envelope plasmid into HEK293T cells. BV-2 stable lines that contain the specific shRNA were obtained by lentivirus infection and selection with 5 μg/ml puromycin. IDE knockdown levels were tested by western blotting with a polyclonal anti-IDE antibody raised against human IDE and affinity purified using an immobilized IDE column. Secreted chemokine level from IDE knockdown BV-2 cells that have the most profound IDE knockdown were measured by ELISA assays (R&D system).
Chemokine response assays in THP-1 cells
Cell migration of THP-1 was measured in 96-well Multiscreen™ filter plate (Millipore) as described previously (Gouwy et al, 2008). For calcium mobilization assay, THP-1 cells were loaded with 2 μM fura-2 AM (Invitrogen). Fura-2 fluorescence was measured with a FluoroMax-3 fluorometer (HORIBA Jobin Yvon Inc.) at excitation wavelengths of 340 and 380 nm and emission wavelength of 510 nm.
Supplementary Material
Acknowledgments
We are grateful to the staff of APS SBC and Biocars for help in data collection. This work was supported by NIH GM81539 to W-J Tang. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Basic Energy Sciences, under contract no. W-31-109-ENG-38. Use of proteomics service facility was supported by Chicago Biomedical Consortium. We thank Drs Peter V Usatyuk and Viswanathan Natarajan for advice on calcium immobilization assay, Dr Angelo Scanu for advices and cell lines in the chemotaxis assays, Dr Tobin Sosnick for the SAXS analysis, Dr Tracy Handel for her insightful comments and Vasilios Kalas for his critical reading of the paper.
Footnotes
The authors declare that they have no conflict of interest.
References
- Allen SJ, Crown SE, Handel TM (2007) Chemokine: receptor structure, interactions, and antagonism. Annu Rev Immunol 25: 787–820 [DOI] [PubMed] [Google Scholar]
- Baltus T, Weber KSC, Johnson Z, Proudfoot AEI, Weber C (2003) Oligomerization of RANTES is required for CCR1-mediated arrest but not CCR5-mediated transmigration of leukocytes on inflamed endothelium. Blood 102: 1985–1988 [DOI] [PubMed] [Google Scholar]
- Bernstein SH, Eaves CJ, Herzig R, Fay J, Lynch J, Phillips GL, Christiansen N, Reece D, Ericson S, Stephan M, Kovalsky M, Hawkins K, Rasmussen H, Devos A, Herzig GP (1997) A randomized phase II study of BB-10010: a variant of human macrophage inflammatory protein-1alpha for patients receiving high-dose etoposide and cyclophosphamide for malignant lymphoma and breast cancer. Br J Haematol 99: 888–895 [DOI] [PubMed] [Google Scholar]
- Bishop JR, Schuksz M, Esko JD (2007) Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 446: 1030–1037 [DOI] [PubMed] [Google Scholar]
- Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P (1995) Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8+ T cells. Science 270: 1811–1815 [DOI] [PubMed] [Google Scholar]
- Cook DN, Beck MA, Coffman TM, Kirby SL, Sheridan JF, Pragnell IB, Smithies O (1995) Requirement of MIP-1 alpha for an inflammatory response to viral infection. Science 269: 1583–1585 [DOI] [PubMed] [Google Scholar]
- Czaplewski LG, McKeating J, Craven CJ, Higgins LD, Appay V, Brown A, Dudgeon T, Howard LA, Meyers T, Owen J, Palan SR, Tan P, Wilson G, Woods NR, Heyworth CM, Lord BI, Brotherton D, Christison R, Craig S, Cribbes S et al. (1999) Identification of amino acid residues critical for aggregation of human CC chemokines macrophage inflammatory protein (MIP)-1alpha, MIP-1beta, and RANTES. Characterization of active disaggregated chemokine variants. J Biol Chem 274: 16077–16084 [DOI] [PubMed] [Google Scholar]
