

Abstract
We model experience-dependent plasticity in the cortical representation of whiskers (the barrel cortex) in normal adult rats, and in adult rats that were prenatally exposed to alcohol. Prenatal exposure to alcohol (PAE) caused marked deficits in experience-dependent plasticity in a cortical barrel-column. Cortical plasticity was induced by trimming all whiskers on one side of the face except two. This manipulation produces high activity from the intact whiskers that contrasts with low activity from the cut whiskers while avoiding any nerve damage. By a computational model, we show that the evolution of neuronal responses in a single barrel-column after this sensory bias is consistent with the synaptic modifications that follow the rules of the Bienenstock, Cooper, and Munro (BCM) theory. The BCM theory postulates that a neuron possesses a moving synaptic modification threshold, θM, that dictates whether the neuron's activity at any given instant will lead to strengthening or weakening of its input synapses. The current value of θM changes proportionally to the square of the neuron's activity averaged over some recent past. In the model of alcohol impaired cortex, the effective θM has been set to a level unattainable by the depressed levels of cortical activity leading to “impaired” synaptic plasticity that is consistent with experimental findings. Based on experimental and computational results, we discuss how elevated θM may be related to (i) reduced levels of neurotransmitters modulating plasticity, (ii) abnormally low expression of N-methyl-d-aspartate receptors (NMDARs), and (iii) the membrane translocation of Ca2+/calmodulin-dependent protein kinase II (CaMKII) in adult rat cortex subjected to prenatal alcohol exposure.
Donald Hebb's conception of activity-dependent neural plasticity (1) has remained central to models of learning and memory in hippocampal and sensory neocortical function over the past decade, particularly in reference to long-term potentiation and depression (LTP and LTD) (2). We have found that circuitry within the adult rat barrel cortex is modified by innocuous changes in whisker experience in accordance with the BCM rules of synaptic plasticity (3–7). Any model for activity-dependent plasticity in cortex must satisfy the central concept of adequate intracellular calcium entry through depolarization-dependent calcium permeable receptors, which in turn induce a cascade of molecular changes leading to LTP or LTD (2). The two major ionotropic postsynaptic glutamate receptors, N-methyl-d-aspartate receptors (NMDARs) and α-amino-3-hydroxy-5-methyl-4-isoxazole propionate receptors (AMPARs), mediate intracortical and thalamocortical sensory relays in rat barrel cortex (8). These same receptors dominate regulation of calcium entry leading to cortical LTP/LTD (2), and are required for experience-dependent plasticity in rat barrel cortex (9).
The mystacial whiskers of rats are aligned in five rows (row A is dorsal and row E is ventral), and the whiskers within a row are numbered from caudal to rostral. Each facial whisker projects via synaptic relays in the trigeminal nuclei and the thalamic ventral posterior medial nucleus (VPM) to a separate barrel, i.e., the cluster of neurons in layer IV of a cortical barrel-column (10, 11). Barrel-columns form the morphological basis of the so-called discrete one whisker-one column organization of barrel cortex (Fig. 1a). To alter the pattern of sensory activity, all whiskers except two were cut close to the fur on one side of the face (12). The principal whisker D2 and one adjacent (surround) whisker in the D row (i.e., D1) were spared. After measured time intervals up to 30 days of whisker pairing (whiskers were reclipped regularly), the activity of single neurons in the D2 barrel-column was measured in response to controlled mechanical deflections of the principal D2 whisker, the intact “D-paired” surround whisker (D1), and three surrounding cut neighbors (“D-cut” = D3, C2, and E2). The same electrophysiological measurements were performed in the barrel cortex of normal adult rats and adult rats that were prenatally exposed to alcohol (PAE rats) (12). Pregnant dams consumed 6.5% ethanol in a nutritionally complete liquid diet during the whole period of pregnancy. Newborn rats were fed like the normal rats (i.e., without alcohol) until they were weaned at 3 weeks and had reached adulthood (P 90). Then alcohol exposed, liquid diet controls, and standard chow fed controls were tested for the ability of their neocortex to undergo plastic changes in response to whisker pairing. Whisker pairing had caused plastic neural changes in both the control adult cortex and the PAE adult cortex. However, the nature of the changes differed markedly (12). (i) Neurons in the normal barrel-column showed potentiation of responses to deflections of spared whiskers in less than 1 day of whisker pairing (5), whereas these responses in the barrel-column of PAE rats required at least 14 days to reach statistically significant elevation compared with pre-whisker-pairing values (12). Most of the response changes occurred in the long latency (10–100 ms poststimulus) response components thought to be mediated by intracortical connections. (ii) Long latency responses to cut whiskers were reduced in magnitude in normal cortex, whereas they remained unaltered in impaired cortex. (iii) The short latency (<10 ms poststimulus), presumably thalamocortical (TC) components of responses were also subject to change. In the normal cortex, the short latency component elevated over days (4), whereas in the impaired cortex it decreased (12).
