

Abstract
Toca-1 (transducer of Cdc42-dependent actin assembly) interacts with the Cdc42·N-WASP and Abi1·Rac·WAVE F-actin branching pathways that function in lamellipodia formation and cell motility. However, the potential role of Toca-1 in these processes has not been reported. Here, we show that epidermal growth factor (EGF) induces Toca-1 localization to lamellipodia, where it co-localizes with F-actin and Arp2/3 complex in A431 epidermoid carcinoma cells. EGF also induces tyrosine phosphorylation of Toca-1 and interactions with N-WASP and Abi1. Stable knockdown of Toca-1 expression by RNA interference has no effect on cell growth, EGF receptor expression, or internalization. However, Toca-1 knockdown cells display defects in EGF-induced filopodia and lamellipodial protrusions compared with control cells. Further analyses reveal a role for Toca-1 in localization of Arp2/3 and Abi1 to lamellipodia. Toca-1 knockdown cells also display a significant defect in EGF-induced motility and invasiveness. Taken together, these results implicate Toca-1 in coordinating actin assembly within filopodia and lamellipodia to promote EGF-induced cell migration and invasion.
Keywords: Actin, Cdc42, Cell Migration, Epithelial Cell, Gene Silencing, Abi1·Rac·WAVE, Cdc42·N-WASP, EGFR, F-BAR Adaptor Proteins, Toca-1
Introduction
Directional cell migration requires that signals from extracellular chemotactic factors be relayed to cytoplasmic proteins that regulate the shape of both membranes and the cytoskeleton. This leads to polarization of the cell with a broad, protrusive leading edge and narrow trailing edge. At the leading edge, the protrusive structures include filopodia containing parallel bundles of F-actin and sheetlike protrusions of membrane called lamellipodia that are supported by a lattice of branched F-actin (1). Although Cdc42 GTPase is key for the formation of filopodia (2), both Cdc42 and Rac are required for formation of lamellipodia (3) and are required for efficient cell migration (4). In lamellipodia, Cdc42 binding to neural Wiskott-Aldrich syndrome protein (N-WASP)2 leads to recruitment of Arp2/3 (actin-related protein 2/3) complex that nucleates new actin filaments on the side of existing filaments that help support the extension of membrane protrusions. Rac GTPase also promotes actin assembly within lamellipodia via the Sra-1·Nap1·Abi·WAVE complex. Abi1 (Abelson-interacting protein-1) promotes Rac activation via SOS-1, which functions as a Rac guanine nucleotide exchange factor when bound to Eps8 and Abi1 (5). Toca-1 is an adaptor protein that may coordinate actin assembly via both the Cdc42 and Rac pathways based on observations that have been made in several model systems (6–8).
Toca-1 (also known as FNBP1L) is an adaptor protein that was first identified as an essential co-factor for Cdc42-induced actin assembly via N-WASP in Xenopus tropicalis (8). Toca-1 is a member of the PCH (Pombe Cdc15 homology) family of proteins that all share phosphoinositide-binding Fer/CIP4 homology-Bin/amphiphysin/Rvs (F-BAR) domains (9–11). In mammals, Toca-1 is part of the CIP4 (Cdc42-interacting protein 4) subfamily of PCH proteins, which also includes CIP4 (12) and FBP17 (formin-binding protein 17) (13). The CIP4 subfamily proteins share similar domain organization, with N-terminal F-BAR, central homology region-1 (HR1), and C-terminal SH3 domains. Unlike FBP17 (14), the HR1 domains of CIP4 and Toca-1 bind to active, GTP-loaded Cdc42 (8, 12). Like other F-BAR proteins, Toca-1 can interact with phosphoinositides via its F-BAR domain and promote tubulation of liposomes in vitro (15) and membranes in vivo (16). The SH3 domain of Toca-1 can interact with many ligands that regulate actin assembly, including N-WASP (8), dynamin (16), diaphanous-related formins (17), and Abi1 (7).
The function of Toca-1 in mammalian cells is not well defined but probably involves the coordination of actin assembly adjacent to the plasma membrane and/or endosomes. Toca-1 is required for Cdc42-induced actin assembly via the N-WASP·WIP complex in vitro, and this requires both Cdc42 and N-WASP binding sites in Toca-1 (8). However, this function of Toca-1 in Cdc42·N-WASP-mediated actin assembly is not likely to be exclusive to Toca-1 in mammals because other PCH proteins that lack Cdc42 binding sites can also promote N-WASP-mediated actin polymerization via SH3-mediated interactions (15, 18–20). Toca-1 is required for actin comet-based propulsion of Shigella flexneri within the cytoplasm of infected mammalian cells, a process that requires Cdc42 and N-WASP (21). In neuronal cells, Toca-1 has been implicated in regulating endocytosis and neurite outgrowth (22, 23). Toca-1 can also modulate trafficking of EGF receptor (EGFR) from Rab5 endosomes (24) and promote anti-bacterial autophagy (25). Recent studies have connected Toca-1 to promoting Rac·WAVE (Wiskott-Aldrich syndrome protein family verprolin-homologous protein) signaling via its interaction with Abi1 (6, 7). In Drosophila, only one CIP4 family protein exists (dCIP4), and this protein functions in both endocytosis and actin-based vesicle motility induced by Cdc42·WASp and Abi1·Rac·WAVE pathways (6, 26). These studies suggest that Toca-1 may function to coordinate both Cdc42·WASp- and Rac·WAVE-induced actin branching, which are required for formation and stabilization of lamellipodia and for cell migration (4). Despite these implications of Toca-1 in actin assembly, the functional role of Toca-1 in mammalian cell migration remains unclear. One recent study demonstrates an inhibitory effect of CIP4 and, to a lesser extent, Toca-1 on platelet-derived growth factor (PDGF)-induced membrane ruffling and migration of fibroblasts (27). This related to the roles of CIP4 and Toca-1 in down-regulating PDGF receptor signaling by promoting its internalization (27). In contrast, we observed no such requirement for CIP4 and Toca-1 for EGFR internalization, but this does require FBP17 (24). Thus, depending on the cell type and mode of receptor internalization, different F-BAR adaptor proteins may function in receptor internalization.
In this study, we show that EGF promotes Toca-1 interactions with N-WASP, Abi1, and Toca-1 localization to both filopodia and lamellipodia. We describe the phenotype of shRNA-driven, stable Toca-1 knockdown in A431 cells, which leads to defects in EGF-induced filopodia and lamellipodia formation, compared with control cells. These defects correlated with reduced Arp2/3 complex and Abi1 staining within lamellipodia of Toca-1 knockdown cells, compared with control cells. Interestingly, we show that Toca-1 is required for EGF-induced migration and invasion of A431 cells. These results suggest that Toca-1 plays a role in recruitment and activation of the actin nucleation machinery within lamellipodia and filopodia to enhance cell migration and invasion.
EXPERIMENTAL PROCEDURES
Reagents and Materials
Non-commercial antibodies included rabbit CIP4 antiserum (24), rabbit Abi1 antiserum (described previously (28)) (kindly provided by Anne-Marie Pendergast, Duke University), and mouse anti-Toca-1 and anti-WAVE hybridoma supernatants (described previously (7, 24)) (kindly provided by Giorgio Scita, Institute of Molecular Oncology Foundation). Commercial sources of antibodies included mouse anti-green fluorescent protein (GFP) from Abcam; rabbit anti-p34-Arc (ArpC2) from Millipore; mouse anti-Abi1 (clone 1B9) from Medical and Biological Laboratories Co. Ltd. (MBL); mouse anti-EGFR and mouse anti-active-EGFR (Tyr(P)-EGFR), R-phycoerythrin-conjugated mouse anti-EGFR (clone EGFR.1 and isotype control clone 27-35), all from BD Biosciences; mouse anti-Tyr(P) (PY99) from Santa Cruz Biotechnology; mouse anti-tubulin from Sigma-Aldrich; and goat anti¬FBP17 from IMGENEX. Secondary antibodies included Alexa Fluor 488- or Alexa Fluor 633-conjugated goat anti-mouse/rabbit IgG, and Alexa633-conjugated goat anti-mouse/rabbit IgG (Molecular Probes). TRITC-conjugated and Alexa Fluor 488-conjugated phalloidin were from Invitrogen and Sigma-Aldrich, respectively. Gelatin, bovine serum albumin (BSA), and normal goat serum were all from Sigma-Aldrich. Recombinant mouse EGF was from Peprotech. Transwell chambers (8-μm pore) and Matrigel were from BD Biosciences.
