

Abstract
Background
Unlike in tomato, little is known about the genetic and molecular control of fleshy fruit development of perennial fruit trees like grapevine (Vitis vinifera L.). Here we present the study of the sequence polymorphism in a 1 Mb grapevine genome region at the top of chromosome 18 carrying the fleshless berry mutation (flb) in order, first to identify SNP markers closely linked to the gene and second to search for possible signatures of domestication.
Results
In total, 62 regions (17 SSR, 3 SNP, 1 CAPS and 41 re-sequenced gene fragments) were scanned for polymorphism along a 3.4 Mb interval (85,127-3,506,060 bp) at the top of the chromosome 18, in both V. vinifera cv. Chardonnay and a genotype carrying the flb mutation, V. vinifera cv. Ugni Blanc mutant. A nearly complete homozygosity in Ugni Blanc (wild and mutant forms) and an expected high level of heterozygosity in Chardonnay were revealed. Experiments using qPCR and BAC FISH confirmed the observed homozygosity. Under the assumption that flb could be one of the genes involved into the domestication syndrome of grapevine, we sequenced 69 gene fragments, spread over the flb region, representing 48,874 bp in a highly diverse set of cultivated and wild V. vinifera genotypes, to identify possible signatures of domestication in the cultivated V. vinifera compartment. We identified eight gene fragments presenting a significant deviation from neutrality of the Tajima's D parameter in the cultivated pool. One of these also showed higher nucleotide diversity in the wild compartments than in the cultivated compartments. In addition, SNPs significantly associated to berry weight variation were identified in the flb region.
Conclusions
We observed the occurrence of a large homozygous region in a non-repetitive region of the grapevine otherwise highly-heterozygous genome and propose a hypothesis for its formation. We demonstrated the feasibility to apply BAC FISH on the very small grapevine chromosomes and provided a specific probe for the identification of chromosome 18 on a cytogenetic map. We evidenced genes showing putative signatures of selection and SNPs significantly associated with berry weight variation in the flb region. In addition, we provided to the community 554 SNPs at the top of chromosome 18 for the development of a genotyping chip for future fine mapping of the flb gene in a F2 population when available.
Background
Berry size is an important trait in relation to both yield (table grapes) and quality (wine grapes). Indeed, the flavor in wine results from the ratio of skin to flesh, the former being the source of most aromatic and tannins compounds, the second providing the organic acids and the sugars [1].
The genetic and molecular basis of fleshy fruit size variation have been studied in depth in tomato during the last two decades, using a large panel of diverse resources that made tomato a model species for fleshy fruit crops [2,3]. Introgression lines between wild and cultivated genotypes [4-6], near isogenic lines (NILs) [7] and artificial or natural mutants [2,8] have been created and used to study the genetic basis of fruit size variation showing that a large part of it is controlled by less than ten loci. The physiological mechanisms involved have been related to the control of (i) the cell number in the pericarp, as for the fw2.2 locus [9,2,10], (ii) the locule number [2,11], (iii) the late endo-reduplication in pericarp cells [12,10] and (iv) the cell wall plasticity in relation to the cell expansion [10]. All these advances in tomato are useful to assist the study of similar trait in other crops with fleshy fruits, less amenable to genetic studies, such as perennial fruit trees. Indeed, encouraging results have already shown syntheny within Solanaceae species for Quantitative Trait Loci (QTL) controlling fruit size [2]. However, the degree of transferability of knowledge from tomato to non-Solanaceae species remains an open question.
Like tomato, grapevine (Vitis vinifera) produces fleshy fruits and a large difference in fruit size between wild and cultivated genotypes can be observed [13]. Indeed the wild V. vinifera genotypes produce mature berries weighting less than 1g while berries of some table grape varieties can weigh 10 g and more [14]. The growth of a grapevine berry roughly follows the same pattern as for tomato fruit: the first phase of fruit development is due to both cell multiplication and cell expansion, followed by a lag phase corresponding to a major cell metabolic shift and a second phase of fruit growth, mostly explained by cell expansion but without evidence of endoreduplication [15]. The genetic analysis of grape berry size variation is more difficult than in tomato, due to the long biological cycle of the plant, to the high level of heterozygosity of the genome and to the large field area usually required for plant growth, which makes experiments in controlled environment more costly [16-19]. In addition, berry size studies have often been performed on population segregating for seedlessness, with a strong negative correlation between the two traits: the seedless berries are in average smaller than the seeded berries [16-19]. Up to now, it has not been possible to establish the relationship between QTL for berry size and processes like cell multiplication or cell enlargement.
A natural mutant of V. vinifera cv. Ugni Blanc, which produces fleshless berries similar to those observed in wild genotypes, was identified as an opportunity to get insights into the control of berry development and berry size [20]. It has been shown that the drastic phenotypic changes observed in berry development are controlled by a dominant mutation in the fleshless berry (flb) gene [21]. Like the fw2-2 gene in tomato, the flb gene impairs cell divisions in the developing ovaries [21]. The closest genetic marker linked to the flb mutation defines a 6 cM region located at the top the chromosome 18 that corresponds to a physical distance of 948 kb according to the last version of the grapevine genome assembly http://urgi.versailles.inra.fr/index.php/urgi/Species/Vitis/Resources; in this region, no homolog to the fw2-2 gene has been identified. Considering the importance of berry size for wine quality, a fine mapping of the flb mutation was thus started for its molecular identification.
Here we describe our efforts in reducing the genome interval of the region carrying the flb mutation. We first started by a classical genetic mapping approach. We showed that the mutation is located on a completely homozygous portion of chromosome 18 in Ugni Blanc mutant. No marker could thus be found in coupling with the mutation and the classical approach was abandoned.
We therefore started another approach similar to the one previously proposed for fw2-2 gene in tomato [9]. Since the berries of Ugni Blanc mutant mimic wild V. vinifera berries (both types of berries have little to no flesh and carry round shaped seeds typical of wild genotypes) [13,20,22], we hypothesized that flb gene could have been one of the genes selected during the domestication process of grapevine. If so, a signature of selection or selective sweep could be found around this gene. Under this assumption, we performed a preliminary scan of the sequence polymorphism of the flb region in a collection cultivated and wild grapevine genotypes.
Methods
Plant material
The genotypes used in the present study were collected in the French National Grapevine Germplasm Collection (Domain of Vassal, Montpellier, France; http://www1.montpellier.inra.fr/vassal/) and are listed in additional file 1. Twenty-six of them were chosen to maximize the genetic diversity of the cultivated Vitis vinifera compartment [23]. Seven other genotypes belonging to the wild Vitis vinifera compartment were chosen because they had well characterized wild-type phenotypes as well as wild-type diverse SSR profiles and because they originated from different countries (8500Mtp3 from Tunisia, 8500Mtp9 and 8500Mtp38 from Germany and the rest from France; [additional file 1]. Five genotypes were added to the sample: the inbred line INRA Colmar lignée PN40024 (PN40024; reference genome; maintained at INRA Colmar, France), Chardonnay, Ugni Blanc, Ugni Blanc mutant and Pinot Noir clone ENTAV-INRA777 (PN777; maintained at the French Institute for Grapevine and Wine; Domaine de l'Espiguette, Le Grau du Roi, France). The average berry weight at maturity was measured from 30 berries cut at the pedicel base 40 days after véraison [additional file 1].
DNA extraction
Total genomic DNA was extracted from 1 g of young leaves according to the DNeasy Plant Maxi Kit (Qiagen) with the following modifications: 1% polyvinylpyrrolidone (PVP 40 000) and 1% (v/v) βmercaptoethanol were added to buffer AP1. The clarified lysate recovered after filtration with the QIA-shredder Maxi spin column (step 12) was extracted with one volume of phenol:chloroform:isoamyl alcohol (25:24:1) and then with one volume of chloroform:isoamyl alcohol (24:1). From this step forward, the supernatant was treated following the Qiagen instructions.
Gene fragments amplification and sequencing
Based on the genome annotation provided by Jaillon et al [24], 86 primer pairs were designed using the Primer 3 software v.0.4.0 [25] in order to amplify every 13 kb in the flb region, a gene fragment of approximately 1300 bp [additional file 2]. In order to estimate the nucleotide diversity at the whole genome scale, seventy-seven other primer pairs were designed on genes chosen randomly along the genome, taking care that each chromosome was represented by three to five fragments [additional file 3]. The amplicon sequences were then aligned on the last 12× version of the genome sequence http://urgi.versailles.inra.fr/cgi-bin/gbrowse/vitis_12x_pub/ and some of them did not correspond to a gene model anymore. Settings for Primer 3 were: optimum Tm = 55°C, minimum Tm = 53°C, maximum Tm = 57°C, max 5' self complementarity = 4, max 3' self complementarity = 1. In order to amplify all the genotypes while at the same time detecting a maximum of polymorphism, all the primers were designed in exons at both sides of introns. Universal primers T7/SP6 extensions were added to the primers to allow sequencing. All PCR amplifications were carried out as described by Philippe et al [26].
Microsatellite, CAPS and SNP genotyping
The markers genotyped are listed and described in additional file 2. Cleaved Amplified Polymorphic Sequence (CAPS) genotyping was performed as described by Salmaso et al [27]. The Australian Genome Research Facility (AGRF) carried out Simple Sequence Repeats (SSR) and Single Nucleotide Polymorphism (SNP) analysis. SNP were scored using the MassARRAY® iPLEX Gold assay with MALDI-TOF MS detection (Sequenom) and SSR analysis was performed as previously described by Thomas et al [28].
Quantitative PCR assay
Two primer pairs were designed to amplify genomic DNA. The first pair (TCTGATGCGATGTTAGTGGT and TCTGGTATTGGCGTTGG) targeted a unique gene (FL) in the flb region (gene ID GSVIVG01013466001). The second pair (AACTGGATTGAAGGGCGTGG and AGGTTCTTGAGCATGTTAAGC) targeted the 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCoA) gene family, which members are respectively located on the chromosomes 4, 3 and 18 (gene id GSVIVG01026444001, GSVIVG011023852001, GSVIVG01013435001). Real-time PCR conditions were conducted as described by Reid et al [29], with half quantity of PCR mix and of DNA. The PCR efficiencies were determined for each gene and were 92.3% and 97.2% for FL and HMGCoA respectively. In order to compare the initial DNA quantity between genotypes in the flb region, the DNA quantity based on FL gene data was normalised using the DNA quantity of HMGCoA genes as a reference.
