


Abstract
Fluctuations in protein abundance among single cells are primarily due to the inherent stochasticity in transcription and translation processes, such stochasticity can often confer phenotypic heterogeneity among isogenic cells. It has been proposed that expression noise can be triggered as an adaptation to environmental stresses and genetic perturbations, and as a mechanism to facilitate gene expression evolution. Thus, elucidating the relationship between expression noise, measured at the single-cell level, and expression variation, measured on population of cells, can improve our understanding on the variability and evolvability of gene expression. Here, we showed that noise levels are significantly correlated with conditional expression variations. We further demonstrated that expression variations are highly predictive for noise level, especially in TATA-box containing genes. Our results suggest that expression variabilities can serve as a proxy for noise level, suggesting that these two properties share the same underlining mechanism, e.g. chromatin regulation. Our work paves the way for the study of stochastic noise in other single-cell organisms.
INTRODUCTION
Many biological systems or processes have stochastic characteristics (1–5), among which the fluctuation in gene expression is perhaps the most studied, where the origin and behavior of such fluctuation have been extensively characterized. In this particular setting, the noise of gene expression is defined as the stochastic fluctuation in transcription and/or translation processes in isogenic cells and under identical experimental condition. Expression noise can contribute to remarkable phenotypic diversities albeit within genetically identical cells (5–7). Analytically, expression noise can be decomposed into two components, i.e. ‘intrinsic’ and ‘extrinsic’ noises. The ‘intrinsic noise’ originates from the fluctuations that are inherent in the system (e.g. fluctuation in transcription initiation or mRNA degradation), whereas ‘extrinsic noises’ originate from variabilities in external factors (such as environment) (5,8). Expression noises are usually experimentally determined by attaching fluorescence reporters to the genes of interest and measuring the cell-to-cell variation of the fluorescence intensities (1,8–14). In this approach, the ‘extrinsic noise’ can usually be filtered out after controlling for cell size or environmental condition, by using cell gating or orthogonal reporters. It has been described that expression noise is influenced by numerous cellular processes, and the intensity and characteristics of expression noise are constrained by cellular networks (12,15,16). For example, signals generated by long transcriptional cascades are generally noisier than those generated by short cascades; negative feedback regulation can reduce the effects of noise (17,18), whereas noise can result in dramatic behavior in the presence of positive feedback regulation (19–22).
It is becoming appreciated that gene expression noise can generate phenotypic variation and diversity among single cells, which can mitigate environmental perturbation or external stresses, and offer benefits to the survival of the species (23–30). For example, expression noise can keep organisms ‘on their toes’, i.e. allowing them to thrive under different environments and to survive harsh conditions (13). Consistent with this proposition, stress-induced genes tend to have noisier characteristics than other genes, which is likely related to their biological function. Furthermore, a growing body of evidences highlighted the essential roles of noise in expression evolvability, and it was even suggested that noise levels could be tuned by the evolution to balance expression divergence (16,31,32).
Parallel to the study of expression noise, extensive research has been done to characterize expression variation of yeast strains. Here, we formally define ‘expression noise’ as fluctuations in gene expression among isogenic cells, and define ‘expression variation’ as changes in expression level of a population of cells upon genetic or environmental perturbations. In this study, utilizing the large amount of yeast genetics and genomics data currently available, we comprehensively studied the relationship between expression noise and expression variation. We attempted to address two major questions: (i) whether stochastic noises are highly correlated with expression variations? (ii) Can expression variations be predictive for noise level? To answer these questions, we compiled 12 budding yeast (Saccharomyces cerevisiae) expression variation data measured under different conditions, and found that noise levels are well correlated with different types of expression variations. Furthermore, we devised a machine learning approach, the support vector regression (SVR), to fit a predictive model to take expression variation data as input and predict expression noise for ∼4000 genes for which expression noise was previously not assayed. The results showed that our model faithfully captured the measured noise level, suggesting that the noise level and gene expression variation are highly correlated and likely determined by common mechanisms. Our method provides a new perspective on the study of expression noises in other single-cell organisms.
MATERIALS AND METHODS
Data
Large-scale ‘expression noise’ data in rich media were obtained from the study by Newman et al. (13), and expression-level adjusted measurements of noise (Distance to median, DM) were used in this work. ‘Transcription plasticity’ was taken from Tirosh and Barkai, which measured yeast transcription profiles under different conditions (33,34). The general ‘responsiveness’ of each gene was calculated from the expression data at different conditional perturbations (28). For ‘stress response’, gene expression variation was measured from a variety of stress conditions (35). The expression variation of responsiveness and stress response data were calculated by averaging the difference between expression level upon environmental perturbation and the normal condition. ‘Mutational variance’ was obtained from mutation accumulation experiments performed by Landry and colleagues (36). ‘Expression variations’ in two yeast strains, BY4716 or RM11-1a, and the ‘expression divergence’ between them were obtained from Brem et al. (23), respectively. Measurements of ‘expression divergence’ between strains (S288c and YJM789) were taken from Gagneur et al. (37). ‘Expression divergence’ among four related species was taken from the measurement under the controlled environmental perturbations (28), and ‘expression difference’ between S. cerevisiae and Saccharomyces paradoxus was taken from Tirosh et al. (26). Changes in expression companying the mutations or deletions of chromatin regulators and transcription factors were compiled from Steinfield et al. (38) and Hu et al. (39), respectively.
A much larger expression data set was used in predicting expression noise using the SVR. We compiled 633 microarray data sets from Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/). We used this expression compendium to calculate five different types of expression variation: (i) expression variation under different environmental conditions; (ii) under genetic perturbations; (iii) expression variations among individuals; (iv) expression divergence between related strains or (v) related species. For type (i), it was calculated as the difference between normal conditions and other conditions; for type (ii), it was calculated as the difference between wild-type isolates and mutation isolates; for type (iii), standard deviation among individuals were measured. Types (iv) and (v) were calculated as Euclidian distance (ED) among different stains and species. All these data are available upon request.