Cell Culture, EGF Stimulation, and Transfection
A431 cells were grown in DMEM supplemented with 10% FBS (Sigma-Aldrich), 1% glutamine (Invitrogen), and antimycotic/antibiotics (Invitrogen) at 37 °C in a humidified atmosphere with 5% CO2. For EGF stimulation, cells were starved overnight in serum-free medium (12–14 h) before treatment with 200 ng/ml EGF. A431 cells were transiently transfected with GFP-Toca-1 expression plasmid (21) (kindly provided by Pietro De Camilli, Yale University) using FuGENE HD reagent according to the manufacturer's instructions (Roche Applied Science). The F-BAR domain 5Q mutation was made by QuikChange mutagenesis (Stratagene) to eliminate conserved basic residues that bind phosphoinositides, as described for CIP4 (24). The HR1 and SH3 domain-inactivating mutations (MGD/IST and W/K, respectively) were described previously (8, 22).
Generation of Stable Toca-1 Knockdown Cell Lines
Lentiviruses encoding empty pLKO.1 vector and five different shRNAs specific for human Toca-1 were obtained from Open Biosystems (RHS4533). Lentiviruses were produced by transfection of HEK293T cells (grown on 100-mm plates) with pLKO.1-based plasmid (15 μg), pCMVΔR8.91 packaging plasmid (15 μg), and pMD.2G envelope plasmid (6 μg) via the calcium phosphate method. Two batches of conditioned medium were collected at 48 and 72 h, filtered through 0.45-μm sterile filters, and stored at −80 °C in aliquots. Viruses were titered according to the manufacturer's instructions (Open Biosystem) using A431 cells. A431 cells were transduced with viral supernatants of similar titer (∼1 × 105 transducing units/ml), and cell pools were selected using puromycin (1.5 μg/ml). After several passages, a vector cell pool and two separate Toca-1-specific shRNA-expressing cell pools were selected based on the efficiency of Toca-1 knockdown (shRNA1 target sequence: 5′-GCCTACGAATGGAAATCCATA-3′, TRCN0000130551, clone D3; shRNA2 target sequence: 5′-GAACAGAACTATGCGAAACAA-3′, TRCN0000130069, clone C12).
Immunofluorescence and Flow Cytometry
A431 cells were fixed with 2% paraformaldehyde; permeabilized with PBS, 0.2% Tween 20; blocked using 5% goat serum (Sigma-Aldrich); and incubated overnight at 4 °C with primary antibodies as follows: rabbit anti-GFP (1:200), rabbit anti-p34-arc (1:50), mouse anti-Abi1 (1:50), mouse anti-Toca-1 (1:20). Fascin staining was performed with mouse anti-fascin (1:50; Chemicon) on methanol-fixed cells. Following extensive washing in PBS, 0.2% Tween 20, coverslips were incubated with the appropriate Alexa Fluor 488-conjugated or Alexa Fluor 633-conjugated goat anti-mouse/rabbit IgG (1:400) and either Alexa Fluor 488-conjugated or TRITC-conjugated phalloidin (1:200) at room temperature for 1 h. After washing five times with PBS, 0.2% Tween 20, coverslips were mounted on slides and analyzed by confocal microscopy (HCX PL APO DIC ×63/1.32 oil CS objective; Leica TCS SP2 Multi Photon; Queen's Protein Function Discovery facility). Surface expression of EGFR was analyzed by flow cytometry, as reported previously (24).
Endocytosis Assay
Cell surface protein biotinylation and endocytosis assays were performed as described previously (29). Briefly, subconfluent A431 vector and Toca-1 knockdown (shRNA2) cells growing on 6-well plates were chilled on ice and washed twice with ice-cold PBS prior to incubation in cold biotinylation buffer (154 mm NaCl, 10 mm Hepes, pH 7.6, 3 mm KCl, 1 mm MgCl2, 0.1 mm CaCl2, 10 mm glucose, 0.6 mg/ml biotin (EZ LINK NHS-SS; Pierce)) for 40 min at 4 °C. For surface EGFR levels, biotinylated cells were lysed, precipitated with streptavidin-conjugated Sepharose beads (Pierce), and subjected to IB with anti-EGFR. For EGFR endocytosis assays, cells were warmed to 37 °C for 15 min in medium containing EGF (100 ng/ml) to allow internalization of biotinylated receptors prior to the addition of cold MESNA buffer (100 mm NaCl, 50 mm Tris-HCl, pH 8.6, 1 mm MgCl2, 0.1 mm CaCl2, 50 mm MESNA (Sigma)) to remove cell surface biotin. As a negative control, cells were treated with MESNA buffer directly (without incubation at 37 °C). Lysates were prepared and subjected to streptavidin-Sepharose pull-downs and IB with anti-EGFR. To normalize for input protein amounts between cell lines, lysates were subjected to IB with anti-tubulin.
Immunoprecipitation and Immunoblotting
A431 cells were serum-starved overnight and stimulated with EGF (200 ng/ml) for the times indicated in the figure legends prior to lysis in Kinase Lysis Buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, 0.5% sodium deoxycholate, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mm Na3VO4, 100 μm phenylmethylsulfonyl fluoride). Cell debris was removed by centrifugation at 12,000 rpm for 10 min at 4 °C, and soluble cell lysates (SCL) were collected. Immunoprecipitation (IP) was performed with rabbit anti-Abi1 (5 μl/sample) or preimmune serum (as control), and with mouse anti-Toca-1 (100 μl/sample), and immune complexes were recovered using 20 μl of GammaBind-Sepharose beads (a 50% (v/v) slurry in PBS; Amersham Biosciences). Following three washes with KLB, bound proteins were recovered by adding 25 μl of 2× SDS sample buffer to the beads. IBs were carried out following the transfer of proteins to Immobilon P membranes (Millipore) using a semidry transfer apparatus (Bio-Rad). Following blocking in 5% milk powder, 10 mm Tris-HCl, pH 8, 150 mm NaCl, 0.1% Tween-20 (TBST), membranes were incubated with primary antibodies as follows: mouse anti-Toca-1 (1:100), mouse anti-Abi1 (1:1000), mouse anti-EGFR (1:2500; BD Biosciences), mouse anti-active EGFR (Tyr(P)-EGFR; 1:1000; BD Biosciences), mouse anti-WAVE (1:100), rabbit anti-p34-Arc (1:1000), rabbit anti-N-WASP (1:1000, Cell Signaling Technology Inc.), goat anti-FBP17 (1:1000, Imgenex) rabbit anti-Erk1/Erk2 (1:1000). Antibody complexes were detected with either horseradish peroxidase (HRP)-conjugated sheep anti-mouse immunoglobulin (1:5000; Amersham Biosciences) or HRP-conjugated goat anti-rabbit immunoglobulin (1:10,000; Amersham Biosciences) or HRP-conjugated donkey anti-goat IgG (1:10,000; Santa Cruz Biotech) and revealed by enhanced chemiluminescence (Applied Biological Materials).
Wound Healing, Scattering, and Invasion Assays
Wound healing assays were performed essentially as described previously (30). Briefly, A431 vector, and Toca-1 shRNA1- or shRNA2-expressing cells were plated on gelatin-coated coverslips within 6-well plates (1 × 106 cells/well) and cultured for 1 day in DMEM complete medium until a uniform monolayer was formed. The cells were serum-starved for 2 days prior to preparing triplicate wounds using 200-μl pipette tips. Following a rinse in serum-free medium to remove the suspended cells, the cells were incubated with DMEM containing EGF (10 ng/ml) for 0 or 24 h. Cells were fixed and permeabilized in 2% paraformaldehyde, 0.2% Tween 20 prior to staining with TRITC-phalloidin. Photomicrographs for three fields of view for each wound area were obtained by epifluorescence microscopy. The cell-free areas were scored (in pixels and then converted to μm2) and presented as means ± S.E. For live cell imaging, cells were plated on 35-mm glass bottom dishes (MatTek, Ashland, MA) and cultured until a uniform monolayer was formed before generating a wound area as described above. Wound closure in EGF (10 ng/ml)-containing medium was imaged over 16 h under an Olympus FV1000 laser-scanning confocal microscope equipped with a 37 °C, 5% CO2 chamber. Movies were made using Fluoview CS3 software (Olympus). Cell velocity was determined using the manual tracking plug-in for ImageJ software. Cell scattering assays were performed as described previously (32) with some modifications. A431 parental cells, vector control, and Toca-1 shRNA-expressing cell lines were plated at 2 × 103/well in a 6-well plate. Cells were cultured in complete medium for 3 days until colonies were formed. Cells were serum-starved for 2 days prior to incubation in DMEM containing EGF (10 ng/ml). Several individual colonies for each cell line were recorded at time 0 and 72 h post-EGF treatment using a phase-contrast microscope equipped with a digital camera. Cell invasion assays were performed essentially as previously described (31). Briefly, A431 vector and Toca-1 knockdown cells (1 × 105 cells; in triplicate) were placed in the upper chamber of Transwell inserts (8-μm pore size) coated with a layer of MatrigelTM (100 μl of 1:5 dilution in DMEM). Cells in the upper chamber were incubated in DMEM supplemented with EGF (10 ng/ml), whereas the lower chamber contained NIH 3T3 cell-conditioned medium. After 48 h, the MatrigelTM and cells were removed from the upper surface, prior to fixation/permeabilization and staining with 4′,6-diamidino-2-phenylindole, dilactate (DAPI; 5 μm). The numbers of invading cell nuclei/field were counted from photomicrographs obtained by epifluorescence microscopy (3 fields/filter).
