

Abstract
RSK2 is a widely expressed serine/threonine kinase, and its activation enhances cell proliferation. Here, we report that ATF1 is a novel substrate of RSK2 and that RSK2-ATF1 signaling plays an important role in EGF-induced neoplastic cell transformation. RSK2 phosphorylated ATF1 at Ser-63 and enhanced ATF1 transcriptional activity. Docking experiments using the crystal structure of the RSK2 N-terminal kinase domain combined with in vitro pulldown assays demonstrated that eriodictyol, a flavanone found in fruits, bound with the N-terminal kinase domain of RSK2 to inhibit RSK2 N-terminal kinase activity. In cells, eriodictyol inhibited phosphorylation of ATF1 but had no effect on the phosphorylation of RSK, MEK1/2, ERK1/2, p38 or JNKs, indicating that eriodictyol specifically suppresses RSK2 signaling. Furthermore, eriodictyol inhibited RSK2-mediated ATF1 transactivation and tumor promoter-induced transformation of JB6 Cl41 cells. Eriodictyol or knockdown of RSK2 or ATF1 also suppressed Ras-mediated focus formation. Overall, these results indicate that RSK2-ATF1 signaling plays an important role in neoplastic cell transformation and that eriodictyol is a novel natural compound for suppressing RSK2 kinase activity.
Keywords: Cancer Tumor Promoter, Protein-Protein Interactions, Serine/Threonine Protein Kinase, Signal Transduction, Transformation, Cell Proliferation, RSK2-ATF1 Signaling, Chemoprevention Target, Eriodictyol
Introduction
RSK2 (ribosomal S6 kinase 2) is a member of the p90RSK protein family that is activated by ERK1/2 and PDK1 (phosphoinositide-dependent kinase 1) (1, 2). RSK2 translocates to the nucleus when activated by growth factors, peptide hormones, or neurotransmitters (3, 4). Numerous proteins, such as the cAMP response element (CRE)3-binding protein (CREB), Elk-1, histones (5–9), ATF4 (activating transcription factor 4) (10), p53 (11), and NFAT3 (12), are phosphorylated by active RSK2. Based on its broad substrate specificity, the RSK2 protein mediates many cellular processes, including proliferation and transformation, as well as the cell cycle. Our recent study provided evidence indicating that RSK2 plays an important role in cell transformation induced by tumor promoters such as EGF and 12-O-tetradecanoylphorbol-13-acetate (13). Furthermore, RSK2 knock-out mice display reduced c-Fos-dependent osteosarcoma formation through the regulation of c-Fos protein stability (14). Thus, RSK2 likely plays a key role in cell proliferation and transformation.
ATF1 is a member of the CREB family, which includes ATF1, CREB1, and the CRE modulator (15). In response to growth factors, stress signals, neurotransmitters, or other agents that elevate intracellular cAMP or Ca2+ levels, CREB family members are activated and promote the expression of numerous cellular target genes that contain CREs in their promoters (16), including proto-oncogenes such as c-fos and c-jun (17, 18) and cell cycle genes such as cyclins D and A and other genes related to cell growth, proliferation, and neuronal activities (19, 20). Phosphorylation of ATF1 at Ser-63 in its kinase-inducible domain by serine/threonine kinases enhances its transactivation activity by promoting recruitment of the coactivator CREB-binding protein/p300 (21). ATF1 is overexpressed in lymphomas and transformed lymphocytes (22), suggesting that ATF1 may contribute to the growth of these tumor cells. ATF1 is up-regulated in metastatic melanoma cells, and inhibition of ATF1 suppresses their tumorigenicity and metastatic potential in nude mice (23). Constitutive activation of ATF1 mediates EWS-ATF1 (Ewing sarcoma protein), transforming phenotypes and unique features of clear cell sarcoma (24). However, the upstream kinases and the role of ATF1 in proliferation and cell transformation have not been completely elucidated.
Flavonoids are ubiquitously found in fruits and vegetables as well as popular beverages, including wine, tea, and coffee (25). Flavonoids also exhibit antioxidant, antitumor, and anti-inflammatory effects (25). In particular, their antitumor activity has attracted much attention as a possible dietary prevention strategy against carcinogenesis (26, 27). Our recent study demonstrated that kaempferol is a natural compound that specifically inhibits RSK2 N-terminal kinase activity (28). Eriodictyol is a major flavonoid extracted from Yerba Santa (Eriodictyon californicum) and has a similar structure to kaempferol. Eriodictyol is found in lemon, lime, sour orange, and peppermint. Research data have shown that eriodictyol also exerts antioxidant, antitumor, and anti-inflammatory effects (29), suggesting that eriodictyol might also inhibit cell proliferation and transformation. However, the underlying mechanism and target protein(s) of eriodictyol have not yet been clarified.
In this study, we show that ATF1 is a novel substrate of RSK2. The phosphorylation of ATF1 at Ser-63 by RSK2 induces ATF1 transactivation and transcriptional activity. Inhibition of RSK2 activity by eriodictyol suppressed ATF1 activities and RSK2-ATF1-mediated cell transformation. These results demonstrate that the inhibition of RSK2 activity by eriodictyol modulates RSK2-ATF1 signaling in cell proliferation and transformation. Therefore, we suggest that eriodictyol is a potential natural compound for chemoprevention.
EXPERIMENTAL PROCEDURES
Materials
Eriodictyol (>95% purity), Tris, NaCl, and SDS were from Sigma. CNBr-Sepharose 4B, glutathione-Sepharose 4B, and the chemiluminescence detection kit were from Amersham Biosciences. The protein assay kit was from Bio-Rad. [γ-32P]ATP was purchased from New England Biolabs. G418 and the luciferase assay substrate were from Promega. Eagle's minimal essential medium was from Invitrogen. Antibodies for Western blot analysis were from Cell Signaling Technology, Santa Cruz Biotechnology, Abcam, and Millipore.