BAC-FISH assay
Roots tips of 0.5-1.5 cm length were treated in the dark with 0.04% 8-hydroxiquinoline for 2 h at 4°C followed by 2 h at room temperature to accumulate metaphases. They were then fixed in 3:1 ethanol-glacial:acetic acid for 12 hours at 4°C and stored in ethanol 70% at -20°C. They were washed in 0.01 M citric acid-sodium citrate pH 4.5 buffer for 15 min and then digested in a solution of 5% Onozuka R-10 cellulase (Sigma), 1% Y23 pectolyase (Sigma) at 37°C for 1 h. Digested root tips were then carefully washed with distilled water for 2 h. One root tip was transferred to a slide and macerated in a drop of 3:1 fixation solution (ethanol-glacial:acetic acid). Chromosome spreads were prepared for hybridization as described by
Leflon et al [30]. VV40024H140P14 Bacterial Artificial Chromosome (BAC) clone (available at http://cnrgv.toulouse.inra.fr) was labelled by random priming with biotin-14-dUTP (Invitrogen). The ribosomal probe used, as a control of hybridation, was pTa-71 which contains a 9 kb EcoRI fragment of ribosomal DNA repeat unit (rDNA 18S-5.8S-26 S genes and spacers) isolated from Triticum aestivum [31]. The probe pTa-71 was labelled with Alexa-488 dUTP (Invitrogen) by random priming. Fluorescence In Situ Hybridization (FISH) experiments and capture of fluorescence images were done as described by Leflon et al [30].
Sequence data analysis, estimation of parameters of diversity and linkage disequilibrium
Raw data were aligned and trimmed using either the Genalys v.2.8.3b software for Macintosh [32] or the Staden software v.2.0.0 [33]. They were manually edited and INsertions/DELetions (INDELs) were added when needed. Single Nucleotide polymorphisms (SNPs) were detected, confirmed, and imported into the SNiPlay database http://sniplay.cirad.fr. Nucleotide diversity (π), number of segregating sites (θ), number of haplotype (H), haplotype diversity (Hd), and Tajima's D test of neutral evolution [34] were obtained for each gene fragment using the DnaSp V5.10 software http://www.ub.edu/dnasp/. Eventually, the total value of each parameter was calculated as a weighted average for the whole data set. As all the gene fragments along the flb region were separated in average by 12 kb (from 3 to 57 kb), it was not possible to reconstitute the haplotypes for the entire flb region in order to estimate the Linkage Disequilibrium (LD). Roger and Huff [35] showed that the genotypic correlation coefficient (based on genotypic data) is a good estimator of the haplotypic correlation coefficient. LD was therefore estimated over the entire studied region as the square of the genotypic Pearson correlation coefficient (r2) together with its p-value using a homemade R program. The results were visualised using in homemade Perl scripts.
Association genetics
A structured association test was carried out using TASSEL software http://www.maizegenetics.net/bioinformatics. The population structure was calculated using STRUCTURE software [36] using the genotypes at 20 SSR markers well spread along the 19 chromosomes (Le Cunff et al, 2008; R. Bacilieri unpublished results; [additional file 1]). A General Linear Model test, which takes into account the structure of the sample, was performed between the SNP markers in the flb region with a allelic frequency >0.05 and the average berry weight at maturity. A Bonferroni correction was applied to control false-positives: a SNP marker was declared significant if its Bonferroni p-value was less than 0.05.
Results
A 1 Mb region at the top of chromosome 18 is homozygous in Ugni Blanc and the fleshless berry mutant
The flb mutation was localised by Fernandez et al [21] at the top of chromosome 18, above the markers VMC2A3 and VMC8B5 on the consensus map of a progeny of Chardonnay by Ugni Blanc mutant. However, the flb locus was mapped indirectly relative to VMC2A3 that segregated in Chardonnay and not in Ugni Blanc mutant. For the purpose of finding polymorphic markers in Ugni Blanc mutant above VMC2A3, we aligned the genetic map to the grapevine reference genome sequence [24] in order to identify SSR and SNP markers segregating in the Ugni Blanc mutant. This region corresponded to 948 kb on chromosome 18 (upper part of scaffold 122; Figure 1) where 100 predicted genes were proposed by the automatic annotation.
Figure 1.
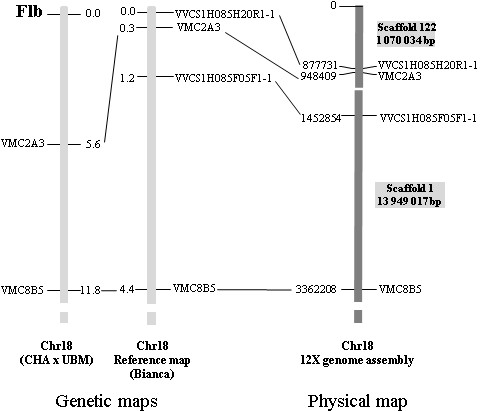
Localization of the region containing the flb locus on the grapevine reference genome sequence. On the left, the map published by Fernandez et al [21] (CHA: Chardonnay, UBM: Ugni Blanc mutant) aligned to one of the informative parental maps used for the genome assembly (A. Canaguier, unpublished results). On the right, alignment to the 12× genome sequence of the top of chromosome 18 http://urgi.versailles.inra.fr/index.php/urgi/Species/Vitis/Resources. The coordinates in kb correspond to the start of the marker sequence on the chromosome sequence. The scaffolds that constitute this part of the chromosome 18 are drawn.
First, 17 SSR, three SNP and one CAPS markers were either developed or retrieved from published genetic maps [37-42] along scaffold 122 and the beginning of scaffold 1, above and below VMC2A3 [additional file 2]. All primer pairs successfully amplified Chardonnay and Ugni Blanc mutant genomic DNAs. One of them (VVS55), not targeting a single locus, was discarded. Chardonnay was heterozygous for ten of the 20 remaining markers, while Ugni Blanc mutant was always homozygous except for VVCS1H085F05F1-1, which is located after VMC2A3 (Table 1).
Table 1.
Marker polymorphism observed between cultivars Chardonnay and Ugni Blanc mutant on the top of the chromosome 18 (12× genome assembly).
| Position on the chromosome 18 (bp) | ||||||
|---|---|---|---|---|---|---|
| Scaffold | Start | End | Marker name | Marker type | Chardonnay$ | Ugni Blanc mutant$ |
| 122 | 212555 | 212699 | VVS50 | SSR | H | h |
| 122 | 213864 | 214083 | VVS51 | SSR | H | h |
| 122 | 226346 | 226575 | VVS52 | SSR | h | h |
| 122 | 230668 | 230769 | VVS53 | SSR | H | h |
| 122 | 308176 | 308256 | 1036L11F | SNP | h | h |
| 122 | 321067 | 321135 | VMC3E5 | SSR | h | h |
| 122 | 388123 | 388423 | VVIN03 | SSR | h | h |
| 122 | 423185 | 423271 | 1038A12F | SNP | h | h |
| 122 | 494374 | 494464 | VVS54 | SSR | H | h |
| 122 | 497723 | 498045 | IN0954 | CAPS | h | h |
| 122 | 670015 | 670200 | VVS56 | SSR | h | h |
| 122 | 804498 | 804634 | VVS57 | SSR | H | h |
| 122 | 877751 | 878077 | VVCS1H085H20R1-1 | SSR | h | h |
| 122 | 895761 | 895846 | 1073P15R | SNP | h | h |
| 122 | 901775 | 901934 | VVS58 | SSR | H | h |
| 122 | 948267 | 948387 | VMC2A3* | SSR | H | h |
| 1 | 1226489 | 1226647 | VVCS1H066N21R1-1 | SSR | H | h |
| 1 | 1297892 | 1298020 | C011 | SSR | H | h |
| 1 | 1452854 | 1453153 | VVCS1H085F05F1-1 | SSR | H | H |
| 1 | 2912753 | 2913088 | VVIB31 | SSR | h | h |
| 1 | 3505999 | 3506060 | VVIV16 | SSR | H | h |
$ H: for heterozygous marker and h: for homozygous marker
* From Fernandez et al [21]
In order to find new heterozygous markers in the flb region, we decided to carry out a re-sequencing approach. Thirty primer pairs were designed along this region [additional file 2], 24 above the SSR marker VMC2A3 and six below. Twenty out of 24 primer pairs (above VMC2A3) successfully amplified the PN40024 genomic DNA and were thus used to sequence the corresponding gene fragments in Chardonnay, Ugni Blanc and Ugni Blanc mutant. We decided to sequence also Ugni Blanc in order to check if the homozygosity of the flb region was specific to the mutant or already present in the wild type.
The 26 fragments of 1300 bp in average were sequenced either only in forward or also in reverse direction, leading to 41 sequences of 161 to 1700 bp long (Table 2), heterozygous INDELs or short repeats leading to the shorter sequences. In total, we analyzed 23,562 bp in Chardonnay and 29,638 bp in Ugni Blanc and Ugni Blanc mutant. This difference was the first observed contrast between Chardonnay and Ugni Blanc, due to a different level of heterozygosity. Comparing the sequences of Chardonnay and Ugni Blanc, 74 polymorphisms were identified (63 SNPs and 11 INDELs). Out of these, 10 differences correspond to homozygous SNPs or INDEL in both samples, while 64 differences correspond to SNPs heterozygous in Chardonnay and homozygous in Ugni Blanc. No heterozygous SNPs or INDELs were observed in Ugni Blanc and its mutant; we deduced that the homozygosity of this region derived from Ugni Blanc. Only Ugni Blanc mutant sequences were considered in the subsequent experiments.