Quantification of Lamellipodia and Filopodia
Filopodia were defined as fascin-containing projections emanating from lamellipodia areas of the cell periphery. Fascin immunofluorescence (IF) staining of A431 vector and Toca-1 knockdown (shRNA2) cells that were either serum-starved or EGF-treated (100 ng/ml for 15 min) were quantified using Image Pro Plus (Media Cybernetics). Filopodia numbers and length from base to tip were measured for 40 cells for each cell line. For scoring of lamellipodia, A431 vector, shRNA1, and shRNA2 cells were serum-starved and treated with or without EGF (100 ng/ml) for 5 min. F-actin staining was performed as described above, and epifluorescence micrographs were analyzed. Cells with more than 25% of their plasma membrane showing lamellipodia were scored as positive. Approximately 30 cells were quantified for each cell line and condition, and the results are presented as the average from three independent experiments. Arc and Abi1 localization to lamellipodia was scored as positive if the ratio of staining intensity within lamella divided by intensity within cytosol was >0.75. On average, 30 cells were scored for each cell line.
Statistical and Image Analysis
Differences between experimental groups were examined for statistical significance (p < 0.05) using a paired Student's t test in Excel (Microsoft, Redmond, WA). In some figures, intensity profiles for green and red channels are provided for specific regions of interest using Leica Lite software (Leica).
RESULTS
EGF Promotes Toca-1 Localization to Lamellipodia and Co-localization with Arp2/3
Toca-1 is a potential regulator of actin assembly via the Cdc42·N-WASP pathways (8) and can promote filopodia formation in neuroblastoma cells (22). In addition, Toca-1 interacts with Abi1 (7), an activator of the Rac·WAVE pathway that drives lamellipodia formation (32, 33). Because EGF treatment of A431 cells causes formation of lamellipodia (30, 34), we tested the effects of EGF treatment on Toca-1 localization. Serum-starved cells were treated with or without EGF for 5 min and processed for IF staining of endogenous Toca-1 and F-actin. In the absence of EGF, Toca-1 localized throughout the cytoplasm with limited co-localization with F-actin or localization at the cell periphery (supplemental Fig. S1). However, upon EGF treatment, A431 cells displayed extensive membrane ruffles and lamellipodial protrusions marked by F-actin (Fig. 1A). Interestingly, Toca-1 localized to sites of lamellipodial membrane protrusions in EGF-treated cells (Fig. 1A). Toca-1 co-localized with F-actin areas within the most F-actin-rich areas of the lamellipodia (Fig. 1B, d–f).
FIGURE 1.

Toca-1 co-localizes with Arp2/3 complex within lamellipodia. A, A431 cells plated on gelatin-coated coverslips were serum-starved overnight and treated with EGF (100 ng/ml) for 5 min prior to fixation and IF staining with mouse anti-Toca-1 (green) and F-actin (red). Representative confocal micrographs are shown for green and red and a merge of both channels. The arrows indicate membrane ruffles, and boxed areas in a–c are shown below at higher magnification (d–f). Scale bar, 30 μm. B, representative confocal microscopy images are shown for IF staining of GFP-Toca-1 (green; a) in transfected A431 cells forming lamellipodia, together with staining for F-actin (red; b) and endogenous p34-Arc (magenta; c). The merged image indicates overlay of green, magenta, and red channels, with co-localization indicated by white color. Higher magnification views (e–h) of the boxed areas of the images are shown below. Scale bar, 30 μm.
To confirm and extend these observations for endogenous Toca-1, we tested GFP-Toca-1 localization relative to F-actin and Arp2/3 complex (p34-Arc subunit) in transiently transfected A431 cells. Confocal microscopy revealed that GFP-Toca-1 localized at lamellipodial membrane protrusions, along with F-actin and Arp2/3 (Fig. 1B, a–h). Some GFP-Toca-1 puncta within the lamella also co-localized with F-actin and Arp2/3 (Fig. 1B, e–h). Taken together, these results show that Toca-1 is recruited to sites of EGF-induced lamellipodial membrane protrusions and are consistent with the proposed role of Toca-1 in promoting actin assembly.
EGF Induces Tyrosine Phosphorylation of Toca-1 and Interactions with N-WASP and Abi1
Considering that EGF treatment led to Toca-1 localization to lamellipodia, we hypothesized that it might also promote Toca-1 interaction with binding partners, such as N-WASP and Abi1. The recent identification of Toca-1 Tyr(P) in lung cancer at Tyr506 (35, 36), which is proximal to the SH3 domain, also prompted us to investigate whether EGF signaling leads to Toca-1 Tyr(P). To test this, we performed Toca-1 IPs from SCL of A431 cells treated with or without EGF. Subsequent IB with anti-Tyr(P) revealed transient tyrosine phosphorylation upon EGF treatment (Fig. 2A, lanes 1–3, top). Subsequent IB with N-WASP antiserum detected N-WASP in the SCL and in the Toca-1 IPs following EGF treatment for 2 min but not at later time points or with a control antibody (Fig. 2A, bottom). Thus, EGF induces rapid Toca-1 Tyr(P) and binding to N-WASP. Potential interactions between Toca-1 and Abi1 were also tested in A431 cells treated with or without EGF. In the absence of EGF treatment, Toca-1 was not detected in Abi1 IPs (Fig. 2B) or weakly detected in some experiments (data not shown). However, EGF treatment of cells for 5 min led to Toca-1 recovery with Abi1 but not with a control antibody (Fig. 2B). Taken together, these results show that Toca-1 is a substrate of EGFR or a downstream protein-tyrosine kinase and that interactions with SH3 domain ligands, such as N-WASP and Abi1, may be regulated by or coincide with tyrosine phosphorylation of Toca-1.
FIGURE 2.
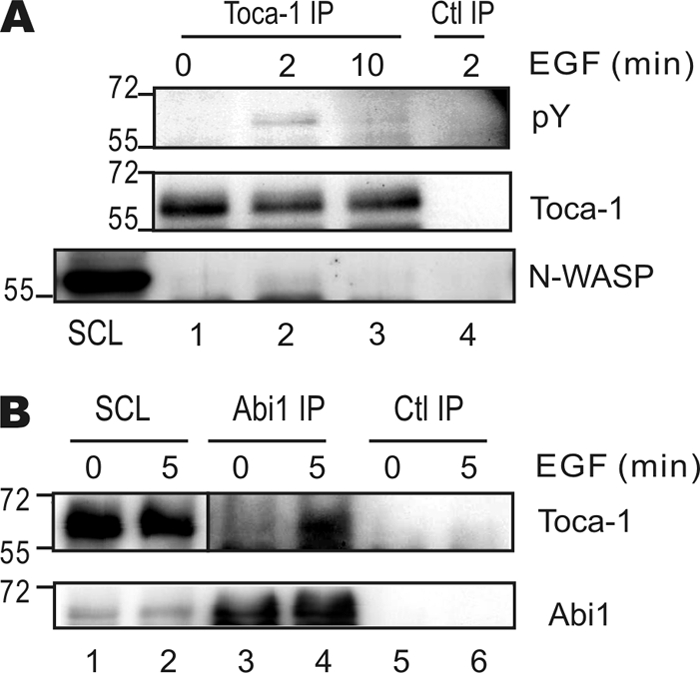
EGF induces Toca-1 phosphorylation and binding to N-WASP and Abi1. A, A431 cells were serum-starved overnight and treated with or without EGF (100 ng/ml) for the times indicated in minutes. SCL were subjected to IP with mouse anti-Toca-1 or a control mouse IgG (Ctl IP) prior to IB with antiserum specific for phosphotyrosine (pY), Toca-1, and N-WASP. Positions of molecular mass markers are shown on the left. B, A431 cells were serum-starved and treated with or without EGF (100 ng/ml) for 5 min. SCL were subjected to IP with rabbit anti-Abi1 or a control rabbit IgG and IB with mouse anti-Toca-1 and mouse and Abi1. Positions of molecular mass markers are shown on the left.