Cell Culture, Transfection, and Treatment with Eriodictyol
Culture of JB6 Cl41 cells and RSK2+/+ and RSK2−/− mouse embryonic fibroblasts (MEFs) was described previously (13). Transfection of the various expression vectors and luciferase reporter plasmids was conducted using jetPEI (Polyplus Transfection, New York, NY) according to the manufacturer's instructions. Eriodictyol was dissolved in dimethyl sulfoxide (DMSO; 50 mm stock solution), aliquoted, and stored at −20 °C. Eriodictyol was mixed with complete cell culture medium at various doses, and the final DMSO concentration was not >0.1% of the total media volume.
MTS Assay
Proliferation of JB6 Cl41 cells and viability of RSK2+/+ and RSK2−/− MEFs were measured using the MTS assay kit (Promega) according to the manufacturer's instructions.
In Vitro Kinase Assay
The plasmids for GST-tagged or His-tagged fusion proteins including ATF1 and ATF1-S63A were purified from BL21 bacteria using glutathione-Sepharose 4B or nickel-nitrilotriacetic acid-agarose beads. The purified fusion proteins (1 μg; ∼90% purity) were used for in vitro kinase assays with 20 ng of active RSK2 (Millipore) and visualized by autoradiography or Western blotting as described (28).
Cell Cycle Analysis
The cell cycle was analyzed by propidium iodide staining using the FACSCalibur flow cytometer (BD Biosciences) as described by Ahmad et al. (30).
Mammalian Two-hybrid Assay
To screen for protein-binding partners, we used the mammalian two-hybrid assay following the Promega CheckMateTM mammalian two-hybrid system protocol as described previously (11). The relative luciferase activity was calculated using the pG5-luciferase basal control by transfection of pG5-luciferase/pACT-mock/pBIND-RSK2 and normalized against Renilla luciferase activity, which included the pBIND vector.
ATF1 Transactivation Assay
JB6 C141 cells (6 × 104) were cultured in 12-well plates for 24 h before transfection. The p5×Gal4-luciferase reporter plasmid was transfected with pcDNA4-RSK2 and the expression vector for Gal4-ATF1 or Gal4-ATF1-S63A. Cells were cultured for 36 h and then disrupted for firefly luciferase activity analysis. The reporter gene vector phRL-SV40 (Promega) was cotransfected into each cell line, and the transfection efficiencies were normalized to the Renilla luciferase activity generated by this vector.
Anchorage-independent Cell Transformation Assay
The effects of eriodictyol on EGF-induced transformation were investigated in JB6 C141 cells as described by Colburn et al. (31). Colonies were counted under a microscope with the Image-Pro Plus software program (Version 6, Media Cybernetics, Silver Spring, MD).
Preparation of Sepharose 4B Beads
Sepharose 4B beads (0.3 g) were washed with 30 ml of 1 mm HCl three times for 5 min each by gentle inversion and then incubated with 3 mg of eriodictyol or DMSO in coupling buffer (0.1 m NaHCO3 and 0.5 m NaCl (pH 8.3)) at 4 °C overnight. The samples were washed five times with coupling buffer and incubated with blocking buffer (0.1 m Tris-HCl (pH 8.0)) at 4 °C overnight. The samples were alternatively washed with 0.1 m acetic acid buffer (pH 4.0) and with 0.1 m Tris-HCl and 0.5 m NaCl (pH 8.0) three times and then resuspended in 1 ml of PBS for use.
Pulldown Assays
For pulldown assays, eriodictyol-Sepharose 4B beads (100 μl, 50% slurry) were combined with purified RSK2 or a cellular supernatant fraction of JB6 C141 cells (500 μg) overnight. Bound RSK2 proteins were visualized by Western blotting as described (28).
Focus Formation Assay
A focus formation assay using NIH3T3 cells was conducted according to standard protocols (24). Foci were fixed and stained with 0.5% crystal violet and counted using the Image-Pro Plus software program (Version 6).
Homology Model of the RSK2 N-terminal Kinase Domain
To build a model of the active form of the RSK2 N-terminal kinase domain, we conducted homology modeling based on the active conformation of the RSK1 N-terminal kinase domain, which was crystallized with the inhibitor staurosporine at a resolution of 2.00 Å (Protein Data Bank code 2z7r). ClustalW was used to align the RSK2 and RSK1 sequences, wherein they were shown to possess an identity of 85% and a similarity of 90% (32). Using Modeller 9v4 (33), we created a template structure of RSK2 based on the active structure of RSK1. To ensure an adequate bound protein form, waters and staurosporine were added from the crystal structure of RSK1 using MAESTRO 9.0 and were allowed to relax with OPLS_2005 force field. This was performed using MacroModel 9.7 following standard minimization procedures (34).
Molecular Docking to a Homology Model of the RSK2 N-terminal Kinase Domain
Kaempferol and eriodictyol were both built and minimized and underwent extensive conformational searches followed by subsequent minimization using MacroModel to determine the lowest energy conformations for docking. Staurosporine as the reference compound was subjected to similar strategies except starting from its crystallized bound orientation. The Induced Fit module was used for docking because both ligand and binding site residue flexibility are provided for, thereby mimicking reality (34). The recommended parameters for induced fit docking were used where the receptor and ligand van der Waals scaling was kept at 0.50 with a total number of poses as 20. Only residues within 5.0 Å were refined. Glide redocked the ligands, using standard precision, into the structures within 30.0 kcal/mol of the best active-site orientation and only within the top 20 structures.