A list of essential genes in S. cerevisiae was downloaded from Mewes et al. (40), and the haploinsufficient genes were taken from Deutschbauer et al. (41). We compiled protein–protein interactions from the BioGrid database in April 2010 (42), which consisted of 4416 proteins and 31 967 binary interactions. We calculated the ratios of non-synonymous to synonymous substitutions (Ka/Ks) to estimate the protein evolutionary rate, and codon-based maximum likelihood method (YN00) nested in PAML package (43) was used.
In vivo nucleosome occupancy for ∼6000 yeast genes (S. cerevisiae) were retrieved from Kaplan et al. (44). In vivo nucleosome occupancy data in S. paradoxus and Saccharomyces. mikatae were obtained from Tsankov et al. (45), respectively. Average nucleosome occupancy at the promoter region (500 bp upstream to 100 bp downstream of the transcription start site) was calculated at every single base pair.
Determination of statistical significance of Gene Ontology terms
We used hypergeometric distribution in calculating statistical significance of Gene Ontology (GO) terms. GO annotations were downloaded from Ensembl database. We performed genome-wide analysis to ensure that it had sufficient power to detect significant GO terms. We use N to denote the total number of genes in yeast that have any GO annotation, and m to denote the number of ‘noisy’ or ‘quiet’ genes. If there are n genes associated with a specific GO term, among which k genes are considered as ‘noisy’ or ‘quiet’, then the P-value is calculated as the following:
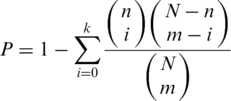 |
The P-values were then corrected for multiple testing using the false discovery rate (FDR) method, which provided an estimate of the fraction of false discoveries among the significant GO terms. We used 0.05 as the cutoff for FDR.
Support vector machine regression
Support vector machine (SVM) was initially introduced for classification, and subsequently it was extended to regression (SVR) after the introduction of an ε-insensitive loss function (46). Given a training data set T = {(x1,y1),(x2,y2), … (xm,ym), x∈Rn, y∈R}, where each xi is labeled by the real-valued yi, and n is the dimension of feature space. Linear SVR aims to find the function:
which has at most ε deviation from the actually obtained targets y and at the same time being as flat as possible (46). It leads to the following convex quadratic programming:
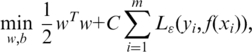 |
where w is the weighted vector for each feature, and b is a bias or offset. The regularization parameter C determines the trade-off between the empirical risk and the regularization term 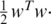
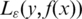 is the ε-insensitive loss function and is defined as:
is the ε-insensitive loss function and is defined as:
A nonlinear SVR projects feature vectors into a high dimensional feature space by using a kernel function, such as a Gaussian kernel:
The linear SVR procedure is then applied to the feature vectors in this feature space. In this work, all SVRs were implemented by LibSVM (47). All the features were normalized by rescaling each feature into [–1,1], and all parameters were selected by grid search (47). Pearson correlation coefficient was used as the measurement to assess the performance of the regression model while the area under receiver operating characteristic (ROC) curve (AUC) was used as the performance measurement of the classification. The scores used in the ROC analysis are the modeled DM values of the optimal SVR models.
Feature selection
Mutual information based minimum redundancy–maximum relevance (mRMR) feature selection method (48) was used to select the most informative features for noise level prediction. This method has been successfully used for gene subset selection from microarray gene expression data (49). Briefly, this method selects features that have the highest relevance with the target class (‘noisy’ and ‘quiet’ genes) and are also minimally redundant, i.e. features that are maximally dissimilar to each other. Thus, we could investigate the contribution of the combination of different features for classification by incrementally using the top m features.
Given fi (representing the feature i) and the class label y, their mutual information is defined in terms of their probabilistic density p(fi), p(y), and p(fi, y) as follows:
To measure the contribution of each feature to discriminate the noise level (‘noisy’ or ‘quiet’ genes), we used the maximum-relevance method to select the top m features in the descent order of I(fi, y), i.e. the best m individual features correlated to the target class:
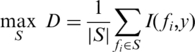 |
where S denotes the subset of the features we are seeking.
Although we can choose the top individual features using maximum-relevance algorithm, it was frequently observed that ‘the m best features are not necessarily the best m features’ because the correlations among those top features might also be high (50). In order to remove the redundancy among features, we used the following minimum-redundancy criteria:
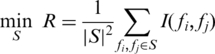 |
where mutual information between each pair of features was taken into consideration. Minimal redundancy will make feature set a better representation of the entire data set.
We simultaneously considered optimization criteria for both of the above two equations, and obtained the mRMR feature selection framework (48). A sequential incremental algorithm to solve the simultaneous optimizations of optimization criteria of the above objects (D and R) is given. Briefly, suppose that set F represents the set of features and we already have Sm–1, the feature set with m–1 features. Then, the task is to select the m-th feature from the set {F – Sm–1}. This feature is selected by maximizing 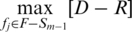 .
.
RESULTS
Expression noise is significantly correlated with expression variations
To gain insight into the relationship between expression noise and expression variation, we considered five categories of gene expression variations in this study: (i) variation of expression level under different environmental conditions; (ii) variation of expression level under genetic perturbation of trans-acting factors; (iii) differences of gene expression among individuals, and among isolates yielded by mutational accumulation; (iv) divergence in expression level between orthologous genes in related strains; and (v) divergence in expression level between orthologous genes in related species. Next, we describe the correlation of each of these five types of expression variations with expression noise (Figure 1).
Figure 1.