Depletion of Toca-1 Leads to Defects in EGF-induced Filopodia and Lamellipodia Formation
To define the potential role of Toca-1 in regulating actin assembly, we transduced A431 cells with control lentivirus (vector) or lentiviruses expressing Toca-1-specific shRNAs (shRNA1 and shRNA2), selected stable cell pools, and tested Toca-1 levels by IB with Toca-1 antiserum. Compared with vector controls, Toca-1 protein levels were reduced by ∼80% and >90% by shRNA1 and shRNA2, respectively (Fig. 3A, top, compare lanes 1–3). Whereas the expression levels of the related proteins CIP4 and FBP17 were unchanged (Fig. 3A). Likewise, no differences in N-WASP, WAVE, Abi1, and p34-Arc were observed between cell lines (Fig. 3A; tubulin served as a loading control). Next, we tested whether Toca-1 knockdown affects EGFR expression levels and found no difference in surface EGFR levels compared with control cells (Fig. 3B). In addition, no differences in EGF-induced tyrosine phosphorylation of EGFR (Tyr(P)-EGFR) were observed between vector control and Toca-1 knockdown cells (Fig. 3C). To test for potential effects of Toca-1 on EGFR endocytosis, we performed surface biotinylation assays and observed no differences in surface or internalized EGFR between vector and Toca-1 knockdown cells (Fig. 3D). Depletion of Toca-1 also had no effect on the growth rate of A431 cells (Fig. 3E).
FIGURE 3.

Toca-1 knockdown in A431 cells does not affect EGFR internalization or cell growth. A, A431 cells transduced with vector control or two Toca-1 shRNA-expressing lentiviruses (shRNA1 and shRNA2) were selected with puromycin. After several passages, cell lysates were prepared and subjected to IB with antiserum specific for Toca-1, CIP4, FBP17, N-WASP, WAVE, Abi1, p34-Arc, and tubulin. Positions of molecular mass markers are shown on the left. B, surface EGFR levels were compared between cell lines by staining with R-phycoerythrin-conjugated anti-EGFR and isotype control followed by flow cytometry. C, A431 vector control and Toca-1 knockdown (shRNA2) cells were serum-starved and treated with or without EGF (100 ng/ml) for 5 or 15 min. Soluble cell lysates were subjected to IB with antiserum specific for active-EGFR (pY-EGFR) and EGFR. Panels for vector and Toca-1 knockdown cells are taken from the same autoradiographs of equal exposure time. D, EGFR endocytosis assays for A431 vector and Toca-1 knockdown (shRNA2) were carried out using surface biotinylation, as described under “Experimental Procedures.” Following biotinylation, cells were either kept at 4 °C (surface) or subjected to quenching with MESNA following incubation at 37 °C for 15 min (internalized). As a negative control, samples were quenched immediately after biotinylation (MESNA control). SCL were subjected to IB with tubulin (input loading control) or pull-down (PD) using streptavidin-Sepharose (Strep-beads) followed by IB with mouse anti-EGFR. E, the graph depicts average cell counts for vector control (solid line) and Toca1 knockdown cells (dashed lines) between 2 and 16 days of plating.
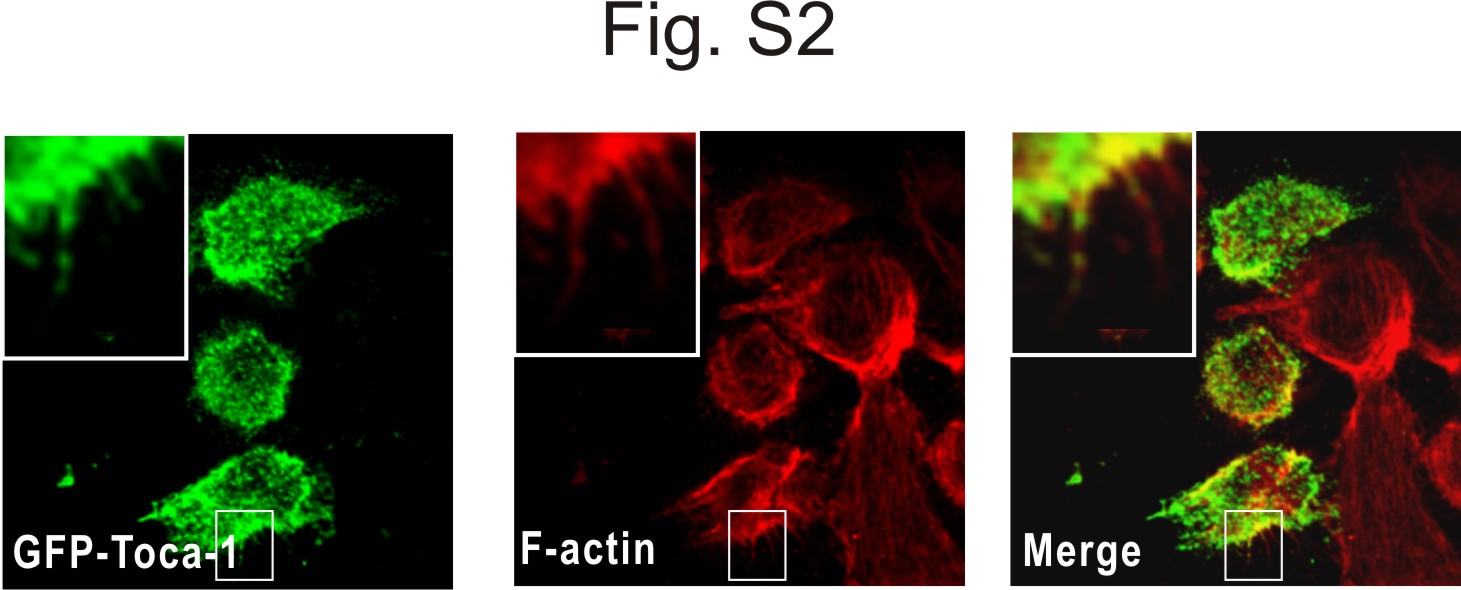
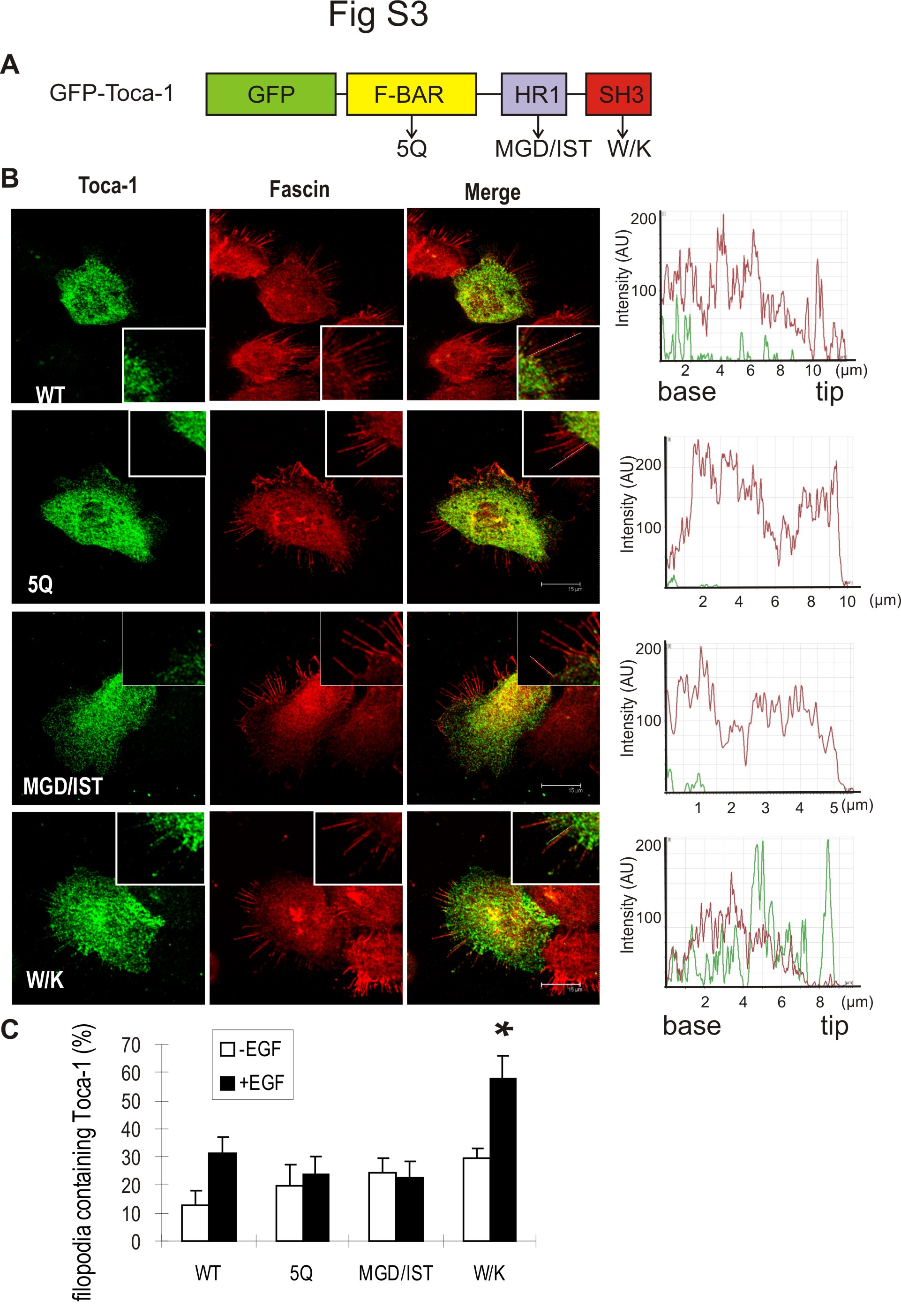
Because Toca-1 overexpression with N-WASP was shown to promote filopodia formation (22), we were interested to test the effects of Toca-1 knockdown on filopodia in A431 cells. A431 cells often display filopodia, but this is further enhanced following EGF treatment for 15 min (34). To characterize filopodia in Toca-1 knockdown cells, we performed IF staining of fascin, an actin-bundling protein and marker of filopodia (37). Fascin staining of vector and Toca-1 knockdown cells revealed that Toca-1 was not required for filopodia formation (Fig. 4A). However, quantification of filopodia number and length revealed a significant increase in the numbers of filopodia, compared with control cells (Fig. 4B). Although EGF treatment did not effect the average number of filopodia in either cell line (Fig. 4B), it caused a significant increase in the average length of filopodia in vector control cells but not in Toca-1 knockdown cells (Fig. 4C). Thus, Toca-1 regulates filopodia extensions but is not required for filopodia formation. This could reflect a direct role for Toca-1 in regulating filopodia dynamics because we observed GFP-Toca-1 localized within filopodia of EGF-treated A431 cells (supplemental Fig. S2). Using a panel of inactivating mutations to the F-BAR, HR1, and SH3 domains of Toca-1 (supplemental Fig. S3A), we found that the SH3 domain mutation (W/K) increased localization to filopodia compared with WT (supplemental Fig. S3, B and C). Mutations to the F-BAR and HR1 domain appeared to reduce filopodia localization slightly (supplemental Fig. S3B), but differences were not significant (supplemental Fig. S3C). In particular, the W/K mutant displayed prominent localization near the tips of filopodia, which was not observed with other mutants or WT (supplemental Fig. S3B). Thus, SH3 domain ligands of Toca-1 may regulate the dynamics of Toca-1 localization within filopodia.
FIGURE 4.