RESULTS
RSK2 Phosphorylates ATF1 at Ser-63 in Vitro
To identify novel substrate(s) of RSK2, we conducted a mammalian two-hybrid assay with 32 transcription factors (11). We found that the luciferase activity representing the binding of RSK2 and ATF1, a member of the CREB/ATF transcription factor family, was ∼7-fold higher than that of the mock control. This binding was slightly higher than RSK2 binding with known protein partners such as ATF4 (∼5-fold) and E2F6 (∼5.5-fold) (Fig. 1A). To confirm the binding of RSK2 and ATF1, pACT-ATF1 and pcDNA4-RSK2 were introduced into 293T cells and immunoprecipitated with anti-VP16 antibody. Co-immunoprecipitated RSK2 was detected with anti-His antibody by Western blotting (Fig. 1B, upper panel). Furthermore, endogenous ATF1 co-precipitated with endogenous RSK2 (Fig. 1B, lower panel), indicating that RSK2 and ATF1 are binding partners. To determine whether RSK2 phosphorylates ATF1, we conducted an in vitro kinase assay with a commercially available active RSK2 protein and purified GST-ATF1 (35) and [32P]ATP. The results indicated that RSK2 phosphorylated ATF1 (Fig. 1C). To identify the amino acid(s) phosphorylated by RSK2, we searched the RSK2 phosphorylation consensus sequence of ATF1 using the Group-based Prediction System (Version 2.1) (Fig. 1D). The data indicated that Ser-63 of ATF1 had a 3.182 score/cutoff value for RSK2 compared with a 1.756 score/cutoff value for MSK1 (mitogen- and stress-activated protein kinase 1), which is a known kinase of ATF1 (Ser-63) (Fig. 1D, table). To confirm this prediction, we conducted an in vitro kinase assay with GST-ATF1 and active RSK2. The results indicated that RSK2 phosphorylated ATF1 at Ser-63 (Fig. 1D, lower left panel). We confirmed that RSK2 phosphorylated ATF1 at Ser-63 using an in vitro kinase assay and a point mutant of ATF1 with Ser-63 replaced with alanine (His-ATF1-S63A) (35) and active RSK2 and [γ-32P]ATP (Fig. 1D, lower right panel). Overall, these results demonstrate that ATF1 is phosphorylated at Ser-63 by RSK2.
FIGURE 1.

RSK2 phosphorylates ATF1 at Ser-63. A, mammalian two-hybrid screening of RSK2. The mammalian two-hybrid assay was conducted in 293T cells to screen for binding partners of RSK2. Activity is expressed as relative luminescence units normalized to a negative control (value for cells transfected with pG5-luciferase (Luc)/pACT-mock/pBIND-RSK2 = 1.0). Firefly luciferase activity was normalized against Renilla luciferase activity. Data are shown as means ± S.D. from triplicate experiments. *, significant (p < 0.05) increase in activity compared with the negative control. B, upper panel, confirmation of RSK2 and ATF1 binding. pACT-ATF1 and pcDNA4-His-RSK2 were introduced into 293T cells, and ATF1 was immunoprecipitated (IP) with anti-VP16 antibody. Co-immunoprecipitated RSK2 was confirmed by Western blotting (WB) with anti-His antibody. H. C., heavy chain. Lower panel, endogenous ATF1 was immunoprecipitated with a specific antibody, and co-immunoprecipitated RSK2 was visualized by Western blotting using an RSK2-specific antibody. C, RSK2 phosphorylates ATF1 in vitro. To determine whether RSK2 can phosphorylate ATF1, purified GST-ATF1 and active RSK2 were combined with [γ-32P]ATP. Phosphorylated ATF1 was visualized by autoradiography. GST-p53 was the positive control. C. B., Coomassie Blue. D, RSK2 phosphorylates ATF1 at Ser-63. Table, putative phosphoamino acid(s) of ATF1 that could be phosphorylated by RSK were identified using the Group-based Prediction System (Version 2.1). Lower left panel, to confirm that ATF1 (Ser-63) is a target of RSK2, samples from an in vitro kinase assay were visualized by Western blotting with anti-ATF1 (Ser-63) antibody. Lower right panel, to confirm that ATF1 (Ser-63) is a target of RSK2, purified His-ATF1-WT or His-ATF1-S63A was used for an in vitro kinase assay with active RSK2 and [γ-32P]ATP and visualized by autoradiography. aa, amino acid.
Eriodictyol Specifically Binds with RSK2
We have demonstrated that RSK2 plays an important role in proliferation and transformation and that RSK2 protein abundance is higher in human skin cancer tissues compared with normal skin (28). Furthermore, we found that kaempferol inhibits RSK2 N-terminal kinase activity (13, 28). Eriodictyol is a flavanone extracted from Yerba Santa, a plant native to North America, and has a structure similar to that of kaempferol (supplemental Fig. 1). We hypothesized that eriodictyol might also inhibit RSK2 N-terminal kinase activity. We built a model of the RSK2 N-terminal kinase domain based on the crystal structure of the active form of RSK1 complexed with staurosporine at a resolution of 2.00 Å (Protein Data Bank code 2z7r). We found that the preference of binding was staurosporine > kaempferol > eriodictyol, with binding affinities of −11.514, −9.216, and −8.816 kcal/mol, respectively. We observed that eriodictyol docked nicely into the ATP-binding active site of RSK2 (Fig. 2A, left panel). Furthermore, we discovered that important hydrogen bonds are formed between the inhibitor and hinge region (i.e. backbone of Asp-148 and Leu-150) and with the aspartic acid of the DFG motif in the back cleft of the active site (Asp-211) (Fig. 2A, right panel). By comparing the docking of kaempferol and eriodictyol, we found that the aromatic substituent group of eriodictyol was rotated out of plane (supplemental Fig. 2). This rotation might be due to the lack of the double bond and hydroxyl group in eriodictyol (supplemental Fig. 1). To verify binding of eriodictyol with RSK2, we conducted a pulldown assay with eriodictyol-conjugated beads and commercially available active RSK2. The results indicated that eriodictyol bound with RSK2 (Fig. 2B, upper panel). To identify the specific binding domain, we used purified truncated and full-length RSK2 proteins (28) and eriodictyol-conjugated beads. The results demonstrated that eriodictyol bound strongly with the N-terminal kinase domain of RSK2 (Fig. 2B, lower panel). Notably, we confirmed in cell lysates that eriodictyol bound with endogenous RSK2 (Fig. 2C). These results demonstrated that eriodictyol is a natural compound that binds with the RSK2 N-terminal kinase domain. To verify the effect of eriodictyol on RSK2 activity, we conducted an in vitro kinase assay with GST-ATF1, active RSK2, [γ-32P]ATP, and different doses of eriodictyol. The autoradiography results demonstrated that eriodictyol inhibited RSK2-mediated ATF1 phosphorylation dose-dependently (Fig. 2D).