- Disanza A, Steffen A, Hertzog M, Frittoli E, Rottner K, Scita G (2005) Actin polymerization machinery: the finish line of signaling networks, the starting point of cellular movement. Cell Mol Life Sci 62: 955–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez R (2009) Actin filament nucleation and elongation factors—structure-function relationships. Crit Rev Biochem Mol Biol 44: 351–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlop DJ, Wright EG, Lorimore S, Graham GJ, Holyoake T, Kerr DJ, Wolpe SD, Pragnell IB (1992) Demonstration of stem cell inhibition and myeloprotective effects of SCI/rhMIP1 alpha in vivo. Blood 79: 2221–2225 [PubMed] [Google Scholar]
- El Khoury J, Luster AD (2008) Mechanisms of microglia accumulation in Alzheimer's disease: therapeutic implications. Trends Pharmacol Sci 29: 626–632 [DOI] [PubMed] [Google Scholar]
- Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, Eckman CB, Tanzi RE, Selkoe DJ, Guenette S (2003) Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci USA 100: 4162–4167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frieden C (2007) Protein aggregation processes: in search of the mechanism. Protein Sci 16: 2334–2344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouwy M, Struyf S, Noppen S, Schutyser E, Springael JY, Parmentier M, Proost P, Van Damme J (2008) Synergy between coproduced CC and CXC chemokines in monocyte chemotaxis through receptor-mediated events. Mol Pharmacol 74: 485–495 [DOI] [PubMed] [Google Scholar]
- Graham G, Wright E, Hewick R, Wolpe S, Wilkie N, Donaldson D, Lorimore S, Pragnell I (1990) Identification and characterization of an inhibitor of haemopoietic stem cell proliferation. Nature 344: 442–444 [DOI] [PubMed] [Google Scholar]
- Graham GJ, MacKenzie J, Lowe S, Tsang ML, Weatherbee JA, Issacson A, Medicherla J, Fang F, Wilkinson PC, Pragnell IB (1994) Aggregation of the chemokine MIP-1 alpha is a dynamic and reversible phenomenon. Biochemical and biological analyses. J Biol Chem 269: 4974–4978 [PubMed] [Google Scholar]
- Guo Q, Manolopoulou M, Bian Y, Schilling AB, Tang W-J (2010) Molecular basis for the recognition and cleavages of IGF-II, TGF-α, and amylin by human insulin-degrading enzyme. J Mol Biol 395: 430–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handel TM, Johnson Z, Crown SE, Lau EK, Proudfoot AE (2005) Regulation of protein function by glycosaminoglycans—as exemplified by chemokines. Annu Rev Biochem 74: 385–410 [DOI] [PubMed] [Google Scholar]
- Handel TM, Johnson Z, Rodrigues DH, Dos Santos AC, Cirillo R, Muzio V, Riva S, Mack M, Déruaz M, Borlat F, Vitte P-A, Wells TNC, Teixeira MM, Proudfoot AEI (2008) An engineered monomer of CCL2 has anti-inflammatory properties emphasizing the importance of oligomerization for chemokine activity in vivo. J Leukoc Biol 84: 1101–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horuk R (2009) Chemokine receptor antagonists: overcoming developmental hurdles. Nat Rev Drug Discov 8: 23–33 [DOI] [PubMed] [Google Scholar]
- Hough RE, Lorigan PC, Poynton C, Newland A, Gupta RK, Foran J, Hancock BW (2003) A phase II protection study of BB-10010 in patients with high grade non-Hodgkin's lymphoma undergoing intensive chemotherapy. Int J Oncol 22: 421–424 [DOI] [PubMed] [Google Scholar]
- Hunter MG, Bawden L, Brotherton D, Craig S, Cribbes S, Czaplewski LG, Dexter TM, Drummond AH, Gearing AH, Heyworth CM, Lord BI, McCourt M, Varley PG, Wood LM, Edwards RM, Lewis PJ (1995) BB-10010: an active variant of human macrophage inflammatory protein-1 alpha with improved pharmaceutical properties. Blood 86: 4400–4408 [PubMed] [Google Scholar]