Table 2.
Sequence polymorphism observed between cultivars Chardonnay and Ugni Blanc mutant on the top of the chromosome 18 (12× genome assembly).
| Position on the 12× genome assembly (bp) | Fragment name | Number of extremities sequenced | Sequence Length | Homozygous polymorphic sites between Chardonnay and Ugni Blanc mutant | Chardonnay: number of heterozygous | Ugni Blanc mutant: number of heterozygous | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Scaffold | Start | End | Chardonnay | Ugni Blanc mutant* | SNP | INDEL | SNP | INDEL | SNP | INDEL | ||
| 122 | 85127 | 85871 | VVC2982A | 2 | 1,553 | 1,553 | 0 | 0 | 2 | 0 | 0 | 0 |
| 122 | 161551 | 161929 | VV05806A | 2 | 1,168 | 1,168 | 0 | 0 | 0 | 0 | 0 | 0 |
| 122 | 211001 | 211674 | VVC2974A | 1 | 969 | 969 | 0 | 0 | 4 | 0 | 0 | 0 |
| 122 | 261445 | 262084 | VV05805A | 2 | 1,562 | 1,562 | 0 | 0 | 10 | 0 | 0 | 0 |
| 122 | 299201 | 299664 | VVC2967B | 2 | 911 | 911 | 0 | 0 | 2 | 0 | 0 | 0 |
| 122 | 321452 | 321822 | VV05803A | 2 | 1,077 | 1,077 | 0 | 0 | 7 | 0 | 0 | 0 |
| 122 | 372496 | 372799 | VV05800A | 2 | 683 | 683 | 1 | 0 | 2 | 0 | 0 | 0 |
| 122 | 382744 | 382940 | VVC2956A | 2 | 452 | 876 | 0 | 0 | 2 | 1 | 0 | 0 |
| 122 | 399382 | 399793 | VVC2953A | 2 | 769 | 1,505 | 0 | 0 | 0 | 1 | 0 | 0 |
| 122 | 429725 | 431077 | VV05799A | 2 | 1,539 | 1,539 | 2 | 0 | 1 | 0 | 0 | 0 |
| 122 | 497378 | 497760 | VVC2942A | 2 | 933 | 1,255 | 0 | 0 | 5 | 1 | 0 | 0 |
| 122 | 510613 | 510723 | VV05798A | 1 | 1,464 | 1,464 | 0 | 0 | 4 | 0 | 0 | 0 |
| 122 | 549494 | 550104 | VV05796A | 2 | 909 | 909 | 1 | 0 | 2 | 0 | 0 | 0 |
| 122 | 615081 | 615296 | VV05793A | 1 | 407 | 1,057 | 0 | 0 | 1 | 1 | 0 | 0 |
| 122 | 668381 | 668534 | VV05788A | 1 | 914 | 1,053 | 0 | 0 | 2 | 1 | 0 | 0 |
| 122 | 702907 | 703637 | VV05785A | 1 | 1,090 | 1,446 | 0 | 0 | 0 | 0 | 0 | 0 |
| 122 | 776756 | 777088 | VV05782A | 1 | 413 | 1,434 | 2 | 0 | 2 | 1 | 0 | 0 |
| 122 | 818661 | 819292 | VV05781A | 1 | 830 | 1,495 | 0 | 0 | 1 | 1 | 0 | 0 |
| 122 | 898379 | 898848 | VV05779A | 1 | 1,565 | 1,565 | 1 | 0 | 1 | 0 | 0 | 0 |
| 122 | 928463 | 929045 | VV05777A | 2 | 1,030 | 1,520 | 1 | 0 | 1 | 1 | 0 | 0 |
| 122 | 949921 | 950653 | VV05775A | 1 | 755 | 1,116 | 1 | 0 | 1 | 0 | 0 | 0 |
| 122 | 1009539 | 1010696 | VVC2869A | 1 | 137 | 498 | 0 | 1 | 0 | 1 | 0 | 0 |
| 122 | 1054896 | 1056021 | VVC2865A | 1 | 136 | 136 | 0 | 0 | 1 | 1 | 0 | 0 |
| 1 | 1084916 | 1085337 | VVC15574A | 2 | 161 | 161 | 0 | 0 | 3 | 0 | 0 | 0 |
| 1 | 1098027 | 1099246 | VVC15572A | 2 | 1,700 | 1,700 | 0 | 0 | 0 | 0 | 0 | 0 |
| 1 | 1104978 | 1106307 | VVC15571A | 2 | 435 | 986 | 0 | 0 | 0 | 0 | 0 | 0 |
| 41 | 23,562 | 29,638 | 9 | 1 | 54 | 10 | 0 | 0 | ||||
* The column Ugni Blanc mutant stands for both Ugni Blanc and Ugni Blanc mutant, as no differences were observed between them.
The table lines in bold characters correspond to the sequence fragments below marker VMC2A3.
In total, 62 regions (17 SSR, three SNP, one CAPS and 41 re-sequenced gene fragments) were scanned for polymorphism both in Chardonnay and Ugni Blanc mutant along a 3.4 Mb interval (85,127-3,506,060 bp) in the flb region (scaffold 122 and the beginning of scaffold 1). This allowed showing a nearly complete homozygosity in Ugni Blanc mutant and as expected, a high level of heterozygosity in Chardonnay.
To discriminate between a complete homozygosity of Ugni Blanc mutant and a large deletion of the flb region, two experiments were realized. First, a quantitative PCR (qPCR) assay was performed on genomic DNA from Ugni Blanc, Ugni Blanc mutant, Chardonnay, PN777 and PN40024 as controls. No difference in the estimation of the initial DNA quantity was observed when amplifying with primer pair FL, which targeted a gene in the flb region and the other primer pair HMGCoA, which targeted three loci elsewhere in the genome (Figure 2a; [additional file 4]). This indicated that this region is homozygous and not deleted in Ugni Blanc or Ugni Blanc mutant. The second experiment consisted in a FISH experiment with a BAC clone (VV40024H140P14) localized specifically in the flb region using mitotic metaphase chromosomes of Ugni Blanc mutant and PN777 as control. Chromosomes were counter stained with DAPI (Figure 2b-e) and FISH signals corresponding to VV40024H140P14 were detected on two homologous chromosomes in both PN777 and Ugni Blanc mutant (Figure 2c and 2e respectively), which confirmed that the flb region was not deleted in Ugni Blanc mutant.
Figure 2.
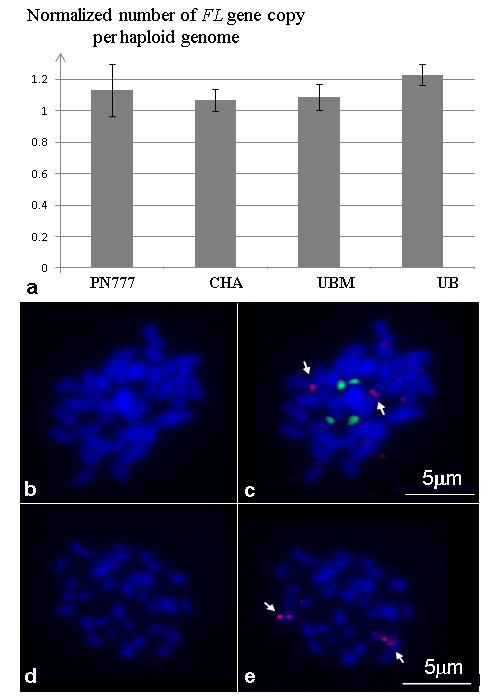
Experimental demonstration of homozygosity of the flb region in Ugni Blanc mutant. (a) Estimation of the number of FL gene copy after normalization in Pinot Noir (PN777), Chardonnay (CHA) Ugni Blanc (UB) and Ugni Blanc mutant (UBM). (b-c) Double fluorescence in situ hybridization (FISH) with BAC clone VV40024H140P14 (red) and pTa-71 (green) as a control, on mitotic metaphase chromosomes of Ugni Blanc mutant and (d-e) FISH signals of BAC clone VV40024H140P14 (red) on mitotic chromosomes of Pinot Noir (PN777) are indicated with arrows. Chromosomes were counterstained with DAPI (blue).
Flb region showed possible signatures of selection in the cultivated V. vinifera compartment
A fragment every ten to 20 kb, in the 948 kb region above marker VMC2A3 was re-sequenced in a highly diverse set of cultivated V. vinifera genotypes [additional file 1], in order to evidence possible traces of selection in the cultivated pool of grapevines.
Sixty-three additional primer pairs were developed; two of them being discarded because they did not amplify in PN40024 [additional file 2]. Eighty-two primer pairs (20 targeting fragments before VMC2A3, one targeting a fragment after VMC2A3 described in the former paragraph and 61 newly developed) were thus used to sequence the corresponding gene fragments in 26 cultivated V. vinifera and the PN40024 as control [additional file 1]. Each fragment was compared to the 12× version of the genome reference sequence, which allowed us to discard the results obtained for eight and three fragments that appeared to be either part of a false duplication in the 8× version of the genome sequence, or to the same gene in the 12× gene annotation, respectively [additional file 2]. The remaining data, from 69 sequenced regions, consisted in a total of 34,355 kb, 61% (21,161 kb) being located in predicted introns or UnTranslated Region (UTR) and 39% (13,194 kb) in exons [additional file 5]. In parallel, 77 random gene fragments spread all over the genome were chosen in order to estimate the nucleotide diversity over the whole genome, and as control for the effect of selection. These gene fragments represented 48,874 kb of total sequence, 55% (27,018 kb) located in predicted introns or UTR and 45% (21,856 kb) in predicted exons [additional file 3].