FIGURE 2.

Eriodictyol binds with RSK2 and inhibits phosphorylation of ATF1 by RSK2 in vitro. A, left panel, surface representation of the N-terminal kinase domain of RSK2 with eriodictyol bound to the ATP-binding site. Red, oxygen; blue, nitrogen; yellow, sulfur. Right panel, N-terminal kinase domain of RSK2 with eriodictyol bound to the ATP-binding site. Hydrogen bonds are formed between eriodictyol and the hinge region (backbone of Asp-148 and Leu-150), with the aspartic acid of the DFG motif in the back cleft of the active site (Asp-211), and finally with the important lysine residue known to anchor the phosphates of ATP. B, eriodictyol binds with RSK2 in vitro. Upper panel, active RSK2 (200 ng) was subjected to a pulldown assay with eriodictyol conjugated with CNBr-Sepharose 4B beads. Eriodictyol binding of RSK2 was visualized by Western blotting (WB) with anti-RSK2 antibody. Lower panel, to identify the domain of RSK2 that binds with eriodictyol, truncated RSK2 proteins were used in the CNBr-eriodictyol pulldown assay, and RSK2 was visualized by Western blotting with anti-His antibody. NTD, N-terminal domain; CTD, C-terminal domain. C, eriodictyol binds with RSK2 ex vivo. The cellular protein fraction (500 μg) of JB6 Cl41 cells was used for the pulldown assay with CNBr-DMSO or CNBr-eriodictyol beads. The RSK2 proteins pulled down were visualized by Western blotting with anti-RSK2 antibody. D, eriodictyol inhibits RSK2 activity. The inhibitory effect of eriodictyol on RSK2 activity was assessed by an in vitro kinase assay using a GST-ATF1 protein and [γ-32P]ATP (upper panel). 32P-Labeled GST-ATF1 was visualized by autoradiography. Band density was measured using the NIH ImageJ computer program (Version 1.41), and the band intensities of active RSK2 and GST-ATF1 (100%) were compared (lower panel). Data are shown as means ± S.D. of values obtained from three independent experiments. *, significant (p < 0.05) decrease in kinase activity.
RSK2 Regulates ATF1 Activity
To determine the significance of ATF1 phosphorylation at Ser-63, we analyzed the transactivation activity of WT and mutant (S63A) ATF1. As expected, ATF1-S63A suppressed transactivation activity (Fig. 3A), indicating that Ser-63 is important in regulating ATF1 activity. To examine the effect of RSK2 on ATF1 transactivation, we rescued RSK2 expression in RSK2−/− MEFs by transduction of pBabe-puro-RSK2-WT and found that phosphorylated and total ATF1 and RSK2 expression was restored (Fig. 3B). We further found that the suppressed ATF1 transactivation in RSK2−/− MEFs was restored in rescued RSK2−/−/RSK2 cells to almost the same level as in RSK2+/+ MEFs (Fig. 3B, lower panel). Furthermore, when we treated JB6 cells with nontoxic doses of eriodictyol (supplemental Fig. 3), EGF-induced transactivation was suppressed (Fig. 3C), and ATF1 transcriptional activity was inhibited (Fig. 3D). Overall, these results indicate that RSK2 plays an important role in the regulation of ATF1 activity by phosphorylating ATF1 at Ser-63.
FIGURE 3.

RSK2 regulates ATF1 activity. A, phosphorylation of ATF1 at Ser-63 plays an important role in ATF1 transactivation. The pGal4-ATF1-WT or pGal4-ATF1-S63A plasmid was cotransfected into JB6 Cl41 cells with the p5×Gal4-luciferase (luc) reporter plasmid. B, RSK2 deficiency attenuates ATF1 transactivation. The pGal4-ATF1-WT and p5×Gal4-luciferase reporter plasmids were transfected into RSK2+/+, RSK2−/−, and RSK2−/−/RSK2-overexpressing MEFs. WB, Western blot. C, EGF-induced ATF1 transactivation is inhibited by eriodictyol treatment. The pGal4-ATF1-WT and p5×Gal4-luciferase reporter plasmids were transfected into JB6 cell, and cells were treated with eriodictyol for 2 h prior to EGF stimulation. D, eriodictyol inhibits ATF1 transcriptional activity. The inhibitory effect of eriodictyol on ATF1 transcriptional activity was measured by introduction of CRE reporter plasmids (pGL4.29-luciferase/CRE) into JB6 cells followed by the indicated dose of eriodictyol. Luciferase activities in A–D were measured, and data are shown as means ± S.D. of values from triplicate experiments. *, significant (p < 0.05) difference in luciferase activity as indicated.
