

Abstract
Premature loss of telomere repeats underlies the pathologies of inherited bone marrow failure syndromes. Over the past decade, researchers have mapped genetic lesions responsible for the accelerated loss of telomere repeats. Haploinsufficiencies in the catalytic core components of the telomere maintenance enzyme telomerase, as well as genetic defects in telomerase holoenzyme components responsible for enzyme stability, have been linked to hematopoietic failure pathologies. Frequencies of these disease-associated alleles in human populations are low. Accordingly, the diseases themselves are rare. On the other hand, single nucleotide polymorphisms of telomerase enzyme components are found with much higher frequencies, with several non-synonymous SNP alleles observed in 2–4% of the general population. Importantly, recent advents of molecular diagnostic techniques have uncovered links between telomere length maintenance deficiencies and an increasing number of pathologies unrelated to the hematopoietic system. In these cases, short telomere length correlates to tissue renewal capacities and predicts clinical progression and disease severity. To the authors of this review, these new discoveries imply that even minor genetic defects in telomere maintenance can culminate in the premature failure of tissue compartments with high renewal rates. In this review, we discuss the biology and molecules of telomere maintenance, and the pathologies associated with an accelerated loss of telomeres, along with their etiologies. We also discuss single nucleotide polymorphisms of key telomerase components and their association with tissue renewal deficiency syndromes and other pathologies. We suggest that inter-individual variability in telomere maintenance capacity could play a significant role in chronic inflammatory diseases, and that this is not yet fully appreciated in the translational research of pharmacogenomics and personalized medicine.
Keywords: Genetic mutations, single nucleotide polymorphisms, telomerase, telomere maintenance, tissue renewal deficiency
INTRODUCTION
Telomeres are nucleoprotein complexes located at the ends of linear chromosomes [1, 2]. Composed of tracts of tandem DNA repeats, telomeric DNA associates with sequence-specific binding proteins, to form a functional telomere. The inability of DNA polymerase to copy through the ends of linear chromosomes, known as the end replication problem [3], causes the loss of telomeric DNA repeats following each cell division. Loss of telomeric DNA accumulates with cell cycles, until a critically short length is reached. At this point, further cell division ceases and cells no longer reproduce [4].
Using primary human fibroblasts from embryos and adult sources, Leonard Hayflick first illustrated the existence of an upper limit for the replicative potential of cultured human cells [5]. Termed the “Hayflick limit”, this describes the number of times normal human cells will divide in culture. This limit in cell proliferation was later shown to be determined by critically short telomere lengths [6].
Telomeric DNA length acts as a mitotic clock, counting down the number of cell cycles before proliferation ceases indefinitely. Hence, the replicative potential of cell lineages are determined by the structural integrity of telomeres and the activities of biological pathways responsible for maintaining the length of these specialized DNA tracts. In the past decade, genetic variations of the components playing critical roles in these biological pathways were found to associate with tissue renewal deficiency syndromes [7–9]. In the following sections, we will discuss the biology of telomere structure and function, biological mechanisms for telomere maintenance, genetic diseases that are associated with telomere dysfunction, and the implications of sub-optimal telomere maintenance in tissue renewal deficiency syndromes. We believe that inter-individual variability in telomere maintenance capacity plays a significant role in several chronic inflammatory conditions, and that this is not yet fully appreciated in the translational research of pharmacogenomics and personalized medicine.
TELOMERE STRUCTURE AND FUNCTION
Telomeres
Telomeres are short tandem DNA repeats found at the ends of linear chromosomes. In humans, they are composed of hexanucleotide TTAGGG repeats, with an average length of 5–15kb [1, 2, 10]. Telomere repeats are associated with sequence-specific binding proteins [2, 11, 12]. Together, telomere repeats and associated binding proteins are organized into specialized chromatin structures known as telomere loops (T-loop), detectable by electron microscopy [13, 14]. T-loops are formed by the resection of the C-rich strand of telomeres, creating a G-rich overhang that loops back and invades the duplex telomeric repeat DNA to form a displacement loop [15, 16]. These structures cap chromosome ends, which allows for epigenetic modifications distinguishing them from normal chromatin [17, 18].
Telomeres provide physical protection against the actions of nucleases and differentiate the natural ends of chromosomes from random DNA breaks [19, 20]. Together, the nuclease-dependent resection of telomeres to create single stranded overhangs of the G-rich strand, and the inability of conventional DNA polymerase to copy through the ends in lagging-strand DNA replication, contribute to the loss of telomeric repeats every time the genome is replicated [21]. An average of 50–100 base pairs are lost from telomeric DNA tracts with each cell division [22]. This attrition accumulates with each replication, until telomeres become critically short. At this point, cellular surveillance mechanisms are activated, and either proliferative senescence or apoptosis is induced [23, 24]. Proliferative arrest induced by short telomeres serves as a tumor-suppressive mechanism: the number of telomere repeats sets the upper limit for the number of times a cell lineage can divide. By limiting the number of cell divisions, one can prevent the propagation of DNA mutations that underlie carcinogenesis [4, 25].
Telomere dysfunction describes the loss of normal capping at chromosome ends. This occurs when proliferation continues past the short telomere checkpoint, or when telomere- binding proteins fail to associate with these repeats [10, 26]. Uncapped chromosome ends are recognized as damaged DNA and set off DNA repair mechanisms [10, 27, 28]. DNA repair of chromosome ends with dysfunctional telomeres has detrimental consequences. In contrast to the repair of actual damaged DNA, the inappropriate repair of telomeric sites results in telomere fusions and gross genomic alterations [1, 29]. In short, the protective role of telomeres is essential, as it promotes survival cues, allows for proliferation, and prevents genomic instability.
Telomere Binding Proteins
A six-protein complex, shelterin, binds to telomeric DNA repeats. Shelterin binding to telomeric DNA is sequence specific; immunofluorescence experiments have shown that components of shelterin localize only to telomeres in normal growth states [20, 30, 31]. Of the six protein members of the shelterin complex, three directly bind to telomeric DNA. These include Telomeric Repeat Binding Factor 1 (TRF1) [32], which binds to the double-stranded area of the telomere, Telomeric Repeat Binding Factor 2 (TRF2) [33], which binds to the double and single-stranded telomeric sequence junctions, and lastly, Protection of Telomeres 1 (POT1) [34], which binds to the single-stranded G-rich overhangs. The other members of the shelterin complex include TRF1-Interacting Nuclear Protein 2 (TIN2) [35], a human version of yeast Repressor/Activator Protein 1 (hRap1) [36] and TPP1 [37, 38] (formerly identified as TINT1, PTOP, or PIP1, encoded by the ACD gene). Other proteins associate with telomeres non-exclusively, mainly through transient or cell cycle dependent protein-protein interactions with members of the shelterin complex [20, 39]. Notably, this list includes a large number of proteins found in DNA damage response pathways. Numerous members of homologous recombination and non-homologous end-joining pathways of double-strand DNA break repair are associated with specific telomere-binding proteins, underscoring the importance of these pathways in telomere function [39–41].
TELOMERASE
Telomerase is a ribonucleoprotein enzyme complex responsible for the de novo synthesis of telomeric DNA repeats [42–44]. A special type of reverse transcriptase, telomerase extends telomere ends by adding T2AG3 repeats templated by its integral RNA subunit [45, 46]. Telomerase activity is rapidly down-regulated following embryonic development, except in germ cells, certain stem cell compartments and specific hematopoietic cell types [47–49]. The downregulation of telomerase activity in somatic cells is considered to be a tumor-suppressive mechanism, limiting cellular lifespan and the accumulation of genetic mutations that could lead to cellular transformation [25, 50, 51].
In vivo, the telomerase holoenzyme is a megadalton complex [52]. Two subunits of this complex are required to reconstitute enzymatic activity in vitro: the telomerase RNA (TER), a ubiquitously expressed non-coding RNA, which provides the template sequence for catalysis [53–55] and the telomerase reverse transcriptase (TERT), responsible for the catalytic activity of the enzyme complex [56, 57]. Enzyme composition is significantly more complex in vivo. TER is found in most human tissues and its in vivo stability is contingent on its stable association with a chaperone, the H/ACA protein complex [58, 59]. Recently, a chain of biogenesis events has been identified: protein factors NAF1 [60, 61] and Shq1 [62, 63] load the core H/ACA protein complexonto their cognate RNA binding partners, followed by Gar1 protein association with the mature H/ACA complex to determine intracellular location [64]. The core H/ACA proteins, dyskerin, Nhp2 and Nop10, form a complex that binds to and remains in association with telomerase RNA throughout its cellular lifespan [63, 65]. TER H/ACA protein complexes are not catalytically active. Enzyme activity is conferred by the association of TERT [57, 66, 67]. Helicases, (reptin, pontin) [68], protein localization/transport factors (nucleolin [69], PinX1 [70], staufen [71], 14-3-3 [72]), chaperone proteins (Hsp90, p23 [73]) and biogenesis accessory factors (TCAB) [74, 75] are required to assemble TERHACA RNP and TERT into the telomerase enzyme [76, 77]. Finally, the newly assembled telomerase enzyme (TERHACA RNP and TERT) needs to associate with transport/accessory factors, including the human Est1 protein [78, 79], hnRNPs [80, 81], TCAB [74, 75], as well as members of the shelterin complex, to be properly localized to the telomere ends for catalysis [82, 83]. Activity-based purification of telomerase holoenzyme in vivo revealed that the active complex has a simple composition, including two copies of TERT, TER and dyskerin [84]. However, the purification scheme was based on a primer extension assay, and this method may not have identified all holoenzyme components that are required to recognize and extend natural telomeres.
In the following sections, we will describe the molecular and functional characteristics of the core telomerase components. In contrast to ciliate telomerase, the human telomerase enzyme shares nearly all of its biogenesis and regulatory components with other biological pathways. Accessory proteins that play a role in the biogenesis of telomerase or regulate its activity are listed in Table 1. The specific roles of these factors have been summarized in several excellent reviews [63, 76, 77].
Table 1.
Transient Telomerase Pathway Components with other Biological Roles.
| Protein Components | Roles in Telomerase Pathways | Other Biological Functions | Effects of changes in expression levels in Telomere maintenance |
|---|---|---|---|
| NAF1 | Early assembly factor for H/ACA RNA (including TER) | H/ACA small RNAs biogenesis | ND |
| Shq1 | Knockdown of SHQ1 causes a decrease in TER accumulation | ||
| hStaufen | TER associating complex | Proper RNA localization | ND |
| L22 | Component of 60S ribosomal subunit | ||
| SmB and SmD3 | TER associating complex, binding to the CAB box of the CR7 region | SMN complex | ND |
| T-CAB | TER associating complex, binding to the CAB box of the CR7 region | Small Cajal body RNAs biogenesis | TCAB knockdown leads to telomere shortening in but no decrease in telomerase activity or amount of TER |
| Pontin and Reptin | Assembly of TER, dyskerin and TERT | AAA+ ATPase Chromatin remodeling, transcription co-regulation, DNA damage responses | Pontin knockdown decreases dyskerin steady state levels and telomerase activity by 10–20%. |
| Hsp90 and p23 | TERT chaperones | Chaperone functions in nuclear and ligand activated receptors | Inhibition of Hsp90 or p23 blocks the assembly of telomerase. |
| 14-3-3 | Nuclear localization of TERT by masking of the TERT nuclear export signal | Controlling intracellular signaling, molecular chaperone functions, and intracellular localization | Mutant 14-3-3 leads to the decrease of hTERT nuclear accumulation but does not reduce telomerase activity in vitro |
| Nucleolin | TERT nucleolar localization | Synthesis and maturation of ribosome RNAs | Nucleolin mutants prevent TERT nucleolar localization but have no effect on telomerase activity |
| PinX1 | TERT nucleolar localization | rRNA and snoRNA maturation | Overexpression of PinX1 inhibits telomerase activity and leads to telomere shortening |
| hnRNPs | Telomere telomerase interaction | Regulation of alternative splicing and mRNA transport and packaging | Overexpression of hnRNP C1 and U leads to a decrease in telomere length |
| NAT10 | Telomerase assembly, disassembly, and localization | Nucleolar assembly, cytokinesis, and microtubule stabilization | NAT10 overexpression causes a decrease in telomere length but not does not affect telomerase activity |
| GNL3L | Telomerase assembly, disassembly, and localization | Nucleolar GTPase | GNL3L overexpression causes a decrease in telomere length but does not affect telomerase activity |
| c-Abl | c-Abl phosphorylates hTERT and inhibits its activity. | Cell differentiation, cell division, cell adhesion, and stress response | Genetic knock-out of c-Abl causes increase in telomere length |
| Akt | Akt phosphorlates hTERT, increasing telomerase activity. | Angiogenesis, cell survival, and cell division | Overexpression of Akt decreases telomere length in HTC cells. |
| MKRN | Regulates ubiquitination of hTERT | Lysine ubiquitination, targeting proteins for proteasome degradation | Overexpression of MKRN1 decreases telomerase activity and telomere length |
| SMURF | SMURF2 activated in presence of shortened telomeres | ubiquitination, targeting proteins for proteasome degradation | SMURF2 is increased by telomere shortening. |
Note: Human telomerase complex shares a large number of its biogenesis, regulatory and localization factors with other biological pathways. Genetic defects and variations in these components may also affect telomere maintenance indirectly.
Telomerase RNA (TER)
Telomerase RNA is a non-coding RNA with a mature length of 451nt [58, 85]. In contrast to many small nuclear RNAs, TER is transcribed by RNA Polymerase II and has a minimal 341nt Pol II-type promoter upstream of the transcription start site [86]. Binding with the proteins NF-Y and Sp1 activates the TER promoter. In contrast, Sp3 binding represses the promoter [87]. Induced mutations in the Sp1 binding sites were found to negatively effect TER transcription. While TER is ubiquitously transcribed in all human tissues, up-regulation of TER transcription is observed in some cancerous cell lines and tumor samples [88–90].