The Tajima's D parameter, was calculated for the 77 random genes and for the 69 genes from the flb region [additional file 3 and 5]. Eight of 69 sequenced fragments in the flb region showed putative traces of selection evidenced by a Tajima's D parameter significantly deviating from neutrality (Table 3). Moreover, for these fragments, the value of Tajima's D parameter was quite divergent from the average calculated for the 77 random genes (-0.1853+/-0.8117; [additional file 3]) and were found in the tails of the distribution of Tajima's D value across the genome (for α = 0.05; Figure 3). A significant negative Tajima's D value, possibly indicative of a purifying selection was observed for four out of the eight gene fragments whereas a significant positive Tajima's D value, possibly indicative of a diversifying selection, was found for the other four (Table 3).
Table 3.
Nucleotide diversity in the wild and cultivated Vitis vinifera genotypes for the gene fragments along the flb region presenting a significant deviation from neutrality of the Tajima's D parameter.
| Wild | Domesticated | Wine | Table | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fragment | Start 12× | π | π standard error | π | π standard error | Tajima's D§ | π | π standard error | Tajima's D§ | π | π standard error | Tajima's D§ | |
| VVC2981A | 94259 | 0.0007 | 0.0001 | 0.0014 | 0.0005 | -2.0* | 0.0013 | 0.0003 | -0.5 | 0.0016 | 0.0008 | -2.2** | |
| VVC2946A | 444180 | 0.0030 | 0.0004 | 0.0043 | 0.0011 | -1.3 | 0.0035 | 0.0017 | -2.2** | 0.0047 | 0.0016 | -1.2 | |
| VV05791A | 638081 | 0 | 0 | 0.0040 | 0.0002 | 0.6 | 0.0034 | 0.0005 | 1.1 | 0.0037 | 0.0003 | 2.4* | |
| VVC2897A | 682572 | 0.0173 | 0.0062 | 0.0044 | 0.0017 | -2.1* | 0.0070 | 0.0042 | -2.1 | 0.0027 | 0.0004 | -0.7 | |
| VV05785A | 702907 | 0.0008 | 0.0004 | 0.0003 | 0.0002 | -1.9* | 0.0003 | 0.0002 | -1.5 | 0.0002 | 0.0001 | -1.5 | |
| VVC2901A | 742593 | 0.0082 | 0.0035 | 0.0156 | 0.0005 | 3.1** | 0.0157 | 0.0011 | 2.4* | 0.0144 | 0.0079 | 2.6** | |
| VVC2885A | 746740 | 0.0115 | 0.0037 | 0.0159 | 0.0007 | 2.7** | 0.0167 | 0.0012 | 2.8** | 0.0145 | 0.0020 | 1.5 | |
| VVC2892A | 808955 | 0.0009 | 0.0003 | 0.0109 | 0.0007 | 2.2 * | 0.0102 | 0.0011 | 2.0 | 0.0119 | 0.0009 | 2.0 | |
§ * 0.01<P-value < 0.05 and ** 0.001<P-value < 0.01
Figure 3.
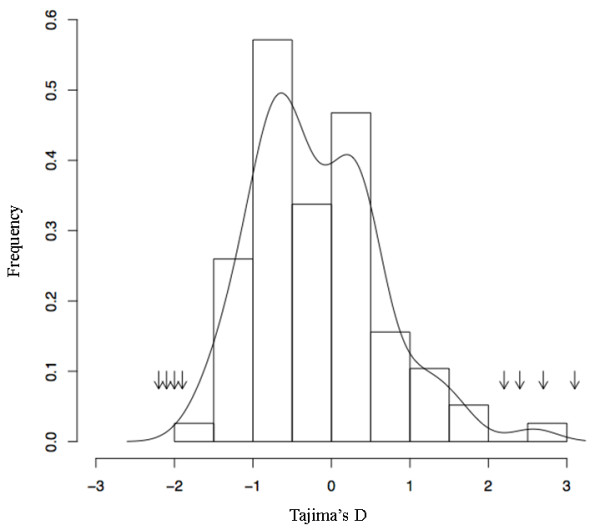
Distribution in cultivated grapevines of the Tajima's D value calculated from the 77 genes randomly distributed across the genome. The arrows correspond to the Tajima's D value from the eight gene fragments in the flb region with a significant deviation of the Tajima's D value from neutrality.
Analysis of the nucleotide diversity along the flb region in a set of cultivated and wild Vitis vinifera genotypes
The 69 gene fragments from the flb region and the 77 random gene fragments spread all over the genome were sequenced in seven diverse wild Vitis vinifera genotypes, in order to compare the nucleotide diversity in the cultivated and wild pools of genotypes. The diversity parameters calculated for each fragment in the two different subsets of individuals, are presented in additional files 3 and 5 and summarized in Table 4. All the indicators of genetic diversity (number of segregating sites, number of haplotypes, and nucleotide diversity: π) were higher in average (roughly doubled pi = 0.0020 vs 0.0041; [additional file 5]) in the whole sample of domesticated genotypes in comparison to the sample of wild genotypes in the flb region. This hold true when each of the wine and table grape sub-compartments of cultivated grapes were compared with the wild compartment, with less unbalanced numbers of individuals in each pairwise comparison (Table 3). Compared to a similar number of re-sequenced fragments spread all over the genome, there was a slightly lower diversity among the wild genotypes in the flb region than in the rest of the genome, which was not the case in the cultivated compartment (Table 4). Moreover, we observed very few specific segregating sites between the wild and the cultivated compartment in the flb region (out of 554 SNP sites, only six were specific to the wild compartment; Figure 4; [additional file 5]). Nucleotide diversity varied along the flb region, also depending on the pool of genotypes considered (Figure 5; [additional file 5; additional file 6] and was locally higher in the cultivated compartment than in the wild compartment (Figure 5). This probably reflected the fact that 18 out of 69 fragments showed no sequence polymorphism among the wild genotypes [additional file 5], whereas only one fragment was monomorphic in the domesticated compartment (VV05795A). This was not the case for the 77 random fragments [additional file 3]. In addition, we found that the wine cultivar Orbois, like Ugni Blanc, was completely homozygous specifically in the flb region (data not shown).
Table 4.
Summary of the sequence polymorphism observed in cultivated and wild V. vinifera genotypes for 69 sequence fragments along 948 kb in the flb region and for 77 sequence fragments spread along the whole genome.
| Average number of genotypes/fragment | Average number of segregating sites/fragment | Average number of haplotypes/fragment | Average and standard deviation of π |
||
|---|---|---|---|---|---|
| Wild (n = 7) | |||||
| Flb region | 5.9 | 2.8 | 2.2 | 0.0020 +/- 0.0006 | |
| Whole genome | 6.7 | 4.8 | 3.6 | 0.0027 +/- 0.0025 | |
| Cultivated (n = 26) | |||||
| Flb region/Wine (n = 15) | 10.5 | 6.2 | 4.3 | 0.0035 +/- 0.0007 | |
| Flb region/Table (n = 11) | 12.5 | 6.6 | 4.7 | 0.0035 +/- 0.0007 | |
| Flb region/Wine + Table | 24.8 | 8 | 5.8 | 0.0041 +/- 0.0004 | |
| Whole genome/Wine + Table | 27.0 | 10.1 | 8.3 | 0.0035 +/- 0.0023 | |
Figure 4.
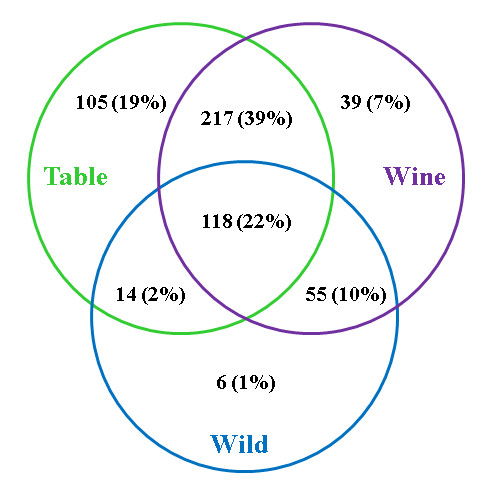
SNP from the flb region in wild and cultivated grapevines. Venn diagram showing the distribution of the 554 non-redundant SNPs found in the 948 kb region at the top of chromosome 18 in the sets of wild and domesticated table and wine V. vinifera genotypes.
Figure 5.
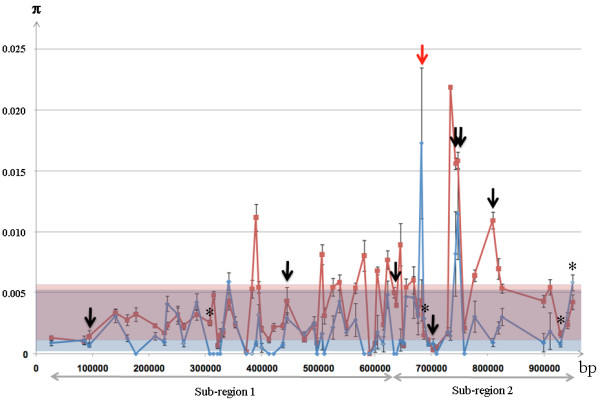
Nucleotide diversity in wild and cultivated grapes along the flb region. Nucleotide diversity (π) in wild (blue line) and cultivated grapes (red line) along the flb region. The standard deviation of the π parameter in the whole genome is represented by a blue and red box for wild and cultivated genotypes respectively. Genes under selection in the cultivated pool of genotypes are indicated with black arrows and the gene under purifying selection showing higer diversity in wild genotypes than in cultivated genotypes with red arrows. The two sub-regions with regard to LD patterns are underlined with grey arrows. Gene fragments with SNP significantly associated with berry weight variation are highlighted with a star.