Correlation between noise level and gene expression variations under different conditions. Each bar represents the Pearson correlation coefficient between noise level and expression variation observed in different types of conditions. The gray bars represent the relationships for all genes, and the black bars represent the relationships for TATA-box containing genes.
Variation under different environmental conditions
In this category, three yeast expression compendiums were considered: expression changes under five different environmental perturbations (28); expression changes under stress response conditions (35); and transcription plasticity calculated based on a variety of conditions (33,34). For each of these data sets, we observed significant positive correlation between noise level and expression changes (Pearson correlation coefficients, R = 0.47, 0.3 and 0.4, respectively, P < 1e-20, Figure 1). This is consistent with previous findings that expression noise can allow cells to thrive under different conditions (51,52).
Genetic perturbations
Next, we used the expression variations accompanied with mutation or deletion of chromatin regulators (38) and transcription factors (39). It was shown that the perturbation effects of both chromatin regulator and transcription factor are positively correlated with expression noise (R = 0.39 and 0.2, respectively, P < 1e-20). Notably, noise level is more significantly correlated with chromatin regulation effects than with transcription factor regulation effect, indicating that chromatin regulation plays a more important role in generating expression variation and noise.
Variation among individuals
We compiled two data sets consisting of expression patterns (23) from a standard laboratory strain (BY4716) and a wild isolate (RM11-1a), respectively. The expression variations among individuals are also well correlated with noise level (R = 0.37 for BY4716, and 0.15 for RM11-1a, respectively, P < 1e-20). Moreover, as previously noted, expression variance among mutational accumulation lines (36) is also highly correlated with noise level (R = 0.27, P < 1e-20).
Variations between strains or species
Finally, we investigated the relationships between noise level and expression divergence in related strains or species. Two expression divergences between related strains (23,37) were measured (BY4716 versus RM11-1a; S288c versus YJM789), and they were both well correlated with noise level (R = 0.41 and 0.2, respectively, P < 1e-20). In addition, we also concluded that the noise level is highly correlated with the expression divergences between yeast species (26,28) (R = 0.34 among four yeast species, P < 1e-20, and R = 0.37 between S. cerevisiae and S. paradoxus, P < 1e-20) (Figure 1).
As TATA-box containing genes in yeast tend to have greater expression variation (28,34), we next treated these genes separately and repeated the above analysis. Figure 1 (dark bars) shows that the noise levels of TATA-box containing genes are more significantly correlated with expression variations, which suggests that TATA-box presence is an important signature to the overall expression variability. In summary, our results further demonstrated that the relationships between noise level and gene expression variations are highly interconnected with each other, especially in TATA-box containing genes.
Expression variations are predictive for noise level
To date, only half of the yeast genes have their expression noise assayed from large-scale fluorescence microscopy measurements (13). The observed significant correlation between expression variation and expression noise motivated us to ask whether expression variations can be used to predict expression noise. In order to do this, we compiled additional yeast gene expression data from NCBI GEO database that were measured under various environmental conditions, and calculated the expression variation for each gene (see ‘Materials and Methods’ section). Using these expression variability measurements, we were able to construct a predictive model to predict expression noise of each gene, taking the previously measured noise level (2126 genes) (13) as training set. In this study, SVR model was used to predict expression noise, taking 633 expression variation features as input; each feature represents variations within an expression data set. In order to evaluate the predictive power of the SVR model, we implemented a 10-fold cross-validation on the training dataset. We randomly divided the training set (2126 genes with assayed noise level) into 10 disjoint sets of equal size. For each run, one set of genes was used as the testing set and the remaining nine data sets were used as the training set. After evaluating different kernels and parameters, we selected the final optimal SVR, which achieved the highest correlation between the measured and modeled noise values (R = 0.52, P ≈ 0, Figure 2A). As suggested by the original paper (13), we separated the genes in the training set into ‘noisy’ genes (DM value ≥ 1) and ‘quiet’ genes (DM value <1), and regarded them as the positive training data (‘noisy’) and the negative training data (‘quiet’), respectively. Based on the modeled noise DM values, we plotted the ROC curve describing the relationship between the false positive rate (FPR) and the true positive rate (TPR) to further verify our performance of the SVR model. The final AUC was 0.72 (Figure 2B), demonstrating that expression variations are predictive for noise level.
Figure 2.

Prediction of expression noise using SVR. (A) Scatter plot of measured noise DM values (x-axis) versus modeled noise DM values (y-axis) of the 2126 genes. (B) ROC curve is generated from the modeled noise values by SVR and the AUC is 0.72. The diagonal dash line represents the ROC curve from randomly guessing. (C) AUC scores (y-axis) from the modeled noise values according to different thresholds for dissecting ‘noisy’ genes and ‘quiet’ genes. Different percentiles of the noise DM values (x-axis) were used as cutoffs when dissecting gene into ‘noisy’ and ‘quiet’ groups. (D) ROC curve is obtained from SVR predicted noise values on TATA-box containing genes, and the AUC is 0.76. (E) Performance of the SVR model with incremental top m features. The selected top 20 features by mRMR method contribute mainly to the discrimination ability.
We further tested different cutoffs to ascertain potential biases in the above described classification process, as we redefined the positive training set (i.e. noisy genes) by incrementally selecting the genes in the top 60th to 95th percentiles of DM values. We observed that the AUC scores concertedly increasing when more stringent cutoffs were used (Figure 2C), which indicates that our predictions were quite robust. This also shows that the correlation between expression variation and expression noise is more pronounced for noisy and variable genes. As it is known that genes that have TATA-box present in their promoter regions tend to have higher expression variation (6), we next investigated the predictive power of our SVR method on these special group of genes. Indeed, our SVR approach had a higher predictive power for TATA-box genes, as the AUC score is 0.76, higher than the entire set of yeast genes (Figure 2D).
