

Abstract
A method for the oxidation of organotrifluoroborates using Oxone® was developed. A variety of aryl-, heteroaryl-, alkenyl-, and alkyltrifluoroborates were converted into the corresponding oxidized products in excellent yields. This method proved to be tolerant of a broad range of functional groups, and in secondary alkyl substrates it was demonstrated to be completely stereospecific.
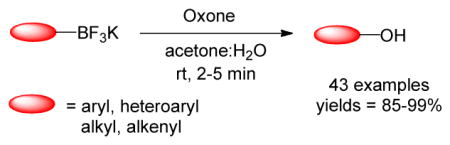
Introduction
Phenols are found in numerous natural products, pharmaceuticals and polymers, and have been widely used as versatile synthetic intermediates.1 Traditional methods for the synthesis of these compounds include various protocols for substitution of aryl halides. Unfortunately, the harsh reaction conditions required for direct nucleophilic substitution make this approach incompatible with a variety of common functional groups.2 Copper- and palladium-catalyzed hydroxylation of aryl halides is reported in the literature and offers an alternative to the former method.3 In this case, however, the use of transition metals with attendant ligands and/or high reaction temperatures leads to processes that are less than ideal. Various boron species can be incorporated into aromatic and heteroaromatic systems in a variety of complementary ways, and recently the oxidation of boronic acids in a copper-catalyzed reaction has been reported.4 Although perhaps useful on small scale in the laboratory, the use of a copper reagent, ligand and excess base significantly reduces its appeal on a larger scale. As an alternative to this method, several non-metal promoted oxidations of boronic acids and their derivatives have been described.5 Among the oxidants used for this transformation, hydrogen peroxide,5a,c,e,f hydroxylamine5b and Oxone®5d,g are most often employed. The first two reagents require long reaction times, and the products are obtained in moderate yields for arylboronic acids containing electron withdrawing groups. On the other hand, the use of Oxone® provides an extremely rapid and generally efficient reaction.5d,g
Over the past years organotrifluoroborates have been used in many contexts as a valuable alternative to boronic acids and boronate esters.6 Because of their tetracoordinate nature, these organoboron species do not undergo undesirable side reactions with commonly employed organic reagents such as bases, organic acids and nucleophiles. Thus, organotrifluoroborates, contrary to most other boron species, can be functionalized to build molecular complexity, while leaving the boron-carbon bond intact (Scheme 1).7 The organotrifluoroborates can then be conveniently converted in a variety of ways depending on the conditions utilized.8 Of further importance, heteroaryltrifluoroborates exhibit enhanced stability, and thus they can be prepared and stored for long periods of time, whereas many of the corresponding boronic acids rapidly decompose.8a,9 Thus, many advantages to using organotrifluroborates as surrogates for boronic acids are beginning to emerge.
SCHEME 1.

To the best of our knowledge, although organotrifluoroborates present all of the benefits mentioned above, reports of the oxidation of these species into the corresponding phenol, alcohol or related derivatives is currently limited.4,10 For example, phenyltrifluoroborate has been oxidized to phenol in a copper-catalyzed procedure requiring 8 h at room temperature.4 In another isolated example, an alkyltrifluoroborate has been oxidized by utilizing an excess of a hypervalent iodonium complex to afford an excellent yield of the desired alcohol in 3 h.10a Finally, during the preparation of this manuscript Fensterbank et al. reported the 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) promoted oxidation of several alkyltrifluoroborates, which required high temperatures and long reaction times (120 ºC, 20 h) with excess TEMPO (1.2 equiv) and copper salts (1.2 equiv) in DMSO, or 3 equiv of TEMPO in the presence of Dess-Martin periodinane in Et2O at room temperature for 1–2 d. The products isolated were the corresponding TEMPO derivatives, which were accessed in modest to good yields depending on the substrates. Importantly, these reactions were demonstrated to proceed via radical intermediates, and thus they would not be expected to be stereospecific for enantiomerically enriched alkyltrifluoroborates, although this latter aspect was not specifically addressed in an unambiguous manner.