- Kim M, Hersh LB, Leissring MA, Ingelsson M, Matsui T, Farris W, Lu A, Hyman BT, Selkoe DJ, Bertram L, Tanzi RE (2007) Decreased catalytic activity of the insulin-degrading enzyme in chromosome 10-linked Alzheimer disease families. J Biol Chem 282: 7825–7832 [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Takahashi H, Sanford AP, Herndon DN, Pollard RB, Suzuki F (2002) An increase in the susceptibility of burned patients to infectious complications due to impaired production of macrophage inflammatory protein 1 alpha. J Immunol 169: 4460–4466 [DOI] [PubMed] [Google Scholar]
- Koopmann W, Ediriwickrema C, Krangel MS (1999) Structure and function of the glycosaminoglycan binding site of chemokine macrophage-inflammatory protein-1 beta. J Immunol 163: 2120–2127 [PubMed] [Google Scholar]
- Kueh HY, Mitchison TJ (2009) Structural plasticity in actin and tubulin polymer dynamics. Science 325: 960–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuschert GS, Hoogewerf AJ, Proudfoot AE, Chung CW, Cooke RM, Hubbard RE, Wells TN, Sanderson PN (1998) Identification of a glycosaminoglycan binding surface on human interleukin-8. Biochemistry 37: 11193–11201 [DOI] [PubMed] [Google Scholar]
- Laurence JS, Blanpain C, Burgner JW, Parmentier M, LiWang PJ (2000) CC chemokine MIP-1 beta can function as a monomer and depends on Phe13 for receptor binding. Biochemistry 39: 3401–3409 [DOI] [PubMed] [Google Scholar]
- Leissring MA, Farris W, Chang AY, Walsh DM, Wu X, Sun X, Frosch MP, Selkoe DJ (2003) Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron 40: 1087–1093 [DOI] [PubMed] [Google Scholar]
- Lindell DM, Standiford TJ, Mancuso P, Leshen ZJ, Huffnagle GB (2001) Macrophage inflammatory protein 1alpha/CCL3 is required for clearance of an acute Klebsiella pneumoniae pulmonary infection. Infect Immun 69: 6364–6369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodi PJ, Garrett DS, Kuszewski J, Tsang ML, Weatherbee JA, Leonard WJ, Gronenborn AM, Clore GM (1994) High-resolution solution structure of the beta chemokine hMIP-1 beta by multidimensional NMR. Science 263: 1762–1767 [DOI] [PubMed] [Google Scholar]
- Lortat-Jacob H, Grosdidier A, Imberty A (2002) Structural diversity of heparan sulfate binding domains in chemokines. Proc Natl Acad Sci USA 99: 1229–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malito E, Ralat LA, Manolopoulou M, Tsay JL, Wadlington NL, Tang W-J (2008) Molecular bases for the recognition of the short peptide substrate and cysteine-directed modifications of human insulin-degrading enzyme. Biochemistry 47: 12822–12834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolopoulou M, Guo Q, Malito E, Schilling AB, Tang WJ (2009) Molecular basis of catalytic chamber-assisted unfolding and cleavage of human insulin by human insulin-degrading enzyme. J Biol Chem 284: 14177–14188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer M, von Stebut E (2004) Macrophage inflammatory protein-1. Int J Biochem Cell Biol 36: 1882–1886 [DOI] [PubMed] [Google Scholar]
- Menten P, Wuyts A, Van Damme J (2002) Macrophage inflammatory protein-1. Cytokine Growth Factor Rev 13: 455–481 [DOI] [PubMed] [Google Scholar]
- Owen-Lynch P, Czaplewski L, Hunter M, Whetton A (1998) The growth inhibitory role and potential clinical value of macrophage inflammatory protein 1 alpha in myeloid leukaemias. Leukemia Lymphoma 30: 41–53 [DOI] [PubMed] [Google Scholar]
- Proost P, Menten P, Struyf S, Schutyser E, De Meester I, Van Damme J (2000) Cleavage by CD26/dipeptidyl peptidase IV converts the chemokine LD78beta into a most efficient monocyte attractant and CCR1 agonist. Blood 96: 1674–1680 [PubMed] [Google Scholar]