Given the good performance of the SVR method, we next investigated which features (e.g. expression data sets) had the highest predictive power. Mutual information is a useful approach to measure the dependency between multiple features, and features with higher mutual information scores were considered to contribute independently to the prediction process. Here, we used mutual information based maximum-relevance method (see ‘Materials and Methods’ section) to select the most informative features. Table 1 lists the 20 most informative features ranked by mutual information scores. Notably, most of these informative features are environmental effects on gene expression variations, such as heat shock, genotoxic stress, stress response, etc. This suggested a strong relationship between expression variation caused by environmental perturbations and gene expression noise. Furthermore, we found that genetic perturbations of chromatin regulators also significantly contributed to the noise prediction.
Table 1.
Most informative features
| GEO id | MI scores | Description |
|---|---|---|
| GSE5608 | 0.043 | Triterpenoid celastrol treatment and heat-shock comparison |
| GSE2224 | 0.039 | Genotoxic stress |
| GSE18 | 0.036 | Hypo-osmotic shock time course |
| GSE15352 | 0.035 | Dynamic transcriptional and metabolic responses in yeast adapting to temperature stress |
| GSE14991 | 0.031 | Time course of Saccharomyces cerevisiae exposed to arsenic under phosphate-limited conditions |
| GSE14761 | 0.031 | Accumulation of sumoylated Rad52 in checkpoint mutants perturbed in DNA replication |
| GSE4709 | 0.03 | Gcn4p-mediated transcriptional stress response |
| GSE9463 | 0.029 | Chemical toxicity of thorium in Saccharomyces cerevisiae |
| GSE2263 | 0.029 | Oxidative stress |
| GSE3406 | 0.029 | Expression patterns in stress conditions |
| GSE3729 | 0.028 | Oxidative stress in stationary-phase cultures |
| GSE1639 | 0.027 | Rpd3 and histone H3 and H4 deletions/mutations |
| GSE1554 | 0.027 | Time course of glycine addition or withdrawal |
| GSE1404 | 0.027 | Exploration of essential gene functions via titratable promoter alleles |
| GSE959 | 0.027 | Global transcriptional response to transient cell wall damage |
| GSE21 | 0.026 | snf/swi mutants |
| GSE20590 | 0.026 | Effects of ethanol stress |
| GSE18456 | 0.025 | Expression patterns in response to zymolyase treatment |
| GSE20749 | 0.025 | Natural selection on cis- and trans-regulation in yeasts |
| GSE2096 | 0.025 | fhl1 and ifh1 deletion mutants |
The 20 most informative features ranked by mutual information scores (MI scores).
For each feature, we list its MI score which represents the relevance of the feature to the classification task (i.e. classifying noisy and quiet genes)
Although we selected informative features according to the mutual information to the target class, simply combining these top informative features might not form a better feature sets. One possible reason is that some of these features could be highly correlated, which raises the issue of ‘redundancy’ of feature set. Here, we used mRMR feature selection method (see ‘Materials and Methods’ section) to choose a comprehensive but non-redundant representation of the characteristics of the noise. Briefly, mRMR used mutual information to select the most relevant features that are minimally redundant. At each cycle, the mRMR method selects a feature which is maximal relevant to the target class and also minimally redundant to the selected features. To check whether all the expression variation features were required to model stochastic noise, we constructed a series of SVRs by incrementally combining the most informative features according to the minimum redundancy-maximum relevance criteria. We added the mth best informative feature to the previously selected m–1 features to run an SVR model at each step. As the mRMR feature selection method selected the non-redundant feature, the combination of the m individual best informative features could be the top m features. We checked the performance of the SVR with the top m features against the number of the best features (m = 1,2, … ). In Figure 2E, it was indicated that not all features were equally important, and the discrimination power of the SVR saturated after the top 20 features were used (the AUC = 0.71). Incorporating additional features do not dramatically improve the performance because of a high degree of redundancy.
Validation of noise prediction by other features
In addition to cross-validation, we next sought to use following lines of evidences to ascertain the predictive power of our SVR method.
Dosage sensitivity and essentiality
It has been documented that noise levels are closely related to gene dosage sensitivity, e.g. essential genes tend to have reduced expression noise (4,31,53). We divided the 3909 yeast genes for which there were no previously assayed noise levels into two groups: the ‘quiet’ group (2065 genes, DM < 1) and the ‘noisy’ group (1844 genes, DM ≥ 1). Indeed, the ‘quiet’ genes contained more haploinsufficient genes and essential genes than the ‘noisy’ genes (Wilcoxon rank sum test, P = 1.2e-5 and P = 4.1e-3 for haploinsufficient genes and essential genes, respectively, Figure S1), which is in agreement with what was previously observed (31).
Protein–protein interactions
It is known that proteins with more interacting partners have lower noise level, and ‘quiet’ genes are more conserved than ‘noisy’ genes at the sequence level (4,16,31). It was shown that hub proteins (degree >10) are highly enriched in the ‘quiet’ genes (Wilcoxon rank sum test, P = 4.2e-4, Figure S1).
GO enrichment
As reported in the original paper by Newman et al. (13), the ‘noisy’ genes are enriched in the following GO categories: ‘heat shock’, ‘stress response’, ‘amino-acid biosynthesis’ and ‘oxidative phosphorylation’, whereas ‘quiet’ genes are enriched in ‘translation initiation’, ‘ribosomal proteins’ and ‘protein degradation’, etc. As now we have made noise predictions on all the yeast genes, we next sought to determine the enriched GO categories for the ‘noise’ and ‘quiet’ genes predicted by our SVR method. Indeed, our results are consistent with previous findings, as ‘noisy’ genes are highly enriched in ‘metabolic process’, ‘stress response’ and ‘biosynthesis process’, and ‘quiet’ genes are mainly involved in ‘protein transport’ and ‘translation proteins’ (Table S1). In terms of cellular component, the protein products of noise genes are enriched in the mitochondria, whereas the protein products of ‘quiet’ genes tend to locate to ribosome and Golgi apparatus. As to our predicted ‘noisy’ and ‘quiet’ groups of genes, most of enriched GO categories are in accordance with previous characterizations, which showed that our genome-wide prediction is of high accuracy.