The lack of examples detailing a practical method for the oxidation of organotrifluoroborates and the limitations associated with other boron derivatives prompted us to investigate a general, efficient, mild, rapid and convenient method for this transformation. Herein, we report the oxidation of a broad range of aryl-, heteroaryl-, alkenyl-, and alkyltrifluoroborates using Oxone® at room temperature.
Results and Discussion
Based on previous research in which a variety of arylboronic acids and pinacol boronates were transformed to the corresponding phenols,5d,g investigations were initiated on the oxidation of potassium naphthalen-1-yltrifluoroborate. Very rapidly it was evident that the protocol developed by Maleczka and Smith for pinacol boronates, using 1 equiv of Oxone® (as a 0.2 M solution in H2O) in acetone at room temperature, open to the air, was also ideal for the rapid (<5 min) oxidation of organotrifluoroborates. The desired phenol was obtained by an aqueous workup and simple filtration in excellent yield (99%). Because Oxone® is an inexpensive,11 widely available and industrially acceptable reagent, all subsequent reactions were performed using this material.
With optimal conditions in hand, the investigation continued to outline the full scope of this process. The requisite potassium organotrifluoroborates needed for the study were readily prepared by previously reported procedures.6,12 Initially, a broad range of electron-rich and electron-neutral aryltrifluoroborates were investigated (Table 1). In all cases, quantitative conversion to the desired products was accomplished in only 2 min. The reaction provided excellent yields for all substrates containing para, meta, or ortho substituents (entries 3 to 5). Sterically hindered compounds also afforded the products in high yields (entry 7). Importantly, we demonstrated that the reaction could be scaled to 55 mmol, providing product 1a in 96% yield (Table 1, entry 1). Contrary to the metal catalyzed processes utilizing other organoboron species, the use of organotrifluoroborates and Oxone® in every case afforded the desired products with little or no formation of organic side products. Therefore, a simple aqueous workup followed, at most, by filtration through a short plug of silica with charcoal afforded the desired products in virtually quantitative yield. Throughout the course of this study, no chromatography was required to obtain pure products.
TABLE 1.
Oxidation of Electron-rich and Electron-neutral Potassium Aryltrifluoroboratesa
 | ||||
|---|---|---|---|---|
| entry | substrate | product | isolated yield (%) | |
| 1 |
|
|
1a | 96b |
| 2 |
|
|
1b | 99 |
| 3 |
|
|
1c | 97 |
| 4 |
|
|
1d | 98 |
| 5 |
|
|
1e | 93 |
| 6 |
|
|
1f | 97 |
| 7 |
|
|
1g | 95 |
| 8 |
|
|
1h | 94 |
| 9 |
|
|
1i | 99 |
| 10 |
|
|
1j | 97 |
All reactions were carried out using 1 mmol of aryltrifluoroborate and Oxone (5 mL, 0.2 M in H2O) in 5 mL of acetone for 2 min at rt.
55 mmol scale.
To demonstrate the ability of organotrifluoroborates to serve in the role detailed in Scheme 1, in which a functionalized substrate could be elaborated and then converted into the phenol in a late synthetic stage, potassium (Z)-(4-(4-cyanobut-1-en-1-yl)phenyl)trifluoroborate was synthesized through a Wittig reaction of 4-formylphenyltrifluoroborate and the nitrile-containing ylide12f (Scheme 2). The organotrifluoroborate was then converted into the phenol, using the oxidation protocol with Oxone®. The desired product was obtained in excellent yield in only 2 minutes without affecting either the nitrile or the double bond of the molecule.13
SCHEME 2.

Next, attention was turned to electron-poor aryltrifluoroborates. The conditions developed worked equally well for these substrates, providing the phenols in excellent yields (Table 2). Substrates such as aldehyde-, ester-, keto-, nitrile-, and nitro-containing aryltrifluoroborates afforded the hydroxylated product without affecting the pendant functional groups (entries 7 to 11). Halogenated trifluoroborates also provide the corresponding phenol in excellent yields (entries 1 to 6).
TABLE 2.