RSK2-ATF1 Signaling Plays an Important Role in EGF-induced Cell Transformation
To examine the role of RSK2-ATF1 signaling in cell proliferation and transformation, we analyzed the level of EGF-induced phosphorylation of ATF1 when treated with different doses of eriodictyol. EGF-induced RSK phosphorylation was not affected by eriodictyol (Fig. 4A). However, EGF-induced phosphorylation of ATF1 at Ser-63 and total c-Fos protein abundance were decreased dose-dependently by eriodictyol (Fig. 4A). In addition, eriodictyol had no effect on phosphorylation of MEK-ERK signaling, including MEK1/2 and ERK1/2 (Fig. 4B). Eriodictyol did not affect phosphorylation of p38, JNKs, or Akt (Fig. 4C). The proliferation of JB6 Cl41 cells was decreased dose-dependently by eriodictyol (Fig. 5A, left panel), an effect that is probably associated with its inhibition of the G1/S cell cycle transition (right panel). We compared the effect of eriodictyol on proliferation of RSK2+/+, RSK2−/−, and rescued RSK2−/−/RSK2 MEFs. We found that eriodictyol inhibited proliferation in RSK2+/+ MEFs (Fig. 5B, left panel). However, eriodictyol had no effect on proliferation of RSK2−/− MEFs (Fig. 5B, middle panel). Importantly, rescued RSK2−/−/RSK2 cells regained sensitivity to the inhibitory effect of eriodictyol on proliferation to the same degree as RSK2+/+ MEFs (Fig. 5B, right panel). We confirmed that eriodictyol was not cytotoxic in MEFs up to 25 μm (supplemental Fig. 3B). Furthermore, additional results indicated that eriodictyol inhibited EGF-induced cell transformation dose-dependently (Fig. 5C). Taken together, these results demonstrate that eriodictyol suppresses RSK2 N-terminal kinase activity, resulting in the suppression of EGF-induced cell proliferation and transformation.
FIGURE 4.
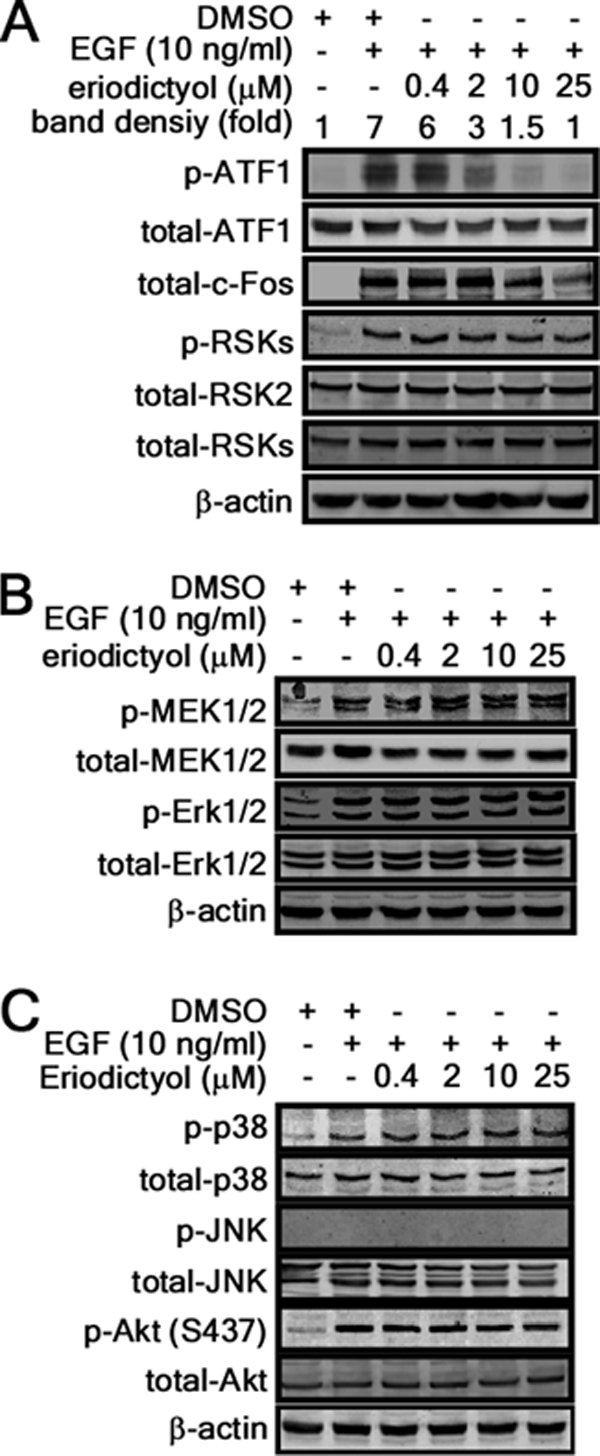
Inhibitory effect of eriodictyol on MAPK signaling. A–C, eriodictyol inhibits RSK2-mediated downstream signaling. JB6 C141 cells were treated with the indicated doses of eriodictyol for 1 h and then stimulated with EGF (10 ng/ml) for 15 min. Individual levels of phosphorylated and total proteins were visualized by Western blotting with specific antibodies. β-Actin was used as an internal control for equal protein loading.
FIGURE 5.

Eriodictyol inhibits proliferation and transformation of JB6 Cl41 cells. A, eriodictyol inhibits cell proliferation by causing arrest at the G0/G1 cell cycle. Left panel, eriodictyol inhibits proliferation. JB6 Cl41 cells (1 × 103 cells/well) were treated with the indicated doses of eriodictyol, and proliferation was measured at the indicated time points using the MTS assay. Data are shown as means ± S.D. from triplicate independent experiments. *, significant (p < 0.05) difference in proliferation compared with the DMSO-treated control. Right panel, JB6 Cl41 cells were treated with EGF and eriodictyol for 18 h, and cell cycle distribution was measured by flow cytometry with propidium iodide. Data are expressed as the percentage of cells in G1/G0, S, or G2/M and are shown as means ± S.D. of values from triplicate experiments. *, significant (p < 0.05) change in G0/G1 and S phase cell cycle distribution compared with the EGF-stimulated group. B, eriodictyol-mediated inhibition of proliferation is attenuated in RSK2 deficiency. RSK2+/+ (left panel), RSK2−/− (middle panel), and WT RSK2-transfected RSK2−/− (right panel) MEFS were treated with the indicated doses of eriodictyol, and viability was assessed at the indicated time points by the MTS assay. Data are shown as means ± S.D. of values from triplicate experiments. *, significant (p < 0.05) change in viability compared with untreated control. C, eriodictyol suppresses EGF-induced anchorage-independent colony formation in soft agar. Cells (8 × 103) were exposed to EGF (10 ng/ml) with various doses of eriodictyol in 1 ml of 0.3% Eagle's basal medium-agar containing 10% FBS. Cells were cultured, and colonies were counted using a microscope and the Image-Pro Plus computer software program (Version 6). Data are shown as means ± S.D. of values from triplicate experiments. *, significant (p < 0.05) decrease in colony number compared with the untreated control.