Figure 1.

(a) Illustration of topography of barrel-columns representing indicated principal whiskers from a view looking down on the cortical surface. To create an activity bias in cortex, whiskers D2 and D1 remain intact, creating greater activity in the gray stippled, “paired” columns than in all other columns when all of the other whiskers are trimmed. After 3, 7, 14, and 30 days of whisker pairing, the response levels of single neurons in the D2 barrel-column were measured in vivo after controlled deflection of whiskers (see ref. 12 for details) (b). In our simple model, the TC relay from the VPM barreloid D2 mediates a major short-latency (0–10 ms poststimulus) response to whisker D2 and a minor short-latency response to whiskers D1, D3, C2, and E2. Long-latency (10–100 ms poststimulus) responses to CRF and SRF whiskers are mediated through intracortical connections, representing the within and between barrel-column relays, respectively. Open triangles denote synaptic pathways that are modifiable in our model. (c) The BCM modification function φ determines the sign of synaptic weight changes, Δm. θM marks the position of the moving synaptic modification threshold along the activity axis, c.
By using the whisker-to-barrel cortex system, our objective in this study is to develop a computational model for cortical modification that is consistent not only with normal activity-dependent plasticity, but is able to account for defective plasticity within a barrel-column. We want to identify the model parameter(s) that make the difference and meaningfully interpret relevant molecular changes arising from maternal alcohol abuse during pregnancy (10). One striking feature of PAE cortex is a 50% reduction in the expression of NMDA receptor subunits in the barrel cortex (12), and we discuss how this and other biochemical factors might influence the model of impaired cortical function.
Methods
Biological Constraints of the Model.
According to electrophysiological studies in urethane-anesthetized rats, the receptive fields (RFs) of cells in barrel-column D2 are constructed from a strong component from the principal whisker D2 (the center RF, CRF) and a weaker component from several surrounding whiskers, e.g., D1, D3, C2, and E2 (surround RF, SRF) (13–15). The RFs of neurons within the VPM barreloid D2 are constructed in a similar way (16). Thus, the TC input into the barrel-column is monosynaptic and multiwhisker in nature, although still dominated by the CRF whisker. When the CRF whisker is deflected, about 20%–30% of the spikes generated in D2 barrel-column cells occur at a short poststimulus latency (0–10 ms) and the remainder at a longer poststimulus latency (10–100 ms) (13, 14). When one of the SRF whiskers (D1, D3, C2, or E2) is stimulated, less than one spike occurs at a short poststimulus latency (0–10 ms). The vast majority of spikes occur at a longer latency (10–100 ms) (13–15). The short latency pathway from VPM to barrel-columns is AMPAR-dependent (8). However, there is some NMDAR-dependent contribution to the short latency response (8). By contrast, local polysynaptic intracortical circuits within the D2 barrel-column itself evoke long latency spikes to the principal whisker input and are mostly dependent on NMDARs (8). Long latency responses to SRF whiskers are produced through lateral intracortical polysynaptic relays from neighboring barrel-columns (17) and are dominated by NMDAR transmission (8).
Thus, our model cell representing the D2 barrel-column, will have (i) one summed TC input, dominated by the principal whisker and (ii) five individual intracortical inputs, corresponding to the principal and four surround whiskers (Fig. 1b). The simulated circuit is an abstraction of anatomically and physiologically recognized excitatory relays (10, 11, 13–17).
Model of the Barrel-Column.
Although the firing rate of a neuron c(t) depends in a complex and nonlinear fashion on the postsynaptic potentials, we will consider that the region between the excitation threshold and saturation may be reasonably approximated by a linear input-output relationship of our model neuron. Thus, if we consider the case of a linear cell, the modification of the ith synapse with the weight mi at time t is proportional to the product of input activity at the ith synapse, di(t), and a function φ, in such a way that (18)
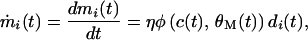 |
1 |
where η is the modification rate, and c(t) = ∑mi(t)di(t). According to Intrator and Cooper (19), φ is a parabolic function of c(t) and the modification threshold θM(t), i.e.,
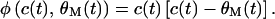 |
2 |
When c(t) > θM(t), all active synapses potentiate. On the other hand, when 0 < c(t) < θM(t), all active synapses weaken (Fig. 1c). The moving modification threshold θM(t) is a whole cell parameter because it is proportional to the square of the postsynaptic response averaged over some recent past time τ, such that
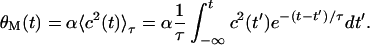 |
3 |
The positive constant α determines how far to the right on the activity axis we can place the actual or effective threshold for synaptic potentiation. We have introduced this constant to reflect the difference between the results obtained from normal and plasticity-impaired cortex.