In addition to providing the template sequence for telomere synthesis, TER has a number of secondary structures that are essential for telomerase activity. TER pseudoknot domain (nt 1–209) contains the telomeric repeat template [91]. Together with the TER CR4-CR5 domain (nt 241–330), the pseudoknot domain is also necessary for the binding of TERT and optimal enzyme processivity [54, 92, 93]. The hairpin-hinge-hairpin-ACA (H/ACA) motif of TER at nt 275–451 is critical for its cellular accumulation, 3′ end processing, and intranuclear trafficking [58, 94]. Through this motif, TER associates with H/ACA proteins dyskerin, Nhp2 and Nop10 at its transcription sites. H/ACA protein binding directs the functional development and endonuclease trimming of TER transcripts into a mature form [95]. The 3′ terminal hairpin (CR7 domain) within the H/ACA motif in TER contains a Cajal body box (CAB box) [96] and a BIO box [97]. The CAB box is essential for the accumulation of TER at Cajal bodies in the cell nucleus, while the BIO box is responsible for transient association of TER with Sm proteins for RNA maturation [97]. Observed deficiencies in the CAB box have indicated that this motif is important not only for TER accumulation at Cajal bodies, but also the association of telomerase with telomeres and subsequent telomere elongation via telomerase [98, 99].
Telomerase Reverse Transcriptase (TERT)
Telomerase reverse transcriptase (TERT) is a single chain polypeptide of 125 kDa [56, 100]. The TERT gene contains 16 exons, spanning more than 37 kb and is found on chromosome 5p15.33 [101, 102]. TERT is functionally divided into four regions [103, 104]. The N-terminal domain (TEN, also known as RID1 or NDAT) is believed to play a role in primer binding and product alignment [105, 106]. Mutagenesis analysis of specific residues from these regions has revealed their involvement in DNA-primer binding specificity [107, 108]. Linker-scan mutations along this region also reveal subdomains that are responsible for the telomere binding and recognition functions of telomerase. Also known as the N-terminal dissociation of telomerase activity domain (NDAT), amino acid mutations in this region prevent the catalytically active telomerase enzyme from elongating telomere substrate in vivo [109]. The high-affinity RNA binding domain (TRBD, also known as RID2) contains several RNA recognition motifs (RRM) and is universally conserved in all known TERT sequences [105, 110–112]. Mutations in RID2 result in a loss of telomerase catalytic activity due to lost interaction with the RNA template TER [111]. The reverse transcriptase (RT) domain is responsible for catalyzing the assembly of the telomeric DNA repeats using the TER template [113, 114]. Mutations in any of its conserved aspartic acid residues ablate telomerase catalytic activity [114]. In contrast to conventional RT, TERT only minimally extends RNA-DNA templates and primer pairs. Catalysis occurs with quantum gains of 6nt repeats per catalytic cycle, followed by dissociation of the product or the translocation of the enzyme towards the ends of newly synthesized telomere repeats, to be positioned for another cycle of catalysis [94, 105]. Finally, studies of the C-terminal domain of TERT (TEC or CDAT) have revealed its role in telomerase enzyme processivity, and that it is essential for telomere substrate recognition and localization [115, 116].
TERT expression is down regulated in most human somatic cells following embryonic development [49, 94]. The TATA-less TERT promoter spans the genomic region from the start codon to 1100 bp upstream [117]. This region exhibits a high GC content, with two CpG islands important for promoter activity [102, 117]. Methylation at the CpG islands correlates with the transcriptional silencing of TERT expression [118]. However, the idea that the TERT promoter is transcriptionally silenced in human somatic cells was questioned when Hahn’s group detected an S-phase specific TERT expression, with levels too low for telomere maintenance, in primary human fibroblasts [119]. Conversely, activation of TERT transcription in cancer is a common phenomenon and has been correlated to the utilization of several oncogenic transcription-enhancer binding sites in the TERT promoter [120, 121].
TERT activity is also regulated via alternative splicing. This regulation is variable, according to developmental stages in human cells [122]. There are eight known TERT alternate transcripts, leading to six TERT protein variants with internal deletions or pre-mature truncations [123]. Three of these variants contain premature translation stops upstream of the essential RT domain. Accordingly, these variants lack polymerase activity. The other three insertional variants terminate upstream of the carboxy-terminal region. TERT’s C-terminal domain is essential for telomere maintenance in vivo [49], suggesting that these variants will not be competent for telomere extension in vivo. Even though none of the six TERT alternate transcripts encode a fully functional protein, they may act in a dominant negative manner by competitively binding to TER. It is known that the proportion of full-length transcripts drops dramatically during the process of carcinogenesis, with some cancer cell lines expressing over 80% of their TERT transcripts as alternate non-functional forms [124, 125].
TERT activity is also regulated by post-translational modifications. TERT phosphorylation by Akt (protein kinase B) increases telomerase activity [126], while phosphorylation by the c-Abl tyrosine kinase decreases enzyme activity [127]. In addition, phosphorylation of TERT is thought to regulate its intracellular localization in T-lymphocytes [128]. Signal transduction events following the activation of CD4+ T lymphocytes phosphorylate TERT, which promotes its translocation into the nucleus, where telomere synthesis can take place. While telomerase has a long biological half-life in human cells [129], following disruption of the structural stability of the enzyme complex, TERT is rapidly degraded through the ubiquitin/proteasome pathway [130].
H/ACA Binding Proteins – Dyskerin, Nhp2, Nop10 and Gar1
The H/ACA proteins, dyskerin, NOP10, NHP2 and Gar1, play important roles in the biogenesis and maturation of noncoding RNA, including ribosomal RNAs and small nuclear RNAs [63, 131, 132]. Dyskerin assembles with NOP10 and NHP2, and at a later stage with the localization factor Gar1, into a ribonucleoprotein complex [133, 134]. In conjunction with H/ACA RNAs acting as guides, this complex mediates the conversion of sequence-specific uridine to pseudouridine in non-coding RNAs [135–137]. Pseudouridylation modifications of these specific non-coding RNA uridine residues are essential for the functioning and biogenesis of mature ribosomes and spliceosomes [63, 138, 139]. In addition to their integral role in non-coding RNA biogenesis, the H/ACA proteins are telomerase holoenzyme components that bind to the H/ACA motif in TER [54, 58, 132]. TER binding to the H/ACA proteins, immediately following transcription, is essential for its processing and in vivo stability [24, 67, 140]. Biochemical studies also show that the association of NHP2, NOP10 and dyskerin with TER is vital to its steady-state accumulation in vivo [141].
Dyskerin (DKC-1)
Dyskerin is a pseudouridinylase that catalyzes the isomerization of specific uridine to pseudouridine [142]. DKC1 is located on chromosome Xq28 and consists of fifteen exons. A phylogenetically conserved housekeeping gene, it is ubiquitously transcribed into a 2.5 kb messenger RNA and translated into a 514 aa (57 kDa) nuclear protein dyskerin [143]. Targeted deletion of DKC1 causes embryonic lethality in mice [144]; dyskerin homologs are also essential in lower eukaryotes such as drosophila and yeast [142, 145]. The pseudouridine synthase activity of dyskerin is conferred by the N-terminal catalytic core, which contains two motifs of tRNA pseudouridine synthase B (TruB, an E. coli. protein). Point mutations of key residues in the TruB domain ablate pseudouridinase functions in yeast and drosophila orthologs of dyskerin. The single aspartic acid residue (D125 in humans) in the second TruB motif forms the enzyme-substrate intermediate during the isomerization of uridine to pseudouridine [146]. The carboxyl-terminal half of dyskerin contains the pseudouridine synthase and archaeosine transglycosylase (PUA) domain [147–149]. Crystallography analysis of the archaea dyskerin ortholog revealed RNA binding and recognition signature motifs that defined its functions. Two surfaces in these dyskerin orthologs were identified to form the main RRM of the PUA domain. These RRMs are able to recognize the minor groove of the lower double-stranded stem of the H/ACA RNA hairpins and the ACA 3′ overhangs that are signature motifs in all H/ACA RNAs [150].
Nhp2 (NHP2, NOLA2) and Nop10 (NOP10, NOLA3)
Together with dyskerin, the H/ACA proteins Nhp2 and Nop10 are presented at the sites of H/ACA RNA transcription and associate with the nascent RNA transcripts from an early assembly stage [151]. NOP10, also known as NOLA3, is a small basic protein of 64 aa (7 kDa), translated from a ubiquitously expressed 558nt mRNA [65]. Crystallography studies of Nop10 in complex with the other H/ACA proteins reveal an extensive interface with dyskerin, and this interaction is crucial for dyskerin’s enzymatic activities [149].
Nhp2, also known as NOLA2, belongs to a family of RNA binding proteins. Nhp2 is translated from a ubiquitously expressed 867nt mRNA transcript into the 153 aa protein (17kDa) [65]. The three H/ACA early assembly proteins, Nhp2, Nop10 and dyskerin, form an RNA binding surface, shown by crystallography to interact with the upper stem of H/ACA RNAs [152]. Together with the PUA domain of dyskerin, which recognizes the lower stem of the H/ACA RNAs [152], these binding surfaces position the H/ACA RNA guide residues in the correct conformation with target RNA sequences.
Gar1 (GAR1, NOLA1)
Gar1 is the late assembly component of the H/ACA complex. It is found only in the mature H/ACA ribonucleoprotein complex [64]. Known also as NOLA1, Gar1 is encoded by a ubiquitously expressed 1.3 kb messenger RNA and translated into a 217 aa (24 kDa) protein [153]. Gar 1 replaces the early H/ACA RNA loading and chaperone factors Naf1 and Shq1 in mature H/ACA RNP complexes, and is postulated to be involved in substrate loading and the intracellular localization of mature H/ACA complexes [64, 154, 155]. Unlike the other three H/ACA proteins, Gar1 deletion does not lead to a decrease in the steady state levels of H/ACA RNAs [156]. In concordance with this, there are currently no known Gar1 mutations associated with telomere maintenance diseases.
GENETIC DISEASES ASSOCIATED WITH TELOMERE MAINTENANCE DYSFUNCTION
Telomere shortening syndromes describe a number of disorders associated with telomere or telomerase dysfunction [157]. These diseases have variable clinical presentation and affect a multitude of systems, with significant overlap. Hematopoietic dysfunctions are frequent clinical presentations of telomere shortening syndromes, underscoring the importance of optimal telomere length maintenance in the homeostasis of blood cell lineages. The term telomere-shortening syndrome reflects the common molecular etiology of shortened telomeres in these disorders. They are increasingly being considered as a continuum of the same disease, rather than being distinct pathologies. With the advent of molecular diagnostic tools, the list of telomere shortening syndromes is rapidly growing and includes several disease states previously considered to be orphan or idiopathic [7, 158]. The genetics and molecular etiologies of genetic diseases associated with telomere maintenance dysfunction are summarized below.
Dyskeratosis Congenita
Dyskeratosis congenita (DC) is a rare, inherited, multisystem disease characterized by premature aging. A triad of epithelial symptoms, including the loss of nails and teeth, reticular skin hyperpigmentation, and oral leukoplakia represent the diagnostic characteristics of DC [159, 160]. Up to 85% of DC patients develop bone marrow failure and this is the primary cause of mortality. In addition to tissue failure, short telomeres prompt the development of genomic instability that precedes cellular transformation and carcinogenesis [161, 162]. Concordantly, an increased risk of cancer is also well documented in DC [163]. There are a number of other symptoms that are characteristic of the disease, including excessive tears in the eye, mental retardation, pulmonary fibrosis, and osteoporosis [164]. However, proliferative deficiencies in the epithelial and hematopoietic compartments are generally considered to be the hallmark features of DC [24, 165].
Autosomal recessive, autosomal dominant, and X-linked recessive forms of the disease have been described and are linked to genes that encode components of telomerase and telomere associating proteins [159, 166]. Of the various forms of DC, X-linked recessive DC (X-DC) accounts for the majority of cases. X-DC is primarily caused by nonsynonymous mutations in the DKC-1 gene, which encodes the protein dyskerin [143]. X-DC associated dyskerin mutations occur in two major clusters, with the first hotspot between amino acids 30 and 50. These residues are located close to the pseudouridinase catalytic domain TruB [167]. A second cluster of X-DC mutations is found in the pseudouridine synthase and archaeosine transglycosylase (PUA) domain of the dyskerin protein, which is responsible for RNA binding functions [150]. Surveys of X-DC patient materials revealed a reduction in steady-state accumulation of telomerase RNA (TER) [24, 168] and the resultant telomere maintenance defects could be rescued with the expression of a TER transgene [169]. A recent study suggested that DC-associated dyskerin mutations affect its association with an RNP accessory factor, Shq1, inferring a compromised abilityto associate with H/ACA RNAs [170].
The autosomal dominant form of DC (AD-DC) has been linked to heterozygous mutations or deletions in TER or TERT [171, 172]. At least eight TER mutations and deletions and ten TERT non-synonymous mutations are known to associate with DC. Mutant TER alleles encode changes in the domains of telomerase RNA thought to be responsible for its inherent stability or enzymatic function [8]. TERT mutations are also associated with reduced telomerase activities in reconstitution assays. Harboring a single mutant allele of TER or TERT is sufficient to precipitate clinical consequences [160, 171, 172]. Telomerase haplo-insufficiency has been confirmed by clinical observations, mouse models and biochemical data from in vivo reconstitution assays [171,173, 174]. Disease anticipation is seen in AD-DC, linked to the progressive telomere shortening evident in successive generations of families with this disease [171, 175].
Recently, mutations in the shelterin protein TIN2 have also been associated with AD-DC [176]. This discovery is the first to implicate mutations of telomere binding proteins (members of shelterin complexes) in DC pathologies and reaffirms that DC is primarily a telomere maintenance disorder [176]. DC patients carrying TIN2 mutations exhibit more severe clinical phenotypes than observed in standard DC pathology [177]. Almost all patients with TIN2 mutations develop bone marrow failure, with over 66% diagnosed before the age of ten – a much earlier disease onset than is generally expected for AD-DC [177].
Autosomal recessive forms of DC (AR-DC) are characterized by some of the most severe clinical phenotypes in the DC spectrum. Homozygous mutations in the H/ACA motif binding Nhp2 and Nop10 components of telomerase have been found to be associated with AR-DC [178, 179]. These two proteins are integral components of the H/ACA ribonucleoprotein complex and a loss of their function, similar to cases of X-linked DC, are associated with decreased telomerase RNA levels and compromised telomere maintenance. Extremely rare, only three families with homozygous mutations in either Nhp2 or Nop10 have been identified [178, 179].