Toca-1 knockdown cells have defects in EGF-induced filopodia formation. A, representative epifluorescence micrographs are shown for A431 vector control (a and c) and Toca-1 knockdown cells (shRNA2; b and d) plated on gelatin-coated coverslips prior to serum starvation (a and b) and treatment with EGF (100 ng/ml for 15 min; c and d). Cells were fixed in methanol and permeabilized, and filopodia were stained using fascin mAb. B, the graph depicts the average number of filopodia per cell ± S.E. (error bars) between cell lines in each condition (n = 20–30 cells). *, statistically significant differences (p < 0.05) between Toca-1 knockdown and vector control cells based on paired Student's t test. C, the graph depicts the average length of filopodia ± S.E. between cell lines in each condition (n = 20 cells for −EGF, and n = 49 for +EGF). *, statistically significant difference (p < 0.05) between Toca-1 knockdown and vector control cells based on paired Student's t test (n = 40 cells).
To test whether Toca-1 regulates lamellipodia, we compared F-actin staining and cell morphology between vector control and Toca-1 knockdown cells that were serum-starved and treated with or without EGF for 5 min. Most vector control cells showed numerous lamellipodial membrane protrusions at the cell periphery in response to EGF (Fig. 5A, a and d). Compared with vector control, Toca-1 knockdown cells formed compact colonies with prominent filopodia upon serum starvation (Fig. 5A, b and c). Upon EGF treatment, Toca-1 knockdown cells showed a more limited number of protrusive lamellipodia than control cells (Fig. 5A, compare d–f). To quantify these phenotypes, the percentage of cells that contained lamellipodia in greater than 25% of their cell periphery was scored for each cell line. Although all cell lines showed an EGF-induced increase in lamellipodia, the percentage of Toca-1 knockdown cells showing this phenotype was significantly reduced compared with vector control cells (Fig. 5B). These data implicate Toca-1 in promoting lamellipodia formation.
FIGURE 5.

Reduced EGF-induced lamellipodia formation in Toca-1 knockdown cells. A, A431 vector, shRNA1, and shRNA2 cell lines were serum-starved, treated with or without EGF (100 ng/ml) for 5 min prior to F-actin staining. Representative epifluorescence micrographs are shown for control and Toca-1 knockdown cells in the absence of EGF (a–c) and with EGF (d–f). B, graph depicts quantification of the percentage of cells showing >25% of the cell periphery comprising lamellipodia ± S.E. (error bars) for vector, shRNA1, and shRNA2 cells. *, statistically significant differences (p < 0.05) between Toca-1 knockdown and vector control cells based on paired Student's t test (n = 30 cells; representative of three separate experiments).
Toca-1 Regulates Recruitment of Arp2/3 and Abi1 to Lamellipodia
To better characterize the lamellipodia in Toca-1 knockdown cells, we performed IF staining of the Arp2/3 complex within lamellipodia of control and Toca-1 knockdown cells treated with EGF for 5 min. As previously shown for parental A431 cells (30), EGF treatment of vector control cells led to formation of prominent lamellipodia containing co-localized F-actin and Arp2/3 complex (Fig. 6A, a–c; see also intensity profiles on the right). Although most Toca-1 knockdown cells formed some lamellipodia in response to EGF treatment, these membrane protrusions stained more weakly for F-actin and contained less Arp2/3 complex (Fig. 6A, d–i; see intensity profiles on the right). Quantification of the relative Arp2/3 staining intensity within lamella, compared with the cytosol of the same cell, revealed a significant reduction in Arp2/3 staining intensity within lamella of Toca-1 knockdown cells compared with control cells (Fig. 6B).
FIGURE 6.

Less Arp2/3 within lamellipodia of Toca-1 knockdown A431 cells. A, A431 vector, shRNA1, and shRNA2 cell lines were serum-starved and treated with EGF (100 ng/ml) for 5 min, prior to staining of F-actin and endogenous Arp2/3 (p34-Arc or Arc-C2 subunit) as described under “Experimental Procedures.” Representative confocal microscopy images for F-actin (green), p34-Arc (red), and a merged image are shown for control (a–c) and Toca-1 knockdown cells (d–i). The insets reflect a higher magnification of the boxed areas, and arrows indicate some examples of lamellipodia. Intensity profiles for green and red channels across a representative lamella (indicated by lines within insets) are shown on the right for each cell line. Scale bar, 30 μm. B, the graph depicts quantification of the relative p34-Arc staining intensity (relative units (RU)) within lamellipodia ± S.E. (error bars) (>30 cells analyzed) for Toca-1 knockdown cells relative to vector control cells, as described under “Experimental Procedures.” *, significant difference between Toca-1 knockdown and vector control cells based on paired Student's t test (p < 0.01).
To gain more insight into the potential molecular mechanism for Toca-1 promoting Arp2/3 recruitment to lamellipodia, we tested for potential effects of Toca-1 depletion on Abi1 localization. Consistent with previous studies (28, 33), Abi1 localized to the tips of lamellipodia and cell-cell junctions in A431 vector cells (Fig. 7A, a–c). In Toca-1 knockdown cells, we detected less Abi1 localized to lamellipodia compared with vector control cells (Fig. 7A, d–i; see intensity profiles on the right). Quantification of the relative Abi1 staining intensity within lamellipodia, compared with the cytosol of the same cell, revealed a significant decrease in Abi1 recruitment in Toca-1 knockdown cells compared with control (Fig. 7B). Taken together, we conclude that during EGF-induced lamellipodia formation, Toca-1 and Abi1 are recruited to promote the formation of actin nucleation sites.