Total postsynaptic activity c of the model barrel-column cell is the sum of the short latency (0–10 ms) and long latency (10–20 ms) poststimulus responses, cSL and cLL, i.e., the number of spikes generated in response to activation of TC and intracortical inputs, respectively, in equal time windows, i.e.,
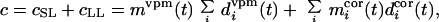 |
4 |
where i = D2, D1, D3, C2, E2. Thus, the summed
input from the thalamic VPM barreloid D2 is weighted by one modifiable
weight, mvpm(t), and each
intracortical input has its own modifiable weight
m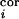 (t) (Fig.
1b). We assume that the thalamic and intracortical input
activities, d
(t) (Fig.
1b). We assume that the thalamic and intracortical input
activities, d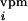 (t)
and d
(t)
and d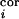 (t),
respectively, relayed from the ith whisker, are comprised of
the sum of the mean evoked response plus random noise, such
that
(t),
respectively, relayed from the ith whisker, are comprised of
the sum of the mean evoked response plus random noise, such
that
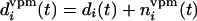 |
5 |
and
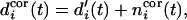 |
6 |
where di(t) and
d′i(t) are equal
to either 1 or 0, depending on whether or not the ith
whisker is deflected. However, when the SRF whisker is deflected, then
for d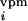 (t),
di(t) = 0.01, because
the thalamic contribution from surround whiskers is much smaller than
from the principal whisker (12). These SRF TC inputs are not crucial;
however, we keep them to be consistent with our previous models (6, 7).
The VPM noise,
n
(t),
di(t) = 0.01, because
the thalamic contribution from surround whiskers is much smaller than
from the principal whisker (12). These SRF TC inputs are not crucial;
however, we keep them to be consistent with our previous models (6, 7).
The VPM noise,
n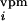 (t), and
intracortical noise,
n
(t), and
intracortical noise,
n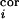 (t), are defined
as independent random variables that are uniformly distributed in the
interval [−Ai,
+Ai], where
Ai < 1 is the noise amplitude.
(t), are defined
as independent random variables that are uniformly distributed in the
interval [−Ai,
+Ai], where
Ai < 1 is the noise amplitude.
Input Environment.
Two input environments were used: one for the bias condition when only
two whiskers were active, and the other one for the control normal
condition when all whiskers were normally active. The inputs from cut
whiskers were assumed to relay noise for 100% of time steps, i.e.,
d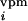 (t) =
n
(t) =
n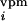 (t) and
d
(t) and
d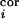 (t) =
n
(t) =
n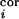 (t) for
i = D-cut (D3), C2, and E2. The level (or amplitude) of
noise plays an important role in synaptic modification according to the
Bienenstock, Cooper, Munro (BCM) theory because it determines the
magnitude of decrease of synaptic weights when the cell response
c is below θM and close to zero (7,
20). The two spared whiskers, D2 and D1, were “stimulated” at
random, either both at once or each one alone. For how often this
happened, see Table 1. Nevertheless,
their inputs relayed noise as if they were unstimulated most of the
time. A similar scenario applies to five intact whiskers. For time
spent in contact when touching an object, we used exact biometric data
for different combinations of three whiskers in one row to simulate the
stimulation of whiskers D1, D2, and D3 (21). Four different instances
of deflection of two additional whiskers, i.e., C2 and E2, were added
at random when at least one whisker of D1, D2, or D3 was stimulated.
(t) for
i = D-cut (D3), C2, and E2. The level (or amplitude) of
noise plays an important role in synaptic modification according to the
Bienenstock, Cooper, Munro (BCM) theory because it determines the
magnitude of decrease of synaptic weights when the cell response
c is below θM and close to zero (7,
20). The two spared whiskers, D2 and D1, were “stimulated” at
random, either both at once or each one alone. For how often this
happened, see Table 1. Nevertheless,
their inputs relayed noise as if they were unstimulated most of the
time. A similar scenario applies to five intact whiskers. For time
spent in contact when touching an object, we used exact biometric data
for different combinations of three whiskers in one row to simulate the
stimulation of whiskers D1, D2, and D3 (21). Four different instances
of deflection of two additional whiskers, i.e., C2 and E2, were added
at random when at least one whisker of D1, D2, or D3 was stimulated.