RSK2-ATF1 Signaling Plays an Important Role in Ras-mediated Cell Transformation
To explore the role of ATF1 in EGF-induced transformation, we established JB6 Cl41 mouse epidermal skin cells stably expressing ATF1 or ATF1 shRNA and conducted an anchorage-independent cell transformation assay in soft agar (Fig. 6A). EGF induced more colony formation in ATF1-overexpressing JB6 Cl41 cells compared with mock-transfected cells (Fig. 6A and supplemental Fig. 4A). In contrast, knockdown of ATF1 inhibited EGF-induced colony formation in soft agar (Fig. 6A and supplemental Fig. 4A), indicating that ATF1 plays an important role in EGF-induced transformation of JB6 Cl41 cells. We confirmed the relationship of colony formation with ATF1 protein level by Western blotting (Fig. 6A). To further examine the role of endogenous RSK2-ATF1 signaling in cell transformation, we conducted a RasG12V-mediated focus formation assay in NIH3T3 cells (13). We introduced various combinations of RasG12V, RSK2, ATF1, ATF1 shRNA, RSK2 siRNA, and eriodictyol into NIH3T3 cells, and focus formation was measured. The results indicated that RasG12V induced focus formation and that RasG12V/RSK2 substantially enhanced focus formation (Fig. 6B and supplemental Fig. 4B). RasG12V/RSK2-induced focus number was increased more by co-introduction of RasG12V/RSK2/ATF1, and knockdown of RSK2 or ATF1 by siRNA or shRNA suppressed focus formation (Fig. 6B and supplemental Fig. 4B). Furthermore, eriodictyol suppressed focus formation in a manner similar to knockdown of RSK2 (Fig. 6B and supplemental Fig. 4B). To examine the role of ATF1 mutation at Ser-63 in cell transformation, we introduced combinations of RasG12V, ATF1, ATF1-S63A, and ATF1 shRNA into NIH3T3 cells, and focus formation was measured. The results indicated that RasG12V/ATF1 enhanced focus formation compared with RasG12V alone, whereas ATF1-S63A or ATF1 shRNA attenuated RasG12V induced focus formation (Fig. 6C). To confirm ATF1 phosphorylation by combinational coexpression of RasG12V, RSK2, and ATF1, we conducted Western blotting using NIH3T3 cells. We confirmed that the transfected expression vectors were well expressed, and the ATF1-S63A mutation inhibited phosphorylation of ATF1 at Ser-63, which was enhanced by RasG12V or RasG12V/RSK2 (supplemental Fig. 4C). Taken together, these results indicate that ATF1 mutated at Ser-63 might act as a dominant-negative mutant. Overall, these results indicate that RSK2-ATF1 signaling plays an important role in neoplastic cell transformation.
FIGURE 6.

RSK2-ATF1 signaling plays an important role in Ras-mediated cell transformation. A, ectopic expression of ATF1 enhances EGF-induced anchorage-independent cell transformation. JB6 Cl41 cells (8 × 103) stabling expressing mock, ATF1 (pBabe-ATF1), and knockdown ATF1 (ATF1 shRNA (sh-ATF1)), were exposed to EGF (10 ng/ml) in 1 ml of 0.3% Eagle's basal medium-agar containing 10% FBS, and colonies were counted using a microscope and the Image-Pro Plus computer software program (Version 6). Data are shown as means ± S.D. of values obtained from two independent triplicate experiments. *, significant (p < 0.05) change as indicated. WB, Western blot. B, RSK2-ATF1 signaling plays an important role in Ras-mediated focus formation. A focus formation assay was performed by introducing combinations of vectors as indicated. Foci were stained with 0.5% crystal violet and counted under a microscope using the Image-Pro Plus software program (Version 6). Data are shown as means ± S.D. of values obtained from two independent triplicate experiments. *, significant (p < 0.05) change in the number of foci as indicated. si-RSK2, RSK2 siRNA. C, ATF1 plays an important role in Ras-mediated focus formation. A focus formation assay was performed by introducing RasG12V combined with ATF1, ATF1-S63A, or ATF1 shRNA vectors as indicated. Foci were stained with 0.5% crystal violet and counted under a microscope using the Image-Pro Plus software program (Version 6). Data are shown as the means ± S.D. of values obtained from triplicate experiments. *, significant (p < 0.05) change in the number of foci as indicated.
DISCUSSION
When cells are stimulated with tumor promoters, EGF or 12-O-tetradecanoylphorbol-13-acetate, phosphorylation of RSK is increased within 5 min, and stable expression of RSK2 in JB6 C141 cells significantly enhances colony formation in either the presence or absence of tumor promoters (13). Moreover, RSK2 is overexpressed in skin tumor tissues and many cancer cell lines compared with normal skin and cells (28). RSK2 plays a critical role in FGFR3-induced hematopoietic transformation (36). Furthermore, proliferation is suppressed in RSK2−/− MEFs compared with RSK2+/+ MEFs, and the effect is associated with an impairment of the G1/S cell cycle transition (13). Our previous study indicated that RSK2 might have multiple substrates involved in cell proliferation and carcinogenesis (11). ATF1 is a transcription factor of the CREB family (15), and tumors highly express the ATF1 protein. Expression of ATF1 is up-regulated in lymphomas (22) and melanomas (23) and contributes to the growth of these cancers. Constitutive activation of ATF1 mediates EWS-ATF1, transforming the phenotype's unique features of clear cell carcinoma (24, 37). In this study, we found that ATF1 is a novel substrate of RSK2 (Fig. 1) and that RSK2 phosphorylated ATF1 at Ser-63, which is a key amino acid for regulation of ATF1 transcriptional and transactivation activities (Figs. 1D and 3). We found that knockdown of RSK2 inhibited RSK2-mediated ATF1 cell transformation (Fig. 6B) and that eriodictyol-mediated proliferation was suppressed in RSK2+/+ MEFs but not in RSK2−/− MEFs (Fig. 5B), indicating that RSK2-ATF1 signaling plays an important role in cell proliferation and transformation.