FIGURE 7.

Toca-1 promotes recruitment of Abi1 to lamellipodia of EGF-treated A431 cells. A, A431 vector, shRNA1, and shRNA2 cell lines were serum-starved and treated with EGF (100 ng/ml) for 5 min, and staining of F-actin and endogenous Abi1 was performed as described under “Experimental Procedures.” Representative confocal microscopy images for F-actin (green), Abi1 (red), and a merged image are shown for control and Toca-1 knockdown cells. The insets reflect a higher magnification of the boxed areas, and arrows indicate some areas of membrane ruffles. Intensity profiles for green and red channels across a representative lamella (indicated by lines within insets) are shown on the right for each cell line. Scale bar, 30 μm. B, the graph depicts quantification of the relative Abi1 staining intensity (relative units (RU)) within lamellipodia ± S.E. (error bars) (>30 cells analyzed) for Toca-1 knockdown cells relative to vector control cells, as described under “Experimental Procedures.” *, significant difference between Toca-1 knockdown and vector control cells based on paired Student's t test (p < 0.05).
Toca-1 Promotes EGF-induced Cell Motility and Invasiveness
To test whether the observed differences in filopodia and lamellipodia in Toca-1 knockdown cells result in cellular defects, we performed EGF-induced cell scattering and wound healing migration assays. As shown previously for A431 cells (30), when vector control and Toca-1 knockdown cells were plated at low density, all cell lines formed colonies (Fig. 8A, a–c). With EGF treatment, the vector control cells showed a robust scattering response (Fig. 8A, d). In contrast, the EGF-induced scattering of Toca-1 knockdown cell colonies was dramatically reduced to that observed for vector control cells (Fig. 8A, d–f). These differences do not reflect defects in cell growth, which was comparable between these cell lines (Fig. 3E).
FIGURE 8.

Toca-1 promotes EGF-induced motility and invasiveness of A431 cells. A, EGF-induced scattering of vector control and Toca-1 knockdown cell colonies was performed as described under “Experimental Procedures.” Representative phase-contrast images of cell colonies before (0 h; a–c) and after incubation in medium containing EGF (10 ng/ml) for 72 h are shown (d–f). The dashed area displays the original colony position. B, representative epifluorescence micrographs of F-actin staining of A431 vector and Toca-1 knockdown cell monolayers at the time of generation of the wound areas (0 h; a–c) and following incubation with EGF (10 ng/ml) for 24 h (d–f). C, the graph depicts the average values for the cell free area ± S.E. (error bars) for each cell line (3 wounds/coverslip, 3 coverslips/cell line) following treatment with EGF (10 ng/ml) for 24 h. *, significant difference between Toca-1 knockdown and vector control cells based on paired Student's t test (p < 0.01). D, Matrigel invasion assays were carried out for A431 vector and Toca-1 knockdown cells, as described under “Experimental Procedures.” The graph depicts the average number of invading cells per field for two experiments performed in triplicate ± S.D. *, significant difference between Toca-1 knockdown and vector control cells based on paired Student's t test (p < 0.05).
In wound healing assays, we observed that EGF treatment of vector control cells led to an almost complete closure of the wound area within 24 h, as shown by F-actin staining and epifluorescence microscopy (Fig. 8B, a and d). In contrast, Toca-1 knockdown cells failed to close the wound area as effectively as control cells at 24 h of EGF treatment (Fig. 8B, compare d–f). Although Toca-1 knockdown cells did partially migrate into the wound area, quantification for multiple wounds from multiple experiments revealed a significant increase (∼3-fold) in the cell-free areas for Toca-1 knockdown cells, compared with vector controls (Fig. 8C). To assess whether Toca-1 knockdown affects the migration speed, we performed time lapse microscopy (supplemental Videos 1 and 2). We tracked individual vector control and Toca-1 knockdown cells, which had average velocities of 2.36 ± 0.59 and 0.97 ± 0.45 nm/s (mean ± S.D.), respectively. Taken together, these results implicate Toca-1 in promoting the velocity and extent of EGF-induced cell motility.
To test whether Toca-1 regulates cell invasion, we compared the ability of A431 vector and Toca-1 knockdown cells to invade through a layer of basement membrane (MatrigelTM). Interestingly, the average number of A431 vector control cells that invaded through the MatrigelTM was twice that observed for Toca-1 knockdown cells (Fig. 8D). Thus, Toca-1 promotes both EGF-induced motility and invasion of A431 cells through a basement membrane.
DISCUSSION
EGF signaling promotes the motility of polarized epithelial cells during the wound healing response and also drives the growth and invasiveness of many epithelial carcinomas (38, 39). Directional cell migration requires filopodia and lamellipodia at the leading edge of motile cells, which is a major site of actin assembly (40). In this study, we provide novel evidence that the F-BAR protein Toca-1 promotes the formation of filopodia, lamellipodia, motility, and invasiveness of A431 cells responding to EGF. Further, we show that Toca-1 interacts with the actin regulatory protein Abi1 and that Abi1 localization to lamellipodia is reduced by depletion of Toca-1 by RNA interference. Abi1 functions upstream of both Cdc42·N-WASP and Rac·WAVE pathways (5) and is known to regulate lamellipodia formation and cell motility (33, 41–43). Thus, altered subcellular localization of Abi1 in Toca-1 knockdown cells may contribute to the defects in EGF-induced lamellipodia and cell motility (this paper). Therefore, targeting of Toca-1 to lamellipodial membranes may allow proper coordination of Cdc42·N-WASP and Rac·WAVE actin nucleation-promoting factors below protruding lamellipodial membranes during cell migration.
For F-BAR proteins like Toca-1 to play a role in dynamic processes in cells, there must exist ways to regulate their scaffolding functions. In one recent study, an acidic domain proximal to the SH3 domain of Toca-1 was found to regulate its activity in promoting actin assembly by N-WASP on liposomes (15). In this paper, we describe EGF-induced Toca-1 phosphorylation in A431 cells. Toca-1 Tyr(P) was previously mapped to Tyr506 in proteomics studies of lung cancers (35, 36). It is tempting to speculate that Toca-1 Tyr(P) at this site, which is proximal to the acidic domain and SH3 or HR1 domains might regulate protein-protein interactions with Toca-1. Indeed, Toca-1 Tyr(P) correlated with its association with N-WASP in A431 cells treated with EGF (this paper). Also, Toca-1·Abi1 complex formation was induced by EGF treatment in our study. In addition, Toca-1 Tyr(P) may lead to interactions with SH2 domain-containing proteins. The recent finding of an autoinhibitory interaction between F-BAR and SH3 domains of Syndapin 1 (44) suggests that these scaffolding adaptors can indeed be regulated in terms of phospholipid binding and protein-protein interactions.
Although F-BAR proteins are generally linked to promoting or stabilizing inward membrane curvature during endocytosis (11), a number of F-BAR proteins, including Toca-1, can also localize to and promote membrane protrusions (22, 45, 46). F-BAR domains were recently linked to lamellipodia formation in a study of the F-BAR domain-containing protein-tyrosine kinase FER (46). In this study, the extended F-BAR (FX) domain of FER was shown to bind phosphatidic acid, and confer localization to lamellipodia. It will be important to define the phospholipid binding preferences of Toca-1 and whether the F-BAR domain of Toca-1 confers localization to lamellipodia. Like our study of Toca-1 (this paper), FER knockdown was shown to result in a defect in cell motility (46). These results are consistent with a previous study showing that mouse embryonic fibroblasts lacking FER kinase activity (47) have reduced tyrosine phosphorylation of cortactin and impaired motility (48). Given the roles for F-BAR adaptors like Toca-1 in recruiting proteins involved in actin branching within the lamellipodia (e.g. N-WASP and Abi1), FER localization to lamellipodia may promote cortactin phosphorylation and branch point stabilization (49). Future studies will be needed to fully understand how different members of the F-BAR protein family may cooperate to regulate the dynamics of membrane protrusions, actin assembly, and cell motility.