Table 1.
List of model parameters and their values
| Parameter | Normal cortex | Impaired cortex | Range |
|---|---|---|---|
| Overall % of time of whisker activity before WP | 20% | 16% | 16–20% |
| Three whiskers out of overall activity | 18% | 18% | |
| Two whiskers out of overall activity | 36% | 36% | |
| One whisker out of overall activity | 46% | 46% | |
| Overall % of time of whisker activity during WP | 16% | 14% | 14–16% |
| Two whiskers out of overall activity | 22% | 22% | 15–25% |
| One whisker out of overall activity | 78% | 78% | 75–85% |
n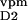 ∶n ∶n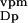 ∶n ∶n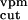 noise amplitudes from
VPM
noise amplitudes from
VPM |
0.1∶0.1∶0.05 | 0.05∶0.05∶0.1 | ≤0.1 |
n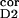 ∶n ∶n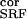 intracortical noise amplitudes before
WP
intracortical noise amplitudes before
WP |
0.4∶0.295 | 0.5∶0.25 | 0.4–0.5∶0.2–0.3 |
n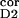 ∶n ∶n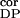 ∶n ∶n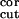 intracortical noise amplitudes during
WP
intracortical noise amplitudes during
WP |
0.4∶0.01∶0.05 | 0.5∶0.2∶0.01 | 0.4–0.5∶≤0.2∶≤0.05 |
| η modification speed | 0.0015 | 0.0015 | 0.0001–0.003 |
| τ iterations | 100 | 100 | 10–103 |
| α (scaling θM) | 16.0 | 32.0 | 16–32 |
Computer Simulations.
We started the simulations with small random initial weights. Because the BCM rule determined synaptic modifications, then after a sufficient time, the system developed to a stable fixed point, as predicted by theory (19, 22). The values of parameters (Table 1) were experimentally adjusted in such a way that, during the control input, after reaching the stable point, the model cell responses oscillated around control values. In previous studies (6, 7), we applied the biased input from the beginning of simulation while calculating the initial weights from Eq. 4. Here, to see the effect on the model dynamics, in particular on θM, we applied either the biased input activity from the outset (whisker pairing condition) or we applied the bias after a period of unbiased (normal) input activity (control before whisker pairing condition). As we will see, in both cases, when the activity bias was introduced, the cell responses achieved qualitative and approximate quantitative match with the experimental whisker pairing data.
At each iteration step, once the activity vector d =
{d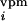 ,
d
,
d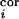 } was generated at random,
the resulting cell response c(t) was calculated
(Eq. 4). For updating synaptic weights, the values of
θM and φ need to be calculated by using Eqs.
2 and 3, with the parameter values listed in
Table 1. At regular intervals, the model cell was “tested” by
calculation of the short- and long-latency responses evoked by
simulated deflection of each of the five whiskers in turn (Eq.
4, noise equal to 0). For comparison of the test
long-latency response component, we took into account the fact that the
experimental value of the long-latency response is in fact the sum of
spikes over the time interval 10–100 ms, which is 9 times greater than
the time interval over which we consider responses in Eq. 4;
thus, we multiplied cLL by 9. We then
multiplied cSL and
cLL by 50 to compare them with the
values obtained in electrophysiological experiments where the average
number of spikes generated by 50 successive deflections of one whisker
was collected (12).
} was generated at random,
the resulting cell response c(t) was calculated
(Eq. 4). For updating synaptic weights, the values of
θM and φ need to be calculated by using Eqs.
2 and 3, with the parameter values listed in
Table 1. At regular intervals, the model cell was “tested” by
calculation of the short- and long-latency responses evoked by
simulated deflection of each of the five whiskers in turn (Eq.
4, noise equal to 0). For comparison of the test
long-latency response component, we took into account the fact that the
experimental value of the long-latency response is in fact the sum of
spikes over the time interval 10–100 ms, which is 9 times greater than
the time interval over which we consider responses in Eq. 4;
thus, we multiplied cLL by 9. We then
multiplied cSL and
cLL by 50 to compare them with the
values obtained in electrophysiological experiments where the average
number of spikes generated by 50 successive deflections of one whisker
was collected (12).