Recently, we found that kaempferol binds and inhibits RSK2 N-terminal kinase activity by occupying the ATP-binding pocket (28). Eriodictyol is a natural chemical compound found in many plants, and its structure is very similar to that of kaempferol (supplemental Fig. 1). We found that eriodictyol also strongly bound with the N-terminal kinase domain of RSK2 (Fig. 2B). Furthermore, ATF1 is involved in skin cancer and melanoma development (23), suggesting that RSK2-ATF1 signaling is involved in skin carcinogenesis. We demonstrated that RSK2 regulated ATF1 activity (Fig. 3B) and that eriodictyol inhibited EGF-induced cell transformation in soft agar (Fig. 5C). These results paralleled the knockdown effect of RSK2 in cell transformation induced by constitutively active RasG12V (Fig. 6B). Furthermore, RasG12V/RSK2/ATF1-mediated focus formation was inhibited by eriodictyol (Fig. 6B), indicating that eriodictyol is a potential chemopreventive agent that can modulate RSK2 kinase activity by binding to the RSK2 N-terminal kinase domain.
Activation of CREB family members is induced by various stimuli, including growth factors, stress signals, neurotransmitters, and other agents (15). The transactivation domain of ATF1 consists of a kinase-inducible domain and a glutamine-rich constitutive activation domain (Q2). Because ATF1 phosphorylation promotes recruitment of CREB-binding protein/p300, a core protein in the transcriptional machinery, leading to enhancement of transactivation and transcriptional activity (21), our finding that RSK2 phosphorylates ATF1 at Ser-63 in the kinase-inducible domain is important. Furthermore, ATF1 can be phosphorylated at Ser-63 by PKA (16) and calmodulin-dependent protein kinase I/II (16, 38). In response to EGF or 12-O-tetradecanoylphorbol-13-acetate stimulation, ATF1 can also be phosphorylated by MSK1/2 (18, 39, 40), but in MSK1/MSK2 double knock-out embryonic fibroblasts, mitogen-induced phosphorylation of ATF1 at Ser-63 is reduced but not totally abolished (40), suggesting phosphorylation by additional kinases. We found that ectopic expression of ATF1 in JB6 Cl41 cells stimulated EGF-induced transformation and ATF1 activation (Figs. 3C and 6A) and that knockdown of ATF1 inhibited cell transformation (Fig. 6A). We also found that ectopic expression of RasG12V/RSK2 enhanced focus formation (Fig. 6B) and that knockdown of ATF1 in RasG12V/RSK2-overexpressing cells suppressed focus formation (Fig. 6B). Furthermore, eriodictyol treatment showed an effect that was similar to knockdown of RSK2 and ATF1 in focus formation (Fig. 6B). Importantly, rescue of RSK2 in RSK2−/− MEFs recovered ATF1 phosphorylation at Ser-63 and ATF1 transactivation activity (Fig. 3B), indicating that the RSK2-ATF1 signaling pathway plays an important role in cell transformation and that eriodictyol is a potential natural chemopreventive compound for targeting RSK2.
Supplementary Material
Acknowledgments
We thank Dr. Margarita L. Malakhova (The Hormel Institute, University of Minnesota) for providing purified RSK2 proteins and Dr. Duo Zheng for technical assistance.
This work was supported, in whole or in part, National Institutes of Health Grants CA111536, CA120388, CA077646, R37CA081064, and ES016548. This work was also supported by The Hormel Foundation.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–4.
- CRE
- cAMP response element
- CREB
- CRE-binding protein
- MEF
- mouse embryonic fibroblast
- DMSO
- dimethyl sulfoxide
- MTS
- [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium].