Although the ability of Toca-1 and related F-BAR adaptors to regulate actin assembly and membrane curvature has been well characterized in recent years (11, 50), the role of Toca-1 in mammalian cells is less well defined. Using RNAi approaches, Toca-1 has been shown to promote actin comet-mediated propulsion of pathogenic bacterium S. flexneri (21), which occurs via a Cdc42·N-WASP-dependent mechanism (51, 52). Interestingly, recent studies of the only Toca-1-like protein in Drosophila (dCIP4) demonstrated a role in actin-based propulsion of Rab5 endosomes (6). We and others have demonstrated that a fraction of the cellular pool of Toca-1 localizes to Rab5 endosomes (22, 24) and may therefore regulate endocytic trafficking of receptors like EGFR (24) or other cargo that regulates filopodia formation (22). The latter study shows that Toca-1 promotes N-WASP-induced filopodia formation in N1E115 neuroblastoma cells (22). In this study, we observed defects in average filopodia length in Toca-1 knockdown cells, compared with control cells. This coincides with a recent study that showed a similar defect in length of filopodia-like structures formed in vitro from lipid bilayers incubated with Toca-1-depleted Xenopus extracts, compared with control extracts (53). Interestingly, they also find Cdc42·Toca-1 localization to the tips of these filopodia-like structures. We report here that Toca-1 can also localize within filopodia of A431 cells and that localization to the tip is enhanced by mutation of the SH3 domain. Thus, SH3 ligands, such as the diaphanous-related formins that undergo retrograde transport with bundled actin in filopodia (54), may regulate Toca-1 trafficking along filopodia. However, future experiments using live cell imaging will be required to fully understand the dynamics and binding partners of Toca-1 that function together in filopodia and lamellipodia protrusions.
Toca-1 may regulate cell motility in part via regulating endocytosis. A recent study using endothelial cells and fibroblasts showed that transient knockdown of Toca-1 or CIP4 caused extended PDGF receptor signaling kinetics, enhanced membrane ruffles, and increased cell motility (27). However, in this study, we show that the levels of surface EGFR were unchanged with knockdown of Toca-1 in A431 cells. Thus, the importance of individual CIP4 family members in receptor internalization probably depends on cell and receptor type and mode of endocytosis. Our previous study showed that only FBP17 is required for early events of EGFR endocytosis, whereas late events in endosomal trafficking are regulated by CIP4 and Toca-1 (24). Recent studies have demonstrated a direct role for endocytic trafficking of activated Rac to the leading edge of motile cells (55, 56). Interestingly, Rac activation occurs on Rab5 endosomes, a compartment that we and others have shown to contain Toca-1 (22, 24). In the future, it will be interesting to test whether Toca-1 knockdown affects Rac activation at lamellipodia or endosomes.
Our results also suggest a unique role for Toca-1, compared with CIP4 and FBP17, in promoting cell motility in A431 cells. Our findings that Toca-1 regulates EGF-induced motility of A431 cells may relate to the “sheetlike” mode of migration that they employ. As described previously for other squamous cell carcinomas (57), A431 cells maintain cell-cell contacts during cell migration and invasion (58). Because Abi1 has also been shown to localize to cell-cell junctions in A431 cells (28), it will be important to characterize whether Toca-1 regulates junctions as recently shown for orthologues in Caenorhabditis elegans and Drosophila (7, 26). Interestingly, Toca-1 knockdown cells tended to retain a colony appearance with extensive cell-cell contacts in our EGF-induced scattering assays, compared with control cells (this paper). Because changes in motility and invasiveness of A431 cells in vitro have also been shown to correlate with altered invasion of A431 cell tumors in vivo (59), it will be important to carry out tumor xenografting studies with Toca-1 knockdown cells to determine if tumor cell migration and invasion in vivo requires Toca-1. It is worth noting that up-regulation of Cdc42 and several downstream effectors has been identified in highly invasive breast tumor cells (60). It will be interesting to determine whether Toca-1 is also up-regulated in highly invasive EGFR-driven tumors, such as lung adenocarcinomas. Interestingly, Toca-1 is significantly up-regulated at the transcript level in non-small cell lung carcinomas (top 2% of genes overexpressed; see the Oncomine Web site) (61). Thus, Toca-1 up-regulation may correlate with enhanced migration and invasive tumor cell phenotypes.
In summary, using a stable cell system for silencing of Toca-1 expression, we provide evidence that Toca-1 coordinates formation of filopodia and lamellipodia to promote the migration and invasion of A431 cells responding to EGF. Loss of Toca-1 results in impaired recruitment of Abi1 and Arp2/3 within lamellipodia. This may explain the positive role of Toca-1 in A431 cell motility and invasiveness.
Supplementary Material
Acknowledgments
We acknowledge the gifts of valuable antisera and plasmids from Giorgio Scita (Institute of Molecular Oncology Foundation), Anne-Marie Pendergast (Duke University), and Pietro De Camilli (Yale University). We thank Chris Nicol and Bruce Banfield for the use of microscopes and the cell imaging facility at Queen's University for help with confocal microscopy and flow cytometry. We also thank Stephanie Everingham for help with mutagenesis and laboratory members for helpful comments on the manuscript.
This work was supported by a grant from the Canadian Cancer Society Research Institute (to A. W. B. C.). Salary support was provided by the Canadian Institutes for Health Research (CIHR) training program in cancer research at Queen's University (to J. H.), Canadian Breast Cancer Foundation-Ontario Chapter (to J. H.), and a CIHR New Investigator award (to A. W. B. C.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3 and Videos 1 and 2.
- N-WASP
- neural Wiskott-Aldrich syndrome protein
- EGFR
- epidermal growth factor receptor
- F-BAR
- Fer/CIP4 homology-Bin/amphiphysin/Rvs
- HR1
- homology region-1
- SH3
- Src homology 3
- IB
- immunoblot
- IP
- immunoprecipitate
- SCL
- soluble cell lysates
- IF
- immunofluorescence
- TRITC
- tetramethylrhodamine isothiocyanate.
REFERENCES
- 1. Mogilner A., Keren K. (2009) Curr. Biol. 19, R762–R771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nobes C. D., Hall A. (1995) Cell 81, 53–62 [DOI] [PubMed] [Google Scholar]
- 3. Kurokawa K., Itoh R. E., Yoshizaki H., Nakamura Y. O., Matsuda M. (2004) Mol. Biol. Cell 15, 1003–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Monypenny J., Zicha D., Higashida C., Oceguera-Yanez F., Narumiya S., Watanabe N. (2009) Mol. Cell. Biol. 29, 2730–2747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Innocenti M., Gerboth S., Rottner K., Lai F. P., Hertzog M., Stradal T. E., Frittoli E., Didry D., Polo S., Disanza A., Benesch S., Di Fiore P. P., Carlier M. F., Scita G. (2005) Nature Cell Biol. 7, 969–976 [DOI] [PubMed] [Google Scholar]
- 6. Fricke R., Gohl C., Dharmalingam E., Grevelhörster A., Zahedi B., Harden N., Kessels M., Qualmann B., Bogdan S. (2009) Curr. Biol. 19, 1429–1437 [DOI] [PubMed] [Google Scholar]
- 7. Giuliani C., Troglio F., Bai Z., Patel F. B., Zucconi A., Malabarba M. G., Disanza A., Stradal T. B., Cassata G., Confalonieri S., Hardin J. D., Soto M. C., Grant B. D., Scita G. (2009) PLoS Genet. 5, e1000675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ho H. Y., Rohatgi R., Lebensohn A. M., Le M., Li J., Gygi S. P., Kirschner M. W. (2004) Cell 118, 203–216 [DOI] [PubMed] [Google Scholar]
- 9. Aspenström P., Fransson A., Richnau N. (2006) Trends Biochem. Sci. 31, 670–679 [DOI] [PubMed] [Google Scholar]