Hoyeraal-Hreidarsson Syndrome
Hoyeraal-Hreidarsson syndrome (HHS) is a multi-system disorder characterized by B cell and NK cell immunodeficiency, developmental retardation, neurological defects, and bone marrow failure. Death usually occurs within the first decade of life [180]. Observations that HHS is more frequent in boys and presents with progressive bone marrow failure, led to the speculation that HHS may be a variant of DC. Subsequent molecular analyses confirmed that individuals diagnosed with HHS showed mutations in DKC1 and TERT [181–184]. Patients were also found to have shortened 3′ telomeric overhangs [185]. Given similarities in clinical presentation, and genetic lesions in the same genes, HHS is now considered to be a severe form of DC [180, 182]. The increased severity of HHS, compared to standard DC, is not currently attributed to the genetic pleiotropy of telomere shortening syndromes, or the involvement of other genetic modifiers, in addition to dyskerin or TERT.
Aplastic Anemia
Aplastic anemia (AA) was first recognized to associate with mutations in the pseudoknot region of TER. Mutations in the TER pseudoknot region reduce telomerase activity in reconstitution assays and disease phenotypes are linked to the haplo-insufficiency of telomerase activity [8, 173]. Subsequent studies of patients with aplastic anemia have revealed linkage to several TERT non-synonymous mutations, which also lead to telomerase activity haplo-insufficiency [186]. Multiple studies have shown that aplastic anemia patients have significantly shorter telomeres compared to age-matched controls [8, 186, 187], establishing the utility of screening for telomere dysfunctions in the molecular diagnosis of AA.
Other Haematopoietic Deficiency Syndromes
TERT and TER heterozygous mutations are also associated with a small number of rare hematopoietic disorders. Myelodysplastic syndrome (MDS) is characterized by the ineffective production of myeloid lineage blood cells and a predisposition to the development of acute myelogenous leukemia [188]. In addition to mutations in two transcription factors, RUNX1 and CEBPA, MDS was reported to associate with a TER nt322 G→A mutation in a single patient in an NIH study and a TER nt323 C→T mutation in a lone Japanese patient [189]. A recent familial study identifying MDS patients harboring TER 212 C→G, 309 G→T and TERT R631Q, P785L strengthened the correlation between telomere maintenance deficiency and the pathology of MDS subtypes [190]. Paroxysmal nocturnal haemoglobinuria (PNH) is a hematopoietic disorder characterized by complement- induced hemolytic anemia. This disorder can be acquired spontaneously, or develop in the context of existing aplastic anemia. A single patient diagnosed with PNH was found to harbor a mutation in the functional Sp1 binding site of the TER promoter (−99 C→G) [191]. Biochemical analysis of this promoter mutation confirmed the disruption of Sp1 binding and dysregulation of TER promoter activity, dependent on cellular context [191]. The extent to which telomere maintenance deficiencies impact PNH pathology will be better understood with further epidemiological and biochemical analysis.
Revesz Syndrome
The Revesz syndrome is characterized by bilateral exudative retinopathy, cerebellar underdevelopment, growth retardation, and nail dystrophy [192]. Many of these clinical features overlap with DC and HHS. Recently, one patient diagnosed with Revesz syndrome has been identified to have a mutation in TIN2 [176], a telomere binding protein previously associated with the AD-DC, further linking this disorder with telomere maintenance dysfunction. Of note, TIN2 mutations associated with the Revesz syndrome are different from those associated with DC or aplastic anemia [177].
Idiopathic Pulmonary Fibrosis
Idiopathic pulmonary fibrosis (IPF) is a form of idiopathic interstitial pneumonia (IIP). It is the most common type of IIP, accounting for about 40% of all cases and affects more men than women [193, 194]. IPF is characterized as the progressive and chronic formation of fibrotic scar tissue in the lungs without any known causative agent. Patients often display chronic dry cough along with exertion-induced breathlessness and 25–50% of patients develop clubbing of the digits. IPF is most commonly found in those between 50 to 70 years of age, with two thirds of all cases diagnosed in people over 60 years old [193, 195]. The average survival time following diagnosis is 2.5 to 3.5 years.
A recent study of familial IPF patients revealed mutations in both TERT and TER indicating that telomere dysfunction could be the underlying cause of the disorder in a significant proportion of patients [7]. Patients with sporadic IIP and IPF have shorter telomeres compared to age-matched controls [196]. Even in IPF patients harboring wildtype sequences for TER or TERT, shorter than normal telomere lengths are highly significant in disease etiology [197]. Given the chronic nature of IPF and the advanced age at diagnosis, telomere attrition could arise from chronic lung inflammation and the increased tissue renewal demands compared to healthy individuals. In individuals with underlying genetic variations that limit starting telomere length or telomerase function, this accelerated telomere erosion could lead to clinical symptoms.
DISEASES WITH INCREASED CELLULAR TURNOVER AND AN ACCELERATED RATE OF TELOMERE ATTRITION
Telomere shortening is one of the molecular phenotypes that define a number of genetic and inflammation-related conditions [197–202]. In contrast to disorders that have defects in telomere maintenance as a primary disease etiology, these disorders are characterized by increased cellular turnover, leading to an accelerated rate of telomere attrition (Fig. 1). Extensive research has shown a relationship between telomere length and disease progression. This section highlights some of the findings that have established this link.
Figure 1. Telomere Length Dynamics.

Telomere length is determined by the sum of various cellular processes. Each one of us inherits a starting telomere length from both parents. Activation of the reverse transcriptase telomerase, by hormonal and other transcriptional activation events, adds telomeric repeats, while genetic or chemical perturbations in this maintenance pathway impairs telomeric DNA addition.
Telomeric DNA repeats are lost during each DNA replication cycle, with the inference that an increased rate of cellular proliferation and tissue renewal will result in an increased telomere attrition rate.
DNA Repair Enzyme Deficiency Syndromes
Genetic diseases with impaired DNA damage repair capacity are frequently associated with an accelerated telomere attrition phenotype [198, 203]. Genomic damage from normal metabolism, exposure to environmental insults, such as ionizing radiation or toxins, together with mistakes accumulated from DNA replication, is detrimental to cell survival. Cellular surveillance and repair mechanisms are in place to restore genomic integrity and prevent the propagation of these errors. Genetic lesions in repair pathway components lead to an unbalanced activation of apoptosis [204]. Increasing the proliferative rate of remaining cell populations compensates for the loss of these under-repaired cells. An increased demand on cellular renewal is mirrored by a loss of telomere repeats in patients’ somatic cells.
In addition to the increased cellular turnover due to the death of unrepaired cells, several DNA damage repair deficiency syndromes impact telomere maintenance directly. Double-stranded DNA repair proteins, such as ataxia telangiectasia mutated (ATM) kinase [201, 205], the Mre11- Rad50-NBS (MRN) complex [41, 206], the Werner helicase [207, 208], the breast cancer 1 (BRCA1) protein [209, 210] and other participants of the double-stranded DNA damage repair pathways (reviewed by Jackson and Bartek [204]) are also known to participate in normal telomere maintenance homeostasis. Genetic deficiencies in these repair pathway components will directly affect telomere length maintenance, in addition to cellular turnover rate changes due to the impairment of DNA repair [201, 206].
Diseases of Chronic inflammation and Infection
Atherosclerosis
Atherosclerosis is the thickening and hardening of arterial walls by the accumulation of lipids, white blood cells, and fibrous materials. The disease is also characterized by the accumulation of immune cells to artery walls, brought about by the inflammation response [211]. The development of atherosclerosis plaques is dependent on an increased proliferation of the endothelial compartment in response to an injury. In agreement with this, patients with coronary heart disease were found to have shorter telomeres in endothelial cells, compared to controls without heart disease [212]. Overexpression of TRF2 mutants, and the ensuing telomere dysfunctions, gave rise to gene expression profiles in endothelial cells very similar to those observed during the genesis of atherosclerosis-related plaques [213]. This data argues that telomere dysfunction may have a prominent role in the pathologies of atherosclerosis.
Hypertension
Increased telomere attrition is also reported in cases of neovascularization and hypertension, two types of cardiovascular dysfunction that are also associated with an active inflammatory response (reviewed in Fuster and Andrés [200]). This led to the idea that telomere length may be useful as a quantitative measure for cardiovascular disease risk. However, it is unclear whether increased cellular proliferation due to inflammation leads to shortened telomeres, or if shorter inherited telomere lengths prompt the development of cardiovascular conditions later in life. In support of the intrinsic model, genetically engineered mice lacking TER expression were found to exhibit higher blood and urinary levels of endothelin-1 and develop hypertension [214]. Given that cardiovascular diseases are complex disease states with a wide range of genetic and environmental causes, variations in telomere reserves and attrition rates between patients warrant epidemiological analysis to illuminate mechanisms.
Hepatitis/Cirrhosis
Hepatitis is characterized by chronic inflammation of the liver, caused by the exposure to chemical toxins or biological agents such as the hepatitis C viruses. Hepatitis, and other chronic liver diseases, can lead to cirrhosis, liver failure characterized by the replacement of normal liver tissue with fibrotic scar tissue [215]. Patients with chronic liver disease have significantly shorter telomeres compared to normal individuals and there is a negative correlation between telomere length and disease progression [216, 217]. In addition, the development of fibrotic scarring in cirrhosis is dependent on telomere shortening in hepatocytes, and their subsequent senescence [218].
Rheumatoid Arthritis
Rheumatoid arthritis (RA) is a disorder of chronic inflammation that manifests in multiple forms [219]. The most common form of RA attacks joints, resulting in the inflammation of the synovium membrane. Immune systems of RA patients show signs of premature senescence, and it is generally associated with an increased CD28 negative T cell population and accelerated telomere loss in peripheral blood mononuclear cells [220]. A recent study examined naïve CD4-positive T cells from RA patients and revealed an impaired ability to activate TERT transcription [221]. Failure of antigen-stimulated induction of TERT expression leads to the premature apoptosis of these naïve T cells, which can be rescued by the ectopic expression of the TERT transgene. Dysfunction of naïve T-cells in RA patients is a further disadvantage as it compels the existing memory T-cell population to mount an immune response. It is unclear whether insufficient TERT transcription induction in naïve T-cells is a manifestation of the pathologies of RA, or, as the authors suggest, an inherent deficiency of TERT induction that precipitates the autoimmune syndrome [221].
Chronic Lung Inflammation Diseases
Chronic obstructive pulmonary disease (COPD) is a respiratory condition with systemic manifestations, including weight loss, muscle dysfunction, atherosclerosis and osteoporosis [222]. COPD is associated with a cumulative burden of noxious fume inhalation, and smoking is a predominant risk factor. In COPD, chronic inflammation of the lower airways and of the lung parenchyma result in the slow destruction of lung tissues and a gradual reduction of airflow [223]. Limitations to airflow are rarely reversible and the prognosis worsens with time. Short telomeres were found in the peripheral blood leukocytes of COPD patients. COPD patients with emphysema have telomere lengths that are significantly shorter than normal controls in alveolar type II cells, endothelial cells, peripheral blood mononuclear cells and fibroblasts [224]. Telomere lengths in COPD patients were also found to correlate positively with PaO2 and negatively with PaCO2, underscoring the role of oxidative stress on telomere maintenance [224].
HIV
Human immunodeficiency virus (HIV) maintains a chronic infection of the CD4+ T lymphocytes. Clinical studies of CD4+ T-lymphocytes have not found a correlation between infection status and residual telomere length. Instead, HIV-positive individuals are found to have short CD8+ telomere lengths, suggesting an increased demand on proliferation of this T-cell lineage to battle the HIV infection [225]. Current therapeutic goals in HIV treatment aim to reduce the viral load and maintain a functional population of CD4+ T-cells. Termed as highly active antiretroviral therapies, these anti-HIV combination therapies usually contain three different anti-viral agents with well-studied nucleoside and non-nucleoside reverse transcriptase inhibitors (NRTIs and NNRTIs) as backbones [226]. The structural similarity between HIV reverse transcriptase and human telomerase reverse transcriptase raises concerns about the adverse effects of long-term administration of these anti-HIV agents on normal telomere length homeostasis [114, 227].
TELOMERASE POLYMORPHISMS AND DISEASE ASSOCIATIONS
Until recently, large-scale screening of TERT and TER polymorphisms in the context of complex diseases has been lacking. This gap in the literature is now being addressed (Table 2). A genome-wide study has uncovered an association between a TERT SNP and sporadic idiopathic pulmonary fibrosis in a Japanese patient group [228]. This SNP (rs2736100) is found in intron 2 of TERT. The same SNP also associates with an increased risk of non small-cell lung cancers (NSCLC) in a Chinese population [229]. Non-coding intronic mutations, through the activation of alternate splicing, or other regulation changes, could conceivably affect the cellular functions of the gene product. However, there have been no reports on possible alterations in telomerase activity, enzyme functionality, or steady-state expression levels with this TERT intron 2 SNP. Alternatively, this SNP could exist in these Asian populations in linkage disequilibrium with another locus that increases susceptibility to lung dysfunction. Further epidemiological or biochemical studies will be needed to differentiate these two possibilities.
Table 2.