Nucleosome positioning
A recent study reported a close association between gene expression variation and the nucleosome positioning in the promoter regions (51). It is known that local nucleosome occupancy in the promoter region affects transcription regulation by modulating the accessibility of transcription factors to their binding sites, and influences the ability of genes to modulate their expression (54). Given these insights, we next examined nucleosome organization over the promoter regions of the noisier genes (genes with top 5% of predicted and measured DM values). As shown in Figure 3, when plotting the average nucleosome occupancy measured by experimental method in vivo (44), we found the measured and predicted noisier genes had significantly higher nucleosome occupancy than the rest of the genes, i.e. their promoters are in a more ‘closed state’. To further quantify the difference in nucleosome occupancy at the nucleosome free regions (–200 to −50 bp upstream of translation start site, TSS) between noisier genes and other genes, we calculated the lowest average nucleosome occupancy (LANO) score in 100-bp sliding windows from the 200 bp upstream of the translation start site, and found that the promoter region of noisier genes reflect a closed (nucleosome-occupied) nucleosome organization (Wilcoxon rank sum test, P = 2.3e-5 for measured noisier genes, and P = 3.8e-4 for modeled noisier genes, respectively). Our results further demonstrated that nucleosome organization in the promoter region plays a dominant role in differential noise pattern (51).
Figure 3.

Nucleosome organizations in the promoter region of noisier genes. Both the measured noisier genes (A, black curve) and predicted noisier genes (B, black curve) have higher nucleosome occupancy in the promoters than the rest of the genes.
In the above discussion, we confirmed that the noise predicted genes share the same characteristics as noise measured genes. We took this as indirect evidences that our predicted noise levels are of sufficient accuracy. However, we must point out that these validations are indirect ways and might result from the strong associations between expression variations and some features. Recently, Li et al. measured the expression noise levels of 40 genes by quantifying fluorescence intensities using high-content screening microscopy (16). We found that our modeled noise values are significantly positive correlated with the variations of fluorescence intensities (R = 0.58, P = 0.005), which further highlights the accuracy of our noise prediction.
Predict noise in other single-cell organisms
We have shown in the above that models incorporating expression variations after environmental perturbations can accurately predict expression noise levels in S. cerevisiae. Next, we asked whether we could apply this model to other single-cell organisms and predict noise level from expression variations in these organisms. We obtained expression variation data (28) under the heat shock, oxidative stress, nitrogen starvation, DNA damage and carbon source switch in three closely related yeast species (S. cerevisiae, S. paradoxus and S. mikatae). We first re-trained our SVR model in S. cerevisiae using only expression data measured under these conditions, under which expression data are available for all three species. We next applied the model to the expression data in the other two yeast species to make noise predictions. Due to the scarcity of measured noise data in other yeast species, the predicted accuracy of noise values cannot be directly validated. To circumvent this, we attempted to use nucleosome occupancy in the promoter regions as a proxy for expression noise, because in S. cerevisiae such occupancy is highly correlated with the measured expression noise (51), and examined whether these genes have distinct nucleosome positioning pattern compared to other genes (45). We found significant differences of LANO scores between noisier genes and other genes (Wilcoxon rank sum test, P = 0.004 for S. paradoxus, and P = 0.01 for S. mikatae, respectively). The result suggests that genes with higher noise levels also have nucleosome-occupied region in their promoter regions (Figure 4A and B). Thus, we indirectly showed that our noise prediction method is also meaningful in other species. One caveat of the above analysis is that expression variation is also correlated with nucleosome occupancy. We therefore sought to compare the evolutionary rate of encoded proteins between the ‘noisy’ genes and the ‘quiet’ genes as no significant relationship between environmental expression variation and evolutionary rate was found (28,55). Consistent with the result in S. cerevisiae (16), we found that noisier genes have lower Ka/Ks ratios than the rest of genes (Wilcoxon rank sum test, P = 1.4e-3 for S. paradoxus, P = 3.8e-4 for S. mikatae).
Figure 4.

Nucleosome organizations in the promoter region of noisier genes in S. paradoxus and S. mikatae. Noisier genes in both S. paradoxus (A, black curve) and S. mikatae (B, black curve) show clear nucleosome-occupied regions in the promoter region gauged by average nucleosome occupancy relative to translation start site of focal genes. (C) Pearson correlation coefficients of modeled noise levels between different yeast species. (D) Impact of nucleosome occupancy changes on noise level divergence. We ordered the genes by noise divergence (x-axis), and y-axis represents the changes of average ‘LANO’ scores in each sliding window of 300 ordered genes. The gray line is the fitted trend line.