- 10. Chitu V., Stanley E. R. (2007) Trends Cell Biol. 17, 145–156 [DOI] [PubMed] [Google Scholar]
- 11. Suetsugu S., Toyooka K., Senju Y. (2010) Semin. Cell Dev. Biol. 21, 340–349 [DOI] [PubMed] [Google Scholar]
- 12. Aspenström P. (1997) Curr. Biol. 7, 479–487 [DOI] [PubMed] [Google Scholar]
- 13. Kamioka Y., Fukuhara S., Sawa H., Nagashima K., Masuda M., Matsuda M., Mochizuki N. (2004) J. Biol. Chem. 279, 40091–40099 [DOI] [PubMed] [Google Scholar]
- 14. Fuchs U., Rehkamp G., Haas O. A., Slany R., Kōnig M., Bojesen S., Bohle R. M., Damm-Welk C., Ludwig W. D., Harbott J., Borkhardt A. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 8756–8761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Takano K., Toyooka K., Suetsugu S. (2008) EMBO J. 27, 2817–2828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Itoh T., Erdmann K. S., Roux A., Habermann B., Werner H., De Camilli P. (2005) Dev. Cell 9, 791–804 [DOI] [PubMed] [Google Scholar]
- 17. Aspenström P., Richnau N., Johansson A. S. (2006) Exp. Cell Res. 312, 2180–2194 [DOI] [PubMed] [Google Scholar]
- 18. Kessels M. M., Qualmann B. (2002) EMBO J. 21, 6083–6094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Qualmann B., Kelly R. B. (2000) J. Cell Biol. 148, 1047–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rodal A. A., Motola-Barnes R. N., Littleton J. T. (2008) J. Neurosci. 28, 8316–8325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Leung Y., Ally S., Goldberg M. B. (2008) Cell Host Microbe 3, 39–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bu W., Chou A. M., Lim K. B., Sudhaharan T., Ahmed S. (2009) J. Biol. Chem. 284, 11622–11636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kakimoto T., Katoh H., Negishi M. (2006) J. Biol. Chem. 281, 29042–29053 [DOI] [PubMed] [Google Scholar]
- 24. Hu J., Troglio F., Mukhopadhyay A., Everingham S., Kwok E., Scita G., Craig A. W. (2009) Cell. Signal. 21, 1686–1697 [DOI] [PubMed] [Google Scholar]
- 25. Huett A., Ng A., Cao Z., Kuballa P., Komatsu M., Daly M. J., Podolsky D. K., Xavier R. J. (2009) J. Immunol. 182, 4917–4930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Leibfried A., Fricke R., Morgan M. J., Bogdan S., Bellaiche Y. (2008) Curr. Biol. 18, 1639–1648 [DOI] [PubMed] [Google Scholar]
- 27. Toguchi M., Richnau N., Ruusala A., Aspenström P. (2010) Biol. Cell 102, 215–230 [DOI] [PubMed] [Google Scholar]
- 28. Ryu J. R., Echarri A., Li R., Pendergast A. M. (2009) Mol. Cell. Biol. 29, 1735–1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu X., Gan B., Yoo Y., Guan J. L. (2005) Dev. Cell 9, 185–196 [DOI] [PubMed] [Google Scholar]
- 30. Malliri A., Symons M., Hennigan R. F., Hurlstone A. F., Lamb R. F., Wheeler T., Ozanne B. W. (1998) J. Cell Biol. 143, 1087–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huang Y. T., Hwang J. J., Lee L. T., Liebow C., Lee P. P., Ke F. C., Lo T. B., Schally A. V., Lee M. T. (2002) Int. J. Cancer 99, 505–513 [DOI] [PubMed] [Google Scholar]
- 32. Innocenti M., Zucconi A., Disanza A., Frittoli E., Areces L. B., Steffen A., Stradal T. E., Di Fiore P. P., Carlier M. F., Scita G. (2004) Nat. Cell Biol. 6, 319–327 [DOI] [PubMed] [Google Scholar]
- 33. Stradal T., Courtney K. D., Rottner K., Hahne P., Small J. V., Pendergast A. M. (2001) Curr. Biol. 11, 891–895 [DOI] [PubMed] [Google Scholar]
- 34. Chinkers M., McKanna J. A., Cohen S. (1979) J. Cell Biol. 83, 260–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Guo A., Villén J., Kornhauser J., Lee K. A., Stokes M. P., Rikova K., Possemato A., Nardone J., Innocenti G., Wetzel R., Wang Y., MacNeill J., Mitchell J., Gygi S. P., Rush J., Polakiewicz R. D., Comb M. J. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 692–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rikova K., Guo A., Zeng Q., Possemato A., Yu J., Haack H., Nardone J., Lee K., Reeves C., Li Y., Hu Y., Tan Z., Stokes M., Sullivan L., Mitchell J., Wetzel R., Macneill J., Ren J. M., Yuan J., Bakalarski C. E., Villen J., Kornhauser J. M., Smith B., Li D., Zhou X., Gygi S. P., Gu T. L., Polakiewicz R. D., Rush J., Comb M. J. (2007) Cell 131, 1190–1203 [DOI] [PubMed] [Google Scholar]
- 37. Kureishy N., Sapountzi V., Prag S., Anilkumar N., Adams J. C. (2002) BioEssays 24, 350–361 [DOI] [PubMed] [Google Scholar]
- 38. Bublil E. M., Yarden Y. (2007) Curr. Opin. Cell Biol. 19, 124–134 [DOI] [PubMed] [Google Scholar]
- 39. Marmor M. D., Skaria K. B., Yarden Y. (2004) Int. J. Radiat. Oncol. Biol. Phys. 58, 903–913 [DOI] [PubMed] [Google Scholar]
- 40. Le Clainche C., Carlier M. F. (2008) Physiol. Rev. 88, 489–513 [DOI] [PubMed] [Google Scholar]
- 41. Funato Y., Terabayashi T., Suenaga N., Seiki M., Takenawa T., Miki H. (2004) Cancer Res. 64, 5237–5244 [DOI] [PubMed] [Google Scholar]
- 42. Wang C., Navab R., Iakovlev V., Leng Y., Zhang J., Tsao M. S., Siminovitch K., McCready D. R., Done S. J. (2007) Mol. Cancer Res. 5, 1031–1039 [DOI] [PubMed] [Google Scholar]
- 43. Yu W., Sun X., Clough N., Cobos E., Tao Y., Dai Z. (2008) Carcinogenesis 29, 1717–1724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rao Y., Ma Q., Vahedi-Faridi A., Sundborger A., Pechstein A., Puchkov D., Luo L., Shupliakov O., Saenger W., Haucke V. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 8213–8218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Guerrier S., Coutinho-Budd J., Sassa T., Gresset A., Jordan N. V., Chen K., Jin W. L., Frost A., Polleux F. (2009) Cell 138, 990–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Itoh T., Hasegawa J., Tsujita K., Kanaho Y., Takenawa T. (2009) Sci. Signal. 2, ra52 [DOI] [PubMed] [Google Scholar]
- 47. Craig A. W., Zirngibl R., Williams K., Cole L. A., Greer P. A. (2001) Mol. Cell. Biol. 21, 603–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sangrar W., Gao Y., Scott M., Truesdell P., Greer P. A. (2007) Mol. Cell. Biol. 27, 6140–6152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Martinez-Quiles N., Ho H. Y., Kirschner M. W., Ramesh N., Geha R. S. (2004) Mol. Cell. Biol. 24, 5269–5280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Suetsugu S. (2009) FEBS Lett. 583, 3401–3404 [DOI] [PubMed] [Google Scholar]
- 51. Lommel S., Benesch S., Rottner K., Franz T., Wehland J., Kühn R. (2001) EMBO Rep. 2, 850–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Snapper S. B., Takeshima F., Antón I., Liu C. H., Thomas S. M., Nguyen D., Dudley D., Fraser H., Purich D., Lopez-Ilasaca M., Klein C., Davidson L., Bronson R., Mulligan R. C., Southwick F., Geha R., Goldberg M. B., Rosen F. S., Hartwig J. H., Alt F. W. (2001) Nat. Cell Biol. 3, 897–904 [DOI] [PubMed] [Google Scholar]
- 53. Lee K., Gallop J. L., Rambani K., Kirschner M. W. (2010) Science 329, 1341–1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Martin S. G., Chang F. (2006) Curr. Biol. 16, 1161–1170 [DOI] [PubMed] [Google Scholar]
- 55. Palamidessi A., Frittoli E., Garré M., Faretta M., Mione M., Testa I., Diaspro A., Lanzetti L., Scita G., Di Fiore P. P. (2008) Cell 134, 135–147 [DOI] [PubMed] [Google Scholar]
- 56. Scita G., Di Fiore P. P. (2010) Nature 463, 464–473 [DOI] [PubMed] [Google Scholar]
- 57. Gaggioli C., Hooper S., Hidalgo-Carcedo C., Grosse R., Marshall J. F., Harrington K., Sahai E. (2007) Nat. Cell Biol. 9, 1392–1400 [DOI] [PubMed] [Google Scholar]
- 58. Macpherson I. R., Hooper S., Serrels A., McGarry L., Ozanne B. W., Harrington K., Frame M. C., Sahai E., Brunton V. G. (2007) Oncogene 26, 5214–5228 [DOI] [PubMed] [Google Scholar]
- 59. Brockbank E. C., Bridges J., Marshall C. J., Sahai E. (2005) Br. J. Cancer 92, 102–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang W., Goswami S., Lapidus K., Wells A. L., Wyckoff J. B., Sahai E., Singer R. H., Segall J. E., Condeelis J. S. (2004) Cancer Res. 64, 8585–8594 [DOI] [PubMed] [Google Scholar]
- 61. Rhodes D. R., Yu J., Shanker K., Deshpande N., Varambally R., Ghosh D., Barrette T., Pandey A., Chinnaiyan A. M. (2004) Neoplasia 6, 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}