Under the hypothesis that flb was one of the genes under selection during grape domestication, we expected to find traces of selection in the cultivated compartment associated with a difference of nucleotide diversity between the cultivated and wild compartments. Eight sequenced gene fragments in the flb region were particularly interesting because they showed such possible traces of selection in the cultivated pool of genotypes (previous paragraph; Table 3). Four out of the eight gene fragments showed differences in nucleotide diversity between the two compartments (VV05791A, VVC2897A, VVC2901A and VVC2901A; Table 3). However, the wild Vitis vinifera sample showing over all the genome a lower diversity than the cultivated Vitis vinifera sample, we could conclude to a significant nucleotide diversity difference between wild and cultivated compartment only in the case where there was a decreasing of nucleotide diversity in the cultivated sample in comparison to the wild sample. Only one out of eight gene fragments (VVC2897A) showed such significant higher nucleotide diversity (π) in the wild compartment compared to the cultivated compartment. This gene encodes a putative glyceraldehyde-3-phospho-dehydrogenase (Table 3). VVC2897A was re-sequenced in Ugni Blanc and Ugni Blanc mutant, showing no polymorphism in the part of the coding region they contained (data not shown).
Flb region showed significant LD and a possible association with berry size variation
In order to check if there was linkage disequilibrium (LD) between the genes possibly under selection, LD was evaluated along the entire flb region. Two sub-regions were highlighted [additional file 7]. The first one, close to the telomere, contained two out of the eight genes possibly under selection (VVC2981A and VVC2946A), showed lower nucleotide diversity (Figure 5) and several gene fragments with no SNP in the wild pool. Moreover, in this sub-region, few significant LD was observed between the different gene fragments in both cultivated and wild pools [additional file 7]. The second sub-region contained six out of eight genes possibly under selection and showed high nucleotide diversity and a significant LD between and within some gene fragments in the cultivated and wild pools (Figure 5 and 6). Most of the SNPs found in the four out of the six genes possibly under selection showed intragenic LD, in the cultivated pool, and for two of them (VVC2901A and VVC2885A), an intergenic LD was found and extended with the adjacent gene fragment VV05782A (Figure 6). In the wild pool, only five out of the six gene fragments possibly under selection were polymorphic and could be used for the estimation of the LD in the second sub-region. Three of them showed intragenic and (excepted for VVC2897A) intergenic LD, together and with the gene fragment VV05782A as for the cultivated pool. Finally, the only gene possibly under selection showing significant nucleotide diversity difference between the two pools (VVC2897A) showed strong intragenic LD and with four adjacent gene fragments (VV05786A, VVC2907A, VV05785A and VVC2903A).
Figure 6.
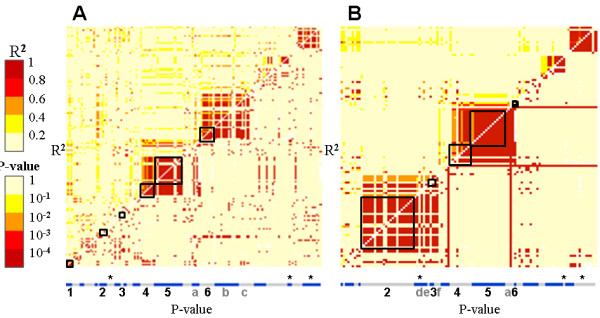
Linkage disequilibrium along the second flb sub-region in the cultivated and wild compartments. LD plots on R2 values (above the diagonal) and associated P-value (below the diagonal) along the second sub-region containing the 4 gene fragments under selection in the cultivated (A) and wild (B) compartments. The gene fragments re-sequenced are represented by alternate grey and blue boxes, which size is proportional to the number of polymorphic SNP used in the LD estimation. The gene fragments under selection are in black boxes and numbered as follows: "1" for VV05791A, "2" for VVC2897A, "3" for VV05785A, "4" for VVC2901A, "5" for VVC2885A and "6" for VVC2892A. The gene fragments in LD with these genes are pointed with small letter "a" for VV05782A, "b" for VV05781A, "c" for VV05780A, "d" for VV05786A, "e" for VVC2907A and "f" for VVC2891A. Gene fragments with SNP significantly associated with berry weight variation are highlighted with a star.
We searched for associations in the set of cultivated genotypes between the average weight of mature berries and the 447 out of 554 SNPs from the flb region with an allelic frequency >0.05. Such significant associations (Figure 7; [additional file 8]) were detected for four SNPs in four gene fragments listed in the Table 5. None of them corresponded to the genes showing a significant deviation from neutrality of the Tajima's D parameter. However, a significant association was found with a non synonymous SNP from a gene fragment (VV05786A; Table 5) showing LD with the only gene fragment possibly under selection with a high nucleotide diversity in the wild pool than in the cultivated pool, VVC2897A.
Figure 7.
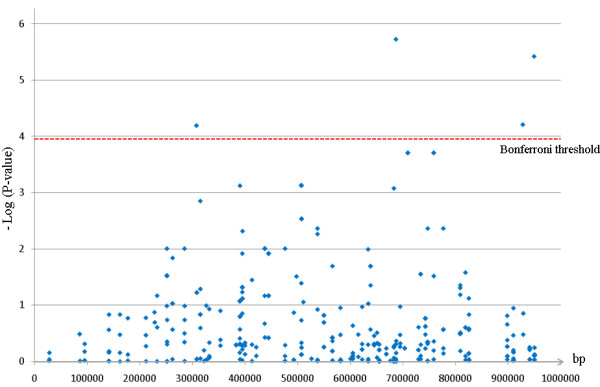
Association tests for berry weight. Level of association between SNP markers and the average berry weight along the flb region, in the cultivated V. vinifera sample. The Bonferroni threshold is equal to 1.12E-4.
Table 5.
SNPs significantly associated with the variation of berry weight in the cultivated and wild pool of Vitis vinifera.
| Fragment | Position of the SNP | P-value$ | Feature | SNP type | SNP frequency | Putative function |
|---|---|---|---|---|---|---|
| VVC2966A | 307,938 | 6.44E-05 | CDS | [A/T] | 5.00% | Unknown |
| VV05786A | 687,166 | 1.87E-06 | CDS | [A/G]* | 9.00% | Protein kinase |
| VV05777A | 928,516 | 6.18E-05 | Intron | [T/G] | 8.00% | Catalase |
| VV05775A | 949,972 | 3.78E-06 | Intron | [A/T] | 9.00% | Ribosome biogenesis protein |
$ Significant P-value after Bonferroni correction (for α = 0.05)
* Non-synonymous mutation
Discussion
With an initial objective to develop markers tightly flanking the flb mutation, 62 genomic regions were scanned for polymorphism along a 1.4 Mb region at the top of chromosome 18, where the mutation was previously located [21]. These regions were either genotyped or sequenced in the genotype carrying the mutation, Ugni Blanc mutant, its wild type (Ugni Blanc) and Chardonnay, which was the other parent of a full sib family segregating for the mutation. The sequenced fragments or markers were completely homozygous in Ugni Blanc and Ugni Blanc mutant, with one marker analyzed each 23 kb in average. Indeed, while analyzing the genome sequence of the heterozygous grapevine cultivar Pinot Noir, Velasco et al [43] showed that, like in other heterozygous species, the frequency of SNPs or INDELs varied along the grapevine genome and found some evidence for scarce quasi-homozygous areas. Here we describe a region of 1 Mb probably completely homozygous that raised two questions. First, as Velasco et al [43] showed that over 65 Mb of sequence are hemizygous in Pinot Noir, we wanted to check if our observations were due to a real homozygosity or to a deletion of a large portion of the top of chromosome 18 in one haplotype of Ugni Blanc. We addressed this issue by two different experiments (Figure 2), a qPCR estimation of the number of copies of a single gene in the homozygous area (FL) compared to genes elsewhere in the genome (three HMGCoA genes). The same number of copies was estimated for FL gene for Ugni Blanc mutant and Chardonnay which is heterozygous in this region. Second, a BAC-FISH hybridization on Ugni Blanc mutant metaphase chromosomes using a BAC clone located in the area was carried out and showed a signal on both homologous chromosomes. We therefore un-ambiguously demonstrated that our observations corresponded to a real homozygosity in Ugni Blanc mutant. This would be consistent with the fact that hemizygous regions identified by Velasco et al [43] would mainly correspond to stretches of repeated sequences, which is not the case of the flb region. These results raised the question whether this high level of homozygosity in Ugni Blanc mutant was restricted to the top of chromosome 18. The scoring of 480 SNPs [44] and 20 SSR ([45], V. Laucou personal communication) regularly spread along the genome showed that whereas this cultivar seems slightly more homozygous in average than for instance Cabernet Sauvignon, Syrah or Chardonnay, the near complete homozygosity observed in the flb region in Ugni Blanc mutant is not the rule on the rest of the genome and may be restricted to this region only. A mechanism which could explain the formation of such large homozygous region in a highly heterozygous out-crosser like grapevine would involve the repair of a DNA double-strand break [46]. When analyzing diversity in the cultivated germplasm, we observed that the cultivar Orbois was also completely homozygous for all fragments re-sequenced at the top of chromosome 18, and confirmed by qPCR assay that it was also due to real homozygosity (data not shown). Whatever its origin, this unexpected result made impossible the fine mapping of the mutation in the available segregating F1 population, which would necessitate the development of a F2 population.
Before having such a population available, we tested another possibility for reducing the interval carrying the flb gene, based on the fact that flb could be a gene selected during grape domestication. Indeed, the berry and seed phenotypes of Ugni Blanc mutant look like the phenotypes of wild V. vinifera seeds and berries [20,22]. We searched for signatures of selection in the flb region in a set of cultivated genotypes. For this purpose, we sequenced in 33 individuals (26 cultivated and 7 wild genotypes) (i) 69 gene fragments for a total of 34,355 kb along 948 kb in the flb region and (ii) 77 additional, totaling 48,874 kb spread along the 19 grapevine chromosomes. As already observed by Vezzulli et al [41], the nucleotide diversity was lower in average in the set of wild genotypes (π = 0.27) than in the set of cultivated genotypes (π = 0.35). This difference was increased in the flb region probably by the fact that a quarter of the fragments showed no sequence polymorphism at all among the wild genotypes whereas only one was in this case in the domesticated compartment. In the present work (but not in Vezzulli et al [42]) the sample of domesticated genotypes was selected after a comprehensive analysis of the world-wide largest collection of domesticated grapevine accessions [23] with the aim to retain a maximum of diversity. Unlike in Vezzulli et al [42], very few specific SNPs were found in the set of wild genotypes (six SNPs out of 554). All these observations, opposite to what has been observed in many other species [47,48], could be due to the fact that only seven to ten wild genotypes were sequenced in both studies and that their choice could not be driven by a maximization of the diversity along their complete natural area of growth. However, several surveys including accessions from the wild V. vinifera germplasm also showed this overall lower genetic diversity compared to the cultivated germplasm [49-51]. Indeed, small population sizes [52,53] as well as dioecy [54] could explain the observed reduced diversity in the wild V. vinifera gene pool, while multiple domestication events [50] and a continuous breeding process the larger diversity in the cultivated gene pool. All these results including ours will have to be confirmed with a larger sample of wild V. vinifera, taking into account all the geographic area where it grows and its overall genetic diversity.