With our predicted noise data, we can examine the difference in noise levels between orthologous genes in three yeast species. The result showed that noise levels in multiple yeast species are highly correlated with each other (Figure 4C), especially between S. paradoxus and S. mikatae (R = 0.75, P ≈0). This indicates that expression noise and expression variation of fungi genes are highly conserved during evolution, at least in the fungi lineage. To detect how nucleosome occupancy influences the variation of noise level among these yeast species, we compared the changes in LANO score with the divergence of noise level among these three species (defined as the standard deviation). Specifically, we first sorted genes by their differences in noise levels among these three species (x-axis in Figure 4D), and then for 300 genes in a sliding window, we calculated the average LANO score differences between the orthologous genes. Figure 4D shows that the genes with diverged noise levels showed much higher changes in LANOs score than genes whose noise levels are conserved among the species, suggesting that the divergence of expression noise is correlated with the divergence in nucleosome organization in the promoter regions (R = 0.35, P < 1e-6). We next investigated the functional enrichments of genes that have divergent expression noise (the top 20% genes sorted by noise differences) and genes that have conserved expression noise (the lowest 50% sorted by noise differences). The noise diverged genes are enriched for ‘protein kinase cascade’ (FDR = 0.017), ‘oxidoreductase activity’ (FDR = 0.008), ‘sterol biosynthetic process’ (FDR = 0.04), etc. In contrast, noise conserved genes are enriched for ‘ubiquitin-dependent protein catabolic process’ (FDR = 0.03) and ‘endopeptidase activity’ (FDR = 0.008) (Supplementaey Table S2). Taken together, we predicted noise level in other species based on the notion of intrinsic expression variation ability. Our work therefore sheds light on the intrinsic expression ability, and can provide a preliminary overview of stochastic noise of gene expression in other single-cell organisms. However, further experimental works need to be done in order to reveal the real patterns of stochastic noise in other taxa.
DISCUSSION AND CONCLUSION
Our aim in this paper is to establish the relationships between stochastic expression noise and expression variations. To this end, we have shown that expression noise in yeast is well correlated with gene expression variation measured under different genetic and environmental perturbations. Gene expression in single-cellular organisms such as S. cerevisiae is highly dynamic (plastic), as the cells are able to adjust their expression program in response to external or internal perturbations (56). In addition to changes in expression program at the population level, isogenic cells also exhibit stochastic expression level (noises) at the single-cell level. It was suggested previously that such stochasticity is an important biological trait that offers adaptive advantages to the organisms, as it provides sufficient phenotypic heterogeneity to survive fluctuating environments (13,31,32). Our findings provided evidences that these two adaptive mechanisms at two population levels are intrinsically linked.
Prior to our work, it was reported that noisy genes are sensitive to the perturbation of chromatin regulators (34,57), suggesting that chromatin regulation plays a pivotal role in generating expression noise during transcription. By investigating nucleosome occupancy in the promoter region, Choi and Kim (51) found that the genes with higher expression variation tend to have higher nucleosome occupancy (i.e. in a more closed state) in a crucial region 50–200 bp upstream from TSS. They further proposed that the plastic nature of gene expression is an intrinsic property of the gene, and nucleosome occupancy plays a dominant role for tuning gene expression to adapt to changing conditions. This is consistent with what was observed experimentally, i.e. the competition between chromatin regulators and transcription factors can influence how a gene response to external stimuli (58). What we showed in this work provided an analytical framework that connected the observations and insights gained from the study of ‘expression variation’ and ‘expression noise’, two related but distinct cell properties. Such connections were captured and represented in our SVR model. TATA box is regarded as one of the most important mechanisms of transcriptional tuning, and presents in ∼20% of S. cerevisiae genes (6). They are characterized as noisy transcription and gene expression evolution control. Furthermore, TATA-box can promote short-term regulatory tuning to environmental changes (6). Our result indicated that TATA-box containing genes tend to have higher variation and noise than the rest of the genes, and are more sensitive to chromatin remodeling.
In summary, we observed that noise levels are highly correlated with expression variations in S. cerevisiae, and we developed a computational model that can be used to predict expression noise, which is a property of individual cells, from expression variation, which is a property associated with populations of cells. Our work offers a new perspective on the origin and behavior of stochastic noise, and serves as a useful tool to study stochastic noise in single-cell organisms.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
A Team Grant from the Canadian Institutes of Health Research (CIHR MOP#82940). Funding for open access charge: Canadian Institutes of Health Research.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We thank Dr. Chris Soon Heng Tan for comments on this manuscript. We would like to thank the Associate Editor and two referees for their constructive comments which have significantly improved the quality of this article.