Single Nucleotide Polymorphism Sites in Key Telomerase Components.
| Component | Mutation | Functional Change | Functional Domain and Biological Roles | Allele Frequency | dbSNP number |
|---|---|---|---|---|---|
| Telomerase Holoenzyme Components | |||||
| TERT | −1327 C→T | C→T | Promoter: affect expression level | 0.31–0.67 | rs2735940 |
| Intron2 G→T | G→T | Intron2: function unknown | 0.42–0.63 | rs2736100 | |
| 630 G→C | S191T | RID1 motif DNA primer interaction |
ND | rs11952056 | |
| 893 G→A | A279T | Linker separating RID1 and RID2 | ND | rs61748181 | |
| 1292 C→T | H412Y | RID2 motif TER binding domain |
0.010 | rs34094720 | |
| 2902 C→A | S948R | Reverse transcriptase Catalytic RT activity |
0.019 | rs34062885 | |
| 3159 G→A | R1034H | C-Terminal Enzyme localization and processivity |
ND | rs62331332 | |
| 3242 G→a | A1062T | C-Terminal Enzyme localization and processivity |
0.012 | rs35719940 | |
| TER | 58 G→A | TER 58G/A | Pseudoknot region 3′ of the template sequence |
0.04 (in African-americans) | |
| 228 G→A | TER 228G/A | hypervariable region | 0.02 (in African-americans) | ||
| 514 G→A | NA | Outside of the mature TER sequence | 0.32 | ||
| DKC1 | 769 T→G | I226S | Trb/PUS Pseudouridine synthase domain | 0.32 | rs2728534 |
| 868 A→C | H259P | ND | rs61757608 | ||
| 945 G→T | V285F | Unknown | ND | rs17850575 | |
| 1053 C→A | L321I | PUA Pseudouridine synthase and archaeosine-specific transglycosylase domain H/ACA RNA binding |
ND | rs2728726 | |
| Nhp2 | 217 A→T | Q25L | N-terminal region, Exon 1 | ND | rs11550513 |
| Nop10 | There are no known non-synonymous SNPs reported for Nop10. | ||||
| Gar1 | There are no known non-synonymous SNPs reported for Gar1. Gar1 is also the only H/ACA protein that did not have any disease associating mutations. |
||||
| Shelterin Components | |||||
| TRF1 | 795 T→C | M257T | TRFH domain Protein heterodimerization |
ND | rs55834252 |
| 1253 C→G | L410V | Myb domain DNA binding |
ND | rs4092743 | |
| TRF2 | 964 C→A | P280Q | Undefined region between dimerization and myb domain | 0.035 | rs3401482 |
| 1254 C→G | L377V | 0.025 | rs13337258 | ||
| 1362 A→G | S413G | 0.010 | rs35874485 | ||
| POT1 | 1681 G→T | G273V | OB fold-2 domain ssDNA binding |
0.025 | rs35536751 |
| 2055 G→A | V398M | POT-1 C-terminal domain TPP1 interacting function |
0.010 | rs34973253 | |
| TINF2 | 469 G→A | A43T | TPP1 interacting domain | 0.010 | rs35653076 |
| 1052 G→A | G237D | TRF1 interacting domain | 0.064 | rs17102313 | |
| 1063 C→T | P241S | 0.009 | rs17102311 | ||
| 1600 A→C | T420P | unknown | ND | rs12883582 | |
| TPP1 | 1239 C→T | T301M | Protein binding domain (PBD) Interaction with POT-1 |
0.010 | rs72547495 |
| 1341 C→T | P335L | TPP-1 C-terminal domain Interaction with Tin2 |
ND | rs34097817 | |
| 1890 T→C | V518A | 0.500 | rs6979 | ||
| Rap1 | 128 C→G | P11A | BRCT domain Recruits negative regulator of telomere maintenance |
ND | rs11556639 |
| 554 C→A | R153S | Myb domain (no direct DNA binding) | ND | rs11556640 | |
| 971 G→A | E292K | RAP-1 C-terminal domain (RCT) Interaction with TRF2 |
ND | rs56261106 | |
| 1067 A→G | K324E | 0.033 | rs4888444 | ||
NOTE: Only selective intronic and regulatory region SNPs are listed due to space limitation, but the full list can be found at the dbSNP website. As well, rare disease-associating mutations are not listed here, but are summarized in several review articles [166,183]. At the time of writing this review, TIN2 is the only member of the shelterin complex identified with disease-associating genetic variations.
A TERT promoter SNP at −1327 (T→C; rs2735940) is associated with decreased telomere length in peripheral blood leukocytes [230]. Biochemical studies using the dual luciferase reporter assay corroborate the hypothesis that −1327C is associated with decreased TERT promoter activity, and theoretically reduces expression of the TERT protein [230]. Homozygous TERT −1327C individuals are also found to harbor an increased risk of coronary artery diseases, presumably due to the accelerated loss of telomere repeats from reduced telomerase activation [231]. TERT −1327C was found with high allele frequencies ranging from 0.31 to 0.67, with higher presentations in Asian, Sub-Saharan African and European populations [232, 233].
TERT H412Y is a nonsynonymous polymorphism (rs34094720) observed in a family with DC. Two recent studies have found that this SNP, in the N-terminal domain of TERT, led to significantly decreased telomerase activity [186, 234]. However, this same H412Y TERT isoform was tested previously and showed only a slight decrease in telomerase activity in vitro, with no measurable differences in vivo [196]. TERT H412Y has been reported with an allelic frequency of up to 2% in a CEPH (Utah residents with ancestry from northern and western Europe) population, which is in direct contrast with the known DC frequency of one case per million, including all different inheritance forms. The moderate reduction in telomerase activity of H412Y TERT suggests that this SNP represents an additional risk factor, rather than a monogenic determinant for disease association.
Epidemiology studies have also linked TERT SNPs to increased incidences of acute myelogenous leukemia (AML) [235]. AML is the uncontrolled replication of white blood cells in the bone marrow that interferes with normal blood cell development. Patients with DC and aplastic anemia have a predisposition for developing AML, presumably due to the increased genomic instability associated with the loss of telomere length protection. AML is therefore considered an alternate progression of disease in bone marrow failure syndromes [236]. A recent linkage study revealed that patients diagnosed with AML had a three times higher prevalence of TERT A1062T SNP (rs35719940) compared to controls and decreased telomerase activity compared to wild type [235]. A subsequent report unveiled a link between this A1062T-TERT and other hematological malignancies including diffuse large B-cell lymphoma (DL-BCL) and chronic lymphocytic leukemia (CLL), but no significant linkage is observed with multiple myeloma (MM) [237]. However, biochemical analyses of reconstituted A1062T TERT telomerase activity by different research groups resulted in contradictory conclusions [196, 235]. The lack of correlation between these studies may be attributed to the variability between laboratory protocols for measuring telomerase activity in vitro. Presumably weaker perturbations of telomerase activity by these SNPs could also approach the sensitivity limit of current assays, leading to a higher variability in activity measurements.
Association to multiple cancers’ susceptibility was shown in several large-scale population studies to 5p15.33, a genomic region where TERT and the CLPTM1-L (Cleft Lip and Palate Transmembrane protein 1-like protein) genes reside [238–240]. In particular, a TERT coding region SNP (rs2736098) corresponding to a synonymous mutation (A305A) has been connected to an increased risk in basal cell carcinoma, as well as a predisposition in lung, bladder, prostate and cervical cancers. It is currently not clear whether this TERT synonymous SNP exists in linkage disequilibrium with other genetic changes [240].
TER nt58 G→A and nt228 G→A were initially reported to associate with aplastic anemia. However, the discovery that these two alleles exist in frequencies of up to 2% and 4% in African American populations weakens this disease association. In addition, contrasting data were obtained using the telomerase reconstitution assays, with reports of weak or no perturbation of in vitro telomerase activities with the introduction of these TER mutations in the reconstituted enzyme. Similarly confusing results were found with other identified TERT nonsynonymous SNPs. TER nt514 GàA SNP was reported to associate with an increased risk of systemic sclerosis, a chronic inflammation disease in connective tissue with unknown etiology [241]. This SNP is found immediately downstream of the mature TER sequence but the functional significance of this genetic variation is not known.
CONCLUSIONS
Autosomal dominant forms of dyskeratosis congenita (DC) have been associated with an earlier onset and an increased severity of disease presentation in subsequent generations. In addition to dysfunctional telomerase enzyme, each successive generation of patients inherits a shorter starting telomere length from affected parents. This makes the case that telomerase dysfunctions precipitate clinical symptoms only when telomere lengths are critically short. The high correlation between residual telomere lengths and the onset of clinical events have greatly changed the diagnosis of DC-like diseases. Due to variability in the clinical presentations of telomere shortening syndromes, recent studies have adopted the use of peripheral blood leukocytes (PBL) telomere length measurement as a diagnostic tool for DC-like conditions. In the case of chronic inflammation diseases, mean telomere length in PBL also serves a predictive role in disease progression and severity. Strong correlations have already been established between the progression of disease and mean telomere length in PBL for chronic disorders such as hepatitis and cirrhosis.
Could SNPs found in telomere pathway genes account for the progression kinetics of these complex diseases? Presumably SNPs in the telomere pathway could either exacerbate or alleviate telomere loss alongside primary disease etiology. Recent discoveries in rheumatoid arthritis provide some clues: the lack of TERT induction in RA patients’ naïve T cells could be an inherent regulatory/transcription activation defect, leading to an exacerbated disease phenotype, by depleting functional T-cell populations and increasing reliance on memory T-cell reactivation for immune function.
We postulate that polymorphisms in telomere pathway genes could alter their function or regulation, in a manner that accelerates the telomere loss phenotype in certain complex disorders (Fig. 2). Weak perturbations of telomere pathway components are not, on their own, sufficient to precipitate clinical symptoms. However, in the presence of other genetic predispositions, such as the loss of DNA repair capacity in xeroderma pigmentosum, or with prolonged exposure to strong environmental stresses such as cigarette smoking in the case of COPD, SNPs with low penetrance could exacerbate the clinical severity of existing conditions, by accelerating the kinetics of telomere loss. Indeed, several TERT polymorphic variants have already been identified as susceptibility factors in IPF and AML. With increased scrutiny and the availability of large-scale epidemiology studies and analytical tools, it is expected that these and other SNPs of telomere pathway components could be identified as risk factors for complex diseases.
Figure 2. Telomere Length Maintenance and Disease Progression.
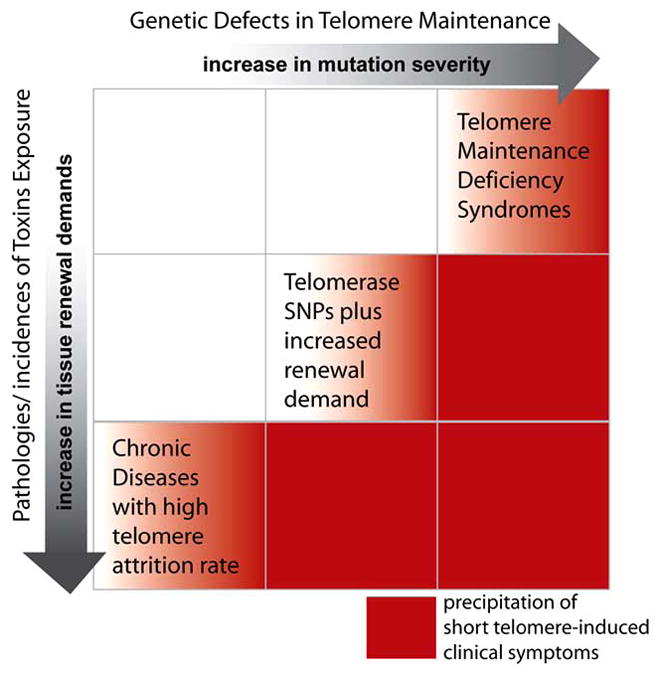
Schematic of genetic perturbations in the telomere maintenance pathway and the presence of disease pathologies. Clinical symptoms correlate with short telomere lengths, by impaired synthesis, or an accelerated loss.
Several studies have illustrated the utility of telomerase replacement strategies to effectively correct DC molecular phenotypes [169, 242, 243]. Hence, telomerase activation strategies using small-molecule transcription activators are being heavily pursued [244, 245]. With the advent of available tools, it is tempting to speculate that personal therapies of telomerase activation/replacement could be tailored to suit individual needs. In addition to reverting clinical phenotypes of severe telomerase deficiency syndromes such as DC, temporally controlled activation of telomerase could also alleviate clinical phenotypes of diseases associated with a high cellular turnover. Currently still being studied in the laboratory, these therapeutic tools will undoubtedly revolutionize the clinical strategies against many disorders with an underlying deficiency in tissue renewal capacity.
Acknowledgments
Research on telomerase deficiency syndromes in the Wong laboratory is supported by the Canadian Institutes of Health Research (MOP-81094) and the Michael Smith Foundation of Health Research (CI-SCH-102). MAT is a recipient of the CIHR Frederick Banting and Charles Best Canada Graduate Scholarship. JMYW is supported by the CRC and the MSFHR Career Award programs. The authors would like to thank fellow members of the Wong laboratory for helpful discussions. We would especially like to thank Helen Fleisig, for providing the illustrations in this manuscript
Footnotes
This manuscript is dedicated to the memory of Werner Kalow.