Putative signatures of selection (Tajima's D parameter significantly deviating from neutrality associated with differences between the cultivated and wild pools in sequence diversity) in the cultivated pool of genotypes and SNPs showing a significant association with berry weight variation were found in the flb region (Figure 5). Moreover, LD was found in this region, the genes under selection presenting intragenic LD and intergenic LD with nearby genes (Figure 6), which strengthened the hypothesis that they might be under selection [48]. Like in Fournier-Level et al [55], no LD was however observed between the six out of eight genes possibly under selection. It is well known that the LD extent varies between the organisms and genomic regions [56]. In grapevine, a recent preliminary genome wide study confirmed the low extent of LD in average (less than 3-10 kb), suggesting a large effective size in the grapevine population at the origin of the current domesticated pool [57]. However, in recent history, vegetative propagation and long intervals between generations may have reduced the impact of recombination, maintaining extensive linkage disequilibrium in some regions under selection [58].
The significant genetic associations found between berry weight variation and SNPs in this region were not found in the fragments putatively under selection. It is still possible that one of these genes is involved into the berry weight variation and that the causative sequence polymorphism was not in the exon fragments sequenced. Indeed, trait variation is often due to sequence variation in regulatory regions (see for instance [9,11,55]) and such variant sites may be more tightly linked to a neighbor gene. Interestingly, the orthologs in Aradidopsis for the six out of eight gene with signature of selection (At1g76540 for VVC2946A, At1g76400 for VV05791A, At1g42970 for VVC2897A, At1g42540 for VV05785A, At5g43820 for VVC2901 and At1g20696 for VVC2892A) and for the two genes with SNPs associated with berry weight variation (At5g24306 for VV05786A and At1g42440 for VV05775A), are all expressed in flowers and showed a peak of expression at the beginning or during the flowering (http://urgv.evry.inra.fr/projects/FLAGdb++/HTML/index.shtml). In tomato, the genes involved into fruit size variation have been shown to be expressed in very early stages of its development, starting at floral development [2,11]. Finally, only one gene fragment, VVC2897A, might be under purifying selection in the cultivated pool with a major haplotype in cultivated pool and a LD extended to a neighbor gene which present a SNP significantly associated with berry weight variation (Figure 5 Figure 6). This would fit with the hypothesis that, like in tomato, the selection of a new haplotype by humans would have ensured the transition from berries with little flesh in wild grapevines to berries with more flesh in cultivated grapevines [2,11]. The real involvement of this gene into berry size variation and in the domestication syndrome remains however to be proven.
Conclusions
While searching for SNP markers in coupling with the fleshless berry mutation, we observed the occurrence of a 1 Mb homozygous region, not associated with repetitive sequences, in the grapevine otherwise highly heterozygous genome. We demonstrated the feasibility to use BAC-FISH on the very small grapevine chromosomes and provided a specific probe for the identification of chromosome 18 on cytogenetic map. Using this method, we showed that the observed homozygosity was not due to a large deletion.
We then searched for signatures of domestication for berry weight along the flb region by re-sequencing 69 gene fragments in 26 domesticated and seven wild V. vinifera genotypes. We found putative signatures of selection associated with significant differences in nucleotide diversity between the cultivated and the wild pool only in one gene (VVC2897A) and also SNPs significantly associated with berry weight variation in three other genes among which one is in DL with VVC2897A. The involvement of these four genes into berry weight variation in grapevine remains to be proved by further functional experiments. In addition, we detected 554 SNPs along the flb region. These polymorphisms could serve to develop a genotyping chip useful for a future fine mapping of the flb gene in a F2 population and for the analysis of genetic diversity in larger sets of wild and cultivated genotypes.
Authors' contributions
CH designed the primers for fragment re-sequencing, participated to their sequencing, analyzed the results, chose the BAC and prepared the root tips for the FISH, draft and corrected the paper. RB, M-CLP and DB were responsible for the sequencing of the fragments. RB and AC participated to the sequence analysis and made the RT-PCR experiment. JC and LT analyzed the SSR and genotyped SNP polymorphism. CG and AD helped with bio-informatics (scripts, database queries...). J-PP and RB generated the set of reference fragment sequences along the genome. PT, TL, and AND provided the DNA for the core-collection and plant phenotypes. M-SV and OC did the BAC-FISH experiments. A-FAB design the experiment, supervised it, drafted and corrected the manuscript. All authors read and approved the final manuscript.
Supplementary Material
supplemental table S1. Plant material. List of the grapevine accessions used in the study, with their average berry weight at maturity.
supplemental table S2. Markers and sequence fragments along the flb region. Description: List and localization on the grapevine genome sequence of the gene fragments sequenced (SEQ) and of the markers used for genotyping (SSR, CAPS, SNP) together with the primers used for amplification.
supplemental table S3. Sequence fragments randomly spread along the genome. List and localization on the grapevine genome sequence of the random gene fragments sequenced spread all over the genome, diversity and Tajima's D parameters obtained in the cultivated and the wild pools of Vitis vinifera.
supplemental table S4. qPCR validation of homozygosity in the flb region in Ugni Blanc mutant. Estimation of the initial number of DNA quantity of the FL gene and of the HMGCoA gene family in Pinot Noir (PN777), Chardonnay (CHA), Ugni Blanc mutant (UBM) and Ugni Blanc (UB), before and after normalization by the result obtained for the HMGCoA genes.
supplemental table S5. Sequence diversity along the flb region in cultivated and wild grapevines. Parameter of diversity obtained for the 69 genome fragments at the top of chromosome 18 in the cultivated and the wild compartment of Vitis vinifera and details of the shared and specific insertions/deletions (INDELs) and segregating SNP sites in wild V. vinifera genotypes and table and wine cultivars of domesticated V. vinifera, numbers of shared and unshared SNPs or INDELs between wild and cultivated genotypes are also indicated.
supplemental figure S1. Nucleotide diversity in the cultivated (table and wine) and wild compartments along the flb region. Nucleotide diversity (π) in the table grapes (green line), the wine grapes (purple line) and the wild grapes (blue line) along the flb region.
supplemental figure S2. Linkage disequilibrium along the flb region in cultivated and wild compartments. LD plots on R2 values (above the diagonal) and associated P-value (below the diagonal) along the entire flb region in cultivated (A) and wild compartments (B). The gene fragments re-sequenced are represented by alternate grey and blue boxes, which size is proportional to the number of polymorphic SNP used in the LD estimation. The black arrow represents the orientation of the region from the telomere (on the left) to the centromere.
supplemental table S6. List of gene fragments and their associated number of SNPs used for the estimation of LD in the flb region, in cultivated and wild V. vinifera pools.
Contributor Information
Cléa Houel, Email: houel@evry.inra.fr.
Rémi Bounon, Email: bounon@evry.inra.fr.
Jamila Chaïb, Email: jamila.chaib@csiro.au.
Cécile Guichard, Email: guichard@evry.inra.fr.
Jean-Pierre Péros, Email: peros@supagro.inra.fr.
Roberto Bacilieri, Email: bacilieri@supagro.inra.fr.
Alexis Dereeper, Email: dereeper@supagro.inra.fr.
Aurélie Canaguier, Email: canaguier@evry.inra.fr.
Thierry Lacombe, Email: lacombe@supagro.inra.fr.
Amidou N'Diaye, Email: ndiaye@supagro.inra.fr.
Marie-Christine Le Paslier, Email: lepaslier@cng.fr.
Marie-Stéphanie Vernerey, Email: vernerey@supagro.inra.fr.
Olivier Coriton, Email: coriton@rennes.inra.fr.
Dominique Brunel, Email: brunel@cng.fr.
Patrice This, Email: this@supagro.inra.fr.
Laurent Torregrosa, Email: torregrosa@supagro.inra.fr.
Anne-Françoise Adam-Blondon, Email: adam@evry.inra.fr.
Aknowledgements
This study was supported by INRA, ANR and the French Ministry of Research. We thank Isabelle Le Clainche, Audrey Weber, Sylvain Santoni, Christophe Lepage and all the team of the Vassal germplasm collection for technical assistance; Stéphane Nicolas, Aurélie Siberchicot, Christine Cierco and Brigitte Mangin for providing a R routine for LD calculation and helpful discussions; Marie-Laure Martin Magniette for help in the statistical analysis, Dr Mark R. Thomas from the CSIRO Plant Industry, Australia for providing unpublished DNA marker information and Loïc Le Cunff for helpful discussions.