REFERENCES
- 1.Blake WJ, Kaern M, Cantor CR, Collins JJ. Noise in eukaryotic gene expression. Nature. 2003;422:633–637. doi: 10.1038/nature01546. [DOI] [PubMed] [Google Scholar]
- 2.Kaern M, Elston TC, Blake WJ, Collins JJ. Stochasticity in gene expression: from theories to phenotypes. Nat. Rev. 2005;6:451–464. doi: 10.1038/nrg1615. [DOI] [PubMed] [Google Scholar]
- 3.Lu T, Shen T, Bennett MR, Wolynes PG, Hasty J. Phenotypic variability of growing cellular populations. Proc. Natl Acad. Sci. USA. 2007;104:18982–18987. doi: 10.1073/pnas.0706115104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raj A, van Oudenaarden A. Nature, nurture, or chance: stochastic gene expression and its consequences. Cell. 2008;135:216–226. doi: 10.1016/j.cell.2008.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raser JM, O'Shea EK. Noise in gene expression: origins, consequences, and control. Science. 2005;309:2010–2013. doi: 10.1126/science.1105891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Basehoar AD, Zanton SJ, Pugh BF. Identification and distinct regulation of yeast TATA box-containing genes. Cell. 2004;116:699–709. doi: 10.1016/s0092-8674(04)00205-3. [DOI] [PubMed] [Google Scholar]
- 7.Rao CV, Wolf DM, Arkin AP. Control, exploitation and tolerance of intracellular noise. Nature. 2002;420:231–237. doi: 10.1038/nature01258. [DOI] [PubMed] [Google Scholar]
- 8.Elowitz MB, Levine AJ, Siggia ED, Swain PS. Stochastic gene expression in a single cell. Science. 2002;297:1183–1186. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- 9.Becskei A, Serrano L. Engineering stability in gene networks by autoregulation. Nature. 2000;405:590–593. doi: 10.1038/35014651. [DOI] [PubMed] [Google Scholar]
- 10.Becskei A, Kaufmann BB, van Oudenaarden A. Contributions of low molecule number and chromosomal positioning to stochastic gene expression. Nat. Genet. 2005;37:937–944. doi: 10.1038/ng1616. [DOI] [PubMed] [Google Scholar]
- 11.Colman-Lerner A, Gordon A, Serra E, Chin T, Resnekov O, Endy D, Pesce CG, Brent R. Regulated cell-to-cell variation in a cell-fate decision system. Nature. 2005;437:699–706. doi: 10.1038/nature03998. [DOI] [PubMed] [Google Scholar]
- 12.Pedraza JM, van Oudenaarden A. Noise propagation in gene networks. Science. 2005;307:1965–1969. doi: 10.1126/science.1109090. [DOI] [PubMed] [Google Scholar]
- 13.Newman JR, Ghaemmaghami S, Ihmels J, Breslow DK, Noble M, DeRisi JL, Weissman JS. Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature. 2006;441:840–846. doi: 10.1038/nature04785. [DOI] [PubMed] [Google Scholar]
- 14.Bar-Even A, Paulsson J, Maheshri N, Carmi M, O'Shea E, Pilpel Y, Barkai N. Noise in protein expression scales with natural protein abundance. Nat. Genet. 2006;38:636–643. doi: 10.1038/ng1807. [DOI] [PubMed] [Google Scholar]
- 15.Rosenfeld N, Young JW, Alon U, Swain PS, Elowitz MB. Gene regulation at the single-cell level. Science. 2005;307:1962–1965. doi: 10.1126/science.1106914. [DOI] [PubMed] [Google Scholar]
- 16.Li J, Min R, Vizeacoumar FJ, Jin K, Xin X, Zhang Z. Exploiting the determinants of stochastic gene expression in Saccharomyces cerevisiae for genome-wide prediction of expression noise. Proc. Natl Acad. Sci. USA. 107:10472–10477. doi: 10.1073/pnas.0914302107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Austin DW, Allen MS, McCollum JM, Dar RD, Wilgus JR, Sayler GS, Samatova NF, Cox CD, Simpson ML. Gene network shaping of inherent noise spectra. Nature. 2006;439:608–611. doi: 10.1038/nature04194. [DOI] [PubMed] [Google Scholar]
- 18.Thattai M, van Oudenaarden A. Intrinsic noise in gene regulatory networks. Proc. Natl Acad. Sci. USA. 2001;98:8614–8619. doi: 10.1073/pnas.151588598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Becskei A, Seraphin B, Serrano L. Positive feedback in eukaryotic gene networks: cell differentiation by graded to binary response conversion. EMBO J. 2001;20:2528–2535. doi: 10.1093/emboj/20.10.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hasty J, Pradines J, Dolnik M, Collins JJ. Noise-based switches and amplifiers for gene expression. Proc. Natl Acad. Sci. USA. 2000;97:2075–2080. doi: 10.1073/pnas.040411297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Isaacs FJ, Hasty J, Cantor CR, Collins JJ. Prediction and measurement of an autoregulatory genetic module. Proc. Natl Acad. Sci. USA. 2003;100:7714–7719. doi: 10.1073/pnas.1332628100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karmakar R, Bose I. Graded and binary responses in stochastic gene expression. Phys. Biol. 2004;1:197–204. doi: 10.1088/1478-3967/1/4/001. [DOI] [PubMed] [Google Scholar]
- 23.Brem RB, Kruglyak L. The landscape of genetic complexity across 5,700 gene expression traits in yeast. Proc. Natl Acad. Sci. USA. 2005;102:1572–1577. doi: 10.1073/pnas.0408709102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brem RB, Storey JD, Whittle J, Kruglyak L. Genetic interactions between polymorphisms that affect gene expression in yeast. Nature. 2005;436:701–703. doi: 10.1038/nature03865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brem RB, Yvert G, Clinton R, Kruglyak L. Genetic dissection of transcriptional regulation in budding yeast. Science. 2002;296:752–755. doi: 10.1126/science.1069516. [DOI] [PubMed] [Google Scholar]
- 26.Tirosh I, Reikhav S, Levy AA, Barkai N. A yeast hybrid provides insight into the evolution of gene expression regulation. Science. 2009;324:659–662. doi: 10.1126/science.1169766. [DOI] [PubMed] [Google Scholar]
- 27.Tirosh I, Weinberger A, Bezalel D, Kaganovich M, Barkai N. On the relation between promoter divergence and gene expression evolution. Mol. Sys. Biol. 2008;4:159. doi: 10.1038/msb4100198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tirosh I, Weinberger A, Carmi M, Barkai N. A genetic signature of interspecies variations in gene expression. Nat. Genet. 2006;38:830–834. doi: 10.1038/ng1819. [DOI] [PubMed] [Google Scholar]