The relation between the number of computer iterations and real time was established as follows: the numerical evolution of short and long latency responses of the model cell under whisker pairing condition was compared with the corresponding values obtained after discrete time intervals in vivo (e.g., after 3, 7, 14, and 30 days; ref. 12). Based on the best mutual numerical match, each day was equated with 104 iterations. This match was the same for simulations of normal and impaired cortex. Thus, one iteration corresponds to 8.64 sec, and the used integration period of τ = 100 iterations corresponds to about 15 min. For comparison, τ used in modeling visual cortex developmental plasticity was equated to 22 min (20).
Results
Evolution of Cell Responses.
In Fig. 2, we plot the evolution of short- (0–10 ms) and long-latency (10–100 ms) responses of a model barrel-column cell and compare the results with experimental data. The simulated changes in cell responses directly reflect underlying weight changes for the corresponding excitatory synaptic inputs (see e.g., Fig. 1b). In normal cortex, the short-latency responses to all whiskers increase monotonically, whereas in the impaired cortex these responses decrease monotonically (Fig. 2a). In Fig. 2a, only the response to D2 is shown. The responses to other whiskers, whether cut or intact, follow the same time course but are ten times smaller in magnitude. In normal cortex, the long latency cell responses to deflections of paired whiskers initially increase and then decrease (Fig. 2 b and c). By contrast, evolution of the later cell responses in impaired cortex is such that they slowly monotonically increase (Fig. 2 b and c). Responses to cut whiskers in normal cortex monotonically decrease, whereas in the impaired cortex they do not change. In Fig. 2d, only the response to D-cut is shown.
Figure 2.

Evolution of short (0–10 ms poststimulus, a) and long latency (10–100 ms poststimulus, b–d) cell responses evoked in the simulated and real barrel-column D2, for the normal cortex (upper curves and squares) and for the impaired cortex (lower curves and diamonds). Discrete points denote average experimental values ± SE (12). We also compare two input environments, i.e., whisker pairing (WP) from the outset (solid lines) and control before WP (dashed lines). Each curve point is an average of 50 different runs.
Evolution of θM.
After about 100 simulated days of control input environment, θM's in the model of normal and impaired cortex reached the control asymptotic values (Fig. 3). After changing the input environment from control to whisker pairing, new asymptotic values were reached. The simulated evolution of cell responses is accompanied by the increase of the mean value of θM in the model of normal cortex and by the decrease in the impaired cortex, respectively (Fig. 3).
Figure 3.

Evolution of the mean value of θM calculated for each simulated day in the model of normal and impaired cortex for whisker pairing (WP) from the outset (solid symbols) and control before WP (open symbols). Each point is an average of 50 different runs.
Other Columns.
To explore how plasticity in the D-paired cortical column would
affect our results, we needed to simulate the ongoing plasticity in the
D-paired column. This result was enabled by employing the course of the
D2 long-latency response, instead of a constant
d′Dp(t) = 1 relayed
intracortically. In the model of normal cortex, the magnitude of
barrel-column D2 cell response to D-paired increased slightly between
the 1st and 7th simulated day, although it was still close to
experimental data. In the model of impaired cortex, the magnitude of
barrel-column D2 cell responses to D-paired increased between the 14th
and 30th simulated day. To compensate for these elevations, it was
sufficient to slightly increase the values of α and
n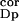 (i.e., the amplitude of noise from
the D-paired column). All other parameters were kept to the same
values. We chose this simple way instead of simulating multicell model
because we do not have experimental data on neighboring columns and we
wanted to avoid introducing additional variables and parameters.
(i.e., the amplitude of noise from
the D-paired column). All other parameters were kept to the same
values. We chose this simple way instead of simulating multicell model
because we do not have experimental data on neighboring columns and we
wanted to avoid introducing additional variables and parameters.
Effect of Model Parameters.
Under control conditions, the values of weights to which the
system converged depended on α, percentage of activity patterns (19,
22), and levels of noise. All these parameters were experimentally
adjusted in such a way that the fixed point solution numerically
matched the pre-whisker pairing values of short and long latency
responses for the normal and impaired cortices. The greatest difference
between values of parameters for the model of normal and impaired
cortex is in the position of the effective threshold for synaptic
potentiation, θM, expressed by the value of
α = 16 or 32, respectively. The percentages of activity patterns
and levels of noise also played an important role. Some were made a
little different to achieve the best numerical match with experimental
data, both for the control and whisker pairing condition (Table 1). For
instance, long-latency responses to cut whiskers in normal cortex
monotonically decrease, whereas in the impaired cortex they do not
change (Fig. 2d). This result has been achieved by the
diminished levels of intracortical noise from cut whiskers in the model
of plasticity-impaired cortex. Further, the monotonic decrease in the
short latency response (Fig. 2a) in the model of impaired
cortex was due to a level of VPM noise,
n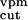 . However, the course and numerical
values of long-latency intracortical responses could have been
reproduced only with different αs. The modification speed η has the
same value for both models. Smaller η in the model of
plasticity-impaired cortex could have reproduced the slow development
of potentiation; however, without large α, the control prewhisker
pairing responses could not have been achieved.