References
- Champagnol F. In: Oenologie, Fondements Scientifiques et Technologiques. Flanzy C, editor. Paris: Lavoisier Tec Do; 1998. Critères de qualité de la vendange; pp. 653–659. [Google Scholar]
- Tanksley SD. The genetic, developmental, and molecular bases of fruit size and shape variation in tomato. Plant Cell. 2004;16:181–189. doi: 10.1105/tpc.018119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano R, Giménez E, Cara B, Capel J, Angosto T. Genetic analysis of reproductive development in tomato. Int J Dev Biol. 2009;53:1635–1648. doi: 10.1387/ijdb.072440rl. [DOI] [PubMed] [Google Scholar]
- Fridman E, Carrari F, liu Y-S, Fernie A, Zamir D. Zooming in on a quantitative trait for for tomato yield using interspecific introgressions. Science. 2004;305:1786–1789. doi: 10.1126/science.1101666. [DOI] [PubMed] [Google Scholar]
- Causse M, Duffe P, Gomez M-C, Buret M, Damidaux R, Zamir D, Gur A, Chevalier C, Lemaire-Chamley M, Rothan C. A genetic map of candidate genes and QTLs involved in tomato fruit size and composition. J Exp Bot. 2004;55:1671–1685. doi: 10.1093/jxb/erh207. [DOI] [PubMed] [Google Scholar]
- Lippman ZB, Semel Y, Zamir D. An integrated view of quantitative trait variation using tomato interspecific introgression lines. Curr Opin Genet & Dev. 2007;17:545–552. doi: 10.1016/j.gde.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Chaïb J, Lecomte L, Buret M, Causse M. Stability over genetic backgrounds, generations and years of quantitative trait locus (QTLs) for organoleptic quality in tomato. Theor Appl Genet. 2006;112:934–944. doi: 10.1007/s00122-005-0197-7. [DOI] [PubMed] [Google Scholar]
- Menda N, Semel Y, Peled D, Eshed Y, Zamir D. In silico screening of a saturated mutation library of tomato. The Plant J. 2004;38:861–872. doi: 10.1111/j.1365-313X.2004.02088.x. [DOI] [PubMed] [Google Scholar]
- Frary A, Nesbitt TC, Frary A, Grandillo S, van der Knaap E, Cong B, Liu J, Meller J, Elber R, Alpert KB, Tanksley SD. fw2.2: A Quantitative Trait Locus key to the evolution of tomato fruit size. Science. 2000;289:85–88. doi: 10.1126/science.289.5476.85. [DOI] [PubMed] [Google Scholar]
- Bertin N, Causse M, Brunel B, Tricon D, Genard M. Identification of growth processes involved in QTLs for tomato fruit size and composition. J Exp Bot. 2009;60:237–248. doi: 10.1093/jxb/ern281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong B, Barrero LS, Tanksley SD. Regulatory change in YABBY-like transcription factor led to evolution of extreme fruit size during tomato domestication. Nat Genet. 2008;40:800–804. doi: 10.1038/ng.144. [DOI] [PubMed] [Google Scholar]
- Cheniclet C, Rong WY, Causse M, Frangne N, Bolling L, Carde J-P, Renaudin J-P. Cell expansion and endoreduplication show a large genetic variability in pericarp and contribute strongly to tomato fruit growth. Plant Physiol. 2005;139:1984–1994. doi: 10.1104/pp.105.068767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- This P, Lacombe T, Thomas MR. Historical origins and genetic diversity of wine grapes. Trends Genet. 2006;22:511–519. doi: 10.1016/j.tig.2006.07.008. [DOI] [PubMed] [Google Scholar]
- Boursiquot JM, Dessup M, Rennes C. Distribution des principaux caractères phénologiques, agronomiques et technologiques chez Vitis vinifera L. Vitis. 1995;34:31–35. [Google Scholar]
- Ollat N, Diakou-Verdin P, Carde J-P, Barrieu F, Gaudillère J-P, Moing A. Grape berry development: a review. J Int Sci Vigne Vin. 2002;36:109–131. [Google Scholar]
- Doligez A, Bouquet A, Danglot Y, Lahogue F, Riaz S, Meredith P, Edwards J, This P. Genetic mapping of grapevine (Vitis vinifera L.) applied to the detection of QTLs for seedlessness and berry weight. Theor Appl Genet. 2002;105:780–795. doi: 10.1007/s00122-002-0951-z. [DOI] [PubMed] [Google Scholar]
- Cabezas JA, Cervera MT, Ruiz-Garcia L, Carreno J, Martinez-Zapater JM. A genetic analysis of seed and berry weight in grapevine. Genome. 2006;49:1572–1585. doi: 10.1139/G06-122. [DOI] [PubMed] [Google Scholar]
- Mejia N, Gebauer M, Muñoz L, Hewstone N, Muñoz C, Hinrichsen P. Identification of QTLs for seedlessness, berry size, and ripening date in a seedless × seedless progeny. Am J Enol Vitic. 2007;58:499–507. [Google Scholar]
- Costantini L, Battilana J, Lamaj F, Fanizza G, Grando M. Berry and phenology-related traits in grapevine (Vitis vinifera L.) From Quantitative Trait Loci to underlying genes. BMC Plant Biology. 2008;17:8–38. doi: 10.1186/1471-2229-8-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez L, Romieu C, Moing A, Bouquet A, Maucourt M, Thomas MR, Torregrosa L. The grapevine fleshless berry mutation. A unique genotype to investigate differences between fleshy and nonfleshy fruit. Plant Physiol. 2006;140:537–547. doi: 10.1104/pp.105.067488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez L, Doligez A, Lopez G, Thomas MR, Bouquet A, Torregrosa L. Somatic chimerism, genetic inheritance, and mapping of the fleshless berry (flb) mutation in grapevine (Vitis vinifera L.) Genome. 2006;49:721–728. doi: 10.1139/G06-034. [DOI] [PubMed] [Google Scholar]
- Terral J-F, Tabard E, Bouby L, Ivorra S, Pastor T, Figueiral I, Picq S, Chevance J-B, Jung C, Fabre L, Tardy C, Compan M, Bacilieri R, Lacombe T, This P. Evolution and history of grapevine (Vitis vinifera) under domestication: new morphometric perspectives to understand seed domestication syndrome and reveal origins of ancient European cultivars. Annals of Botany. 2010;105:443–55. doi: 10.1093/aob/mcp298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecunff L, Fournier-Level A, Laucou V, Vezzulli S, Lacombe T, Adam-Blondon A-F, Boursiquot J-M, This P. Construction of nested core collections to optimize the exploitation of natural diversity in Vitis vinifera L. subsp sativa. BMC Plant Biology. 2008;2:8–31. doi: 10.1186/1471-2229-8-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaillon O, Aury J-M, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C, Vezzi A, Legeai F, Hugueney P, Dasilva C, Horner D, Mica E, Jublot D, Poulain J, Bruyere C, Billault A, Segurens B, Gouyvenoux M, Ugarte E, Cattonaro F, Anthouard V, Vico V, Del Fabbro C, Alaux M, Di Gaspero G, Dumas V, Felice N, Paillard S, Juman I, Moroldo M, Scalabrin S, Canaguier A, Le Clainche I, Malacrida G, Durand E, Pesole G, Laucou V, Chatelet P, Merdinoglu D, Delledonne M, Pezzotti M, Lecharny A, Scarpelli C, Artiguenave F, Pé E, Valle G, Morgante M, Caboche M, Adam-Blondon A-F, Weissenbach J, Quétier F, Wincker P. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007;449:463–468. doi: 10.1038/nature06148. [DOI] [PubMed] [Google Scholar]
- Rozen S, Skaletsky HJ. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–86. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- Philippe R, Courtois B, McNally KL, Mournet P, El-Malki R, Le Paslier MC, Fabre D, Billot C, Brunel D, Glaszmann JC, This D. Structure, allelic diversity and selection of Asr genes, candidate for drought tolerance, in Oryza sativa L. and wild relatives. Theor Appl Genet. 2010;121:769–787. doi: 10.1007/s00122-010-1348-z. [DOI] [PubMed] [Google Scholar]
- Salmaso M, Malacarne G, Troggio M, Faes G, Stefanini M, Grando MS, Velasco R. A grapevine (Vitis vinifera L.) genetic map integrating the position of 139 expressed genes. Theor Appl Genet. 2008;116:1129–1143. doi: 10.1007/s00122-008-0741-3. [DOI] [PubMed] [Google Scholar]
- Thomas MR, Cain P, Scott NS. DNA Typing of Grapevines. A universal methodology and database for describing cultivars and evaluating genetic relatedness. Plant Mol Biol. 1994;25:939–949. doi: 10.1007/BF00014668. [DOI] [PubMed] [Google Scholar]
- Reid KE, Olsson N, Schlosser J, Peng F, Lund ST. An optimized grapevine RNA isolation procedure and statistical determination of reference genes for real-time RT-PCR during berry development. BMC Plant Biology. 2006;14:6–27. doi: 10.1186/1471-2229-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leflon M, Eber F, Letanneur JC, Chelysheva L, Coriton O, Huteau V, Ryder CD, Barker G, Jenczewski E, Chevre AM. Pairing and recombination at meiosis of Brassica rapa (AA) × Brassica napus (AACC) hybrids. Theor Appl Genet. 2006;113:1467–1480. doi: 10.1007/s00122-006-0393-0. [DOI] [PubMed] [Google Scholar]
- Gerlach W, Bedbrook J. Cloning and characterization of ribosomal RNA genes from wheat and barley. Nucl Acids Res. 1979;7:1869–1885. doi: 10.1093/nar/7.7.1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Matsuda F, Margetic N, Lathrop M. Automated identification of single nucleotide polymorphisms from sequencing data. Journal of Bioinformatics and Computational Biology. 2003;1:253–265. doi: 10.1142/S021972000300006X. [DOI] [PubMed] [Google Scholar]
- Staden R. The Staden Sequence Analysis Package. Molecular Biotechnology. 1996;5:233–241. doi: 10.1007/BF02900361. [DOI] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roger AR, Huff C. Linkage disequilibrium between loci with unknown phase. Genetics. 2009;182:839–844. doi: 10.1534/genetics.108.093153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam-Blondon AF, Roux C, Claux D, Butterlin G, Merdinoglu D, This P. Mapping 245 SSR markers on the Vitis vinifera genome: a tool for grape genetics. Theor Appl Genet. 2004;109:1017–1027. doi: 10.1007/s00122-004-1704-y. [DOI] [PubMed] [Google Scholar]