- 29.Townsend JP, Cavalieri D, Hartl DL. Population genetic variation in genome-wide gene expression. Mol. Biol. Evol. 2003;20:955–963. doi: 10.1093/molbev/msg106. [DOI] [PubMed] [Google Scholar]
- 30.Yvert G, Brem RB, Whittle J, Akey JM, Foss E, Smith EN, Mackelprang R, Kruglyak L. Trans-acting regulatory variation in Saccharomyces cerevisiae and the role of transcription factors. Nat. Genet. 2003;35:57–64. doi: 10.1038/ng1222. [DOI] [PubMed] [Google Scholar]
- 31.Lehner B. Selection to minimise noise in living systems and its implications for the evolution of gene expression. Mol. Sys. Biol. 2008;4:170. doi: 10.1038/msb.2008.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Z, Qian W, Zhang J. Positive selection for elevated gene expression noise in yeast. Mol. Sys. Biol. 2009;5:299. doi: 10.1038/msb.2009.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ihmels J, Friedlander G, Bergmann S, Sarig O, Ziv Y, Barkai N. Revealing modular organization in the yeast transcriptional network. Nat. Genet. 2002;31:370–377. doi: 10.1038/ng941. [DOI] [PubMed] [Google Scholar]
- 34.Tirosh I, Barkai N. Two strategies for gene regulation by promoter nucleosomes. Genome Res. 2008;18:1084–1091. doi: 10.1101/gr.076059.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Landry CR, Lemos B, Rifkin SA, Dickinson WJ, Hartl DL. Genetic properties influencing the evolvability of gene expression. Science. 2007;317:118–121. doi: 10.1126/science.1140247. [DOI] [PubMed] [Google Scholar]
- 37.Gagneur J, Sinha H, Perocchi F, Bourgon R, Huber W, Steinmetz LM. Genome-wide allele- and strand-specific expression profiling. Mol. Sys. Biol. 2009;5:274. doi: 10.1038/msb.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Steinfeld I, Shamir R, Kupiec M. A genome-wide analysis in Saccharomyces cerevisiae demonstrates the influence of chromatin modifiers on transcription. Nat. Genet. 2007;39:303–309. doi: 10.1038/ng1965. [DOI] [PubMed] [Google Scholar]
- 39.Hu Z, Killion PJ, Iyer VR. Genetic reconstruction of a functional transcriptional regulatory network. Nat. Genet. 2007;39:683–687. doi: 10.1038/ng2012. [DOI] [PubMed] [Google Scholar]
- 40.Mewes HW, Frishman D, Mayer KF, Munsterkotter M, Noubibou O, Pagel P, Rattei T, Oesterheld M, Ruepp A, Stumpflen V. MIPS: analysis and annotation of proteins from whole genomes in 2005. Nucleic Acids Res. 2006;34:D169–D172. doi: 10.1093/nar/gkj148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deutschbauer AM, Jaramillo DF, Proctor M, Kumm J, Hillenmeyer ME, Davis RW, Nislow C, Giaever G. Mechanisms of haploinsufficiency revealed by genome-wide profiling in yeast. Genetics. 2005;169:1915–1925. doi: 10.1534/genetics.104.036871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, Tyers M. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006;34:D535–D539. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- 44.Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, LeProust EM, Hughes TR, Lieb JD, Widom J, et al. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature. 2009;458:362–366. doi: 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsankov AM, Thompson DA, Socha A, Regev A, Rando OJ. The role of nucleosome positioning in the evolution of gene regulation. PLoS Biol. 8:e1000414. doi: 10.1371/journal.pbio.1000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smola AJ, Scholkopf B. A tutorial on support vector regression. Stat. Comput. 2004;14:199–222. [Google Scholar]
- 47.Chang CC, Lin CJ. LIBSVM: a library for support vector machines. 2001 Available at http://www.csie.ntu.edu.tw/∼cjlin/libsvm/ (15 September 2010, date last accessed) [Google Scholar]
- 48.Peng H, Long F, Ding C. Feature selection based on mutual information: criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 2005;27:1226–1238. doi: 10.1109/TPAMI.2005.159. [DOI] [PubMed] [Google Scholar]
- 49.Zhang Y, Ding C, Li T. Gene selection algorithm by combining reliefF and mRMR. BMC Genomics. 2008;9(Suppl. 2):S27. doi: 10.1186/1471-2164-9-S2-S27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ding C, Peng H. Minimum redundancy feature selection from microarray gene expression data. J. Bioinform. Computat. Biol. 2005;3:185–205. doi: 10.1142/s0219720005001004. [DOI] [PubMed] [Google Scholar]
- 51.Choi JK, Kim YJ. Intrinsic variability of gene expression encoded in nucleosome positioning sequences. Nat. Genet. 2009;41:498–503. doi: 10.1038/ng.319. [DOI] [PubMed] [Google Scholar]
- 52.Lopez-Maury L, Marguerat S, Bahler J. Tuning gene expression to changing environments: from rapid responses to evolutionary adaptation. Nat. Rev. Genet. 2008;9:583–593. doi: 10.1038/nrg2398. [DOI] [PubMed] [Google Scholar]
- 53.Batada NN, Hurst LD. Evolution of chromosome organization driven by selection for reduced gene expression noise. Nat. Genet. 2007;39:945–949. doi: 10.1038/ng2071. [DOI] [PubMed] [Google Scholar]
- 54.Komili S, Silver PA. Coupling and coordination in gene expression processes: a systems biology view. Nat. Rev. Genet. 2008;9:38–48. doi: 10.1038/nrg2223. [DOI] [PubMed] [Google Scholar]
- 55.Tirosh I, Barkai N. Evolution of gene sequence and gene expression are not correlated in yeast. Trends Genet. 2008;24:109–113. doi: 10.1016/j.tig.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 56.Lopez-Maury L, Marguerat S, Bahler J. Tuning gene expression to changing environments: from rapid responses to evolutionary adaptation. Nat. Rev. 2008;9:583–593. doi: 10.1038/nrg2398. [DOI] [PubMed] [Google Scholar]
- 57.Choi JK, Kim YJ. Epigenetic regulation and the variability of gene expression. Nat. Genet. 2008;40:141–147. doi: 10.1038/ng.2007.58. [DOI] [PubMed] [Google Scholar]
- 58.Lam FH, Steger DJ, O'Shea EK. Chromatin decouples promoter threshold from dynamic range. Nature. 2008;453:246–250. doi: 10.1038/nature06867.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.