. However, the course and numerical
values of long-latency intracortical responses could have been
reproduced only with different αs. The modification speed η has the
same value for both models. Smaller η in the model of
plasticity-impaired cortex could have reproduced the slow development
of potentiation; however, without large α, the control prewhisker
pairing responses could not have been achieved.
Discussion
The fundamental problem addressed by our simple model was to detect which model parameter makes a difference in accounting for the impaired experience-dependent plasticity in a cortical barrel-column. In turn, manipulations with this parameter would lead toward recovery of normal whisker pairing plasticity in rats exposed prenatally to alcohol. Identifying important parameters might predict which cellular and molecular mechanisms may be involved. In the experimental studies, the effect of enriched postnatal environment was to increase the NMDAR-mediated, long-latency response magnitude for each whisker, and to shorten the period of significant response potentiation during whisker pairing (12). When we simulated our “impaired” model with corresponding increase in magnitude of long-latency responses and high effective threshold (α = 32.0), the potentiation remained slow. To obtain faster potentiation, the effective threshold had to be lowered accordingly, down to the value of α = 16.0 used in the model of normal plasticity. These manipulations with the scaling constant α suggest that, in addition to the averaged squared activity, the effective (or mean) position of θM depends also on some other whole cell properties that are subject to influence by endogenous and exogenous factors.
Cooper (23) suggested that the BCM modification function may depend also on the so-called global variables, which would represent “enabling factors” like presence or absence of neurotransmitters such as norepinephrine (NE), acetylcholine (ACh), etc. For instance, after ACh depletion in adult rats, the whisker-pairing potentiation of long-latency responses does not occur at all (24, 25). We modeled the data of Sachdev et al. (25) with our “impaired” model with adequately increased α, and it simulated this case, also. Indeed, a reduction of ACh axons in the cortex of PAE animals has been reported (26). It is known that ACh increases neuronal excitability (27) and potentiates the entry of Ca2+ through NMDARs (28). Thus, reduction in ACh levels, and perhaps also in NE levels, might explain at least in part the need for large α. However, in adult cortices depleted of ACH, there is only a slight reduction of control pre-whisker pairing (WP) cortical cell responses, as compared with the PAE animals. Thus, in addition, something else must be affected by alcohol.
Theoretical and experimental results indicate that the search for the mechanism of θM should be focused on the excitatory synapses that are formed on dendritic spines with NMDAR ion channels, which provide for activity-dependent influx of Ca2+ (29, 30). When the amplitude of the evoked Ca2+ signal falls above (or below) a certain threshold, then active synapses will be strengthened (or weakened) over time, respectively (see also ref. 2). Although, the biochemical steps occurring after the increase in the postsynaptic concentration of Ca2+ have not yet been completely worked out, Mayford et al. (31) have demonstrated that the frequency-response function for the production of LTD and LTP (i.e., the LTD/LTP threshold) is closely regulated by Ca2+/calmodulin-dependent protein kinase II (CaMKII). Strong evidence has shown that the α-isoform of CaMKII must be present to induce experience-dependent plasticity in the adult barrel cortex (32) and similarly that the whisker pairing plasticity requires local NMDAR activation (9). Increased intracellular Ca2+ results in autophosphorylation of CaMKII, thereby converting the enzyme to a Ca2+-independent (autonomous) form (33). It has been proposed that the level of Ca2+-independent CaMKII sets the value of θM (30, 31). Thus, the value of θM is raised when a surge of Ca2+ results in autophosphorylation of CaMKII, which switches the enzyme to the Ca2+-independent state. This new value of θM would be expected to persist as long as CaMKII remains Ca2+ independent.