- Di Gaspero G, Cipriani G, Adam-Blondon AF, Testolin R. Linkage maps of grapevine displaying the chromosomal locations of 420 microsatellite markers and 82 markers for R-gene candidates. Theor Appl Genet. 2007;114:1249–1263. doi: 10.1007/s00122-007-0516-2. [DOI] [PubMed] [Google Scholar]
- Lamoureux D, Bernole A, Le Clainche I, Tual S, Thareau V, Paillard S, Legeai F, Dossat C, Wincker P, Oswald M, Merdinoglu D, Vignault C, Delrot S, Caboche M, Chalhoub B, Adam-Blondon AF. Anchoring of a large set of markers onto a BAC library for the development of a draft physical map of the grapevine genome. Theor Appl Genet. 2006;113:344–356. doi: 10.1007/s00122-006-0301-7. [DOI] [PubMed] [Google Scholar]
- Riaz S, Dangl GS, Edwards KJ, Meredith CP. A microsatellite marker based framework linkage map of Vitis vinifera L. Theor Appl Genet. 2004;108:864–872. doi: 10.1007/s00122-003-1488-5. [DOI] [PubMed] [Google Scholar]
- Troggio M, Malacarne G, Coppola G, Segala C, Cartwright DA, Pindo M, Stefanini M, Mank R, Moroldo M, Morgante M, Grando MS, Velasco R. A dense single-nucleotide polymorphism-based genetic linkage map of grapevine (Vitis vinifera L.). Anchoring Pinot Noir Bacterial Artificial Chromosome Contigs. Genetics. 2007;176:2637–2650. doi: 10.1534/genetics.106.067462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzulli S, Micheletti D, Riaz S, Pindo M, Viola R, This P, Walker MA, Troggio M, Velasco R. A SNP transferability survey within the genus Vitis. BMC Plant Biology. 2008;16:8–128. doi: 10.1186/1471-2229-8-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco R, Zharkikh A, Troggio M, Cartwright DA, Cestaro A, Pruss D, Pindo M, Fitzgerald LM, Vezzulli S, Reid J, Malacarne G, Iliev D, Coppola G, Wardell B, Micheletti D, Macalma T, Facci M, Mitchell JT, Perazzolli M, Eldredge G, Gatto P, Oyzerski R, Moretto M, Gutin N, Stefanini M, Chen Y, Segala C, Davenport C, Demattè L, Mraz A, Battilana J, Stormo K, Costa F, Tao Q, Si-Ammour A, Harkins T, Lackey A, Perbost C, Taillon B, Stella A, Solovyev V, Fawcett JA, Sterck L, Vandepoele K, Grando SM, Toppo S, Moser C, Lanchbury J, Bogden R, Skolnick M, Sgaramella V, Bhatnagar SK, Fontana P, Gutin A, Van de Peer Y, Salamini F, Viola R. A high quality draft consensus sequence of the genome of a heterozygous grapevine variety. PLoS ONE. 2007;2:e1326. doi: 10.1371/journal.pone.0001326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaïb J, Torregrosa L, Mackenzie D, Corena1 P, Bouquet A, Thomas MR. The grape microvine - a model system for rapid forward and reverse genetics of grapevines. The Plant J. 2010;62:1083–1092. doi: 10.1111/j.1365-313X.2010.04219.x. [DOI] [PubMed] [Google Scholar]
- Di Vecchi-Staraz M, Bandinelli R, Boselli M, This P, Boursiquot JM, Laucou V, Lacombe T, Vares D. Genetic structuring and parentage analysis for evolutionary studies in grapevine: Kin group and origin of the cultivar Sangiovese revealed. Journal of the American Society for Horticultural Science. 2007;132:514–524. [Google Scholar]
- Puchta H. The repair of double-strand breaks in plants: mechanisms and consequences for genome evolution. J Exp Bot. 2005;56:1–14. doi: 10.1093/jxb/eri123. [DOI] [PubMed] [Google Scholar]
- Miller JC, Tankley SD. RFLP analysis of phylogenetic relationships and genetic variation in the genus Lycopersicon. Theor Appl Genet. 1990;80:437–448. doi: 10.1007/BF00226743. [DOI] [PubMed] [Google Scholar]
- Palaisa KA, Morgante M, Williams M, Rafalski A. Long-range patterns of diversity and linkage disequilibrium surrounding the maize Y1 gene are indicative of an asymmetric selective sweep. PNAS. 2004;101:9885–9890. doi: 10.1073/pnas.0307839101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aradhya MK, Dangl GS, Prins BH, Boursiquot J-M, Walker MA, Meredith CP, Simon CJ. Genetic structure and differentiation in cultivated grape, Vitis vinifera L. Genet Res Camb. 2003;81:179–192. doi: 10.1017/S0016672303006177. [DOI] [PubMed] [Google Scholar]
- Arroyo-Garcia R, Ruiz-Garcia L, Bolling L, Ocete R, Lopez MA, Arnold C, Ergul A, Söylemezoglu G, Uzun HJ, Cabello F, Ibanez J, Aradhya MK, Atanassov I, Balint S, Cenis JL, Costantini L, Gorislavets S, Grando MS, Klein BY, Govern PEM, Merdinoglu D, Pejic I, Pelsy F, Primikirios N, Risovannaya V, Roubelakis-Angelakis KA, Snoussi H, Sotiri P, Tamhankar S, This P, Troshin L, Malpica JM, Lefort F, Martinez-Zapater JM. Multiple origins of cultivated grapevine (Vitis vinifera L. ssp. sativa) based on chloroplast DNA polymorphisms. Molecular Ecology. 2006;15:3707–3714. doi: 10.1111/j.1365-294X.2006.03049.x. [DOI] [PubMed] [Google Scholar]
- Grassi F, De Mattia F, Zecca G, Sala F, Labra M. Historical isolation and Quaternary range expansion of divergent lineages in wild grapevine. Biological Journal of the Linnean Society. 2008;95:611–619. doi: 10.1111/j.1095-8312.2008.01081.x. [DOI] [Google Scholar]
- Levadoux L. Les Populations sauvages et cultivées de Vitis vinifera L. Annales de l'Amélioration de Plantes. 1956;6:59–118. [Google Scholar]
- Arnold C, Gillet F, Gobat JM. Occurrence of the wild vine Vitis vinifera ssp. silvestris in Europe. Vitis. 1998;37:159–170. [Google Scholar]
- Di Vecchi-Staraz M, Laucou V, Bruno G, Lacombe T, Gerber S, Bourse T, Boselli M, This P. Low level of pollen-mediated gene flow from cultivated to wild grapevine: consequences for the evolution of the endangered subspecies Vitis vinifera L. subsp. silvestris. J Heredity. 2009;100:66–75. doi: 10.1093/jhered/esn084. [DOI] [PubMed] [Google Scholar]
- Fournier-Level A, LeCunff L, Gomez C, Doligez A, Ageorges A, Roux C, Bertrand Y, Souquet JM, Cheynier V, This P. Quantitative genetic bases od anthocyanin variation in grape (Vitis vinifera L. ssp. sativa) berry: a quantitative trait locus to quantitative trait nucleotide integrated study. Genetics. 2009;183:1127–1139. doi: 10.1534/genetics.109.103929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta PK, Rustgir S, Kulwal PL. Linkage disequilibrium and association studies in higher plants: Present status and future prospects. Plant Molecular Biology. 2005;57:461–485. doi: 10.1007/s11103-005-0257-z. [DOI] [PubMed] [Google Scholar]
- Myles S, Chia J-M, Hurwitz B, Simon C, Zhong GY, Buckler E, Ware D. Rapid genomic gharacterization of the genus Vitis. Plos One. 2010;5:e8219. doi: 10.1371/journal.pone.0008219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnaud A, Lacombe T, Doligez A. Linkage disequilibrium in cultivated grapevine, Vitis vinifera L. Theor Appl Genet. 2009;112:708–716. doi: 10.1007/s00122-005-0174-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplemental table S1. Plant material. List of the grapevine accessions used in the study, with their average berry weight at maturity.
supplemental table S2. Markers and sequence fragments along the flb region. Description: List and localization on the grapevine genome sequence of the gene fragments sequenced (SEQ) and of the markers used for genotyping (SSR, CAPS, SNP) together with the primers used for amplification.
supplemental table S3. Sequence fragments randomly spread along the genome. List and localization on the grapevine genome sequence of the random gene fragments sequenced spread all over the genome, diversity and Tajima's D parameters obtained in the cultivated and the wild pools of Vitis vinifera.
supplemental table S4. qPCR validation of homozygosity in the flb region in Ugni Blanc mutant. Estimation of the initial number of DNA quantity of the FL gene and of the HMGCoA gene family in Pinot Noir (PN777), Chardonnay (CHA), Ugni Blanc mutant (UBM) and Ugni Blanc (UB), before and after normalization by the result obtained for the HMGCoA genes.
supplemental table S5. Sequence diversity along the flb region in cultivated and wild grapevines. Parameter of diversity obtained for the 69 genome fragments at the top of chromosome 18 in the cultivated and the wild compartment of Vitis vinifera and details of the shared and specific insertions/deletions (INDELs) and segregating SNP sites in wild V. vinifera genotypes and table and wine cultivars of domesticated V. vinifera, numbers of shared and unshared SNPs or INDELs between wild and cultivated genotypes are also indicated.
supplemental figure S1. Nucleotide diversity in the cultivated (table and wine) and wild compartments along the flb region. Nucleotide diversity (π) in the table grapes (green line), the wine grapes (purple line) and the wild grapes (blue line) along the flb region.
supplemental figure S2. Linkage disequilibrium along the flb region in cultivated and wild compartments. LD plots on R2 values (above the diagonal) and associated P-value (below the diagonal) along the entire flb region in cultivated (A) and wild compartments (B). The gene fragments re-sequenced are represented by alternate grey and blue boxes, which size is proportional to the number of polymorphic SNP used in the LD estimation. The black arrow represents the orientation of the region from the telomere (on the left) to the centromere.
supplemental table S6. List of gene fragments and their associated number of SNPs used for the estimation of LD in the flb region, in cultivated and wild V. vinifera pools.