References
- 1.Blackburn EH. Switching and signaling at the telomere. Cell. 2001;106(6):661–73. doi: 10.1016/s0092-8674(01)00492-5. [DOI] [PubMed] [Google Scholar]
- 2.Collins K. Mammalian telomeres and telomerase. Curr Opin Cell Biol. 2000;12:378–83. doi: 10.1016/s0955-0674(00)00103-4. [DOI] [PubMed] [Google Scholar]
- 3.Levy MZ, Allsopp RC, Futcher AB, et al. Telomere end-replication problem and cell aging. J Mol Biol. 1992;225:951–60. doi: 10.1016/0022-2836(92)90096-3. [DOI] [PubMed] [Google Scholar]
- 4.Campisi J. From cells to organisms: can we learn about aging from cells in culture? Exp Gerontol. 2001;36(4–6):607–18. doi: 10.1016/s0531-5565(00)00230-8. [DOI] [PubMed] [Google Scholar]
- 5.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 6.Allsopp RC, Chang E, Kashefi-Aazam M, et al. Telomere shortening is associated with cell division in vitro and in vivo. Exp Cell Res. 1995;220:194–200. doi: 10.1006/excr.1995.1306. [DOI] [PubMed] [Google Scholar]
- 7.Armanios MY, Chen JJ, Cogan JD, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356(13):1317–26. doi: 10.1056/NEJMoa066157. [DOI] [PubMed] [Google Scholar]
- 8.Marrone A, Stevens D, Vulliamy T, et al. Heterozygous telomerase RNA mutations found in dyskeratosis congenita and aplastic anemia reduce telomerase activity via haploinsufficiency. Blood. 2004;104(13):3936–42. doi: 10.1182/blood-2004-05-1829. [DOI] [PubMed] [Google Scholar]
- 9.Vulliamy T, Dokal I. Dyskeratosis congenita. Semin Hematol. 2006;43(3):157–66. doi: 10.1053/j.seminhematol.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 10.de Lange T. Protection of mammalian telomeres. Oncogene. 2002;21(4):532–40. doi: 10.1038/sj.onc.1205080. [DOI] [PubMed] [Google Scholar]
- 11.Greider CW, Collins K, Autexier C. DNA Replication. Cold Spring Harbor Laboratory Press; 1996. DNA telomerases; pp. 619–38. [Google Scholar]
- 12.McEachern MJ, Krauskopf A, Blackburn EH. Telomeres and their control. Annu Rev Gen. 2000;34:331–58. doi: 10.1146/annurev.genet.34.1.331. [DOI] [PubMed] [Google Scholar]
- 13.de Lange T. T-loops and the origin of telomeres. Nat Rev Mol Cell Biol. 2004;5(4):323–9. doi: 10.1038/nrm1359. [DOI] [PubMed] [Google Scholar]
- 14.Griffith JD, Comeau L, Rosenfield S, et al. Mammalian telomeres end in a large duplex loop. Cell. 1999;97(4):503–14. doi: 10.1016/s0092-8674(00)80760-6. [DOI] [PubMed] [Google Scholar]
- 15.Makarov VL, Hirose Y, Langmore JP. Long G tails at both ends of human chromosomes suggest a C strand degradation mechanism for telomere shortening. Cell. 1997;88(5):657–66. doi: 10.1016/s0092-8674(00)81908-x. [DOI] [PubMed] [Google Scholar]
- 16.McElligott R, Wellinger RJ. The terminal DNA structure of mammalian chromosomes. EMBO J. 1997;16(12):3705–14. doi: 10.1093/emboj/16.12.3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garcia-Cao M, O’Sullivan R, Peters AH, et al. Epigenetic regulation of telomere length in mammalian cells by the Suv39h1 and Suv39h2 histone methyltransferases. Nat Genet. 2004;36(1):94–9. doi: 10.1038/ng1278. [DOI] [PubMed] [Google Scholar]
- 18.Schoeftner S, Blasco MA. Developmentally regulated transcription of mammalian telomeres by DNA-dependent RNA polymerase II. Nat Cell Biol. 2008;10(2):228–36. doi: 10.1038/ncb1685. [DOI] [PubMed] [Google Scholar]
- 19.Maser RS, DePinho RA. Telomeres and the DNA damage response: why the fox is guarding the henhouse. DNA Repair (Amst) 2004;3(8–9):979–88. doi: 10.1016/j.dnarep.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 20.de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19(18):2100–10. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- 21.Shay JW, Wright WE. Hayflick, his limit, and cellular ageing. Nat Rev Mol Cell Biol. 2000;1(1):72–6. doi: 10.1038/35036093. [DOI] [PubMed] [Google Scholar]
- 22.Huffman KE, Levene SD, Tesmer VM, et al. Telomere shortening is proportional to the size of the G-rich telomeric 3′-overhang. J Biol Chem. 2000;26:19719–22. doi: 10.1074/jbc.M002843200. [DOI] [PubMed] [Google Scholar]
- 23.Stewart SA, Weinberg RA. Telomerase and human tumorigenesis. Sem in Cancer Biol. 2000;10:399–406. doi: 10.1006/scbi.2000.0339. [DOI] [PubMed] [Google Scholar]
- 24.Wong JM, Collins K. Telomere maintenance and disease. Lancet. 2003;362(9388):983–8. doi: 10.1016/S0140-6736(03)14369-3. [DOI] [PubMed] [Google Scholar]
- 25.Rodier F, Kim SH, Nijjar T, et al. Cancer and aging: the importance of telomeres in genome maintenance. Int J Biochem Cell Biol. 2005;37(5):977–90. doi: 10.1016/j.biocel.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 26.Karlseder J, Smogorzewska A, de Lange T. Senescence induced by altered telomere state, not telomere loss. Science. 2002;295(5564):2446–9. doi: 10.1126/science.1069523. [DOI] [PubMed] [Google Scholar]
- 27.d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426(6963):194–8. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 28.Karlseder J, Broccoli D, Dai Y, et al. p53- and ATM- dependent apoptosis induced by telomeres lacking TRF2. Science. 1999;283:1321–5. doi: 10.1126/science.283.5406.1321. [DOI] [PubMed] [Google Scholar]
- 29.Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol. 2003;13(17):1549–56. doi: 10.1016/s0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]
- 30.Liu D, O’Connor MS, Qin J, et al. Telosome, a mammalian telomere- associated complex formed by multiple telomeric proteins. J Biol Chem. 2004;279(49):51338–42. doi: 10.1074/jbc.M409293200. [DOI] [PubMed] [Google Scholar]
- 31.Xin H, Liu D, Songyang Z. The telosome/shelterin complex and its functions. Genome Biol. 2008;9(9):232. doi: 10.1186/gb-2008-9-9-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chong L, van Steensel B, Broccoli D, et al. A human telomeric protein. Science. 1995;270:1663–7. doi: 10.1126/science.270.5242.1663. [DOI] [PubMed] [Google Scholar]
- 33.Broccoli D, Smogorzewska A, Chong L, et al. Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2. Nat Genet. 1997;17(2):231–5. doi: 10.1038/ng1097-231. [DOI] [PubMed] [Google Scholar]
- 34.Baumann P, Cech TR. Pot1, the putative telomere end-binding protein in fission yeast and humans. Science. 2001;292:1171–5. doi: 10.1126/science.1060036. [DOI] [PubMed] [Google Scholar]
- 35.Kim S, Kaminker P, Campisi J. TIN2, a new regulator of telomere length in human cells. Nat Genet. 1999;23:405–12. doi: 10.1038/70508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li B, Oestreich S, de Lange T. Identification of human Rap1: implications for telomere evolution. Cell. 2000;101:471–83. doi: 10.1016/s0092-8674(00)80858-2. [DOI] [PubMed] [Google Scholar]
- 37.O’Connor MS, Safari A, Xin H, et al. A critical role for TPP1 and TIN2 interaction in high-order telomeric complex assembly. Proc Natl Acad Sci USA. 2006;103(32):11874–9. doi: 10.1073/pnas.0605303103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ye JZ, Hockemeyer D, Krutchinsky AN, et al. POT1-interacting protein PIP1: a telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev. 2004;18(14):1649–54. doi: 10.1101/gad.1215404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dimitrova N. Telomere-binding proteins in humans. In: Hiyama K, editor. Telomeres and Telomerase in Cancer. Humana Press; 2009. pp. 23–46. [Google Scholar]
- 40.d’Adda di Fagagna F, Teo SH, Jackson SP. Functional links between telomeres and proteins of the DNA-damage response. Genes Dev. 2004;18(15):1781–99. doi: 10.1101/gad.1214504. [DOI] [PubMed] [Google Scholar]
- 41.Zhu X, Küster B, Mann M, et al. Cell-cycle-regulated association of RAD50/MRE11/NBS1 with TRF2 and human telomeres. Nat Genet. 2000;25:347–52. doi: 10.1038/77139. [DOI] [PubMed] [Google Scholar]
- 42.Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985;43:405–13. doi: 10.1016/0092-8674(85)90170-9. [DOI] [PubMed] [Google Scholar]
- 43.Blackburn EH. The end of the (DNA) line. Nat Struct Biol. 2000;7:847–850. doi: 10.1038/79594. [DOI] [PubMed] [Google Scholar]
- 44.Collins K. Structure and function of telomerase. Curr Opin Cell Biol. 1996;8:374–80. doi: 10.1016/s0955-0674(96)80013-5. [DOI] [PubMed] [Google Scholar]
- 45.Greider CW, Blackburn EH. The telomere terminal transferase of Tetrahymena is a ribonucleoprotein enzyme with two kinds of primer specificity. Cell. 1987;51:887–98. doi: 10.1016/0092-8674(87)90576-9. [DOI] [PubMed] [Google Scholar]
- 46.Greider CW, Blackburn EH. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature. 1989;337:331–7. doi: 10.1038/337331a0. [DOI] [PubMed] [Google Scholar]
- 47.Holt SE, Shay JW. Role of telomerase in cellular proliferation and cancer. J Cell Physiol. 1999;180:10–8. doi: 10.1002/(SICI)1097-4652(199907)180:1<10::AID-JCP2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 48.Weng NP, Hathcock KS, Hodes RJ. Regulation of telomere length and telomerase in T and B cells: a mechanism for maintaining replicative potential. Immunity. 1998;9(2):151–7. doi: 10.1016/s1074-7613(00)80597-x. [DOI] [PubMed] [Google Scholar]
- 49.Wright WE, Piatyszek MA, Rainey WE, et al. Telomerase activity in human germline and embryonic tissues and cells. Dev Genet. 1996;18:173–9. doi: 10.1002/(SICI)1520-6408(1996)18:2<173::AID-DVG10>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 50.Kim SH, Kaminker P, Campisi J. Telomeres, aging and cancer: in search of a happy ending. Oncogene. 2002;21(4):503–11. doi: 10.1038/sj.onc.1205077. [DOI] [PubMed] [Google Scholar]
- 51.Fleisig HB, Wong JM. Telomerase as a clinical target: current strategies and potential applications. Exp Gerontol. 2007;42(1–2):102–12. doi: 10.1016/j.exger.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 52.Schnapp G, Rodi H-P, Rettig WJ, et al. One-step affinity purification protocol for human telomerase. Nucleic Acids Res. 1998;26(13):3311–3. doi: 10.1093/nar/26.13.3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beattie TL, Zhou W, Robinson MO, et al. Reconstitution of human telomerase activity in vitro. Curr Biol. 1998;8:177–80. doi: 10.1016/s0960-9822(98)70067-3. [DOI] [PubMed] [Google Scholar]
- 54.Theimer CA, Blois CA, Feigon J. Structure of the human telomerase RNA pseudoknot reveals conserved tertiary interactions essential for function. Mol Cell. 2005;17(5):671–82. doi: 10.1016/j.molcel.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 55.Autexier C, Pruzan R, Funk W, et al. Reconstitution of human telomerase activity and identification of a minimal functional region of the human telomerase RNA. EMBO. 1996;15(21):5928–35. [PMC free article] [PubMed] [Google Scholar]
- 56.Meyerson M, Counter CM, Eaton EN, et al. hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell. 1997;90:785–95. doi: 10.1016/s0092-8674(00)80538-3. [DOI] [PubMed] [Google Scholar]
- 57.Morin GB. The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell. 1989;59:521–9. doi: 10.1016/0092-8674(89)90035-4. [DOI] [PubMed] [Google Scholar]
- 58.Mitchell JR, Cheng J, Collins K. A box H/ACA small nucleolar RNA-like domain at the human telomerase RNA 3′ end. Mol Cell Biol. 1999;19:567–76. doi: 10.1128/mcb.19.1.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mitchell JR, Wood E, Collins K. A telomerase component is defective in the human disease dyskeratosis congenita. Nature. 1999;402:551–5. doi: 10.1038/990141. [DOI] [PubMed] [Google Scholar]
- 60.Fatica A, Dlakic M, Tollervey D. Naf1 p is a box H/ACA snoRNP assembly factor. RNA. 2002;8(12):1502–14. [PMC free article] [PubMed] [Google Scholar]
- 61.Trahan C, Dragon F. Dyskeratosis congenita mutations in the H/ACA domain of human telomerase RNA affect its assembly into a pre-RNP. RNA. 2009;15(2):235–43. doi: 10.1261/rna.1354009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grozdanov PN, Roy S, Kittur N, et al. SHQ1 is required prior to NAF1 for assembly of H/ACA small nucleolar and telomerase RNPs. RNA. 2009;15(6):1188–97. doi: 10.1261/rna.1532109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Meier UT. The many facets of H/ACA ribonucleoproteins. Chromosoma. 2005;114(1):1–14. doi: 10.1007/s00412-005-0333-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Darzacq X, Kittur N, Roy S, et al. Stepwise RNP assembly at the site of H/ACA RNA transcription in human cells. J Cell Biol. 2006;173(2):207–18. doi: 10.1083/jcb.200601105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pogacic V, Dragon F, Filipowicz W. Human H/ACA small nucleolar RNPs and telomerase share evolutionarily conserved proteins NHP2 and NOP10. Mol Cell Biol. 2000;20:9028–40. doi: 10.1128/mcb.20.23.9028-9040.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Harrington L. Biochemical aspects of telomerase function. Cancer Lett. 2003;194:139–54. doi: 10.1016/s0304-3835(02)00701-2. [DOI] [PubMed] [Google Scholar]
- 67.Mitchell JR, Collins K. Human telomerase activation requires two independent interactions between telomerase RNA and telomerase reverse transcriptase in vivo and in vitro. Mol Cell. 2000;6:361–71. doi: 10.1016/s1097-2765(00)00036-8. [DOI] [PubMed] [Google Scholar]
- 68.Venteicher AS, Meng Z, Mason PJ, et al. Identification of ATPases pontin and reptin as telomerase components essential for holoenzyme assembly. Cell. 2008;132(6):945–57. doi: 10.1016/j.cell.2008.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Khurts S, Masutomi K, Delgermaa L, et al. Nucleolin interacts with telomerase. J Biol Chem. 2004;279(49):51508–15. doi: 10.1074/jbc.M407643200. [DOI] [PubMed] [Google Scholar]