REFERENCES
- 1. Jones S. W., Erikson E., Blenis J., Maller J. L., Erikson R. L. (1988) Proc. Natl. Acad. Sci. U.S.A. 85, 3377–3381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Frödin M., Jensen C. J., Merienne K., Gammeltoft S. (2000) EMBO J. 19, 2924–2934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Frödin M., Gammeltoft S. (1999) Mol. Cell. Endocrinol. 151, 65–77 [DOI] [PubMed] [Google Scholar]
- 4. Nebreda A. R., Gavin A. C. (1999) Science 286, 1309–1310 [DOI] [PubMed] [Google Scholar]
- 5. Xing J., Ginty D. D., Greenberg M. E. (1996) Science 273, 959–963 [DOI] [PubMed] [Google Scholar]
- 6. Sassone-Corsi P., Mizzen C. A., Cheung P., Crosio C., Monaco L., Jacquot S., Hanauer A., Allis C. D. (1999) Science 285, 886–891 [DOI] [PubMed] [Google Scholar]
- 7. Blenis J. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 5889–5892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Davis R. J. (1995) Mol. Reprod. Dev 42, 459–467 [DOI] [PubMed] [Google Scholar]
- 9. Ward G. E., Kirschner M. W. (1990) Cell 61, 561–577 [DOI] [PubMed] [Google Scholar]
- 10. Yang X., Matsuda K., Bialek P., Jacquot S., Masuoka H. C., Schinke T., Li L., Brancorsini S., Sassone-Corsi P., Townes T. M., Hanauer A., Karsenty G. (2004) Cell 117, 387–398 [DOI] [PubMed] [Google Scholar]
- 11. Cho Y. Y., He Z., Zhang Y., Choi H. S., Zhu F., Choi B. Y., Kang B. S., Ma W. Y., Bode A. M., Dong Z. (2005) Cancer Res. 65, 3596–3603 [DOI] [PubMed] [Google Scholar]
- 12. Cho Y. Y., Yao K., Bode A. M., Bergen H. R., 3rd, Madden B. J., Oh S. M., Ermakova S., Kang B. S., Choi H. S., Shim J. H., Dong Z. (2007) J. Biol. Chem. 282, 8380–8392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cho Y. Y., Yao K., Kim H. G., Kang B. S., Zheng D., Bode A. M., Dong Z. (2007) Cancer Res. 67, 8104–8112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. David J. P., Mehic D., Bakiri L., Schilling A. F., Mandic V., Priemel M., Idarraga M. H., Reschke M. O., Hoffmann O., Amling M., Wagner E. F. (2005) J. Clin. Invest. 115, 664–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mayr B., Montminy M. (2001) Nat. Rev. Mol. Cell Biol. 2, 599–609 [DOI] [PubMed] [Google Scholar]
- 16. Liu F., Thompson M. A., Wagner S., Greenberg M. E., Green M. R. (1993) J. Biol. Chem. 268, 6714–6720 [PubMed] [Google Scholar]
- 17. Wang Y., Prywes R. (2000) Oncogene 19, 1379–1385 [DOI] [PubMed] [Google Scholar]
- 18. Gupta P., Prywes R. (2002) J. Biol. Chem. 277, 50550–50556 [DOI] [PubMed] [Google Scholar]
- 19. Zhang X., Odom D. T., Koo S. H., Conkright M. D., Canettieri G., Best J., Chen H., Jenner R., Herbolsheimer E., Jacobsen E., Kadam S., Ecker J. R., Emerson B., Hogenesch J. B., Unterman T., Young R. A., Montminy M. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 4459–4464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Impey S., McCorkle S. R., Cha-Molstad H., Dwyer J. M., Yochum G. S., Boss J. M., McWeeney S., Dunn J. J., Mandel G., Goodman R. H. (2004) Cell 119, 1041–1054 [DOI] [PubMed] [Google Scholar]
- 21. Chrivia J. C., Kwok R. P., Lamb N., Hagiwara M., Montminy M. R., Goodman R. H. (1993) Nature 365, 855–859 [DOI] [PubMed] [Google Scholar]
- 22. Hsueh Y. P., Lai M. Z. (1995) J. Immunol. 154, 5675–5683 [PubMed] [Google Scholar]
- 23. Jean D., Tellez C., Huang S., Davis D. W., Bruns C. J., McConkey D. J., Hinrichs S. H., Bar-Eli M. (2000) Oncogene 19, 2721–2730 [DOI] [PubMed] [Google Scholar]
- 24. Brown A. D., Lopez-Terrada D., Denny C., Lee K. A. (1995) Oncogene 10, 1749–1756 [PubMed] [Google Scholar]
- 25. Hollman P. C., Katan M. B. (1999) Food Chem. Toxicol. 37, 937–942 [DOI] [PubMed] [Google Scholar]
- 26. Zhou Q., Yan B., Hu X., Li X. B., Zhang J., Fang J. (2009) Mol. Cancer Ther. 8, 1684–1691 [DOI] [PubMed] [Google Scholar]
- 27. Li Z. D., Hu X. W., Wang Y. T., Fang J. (2009) FEBS Lett. 583, 1999–2003 [DOI] [PubMed] [Google Scholar]
- 28. Cho Y. Y., Yao K., Pugliese A., Malakhova M. L., Bode A. M., Dong Z. (2009) Cancer Res. 69, 4398–4406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Clavin M., Gorzalczany S., Macho A., Muñoz E., Ferraro G., Acevedo C., Martino V. (2007) J. Ethnopharmacol. 112, 585–589 [DOI] [PubMed] [Google Scholar]
- 30. Ahmad N., Feyes D. K., Nieminen A. L., Agarwal R., Mukhtar H. (1997) J. Natl. Cancer Inst. 89, 1881–1886 [DOI] [PubMed] [Google Scholar]
- 31. Colburn N. H., Wendel E. J., Abruzzo G. (1981) Proc. Natl. Acad. Sci. U.S.A. 78, 6912–6916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 33. Sali A., Blundell T. L. (1993) J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 34. Schroginger Suite (2009) Induced Fit Docking Protocol, Glid Version 5.5, Prime version 2.1., Schrondinger, LLC, New York [Google Scholar]
- 35. Zheng D., Cho Y. Y., Lau A. T., Zhang J., Ma W. Y., Bode A. M., Dong Z. (2008) Cancer Res. 68, 7650–7660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kang S., Elf S., Dong S., Hitosugi T., Lythgoe K., Guo A., Ruan H., Lonial S., Khoury H. J., Williams I. R., Lee B. H., Roesel J. L., Karsenty G., Hanauer A., Taunton J., Boggon T. J., Gu T. L., Chen J. (2009) Mol. Cell. Biol. 29, 2105–2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zucman J., Delattre O., Desmaze C., Epstein A. L., Stenman G., Speleman F., Fletchers C. D., Aurias A., Thomas G. (1993) Nat. Genet. 4, 341–345 [DOI] [PubMed] [Google Scholar]
- 38. Shimomura A., Ogawa Y., Kitani T., Fujisawa H., Hagiwara M. (1996) J. Biol. Chem. 271, 17957–17960 [DOI] [PubMed] [Google Scholar]
- 39. Arthur J. S., Cohen P. (2000) FEBS Lett. 482, 44–48 [DOI] [PubMed] [Google Scholar]
- 40. Wiggin G. R., Soloaga A., Foster J. M., Murray-Tait V., Cohen P., Arthur J. S. (2002) Mol. Cell. Biol. 22, 2871–2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.