Two crucial biochemical characteristics of the adult rat somatosensory cortex, which may be related to θM, are significantly altered after prenatal alcohol exposure. First, we have determined that the densities of the three most common NMDAR subunits in cortex (NMDAR1 + 2A + 2B) are reduced by 50%, 40%, and 30%, respectively, for over 4 months (i.e., into rat adulthood), as judged qualitatively by immunocytochemistry and quantitatively by Western blot analysis (12). Second, we have preliminary results suggesting that the membrane-enriched fraction from the impaired somatosensory cortex before whisker pairing contains roughly twice as much CaMKII-α as samples from the normal cortex (unpublished data). What might account for this excess? CaMKII-α is translocated to the membrane on becoming Ca2+ independent (34) where it is bound to the membrane by the NMDAR2B subunit (35). During life, the PAE rats display a marked reduction of cortical glucose utilization (36). Hypoglycemia is known to lead to persistent translocation of CaMKII-α to the membrane (37) and may explain our data on CaMKII-α.
Thus, consistent with the reduction of NMDAR levels, increase of the levels of bounded CaMKII, and presumably reduced levels of modulatory neurotransmitters, we have set up the effective θM in the model of plasticity-impaired cortex to a higher value than would be predicted by the small average cell activity. Relation of the mean θM to these biochemical factors is the main prediction of our model and can be tested by its application to biochemically altered cortices. Also, a more detailed neuronal model that includes the relevant biochemistry of modifiable synapses may identify more clearly the putative mechanisms of θM.
Acknowledgments
We thank L. N. Cooper, H. Z. Shouval, and, in particular, B. Blais for valuable comments on our model. The criticism of two anonymous reviewers also helped to improve the paper. The work was supported by the U.S.-Slovak Science and Technology Joint Fund in cooperation with the Department of Health and Human Services in the United States and the Ministry of Education in Slovakia, under Project No. 015-95, and by Vedecká Grantová Agentúra (VEGA) Grants 2/6018/99 and 1/7611/20.
Abbreviations
- BCM
Bienenstock, Cooper, Munro
- NMDAR
N-methyl-d-aspartate receptor
- AMPAR
α-amino-3-hydroxy-5-methyl-4-isoxazole propionate receptor
- CaMKII
Ca2+/calmodulin-dependent protein kinase II
- LTP
long-term potentiation
- LTD
long-term depression
- RF
receptive field
- CRF
center RF
- SRF
surround RF
- VPM
ventral posterior medial nucleus
- TC
thalamocortical
- WP
whisker-pairing
- PAE
prenatal exposure to alcohol
- ACh
acetylcholine
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Hebb D O. The Organization of Behaviour. New York: Wiley; 1949. [Google Scholar]
- 2.Artola A, Singer W. Trends Neurosci. 1993;16:480–487. doi: 10.1016/0166-2236(93)90081-v. [DOI] [PubMed] [Google Scholar]
- 3.Diamond M E, Armstrong-James M, Ebner F F. Proc Natl Acad Sci USA. 1993;90:2082–2086. doi: 10.1073/pnas.90.5.2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Armstrong-James M, Diamond M E, Ebner F F. J Neurosci. 1994;14:6978–6991. doi: 10.1523/JNEUROSCI.14-11-06978.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Diamond M E, Huang W, Ebner F F. Science. 1994;265:1885–1888. doi: 10.1126/science.8091215. [DOI] [PubMed] [Google Scholar]
- 6.Beňušková L, Diamond M E, Ebner F F. Proc Natl Acad Sci USA. 1994;91:4791–4795. doi: 10.1073/pnas.91.11.4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beňušková L, Ebner F F, Diamond M E, Armstrong-James M. Network: Comput Neural Syst. 1999;10:303–323. [PubMed] [Google Scholar]