- 70.Zhou XZ, Lu KP. The Pin2/TRF1-interacting protein PinX1 is a potent telomerase inhibitor. Cell. 2001;107(3):347–59. doi: 10.1016/s0092-8674(01)00538-4. [DOI] [PubMed] [Google Scholar]
- 71.Le S, Sternglanz R, Greider CW. Identification of two RNA binding proteins associated with human telomerase RNA. Mol Biol Cell. 2000;11(3):999–1010. doi: 10.1091/mbc.11.3.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Seimiya H, Sawada H, Muramatsu Y, et al. Involvement of 14-3-3 proteins in nuclear localization of telomerase. EMBO J. 2000;19:2652–61. doi: 10.1093/emboj/19.11.2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Holt SE, Aisner DL, Baur J, et al. Functional requirement of p23 and Hsp90 in telomerase complexes. Genes Dev. 1999;13:817–26. doi: 10.1101/gad.13.7.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tycowski KT, Shu MD, Kukoyi A, et al. A conserved WD40 protein binds the Cajal body localization signal of scaRNP particles. Mol Cell. 2009;34(1):47–57. doi: 10.1016/j.molcel.2009.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Venteicher AS, Abreu EB, Meng Z, et al. A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science. 2009;323(5914):644–8. doi: 10.1126/science.1165357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Collins K. The biogenesis and regulation of telomerase holoenzymes. Nat Rev Mol Cell Biol. 2006;7(7):484–94. doi: 10.1038/nrm1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Collins K. Physiological assembly and activity of human telomerase complexes. Mech Ageing Dev. 2008;129(1–2):91–8. doi: 10.1016/j.mad.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Reichenbach P, Hoss M, Azzalin CM, et al. A human homolog of yeast Est1 associates with telomerase and uncaps chromosome ends when overexpressed. Curr Biol. 2003;13(7):568–74. doi: 10.1016/s0960-9822(03)00173-8. [DOI] [PubMed] [Google Scholar]
- 79.Snow BE, Erdmann N, Cruickshank J, et al. Functional conservation of the telomerase protein Est1p in humans. Curr Biol. 2003;13(8):698–704. doi: 10.1016/s0960-9822(03)00210-0. [DOI] [PubMed] [Google Scholar]
- 80.LaBranche H, Dupuis S, Ben-David Y, et al. Telomere elongation by hnRNP A1 and a derivative that interacts with telomeric repeats and telomerase. Nat Genet. 1998;19:199–202. doi: 10.1038/575. [DOI] [PubMed] [Google Scholar]
- 81.Fiset S, Chabot B. hnRNP A1 may interact simultaneously with telomeric DNA and the human telomerase RNA in vitro. Nucl Acids Res. 2001;29:2268–75. doi: 10.1093/nar/29.11.2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Loayza D, De Lange T. POT1 as a terminal transducer of TRF1 telomere length control. Nature. 2003;423(6943):1013–8. doi: 10.1038/nature01688. [DOI] [PubMed] [Google Scholar]
- 83.Xin H, Liu D, Wan M, et al. TPP1 is a homologue of ciliate TEBPbeta and interacts with POT1 to recruit telomerase. Nature. 2007;445(7127):559–62. doi: 10.1038/nature05469. [DOI] [PubMed] [Google Scholar]
- 84.Cohen SB, Graham ME, Lovrecz GO, et al. Protein composition of catalytically active human telomerase from immortal cells. Science. 2007;315(5820):1850–3. doi: 10.1126/science.1138596. [DOI] [PubMed] [Google Scholar]
- 85.Mitchell JR, Collins K. Telomerase in the human organism. Oncogene. 2002;21(4):564–79. doi: 10.1038/sj.onc.1205083. [DOI] [PubMed] [Google Scholar]
- 86.Zhao JQ, Hoare SF, McFarlane R, et al. Cloning and characterization of human and mouse telomerase RNA gene promoter sequences. Oncogene. 1998;16(10):1345–50. doi: 10.1038/sj.onc.1201892. [DOI] [PubMed] [Google Scholar]
- 87.Zhao JQ, Glasspool RM, Hoare SF, et al. Activation of telomerase rna gene promoter activity by NF-Y, Sp1, and the retinoblastoma protein and repression by Sp3. Neoplasia. 2000;2(6):531–9. doi: 10.1038/sj.neo.7900114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Blasco MA, Rizen M, Greider CW, et al. Differential regulation of telomerase activity and telomerase RNA during multi-stage tumorigenesis. Nat Genet. 1996;12:200–4. doi: 10.1038/ng0296-200. [DOI] [PubMed] [Google Scholar]
- 89.Park TW, Riethdorf S, Riethdorf L, et al. Differential telomerase activity, expression of the telomerase catalytic sub-unit and telomerase-RNA in ovarian tumors. Int J Cancer. 1999;84(4):426–31. doi: 10.1002/(sici)1097-0215(19990820)84:4<426::aid-ijc17>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 90.Rathi A, Hur K, Gazdar AF, et al. Telomerase RNA expression during progression of gastric cancer. Hum Pathol. 1999;30(11):1302–8. doi: 10.1016/s0046-8177(99)90060-6. [DOI] [PubMed] [Google Scholar]
- 91.Chen JL, Greider CW. Determinants in mammalian telomerase RNA that mediate enzyme processivity and cross-species incompatibility. EMBO J. 2003;22(2):304–14. doi: 10.1093/emboj/cdg024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kim NK, Zhang Q, Zhou J, et al. Solution structure and dynamics of the wild-type pseudoknot of human telomerase RNA. J Mol Biol. 2008;384(5):1249–61. doi: 10.1016/j.jmb.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ly H, Blackburn EH, Parslow TG. Comprehensive structure function analysis of the core domain of human telomerase RNA. Mol Cell Biol. 2003;23(19):6849–56. doi: 10.1128/MCB.23.19.6849-6856.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Collins K, Mitchell JR. Telomerase in the human organism. Oncogene. 2002;21:564–79. doi: 10.1038/sj.onc.1205083. [DOI] [PubMed] [Google Scholar]
- 95.Fu D, Collins K. Distinct biogenesis pathways for human telomerase RNA and H/ACA small nucleolar RNAs. Mol Cell. 2003;11:1361–72. doi: 10.1016/s1097-2765(03)00196-5. [DOI] [PubMed] [Google Scholar]
- 96.Jady BE, Bertrand E, Kiss T. Human telomerase RNA and box H/ACA scaRNAs share a common Cajal body-specific localization signal. J Cell Biol. 2004;164(5):647–52. doi: 10.1083/jcb.200310138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fu D, Collins K. Human telomerase and Cajal body ribonucleoproteins share a unique specificity of Sm protein association. Genes Dev. 2006;20(5):531–6. doi: 10.1101/gad.1390306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cristofari G, Adolf E, Reichenbach P, et al. Human telomerase RNA accumulation in Cajal bodies facilitates telomerase recruitment to telomeres and telomere elongation. Mol Cell. 2007;27(6):882–9. doi: 10.1016/j.molcel.2007.07.020. [DOI] [PubMed] [Google Scholar]
- 99.Theimer CA, Jady BE, Chim N, et al. Structural and functional characterization of human telomerase RNA processing and cajal body localization signals. Mol Cell. 2007;27(6):869–81. doi: 10.1016/j.molcel.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 100.Bodnar AG, Ouellette M, Frolkis M, et al. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279(5349):349–52. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 101.Cong YS, Wen J, Bacchetti S. The human telomerase catalytic subunit hTERT: organization of the gene and characterization of the promoter. Hum Mol Genet. 1999;8(1):137–42. doi: 10.1093/hmg/8.1.137. [DOI] [PubMed] [Google Scholar]
- 102.Horikawa I, Cable PL, Afshari C, et al. Cloning and characterization of the promoter region of human telomerase reverse transcriptase gene. Cancer Res. 1999;59(4):826–30. [PubMed] [Google Scholar]
- 103.Legassie JD, Jarstfer MB. The unmasking of telomerase. Structure. 2006;14(11):1603–9. doi: 10.1016/j.str.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 104.O’Connor CM, Collins K. A novel RNA binding domain in tetrahymena telomerase p65 initiates hierarchical assembly of telomerase holoenzyme. Mol Cell Biol. 2006;26(6):2029–36. doi: 10.1128/MCB.26.6.2029-2036.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Autexier C, Lue NF. The structure and function of telomerase reverse transcriptase. Annu Rev Biochem. 2006;75:493–517. doi: 10.1146/annurev.biochem.75.103004.142412. [DOI] [PubMed] [Google Scholar]
- 106.Lue NF, Bosoy D, Moriarty TJ, et al. Telomerase can act as a template- and RNA-independent terminal transferase. Proc Natl Acad Sci USA. 2005;102(28):9778–83. doi: 10.1073/pnas.0502252102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lee SR, Wong JM, Collins K. Human telomerase reverse transcriptase motifs required for elongation of a telomeric substrate. J Biol Chem. 2003;278:52531–6. doi: 10.1074/jbc.M311359200. [DOI] [PubMed] [Google Scholar]
- 108.Wyatt HD, Tsang AR, Lobb DA, et al. Human telomerase reverse transcriptase (hTERT) Q169 is essential for telomerase function in vitro and in vivo. PLoS One. 2009;4(9):e7176. doi: 10.1371/journal.pone.0007176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Armbruster BN, Banik SSR, Guo C, et al. N-terminal domains of the human telomerase catalytic subunit required for enzyme activity in vivo. Mol Cell Biol. 2001;22:7775–86. doi: 10.1128/MCB.21.22.7775-7786.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Beattie TL, Zhou W, Robinson MO, et al. Functional multimerization of the human telomerase reverse transcriptase. Mol Cell Biol. 2001;21:6151–60. doi: 10.1128/MCB.21.18.6151-6160.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lai CK, Mitchell JR, Collins K. RNA binding domain of telomerase reverse transcriptase. Mol Cell Biol. 2001;21:990–1000. doi: 10.1128/MCB.21.4.990-1000.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rouda S, Skordalakes E. Structure of the RNA-binding domain of telomerase: implications for RNA recognition and binding. Structure. 2007;15(11):1403–12. doi: 10.1016/j.str.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 113.Bosoy D, Lue NF. Functional analysis of conserved residues in the putative “finger” domain of telomerase reverse transcriptase. J Biol Chem. 2001;276:46305–12. doi: 10.1074/jbc.M108168200. [DOI] [PubMed] [Google Scholar]
- 114.Gillis AJ, Schuller AP, Skordalakes E. Structure of the Tribolium castaneum telomerase catalytic subunit TERT. Nature. 2008;455(7213):633–7. doi: 10.1038/nature07283. [DOI] [PubMed] [Google Scholar]
- 115.Banik SSR, Guo C, Smith AC, et al. C-terminal regions of the human telomerase catalytic subunit essential for in vivo enzyme activity. Mol Cell Biol. 2002;22:6234–46. doi: 10.1128/MCB.22.17.6234-6246.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Huard S, Moriarty TJ, Autexier C. The C terminus of the human telomerase reverse transcriptase is a determinant of enzyme processivity. Nucleic Acids Res. 2003;31(14):4059–70. doi: 10.1093/nar/gkg437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wick M, Zubov D, Hagen G. Genomic organization and promoter characterization of the gene encoding the human telomerase reverse transcriptase (hTERT) Gene. 1999;232(1):97–106. doi: 10.1016/s0378-1119(99)00108-0. [DOI] [PubMed] [Google Scholar]
- 118.Bechter OE, Eisterer W, Dlaska M, et al. CpG island methylation of the hTERT promoter is associated with lower telomerase activity in B-cell lymphocytic leukemia. Exp Hematol. 2002;30(1):26–33. doi: 10.1016/s0301-472x(01)00760-3. [DOI] [PubMed] [Google Scholar]
- 119.Masutomi K, Yu EY, Khurts S, et al. Telomerase maintains telomere structure in normal human cells. Cell. 2003;114(2):241–53. doi: 10.1016/s0092-8674(03)00550-6. [DOI] [PubMed] [Google Scholar]
- 120.Horikawa I, Barrett JC. Transcriptional regulation of the telomerase hTERT gene as a target for cellular and viral oncogenic mechanisms. Carcinogenesis. 2003;24(7):1167–76. doi: 10.1093/carcin/bgg085. [DOI] [PubMed] [Google Scholar]
- 121.Lin SY, Elledge SJ. Multiple tumor suppressor pathways negatively regulate telomerase. Cell. 2003;113(7):881–9. doi: 10.1016/s0092-8674(03)00430-6. [DOI] [PubMed] [Google Scholar]
- 122.Yi X, White DM, Aisner DL, et al. An alternate splicing variant of the human telomerase catalytic subunit inhibits telomerase activity. Neoplasia. 2000;2:433–40. doi: 10.1038/sj.neo.7900113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Keith WN, Hoare SF. Detection of telomerase hTERT gene expression and its splice variants by RT-PCR. Methods Mol Med. 2004;97:297–309. doi: 10.1385/1-59259-760-2:297. [DOI] [PubMed] [Google Scholar]
- 124.Lincz LF, Mudge LM, Scorgie FE, et al. Quantification of hTERT splice variants in melanoma by SYBR green real-time polymerase chain reaction indicates a negative regulatory role for the beta deletion variant. Neoplasia. 2008;10(10):1131–7. doi: 10.1593/neo.08644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yi X, Shay JW, Wright WE. Quantitation of telomerase components and hTERT mRNA splicing patterns in immortal human cells. Nucleic Acids Res. 2001;29:4818–25. doi: 10.1093/nar/29.23.4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kang SS, Kwon T, Kwon DY, et al. Akt protein kinase enhances human telomerase activity through phosphorylation of telomerase reverse transcriptase subunit. J Biol Chem. 1999;274:13085–90. doi: 10.1074/jbc.274.19.13085. [DOI] [PubMed] [Google Scholar]
- 127.Kharbanda S, Kumar V, Dhar S, et al. Regulation of the hTERT telomerase catalytic subunit by the c-Abl tyrosine kinase. Curr Biol. 2000;10(10):568–75. doi: 10.1016/s0960-9822(00)00483-8. [DOI] [PubMed] [Google Scholar]
- 128.Liu K, Hodes RJ, Weng N. Cutting edge: telomerase activation in human T lymphocytes does not require increase in telomerase reverse transcriptase (hTERT) protein but is associated with hTERT phosphorylation and nuclear translocation. J Immunol. 2001;166:4826–30. doi: 10.4049/jimmunol.166.8.4826. [DOI] [PubMed] [Google Scholar]