- Proudfoot AEI, Handel TM, Johnson Z, Lau EK, LiWang P, Clark-Lewis I, Borlat F, Wells TNC, Kosco-Vilbois MH (2003) Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc Natl Acad Sci USA 100: 1885–1890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralat LA, Ren M, Schilling AB, Tang W-J (2009) Protective role of Cys-178 against the inactivation and oligomerization of human insulin-degrading enzyme by oxidation and nitrosylation. J Biol Chem 284: 34005–34018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallusto F, Palermo B, Lenig D, Miettinen M, Matikainen S, Julkunen I, Forster R, Burgstahler R, Lipp M, Lanzavecchia A (1999) Distinct patterns and kinetics of chemokine production regulate dendritic cell function. Eur J immunol 29: 1617–1625 [DOI] [PubMed] [Google Scholar]
- Sato N, Kuziel WA, Melby PC, Reddick RL, Kostecki V, Zhao W, Maeda N, Ahuja SK, Ahuja SS (1999) Defects in the generation of IFN-gamma are overcome to control infection with Leishmania donovani in CC chemokine receptor (CCR) 5-, macrophage inflammatory protein-1 alpha-, or CCR2-deficient mice. J Immunol 163: 5519–5525 [PubMed] [Google Scholar]
- Shen Y, Joachimiak A, Rosner MR, Tang WJ (2006) Structures of human insulin-degrading enzyme reveal a new substrate recognition mechanism. Nature 443: 870–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, Boutin P, Vincent D, Belisle A, Hadjadj S, Balkau B, Heude B, Charpentier G, Hudson TJ, Montpetit A, Pshezhetsky AV, Prentki M, Posner BI, Balding DJ, Meyre D et al. (2007) A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 445: 881–885 [DOI] [PubMed] [Google Scholar]
- Svergun DI (1991) Mathematical methods in small-angle scattering data analysis. J Appl Cryst 24: 485–492 [Google Scholar]
- Tanaka Y, Adams DH, Hubscher S, Hirano H, Siebenlist U, Shaw S (1993) T-cell adhesion induced by proteoglycan-immobilized cytokine MIP-1 beta. Nature 361: 79–82 [DOI] [PubMed] [Google Scholar]
- Teng MS, Shadbolt P, Fraser AG, Jansen G, McCafferty J (2008) Control of feeding behavior in C. elegans by human G protein-coupled receptors permits screening for agonist-expressing bacteria. Proc Natl Acad Sci USA 105: 14826–14831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dongen PGJ, Ernst MH (1984) Kinetics of reversible polymerization. J Stat Phys 37: 301–324 [Google Scholar]
- Wagner L, Yang OO, Garcia-Zepeda EA, Ge Y, Kalams SA, Walker BD, Pasternack MS, Luster AD (1998) Beta-chemokines are released from HIV-1-specific cytolytic T-cell granules complexed to proteoglycans. Nature 391: 908–911 [DOI] [PubMed] [Google Scholar]
- Wells TNC, Power CA, Shaw JP, Proudfoot AEI (2006) Chemokine blockers—therapeutics in the making? Trends Pharmacol Sci 27: 41–47 [DOI] [PubMed] [Google Scholar]
- Wolf M, Albrecht S, Märki C (2008) Proteolytic processing of chemokines: implications in physiological and pathological conditions. Inter J Biochem Cell Biol 40: 1185–1198 [DOI] [PubMed] [Google Scholar]
- Wolf M, Clark-Lewis I, Buri C, Langen H, Lis M, Mazzucchelli L (2003) Cathepsin D specifically cleaves the chemokines macrophage inflammatory protein-1 alpha, macrophage inflammatory protein-1 beta, and SLC that are expressed in human breast cancer. Am J Pathol 162: 1183–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolpe SD, Davatelis G, Sherry B, Beutler B, Hesse DG, Nguyen HT, Moldawer LL, Nathan CF, Lowry SF, Cerami A (1988) Macrophages secrete a novel heparin-binding protein with inflammatory and neutrophil chemokinetic properties. J Exp Med 167: 570–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.