- 8.Armstrong-James M, Welker E, Callahan C A. J Neurosci. 1993;13:2149–2160. doi: 10.1523/JNEUROSCI.13-05-02149.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rema V, Armstrong-James M, Ebner F F. J Neurosci. 1998;18:10196–10206. doi: 10.1523/JNEUROSCI.18-23-10196.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Killackey H P. Brain Res. 1973;51:326–331. doi: 10.1016/0006-8993(73)90383-1. [DOI] [PubMed] [Google Scholar]
- 11.Jensen K F, Killackey H P. J Neurosci. 1987;7:3529–3543. doi: 10.1523/JNEUROSCI.07-11-03529.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rema V, Ebner F F. J Neurosci. 1999;19:10993–11006. doi: 10.1523/JNEUROSCI.19-24-10993.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Armstrong-James M, Fox K. J Comp Neurol. 1987;263:265–281. doi: 10.1002/cne.902630209. [DOI] [PubMed] [Google Scholar]
- 14.Armstrong-James M, Callahan C A, Friedman M A. J Comp Neurol. 1991;303:193–210. doi: 10.1002/cne.903030203. [DOI] [PubMed] [Google Scholar]
- 15.Fox K. J Neurosci. 1994;14:7665–7679. doi: 10.1523/JNEUROSCI.14-12-07665.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diamond M E, Armstrong-James M, Ebner F F. J Comp Neurol. 1992;318:462–476. doi: 10.1002/cne.903180410. [DOI] [PubMed] [Google Scholar]
- 17.Kim U, Ebner F F. J Comp Neurol. 1999;408:489–505. [PubMed] [Google Scholar]
- 18.Bienenstock E L, Cooper L N, Munro P W. J Neurosci. 1982;2:32–48. doi: 10.1523/JNEUROSCI.02-01-00032.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Intrator N, Cooper LN. Neural Networks. 1992;5:3–17. [Google Scholar]
- 20.Clothiaux E, Bear M F, Cooper L N. J Neurophysiol. 1991;66:1785–1804. doi: 10.1152/jn.1991.66.5.1785. [DOI] [PubMed] [Google Scholar]
- 21.Carvell G E, Simons D J. J Neurosci. 1990;10:2638–2648. doi: 10.1523/JNEUROSCI.10-08-02638.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Castellani G C, Intrator N, Shouval H, Cooper L N. Network: Comput Neural Syst. 1999;10:111–121. [PubMed] [Google Scholar]
- 23.Cooper L N. In: Imprinting and Cortical Plasticity. Rauschecker J P, Marler P, editors. New York: Wiley; 1987. pp. 177–192. [Google Scholar]
- 24.Baskerville K A, Schweitzer J B, Herron P. Neuroscience. 1997;80:1159–1169. doi: 10.1016/s0306-4522(97)00064-x. [DOI] [PubMed] [Google Scholar]
- 25.Sachdev R S, Lu S-M, Wiley R G, Ebner FF. J Neurophysiol. 1998;79:3216–3228. doi: 10.1152/jn.1998.79.6.3216. [DOI] [PubMed] [Google Scholar]
- 26.Miller M W, Rieck R W. Brain Res. 1993;627:104–112. doi: 10.1016/0006-8993(93)90753-a. [DOI] [PubMed] [Google Scholar]
- 27.McCormick D A. Prog Neurobiol. 1992;39:337–388. doi: 10.1016/0301-0082(92)90012-4. [DOI] [PubMed] [Google Scholar]
- 28.Auerbach J M, Segal M. Brain Res. 1992;587:83–87. doi: 10.1016/0006-8993(92)91430-m. [DOI] [PubMed] [Google Scholar]
- 29.Bear M F, Cooper L N, Ebner F F. Science. 1987;237:42–48. doi: 10.1126/science.3037696. [DOI] [PubMed] [Google Scholar]
- 30.Bear M F. Neuron. 1995;15:1–4. doi: 10.1016/0896-6273(95)90056-x. [DOI] [PubMed] [Google Scholar]
- 31.Mayford M, Wang J, Kandel E R, O'Dell T J. Cell. 1995;81:891–904. doi: 10.1016/0092-8674(95)90009-8. [DOI] [PubMed] [Google Scholar]
- 32.Glazewski S, Chen C-M, Silva A, Fox K. Science. 1996;272:421–423. doi: 10.1126/science.272.5260.421. [DOI] [PubMed] [Google Scholar]
- 33.Braun A P, Schulman H. Annu Rev Neurosci. 1995;57:417–445. doi: 10.1146/annurev.ph.57.030195.002221. [DOI] [PubMed] [Google Scholar]
- 34.Strack S, Choi S, Lovinger D M, Colbran R J. J Biol Chem. 1997;272:13467–13470. doi: 10.1074/jbc.272.21.13467. [DOI] [PubMed] [Google Scholar]
- 35.Strack S, Colbran R J. J Biol Chem. 1998;273:20689–20692. doi: 10.1074/jbc.273.33.20689. [DOI] [PubMed] [Google Scholar]
- 36.Miller M W, Dow-Edwards D L. Brain Res. 1988;474:316–326. doi: 10.1016/0006-8993(88)90445-3. [DOI] [PubMed] [Google Scholar]
- 37.Hu B-R, Kurihara J, Wieloch T. J Neurochem. 1995;64:1361–1369. doi: 10.1046/j.1471-4159.1995.64031361.x. [DOI] [PubMed] [Google Scholar]