- 129.Yi X, Tesmer VM, Savre-Train I, et al. Both transcriptional and posttranscriptional mechanisms regulate human telomerase template RNA levels. Mol Cell Biol. 1999;19:3989–97. doi: 10.1128/mcb.19.6.3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kim JH, Park SM, Kang MR, et al. Ubiquitin ligase MKRN1 modulates telomere length homeostasis through a proteolysis of hTERT. Genes Dev. 2005;19(7):776–81. doi: 10.1101/gad.1289405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Dragon F, Gallagher JE, Compagnone-Post PA, et al. A large nucleolar U3 ribonucleoprotein required for 18S ribosomal RNA biogenesis. Nature. 2002;417(6892):967–70. doi: 10.1038/nature00769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Terns M, Terns R. Noncoding RNAs of the H/ACA family. Cold Spring Harb Symp Quant Biol. 2006;71:395–405. doi: 10.1101/sqb.2006.71.034. [DOI] [PubMed] [Google Scholar]
- 133.Filipowicz W, Pelczar P, Pogacic V, et al. Structure and biogenesis of small nucleolar RNAs acting as guides for ribosomal RNA modification. Acta Biochim Pol. 1999;46:377–89. [PubMed] [Google Scholar]
- 134.Wang C, Meier UT. Architecture and assembly of mammalian H/ACA small nucleolar and telomerase ribonucleoproteins. EMBO J. 2004;23(8):1857–67. doi: 10.1038/sj.emboj.7600181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ganot P, Bortolin M-L, Kiss T. Site-specific pseudouridine formation in preribosomal RNA is guided by small nucleolar RNAs. Cell. 1997;89:799–809. doi: 10.1016/s0092-8674(00)80263-9. [DOI] [PubMed] [Google Scholar]
- 136.Koonin EV. Pseudouridine synthases: four families of enzymes containing a putative uridine-binding motif also conserved in dUTPases and dCTP deaminases. Nucleic Acids Res. 1996;24:2411–5. doi: 10.1093/nar/24.12.2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tollervey D, Kiss T. Function and synthesis of small nucleolar RNAs. Curr Opin Cell Biol. 1997;9:337–42. doi: 10.1016/s0955-0674(97)80005-1. [DOI] [PubMed] [Google Scholar]
- 138.Charette M, Gray MW. Pseudouridine in RNA: what, where, how, and why. IUBMB Life. 2000;49(5):341–51. doi: 10.1080/152165400410182. [DOI] [PubMed] [Google Scholar]
- 139.Ofengand J. Ribosomal RNA pseudouridines and pseudouridine synthases. FEBS Lett. 2002;514(1):17–25. doi: 10.1016/s0014-5793(02)02305-0. [DOI] [PubMed] [Google Scholar]
- 140.Kiss T. Small nucleolar RNAs: an abundant group of noncoding RNAs with diverse cellular functions. Cell. 2002;109(2):145–8. doi: 10.1016/s0092-8674(02)00718-3. [DOI] [PubMed] [Google Scholar]
- 141.Dez C, Henras A, Faucon B, et al. Stable expression in yeast of the mature form of human telomerase RNA depends of its association with the box H/ACA small nucleolar RNP proteins Cbf5p, Nhp2p and Nop10p. Nucleic Acids Res. 2001;29:598–603. doi: 10.1093/nar/29.3.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lafontaine DLJ, Bousquet-Antonelli C, Henry Y, et al. The box HACA snoRNAs carry Cbf5p, the putative rRNA pseudouridinesynthase. Genes Dev. 1998;12:527–37. doi: 10.1101/gad.12.4.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Heiss NS, Knight SW, Vulliamy TJ, et al. X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat Genet. 1998;19(1):32–8. doi: 10.1038/ng0598-32. [DOI] [PubMed] [Google Scholar]
- 144.He J, Navarrete S, Jasinski M, et al. Targeted disruption of Dkc1, the gene mutated in X-linked dyskeratosis congenita, causes embryonic lethality in mice. Oncogene. 2002;21(50):7740–4. doi: 10.1038/sj.onc.1205969. [DOI] [PubMed] [Google Scholar]
- 145.Phillips B, Billin AN, Cadwell C, et al. The Nop60B gene of Drosophila encodes an essential nucleolar protein that functions in yeast. Mol Gen Genet. 1998;260(1):20–9. doi: 10.1007/s004380050866. [DOI] [PubMed] [Google Scholar]
- 146.Ramamurthy V, Swann SL, Spedaliere CJ, et al. Role of cysteine residues in pseudouridine synthases of different families. Biochemistry. 1999;38(40):13106–11. doi: 10.1021/bi9913911. [DOI] [PubMed] [Google Scholar]
- 147.Hamma T, Reichow SL, Varani G, et al. The Cbf5-Nop10 complex is a molecular bracket that organizes box H/ACA RNPs. Nat Struct Mol Biol. 2005;12(12):1101–7. doi: 10.1038/nsmb1036. [DOI] [PubMed] [Google Scholar]
- 148.Manival X, Charron C, Fourmann JB, et al. Crystal structure determination and site-directed mutagenesis of the Pyrococcus abyssi a CBF5-aNOP10 complex reveal crucial roles of the C-terminal domains of both proteins in H/ACA sRNP activity. Nucleic Acids Res. 2006;34(3):826–39. doi: 10.1093/nar/gkj482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Rashid R, Liang B, Baker DL, et al. Crystal structure of a Cbf5-Nop10-Gar1 complex and implications in RNA-guided pseudouridylation and dyskeratosis congenita. Mol Cell. 2006;21(2):249–60. doi: 10.1016/j.molcel.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 150.Perez-Arellano I, Gallego J, Cervera J. The PUA domain – a structural and functional overview. FEBS J. 2007;274(19):4972–84. doi: 10.1111/j.1742-4658.2007.06031.x. [DOI] [PubMed] [Google Scholar]
- 151.Henras A, Henry Y, Bousquet-Antonelli C, et al. Nhp2p and Nop10p are essential for the function of H/ACA snoRNPs. EMBO J. 1998;17:7078–90. doi: 10.1093/emboj/17.23.7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Li L, Ye K. Crystal structure of an H/ACA box ribonucleoprotein particle. Nature. 2006;443(7109):302–7. doi: 10.1038/nature05151. [DOI] [PubMed] [Google Scholar]
- 153.Girard J-P, Lehtonen H, Caizergues-Ferrer M, et al. GAR1 is an essential small nucleolar RNP protein required for pre-rRNA processing in yeast. EMBO J. 1992;11:673–82. doi: 10.1002/j.1460-2075.1992.tb05099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tremblay A, Lamontagne B, Catala M, et al. A physical interaction between Gar1p and Rnt1p is required for the nuclear import of H/ACA small nucleolar RNA-associated proteins. Mol Cell Biol. 2002;22(13):4792–802. doi: 10.1128/MCB.22.13.4792-4802.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Leulliot N, Godin KS, Hoareau-Aveilla C, et al. The box H/ACA RNP assembly factor Naf1p contains a domain homologous to Gar1p mediating its interaction with Cbf5p. J Mol Biol. 2007;371(5):1338–53. doi: 10.1016/j.jmb.2007.06.031. [DOI] [PubMed] [Google Scholar]
- 156.Bousquet-Antonelli C, Henry Y, G’Elugne JP, et al. A small nucleolar RNP protein is required for pseudouridylation of eukaryotic ribosomal RNAs. EMBO J. 1997;16(15):4770–6. doi: 10.1093/emboj/16.15.4770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Armanios M. Syndromes of telomere shortening. Annu Rev Genomics Hum Genet. 2009;10:45–61. doi: 10.1146/annurev-genom-082908-150046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Vulliamy TJ, Marrone A, Dokal I, et al. Association between aplastic anemia and mutations in telomerase RNA. Lancet. 2002;359:2168–70. doi: 10.1016/S0140-6736(02)09087-6. [DOI] [PubMed] [Google Scholar]
- 159.Dokal I. Dyskeratosis congenita in all its forms. Br J Haematol. 2000;110(4):768–79. doi: 10.1046/j.1365-2141.2000.02109.x. [DOI] [PubMed] [Google Scholar]
- 160.Kirwan M, Dokal I. Dyskeratosis congenita: a genetic disorder of many faces. Clin Genet. 2008;73(2):103–12. doi: 10.1111/j.1399-0004.2007.00923.x. [DOI] [PubMed] [Google Scholar]
- 161.Artandi SE, Chang S, Lee S, et al. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature. 2000;406:641–5. doi: 10.1038/35020592. [DOI] [PubMed] [Google Scholar]
- 162.Rudolph KL, Millard M, Bosenberg MW, et al. Telomere dysfunction and evolution of intestinal carcinoma in mice and humans. Nat Genet. 2001;28:155–9. doi: 10.1038/88871. [DOI] [PubMed] [Google Scholar]
- 163.Alter BP, Giri N, Savage SA, et al. Cancer in dyskeratosis congenita. Blood. 2009;113(26):6549–57. doi: 10.1182/blood-2008-12-192880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kirwan M, Dokal I. Dyskeratosis congenita, stem cells and telomeres. Biochim Biophys Acta. 2009;1792(4):371–9. doi: 10.1016/j.bbadis.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Knight S, Vulliamy T, Copplestone A, et al. Dyskeratosis congenita (DC) registry: identification of new features of DC. Br J Haematol. 1998;103:990–6. doi: 10.1046/j.1365-2141.1998.01103.x. [DOI] [PubMed] [Google Scholar]
- 166.Vulliamy TJ, Marrone A, Knight SW, et al. Mutations in dyskeratosis congenita: their impact on telomere length and the diversity of clinical presentation. Blood. 2006;107(7):2680–5. doi: 10.1182/blood-2005-07-2622. [DOI] [PubMed] [Google Scholar]
- 167.Zucchini C, Strippoli P, Biolchi A, et al. The human TruB family of pseudouridine synthase genes, including the Dyskeratosis Congenita 1 gene and the novel member TRUB1. Int J Mol Med. 2003;11(6):697–704. [PubMed] [Google Scholar]
- 168.Wong JM, Kyasa MJ, Hutchins L, et al. Telomerase RNA deficiency in peripheral blood mononuclear cells in X-linked dyskeratosis congenita. Hum Genet. 2004;115(5):448–55. doi: 10.1007/s00439-004-1178-7. [DOI] [PubMed] [Google Scholar]
- 169.Wong JM, Collins K. Telomerase RNA level limits telomere maintenance in X-linked dyskeratosis congenita. Genes Dev. 2006;20(20):2848–58. doi: 10.1101/gad.1476206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Grozdanov PN, Fernandez-Fuentes N, Fiser A, et al. Pathogenic Nap57 Mutations Decrease Ribonucleoprotein Assembly in Dyskeratosis Congenita. Hum Mol Genet. 2009;18(23):4546–51. doi: 10.1093/hmg/ddp416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Armanios M, Chen JL, Chang YP, et al. Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita. Proc Natl Acad Sci USA. 2005;102(44):15960–4. doi: 10.1073/pnas.0508124102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Vulliamy T, Marrone A, Goldman F, et al. The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature. 2001;413(6854):432–5. doi: 10.1038/35096585. [DOI] [PubMed] [Google Scholar]
- 173.Goldman F, Bouarich R, Kulkarni S, et al. The effect of TERC haploinsufficiency on the inheritance of telomere length. Proc Natl Acad Sci USA. 2005;102(47):17119–24. doi: 10.1073/pnas.0505318102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Errington TM, Fu D, Wong JM, et al. Disease-associated human telomerase RNA variants show loss of function for telomere synthesis without dominant-negative interference. Mol Cell Biol. 2008;28(20):6510–20. doi: 10.1128/MCB.00777-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Vulliamy T, Marrone A, Szydlo R, et al. Disease anticipation is associated with progressive telomere shortening in families with dyskeratosis congenita due to mutations in TERC. Nat Genet. 2004;36(5):447–9. doi: 10.1038/ng1346. [DOI] [PubMed] [Google Scholar]
- 176.Savage SA, Giri N, Baerlocher GM, et al. TINF2, a component of the shelterin telomere protection complex, is mutated in dyskeratosis congenita. Am J Hum Genet. 2008;82(2):501–9. doi: 10.1016/j.ajhg.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Walne AJ, Vulliamy T, Beswick R, et al. TINF2 mutations result in very short telomeres: analysis of a large cohort of patients with dyskeratosis congenita and related bone marrow failure syndromes. Blood. 2008;112(9):3594–3600. doi: 10.1182/blood-2008-05-153445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Vulliamy T, Beswick R, Kirwan M, et al. Mutations in the telomerase component NHP2 cause the premature ageing syndrome dyskeratosis congenita. Proc Natl Acad Sci USA. 2008;105(23):8073–8. doi: 10.1073/pnas.0800042105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Walne AJ, Vulliamy T, Marrone A, et al. Genetic heterogeneity in autosomal recessive dyskeratosis congenita with one subtype due to mutations in the telomerase-associated protein NOP10. Hum Mol Genet. 2007;16(13):1619–29. doi: 10.1093/hmg/ddm111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Yaghmai R, Kimyai-Asadi A, Rostamiani K, et al. Overlap of dyskeratosis congenita with the Hoyeraal-Hreidarsson syndrome. J Pediatr. 2000;136(3):390–3. doi: 10.1067/mpd.2000.104295. [DOI] [PubMed] [Google Scholar]
- 181.Cossu F, Vulliamy TJ, Marrone A, et al. A novel DKC1 mutation, severe combined immunodeficiency (T+B-NK-SCID) and bone marrow transplantation in an infant with Hoyeraal-Hreidarsson syndrome. Br J Haematol. 2002;119:765–8. doi: 10.1046/j.1365-2141.2002.03822.x. [DOI] [PubMed] [Google Scholar]
- 182.Knight SW, Heiss NS, Vulliamy TJ, et al. Unexplained aplastic anaemia, immunodeficiency, and cerebellar hypoplasia (Hoyeraal-Hreidarsson syndrome) due to mutations in the dyskeratosis congenita gene, DKC1. Br J Haematol. 1999;107:335–9. doi: 10.1046/j.1365-2141.1999.01690.x. [DOI] [PubMed] [Google Scholar]
- 183.Marrone A, Walne A, Tamary H, et al. Telomerase reverse transcriptase homozygous mutations in autosomal recessive dyskeratosis congenita and Hoyeraal-Hreidarsson syndrome. Blood. 2007;110(13):4198–205. doi: 10.1182/blood-2006-12-062851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Pearson T, Curtis F, Al-Eyadhy A, et al. An intronic mutation in DKC1 in an infant with Hoyeraal-Hreidarsson syndrome. Am J Med Genet A. 2008;146A(16):2159–61. doi: 10.1002/ajmg.a.32412. [DOI] [PubMed] [Google Scholar]
- 185.Lamm N, Ordan E, Shponkin R, et al. Diminished telomeric 3′ overhangs are associated with telomere dysfunction in hoyeraalhreidarsson syndrome. PLoS One. 2009;4(5):e5666. doi: 10.1371/journal.pone.0005666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Yamaguchi H, Calado RT, Ly H, et al. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N Engl J Med. 2005;352(14):1413–24. doi: 10.1056/NEJMoa042980. [DOI] [PubMed] [Google Scholar]
- 187.Ball SE, Gibson FM, Rizzo S, et al. Progressive telomere shortening in aplastic anemia. Blood. 1998;91(10):3582–92. [PubMed] [Google Scholar]
- 188.Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108(8):2509–19. doi: 10.1182/blood-2006-03-010777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Takeuchi J, Ly H, Yamaguchi H, et al. Identification and functional characterization of novel telomerase variant alleles in Japanese patients with bone-marrow failure syndromes. Blood Cells Mol Dis. 2008;40(2):185–91. doi: 10.1016/j.bcmd.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kirwan M, Vulliamy T, Marrone A, et al. Defining the pathogenic role of telomerase mutations in myelodysplastic syndrome and acute myeloid leukemia. Hum Mutat. 2009;30(11):1567–73. doi: 10.1002/humu.21115. [DOI] [PubMed] [Google Scholar]
- 191.Keith WN, Vulliamy T, Zhao J, et al. A mutation in a functional Sp1 binding site of the telomerase RNA gene (hTERC) promoter in a patient with Paroxysmal Nocturnal Haemoglobinuria. BMC Blood Disord. 2004;4(1):3. doi: 10.1186/1471-2326-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Revesz T, Fletcher S, al-Gazali LI, et al. Bilateral retinopathy, aplastic anaemia, and central nervous system abnormalities: a new syndrome? J Med Genet. 1992;29(9):673–5. doi: 10.1136/jmg.29.9.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med. 2002;165(2):277–304. doi: 10.1164/ajrccm.165.2.ats01. [DOI] [PubMed] [Google Scholar]
- 194.King TE., Jr Clinical advances in the diagnosis and therapy of the interstitial lung diseases. Am J Respir Crit Care Med. 2005;172(3):268–79. doi: 10.1164/rccm.200503-483OE. [DOI] [PubMed] [Google Scholar]
- 195.Meltzer EB, Noble PW. Idiopathic pulmonary fibrosis. Orphanet J Rare Dis. 2008;3:8. doi: 10.1186/1750-1172-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Alder JK, Chen JJ, Lancaster L, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci USA. 2008;105(35):13051–6. doi: 10.1073/pnas.0804280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Cronkhite JT, Xing C, Raghu G, et al. Telomere shortening in familial and sporadic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178(7):729–37. doi: 10.1164/rccm.200804-550OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Leteurtre F, Li X, Guardiola P, et al. Accelerated telomere shortening and telomerase activation in Fanconi’s anaemia. Br J of Haematol. 1999;105:883–93. doi: 10.1046/j.1365-2141.1999.01445.x. [DOI] [PubMed] [Google Scholar]
- 199.Steer SE, Williams FM, Kato B, et al. Reduced telomere length in rheumatoid arthritis is independent of disease activity and duration. Ann Rheum Dis. 2007;66(4):476–80. doi: 10.1136/ard.2006.059188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Fuster JJ, Andres V. Telomere biology and cardiovascular disease. Circ Res. 2006;99(11):1167–80. doi: 10.1161/01.RES.0000251281.00845.18. [DOI] [PubMed] [Google Scholar]
- 201.Pandita TK. The role of ATM in telomere structure and function. Radiat Res. 2001;156(5 Pt 2):642–7. doi: 10.1667/0033-7587(2001)156[0642:troait]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 202.Ting AP, Low GK, Gopalakrishnan K, et al. Telomere attrition and genomic instability in xeroderma pigmentosum type-B deficient fibroblasts under oxidative stress. J Cell Mol Med. 2009 doi: 10.1111/j.1582-4934.2009.00945.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Metcalfe JA, Parkhill J, Campbell L, et al. Accelerated telomere shortening in ataxia telangiectasia. Nat Genet. 1996;13:350–3. doi: 10.1038/ng0796-350. [DOI] [PubMed] [Google Scholar]
- 204.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071–8. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Pandita TK. ATM function and telomere stability. Oncogene. 2002;21(4):611–8. doi: 10.1038/sj.onc.1205060. [DOI] [PubMed] [Google Scholar]
- 206.Ranganathan V, Heine WF, Ciccone DN, et al. Rescue of a telomere length defect of Nijmegen breakage syndrome cells requires NBS and telomerase catalytic subunit. Curr Biol. 2001;11:962–6. doi: 10.1016/s0960-9822(01)00267-6. [DOI] [PubMed] [Google Scholar]
- 207.Opresko PL, Otterlei M, Graakjaer J, et al. The Werner syndrome helicase and exonuclease cooperate to resolve telomeric D loops in a manner regulated by TRF1 and TRF2. Mol Cell. 2004;14(6):763–74. doi: 10.1016/j.molcel.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 208.Chang S, Multani AS, Cabrera NG, et al. Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nat Genet. 2004;36(8):877–82. doi: 10.1038/ng1389. [DOI] [PubMed] [Google Scholar]
- 209.McPherson JP, Hande MP, Poonepalli A, et al. A role for Brca1 in chromosome end maintenance. Hum Mol Genet. 2006;15(6):831–8. doi: 10.1093/hmg/ddl002. [DOI] [PubMed] [Google Scholar]
- 210.Ballal RD, Saha T, Fan S, et al. BRCA1 localization to the telomere and its loss from the telomere in response to DNA damage. J Biol Chem. 2009;284:36083–98. doi: 10.1074/jbc.M109.025825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Lusis AJ. Atherosclerosis. Nature. 2000;407(6801):233–41. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Ogami M, Ikura Y, Ohsawa M, et al. Telomere shortening in human coronary artery diseases. Arterioscler Thromb Vasc Biol. 2004;24(3):546–50. doi: 10.1161/01.ATV.0000117200.46938.e7. [DOI] [PubMed] [Google Scholar]
- 213.Minamino T, Miyauchi H, Yoshida T, et al. Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation. 2002;105(13):1541–4. doi: 10.1161/01.cir.0000013836.85741.17. [DOI] [PubMed] [Google Scholar]
- 214.Perez-Rivero G, Ruiz-Torres MP, Rivas-Elena JV, et al. Mice deficient in telomerase activity develop hypertension because of an excess of endothelin production. Circulation. 2006;114(4):309–17. doi: 10.1161/CIRCULATIONAHA.105.611111. [DOI] [PubMed] [Google Scholar]
- 215.Alcolado R, Arthur MJ, Iredale JP. Pathogenesis of liver fibrosis. Clin Sci (Lond) 1997;92(2):103–12. doi: 10.1042/cs0920103. [DOI] [PubMed] [Google Scholar]
- 216.Aikata H, Takaishi H, Kawakami Y, et al. Telomere reduction in human liver tissues with age and chronic inflammation. Exp Cell Res. 2000;256(2):578–82. doi: 10.1006/excr.2000.4862. [DOI] [PubMed] [Google Scholar]
- 217.Kitada T, Seki S, Kawakita N, et al. Telomere shortening in chronic liver diseases. Biochem Biophys Res Commun. 1995;211(1):33–9. doi: 10.1006/bbrc.1995.1774. [DOI] [PubMed] [Google Scholar]
- 218.Wiemann SU, Satyanarayana A, Tsahuridu M, et al. Hepatocyte telomere shortening and senescence are general markers of human liver cirrhosis. FASEB J. 2002;16(9):935–42. doi: 10.1096/fj.01-0977com. [DOI] [PubMed] [Google Scholar]
- 219.Goronzy JJ, Weyand CM. Aging, autoimmunity and arthritis: T cell senescence and contraction of T-cell repertoire diversity – catalysts of autoimmunity and chronic inflammation. Arthritis Res Ther. 2003;5(5):225–34. doi: 10.1186/ar974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Wagner UG, Koetz K, Weyand CM, et al. Perturbation of the T cell repertoire in rheumatoid arthritis. Proc Natl Acad Sci USA. 1998;95(24):14447–52. doi: 10.1073/pnas.95.24.14447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Fujii H, Shao L, Colmegna I, et al. Telomerase insufficiency in rheumatoid arthritis. Proc Natl Acad Sci USA. 2009;106(11):4360–5. doi: 10.1073/pnas.0811332106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Buist AS, McBurnie MA, Vollmer WM, et al. International variation in the prevalence of COPD (the BOLD Study): a population based prevalence study. Lancet. 2007;370(9589):741–50. doi: 10.1016/S0140-6736(07)61377-4. [DOI] [PubMed] [Google Scholar]
- 223.Ito K, Barnes PJ. COPD as a disease of accelerated lung aging. Chest. 2009;135(1):173–80. doi: 10.1378/chest.08-1419. [DOI] [PubMed] [Google Scholar]
- 224.Savale L, Chaouat A, Bastuji-Garin S, et al. Shortened telomeres in circulating leukocytes of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2009;179(7):566–71. doi: 10.1164/rccm.200809-1398OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Effros RB, Allsopp R, Chiu CP, et al. Shortened telomeres in the expanded CD28-CD8+ cell subset in HIV disease implicate replicative senescence in HIV pathogenesis. AIDS. 1996;10(8):F17–22. doi: 10.1097/00002030-199607000-00001. [DOI] [PubMed] [Google Scholar]
- 226.Prajapati DG, Ramajayam R, Yadav MR, et al. The search for potent, small molecule NNRTIs: A review. Bioorg Med Chem. 2009;17(16):5744–62. doi: 10.1016/j.bmc.2009.06.060. [DOI] [PubMed] [Google Scholar]
- 227.Peng Y, Mian IS, Lue NF. Analysis of telomerase activity processivity: mechanistic similarity to HIV-1 reverse transcriptase and role in telomere maintenance. Mol Cell. 2001;7:1201–11. doi: 10.1016/s1097-2765(01)00268-4. [DOI] [PubMed] [Google Scholar]
- 228.Mushiroda T, Wattanapokayakit S, Takahashi A, et al. A genome wide association study identifies an association of a common variant in TERT with susceptibility to idiopathic pulmonary fibrosis. J Med Genet. 2008;45(10):654–6. doi: 10.1136/jmg.2008.057356. [DOI] [PubMed] [Google Scholar]
- 229.Jin G, Xu L, Shu Y, et al. Common genetic variants on 5p15.33 contribute to risk of lung adenocarcinoma in a Chinese population. Carcinogenesis. 2009;30(6):987–90. doi: 10.1093/carcin/bgp090. [DOI] [PubMed] [Google Scholar]
- 230.Matsubara Y, Murata M, Yoshida T, et al. Telomere length of normal leukocytes is affected by a functional polymorphism of hTERT. Biochem Biophys Res Commun. 2006;341(1):128–31. doi: 10.1016/j.bbrc.2005.12.163. [DOI] [PubMed] [Google Scholar]
- 231.Matsubara Y, Murata M, Watanabe K, et al. Coronary artery disease and a functional polymorphism of hTERT. Biochem Biophys Res Commun. 2006;348(2):669–72. doi: 10.1016/j.bbrc.2006.07.103. [DOI] [PubMed] [Google Scholar]
- 232.Packer BR, Yeager M, Staats B, et al. SNP500Cancer: a public resource for sequence validation and assay development for genetic variation in candidate genes. Nucleic Acids Res. 2004;32(Database issue):D528–32. doi: 10.1093/nar/gkh005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Rieder MJ, Livingston RJ, Stanaway IB, et al. The environmental genome project: reference polymorphisms for drug metabolism genes and genome-wide association studies. Drug Metab Rev. 2008;40(2):241–61. doi: 10.1080/03602530801952880. [DOI] [PubMed] [Google Scholar]
- 234.Du HY, Pumbo E, Manley P, et al. Complex inheritance pattern of dyskeratosis congenita in two families with 2 different mutations in the telomerase reverse transcriptase gene. Blood. 2008;111(3):1128–30. doi: 10.1182/blood-2007-10-120907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Calado RT, Regal JA, Hills M, et al. Constitutional hypomorphic telomerase mutations in patients with acute myeloid leukemia. Proc Natl Acad Sci USA. 2009;106(4):1187–92. doi: 10.1073/pnas.0807057106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Bennett JM, Komrokji RS. The myelodysplastic syndromes: diagnosis, molecular biology and risk assessment. Hematology. 2005;10(Suppl 1):258–69. doi: 10.1080/10245330512331390311. [DOI] [PubMed] [Google Scholar]
- 237.Hills M, Lansdorp PM. Short telomeres resulting from heritable mutations in the telomerase reverse transcriptase gene predispose for a variety of malignancies. Ann N Y Acad Sci. 2009;1176:178–90. doi: 10.1111/j.1749-6632.2009.04565.x. [DOI] [PubMed] [Google Scholar]
- 238.McKay JD, Hung RJ, Gaborieau V, et al. Lung cancer susceptibility locus at 5p15.33. Nat Genet. 2008;40(12):1404–6. doi: 10.1038/ng.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Wang Y, Broderick P, Webb E, et al. Common 5p15.33 and 6p21.33 variants influence lung cancer risk. Nat Genet. 2008;40(12):1407–9. doi: 10.1038/ng.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Rafnar T, Sulem P, Stacey SN, et al. Sequence variants at the TERT-CLPTM1L locus associate with many cancer types. Nat Genet. 2009;41(2):221–7. doi: 10.1038/ng.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Ohtsuka T, Yamakage A, Yamazaki S. The polymorphism of telomerase RNA component gene in patients with systemic sclerosis. Br J Dermatol. 2002;147(2):250–4. doi: 10.1046/j.1365-2133.2002.04937.x. [DOI] [PubMed] [Google Scholar]
- 242.Westin ER, Chavez E, Lee KM, et al. Telomere restoration and extension of proliferative lifespan in dyskeratosis congenita fibroblasts. Aging Cell. 2007;6(3):383–94. doi: 10.1111/j.1474-9726.2007.00288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Gourronc FA, Robertson MM, Herrig AK, et al. Proliferative defects in dyskeratosis congenita skin keratinocytes are corrected by expression of the telomerase reverse transcriptase, TERT, or by activation of endogenous telomerase through expression of papillomavirus E6/E7 or the telomerase RNA component, TERC. ExpDermatol. 2009 doi: 10.1111/j.1600-0625.2009.00916.x. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Fauce SR, Jamieson BD, Chin AC, et al. Telomerase-based pharmacologic enhancement of antiviral function of human CD8+ T lymphocytes. J Immunol. 2008;181(10):7400–6. doi: 10.4049/jimmunol.181.10.7400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Harley CB. Telomerase therapeutics for degenerative diseases. Curr Mol Med. 2005;5(2):205–11. doi: 10.2174/1566524053586671. [DOI] [PubMed] [Google Scholar]