Oxidation of Electron-poor Potassium Aryltrifluoroboratesa
 | ||||
|---|---|---|---|---|
| entry | substrate | product | isolated yield (%) | |
| 1 |
|
|
2a | 85 |
| 2 |
|
|
2b | 98 |
| 3 |
|
|
2c | 99 |
| 4 |
|
|
2d | 99 |
| 5 |
|
|
2e | 97 |
| 6 |
|
|
2f | 98 |
| 7 |
|
|
2g | 98 |
| 8 |
|
|
2h | 99 |
| 9 |
|
|
2i | 99 |
| 10 |
|
|
2j | 99 |
| 11 |
|
|
2k | 98 |
All reactions were carried out using 1 mmol of aryltrifluoroborate and Oxone (5 mL, 0.2 M in H2O) in 5 mL of acetone for 2 min at rt.
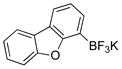
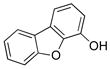
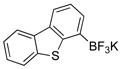
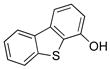
To expand the utility of the developed conditions further, heteroaryl systems were investigated, including dibenzothiophenyl-, dibenzofuranyl-, pyridinyl-, benzothiophenyl-, benzofuranyl-, thiophenyl- and furanyl derivatives (Table 3). Heteroaryls, such as dibenzo[b,d]furan-4-yl-, dibenzo[b,d]thiophen-4-yl-, 6-chloropyridin-3-yl- and 6-fluoro-5-methylpyridin-3-yltrifluoroborate, afforded the desired phenol in excellent yields in only 5 min (entries 1 to 4). For substrates where the most stable tautomers are the carbonyl isomer,14 only the keto-tautomer was observed (entries 5 to 8). 2-Trifluoroboratofuran afforded the β,γ-unsaturated lactone (entry 9), along with 10% of the α,β-unsaturated product. Interestingly, under the reaction conditions, no oxidation of the nitrogen or sulfur atom of these heterocycles was observed.15
TABLE 3.
Hydroxylation of Potassium Heteroaryltrifluoroboratesa
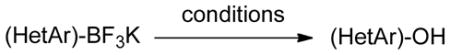 | ||||
|---|---|---|---|---|
| entry | substrate | product | isolated yield (%) | |
| 1 |
|
|
3a | 97 |
| 2 |
|
|
3b | 95 |
| 3 |
|
|
3c | 91 |
| 4 |
|
|
3d | 94 |
| 5 |
|
|
3e | 99 |
| 6 |
|
|
3f | 97 |
| 7 |
|
|
3g | 94 |
| 8 |
|
|
3h | 89 |
| 9 |
|
|
3i | 97 |
All reactions were carried out using 1 mmol of aryltrifluoroborate and Oxone (5 mL, 0.2 M in H2O) in 5 mL of acetone for 2 min at rt.
The scope of the general reaction conditions was further extended to alkyl- and alkenyltrifluoroborates (Table 4). We were pleased to find that the desired alcohol was obtained in excellent yields from primary, secondary and benzylic alkyltrifluoroborates (entries 1 to 8). Primary alcohols containing halogens or ester groups were generated without affecting these incorporated functional groups. Alkenyltrifluoroborates were converted into the corresponding aldehydes, also in excellent yields (entries 9 to 11). However, under the reaction conditions, minor amounts of the hydrated aldehyde were observed (entries 9 and 10). Oxidation of both alkyltrifluoroborates and alkenyltrifluoroborates was complete in only 2 minutes.
TABLE 4.
Hydroxylation of Potassium Alkyl- and Alkenyltrifluoroboratesa
 | ||||
|---|---|---|---|---|
| entry | substrate | product | isolated yield (%) | |
| 1 |
|
|
4a | 99 |
| 2 |
|
|
4b | 99 |
| 3 |
|
|
4c | 98 |
| 4 |
|
|
4d | 95 |
| 5 |
|
|
4e | 96 |
| 6 |
|
|
4f | 98 |
| 7 |
|
|
4g | 99 |
| 8 |
|
|
4h | 99 |
| 9 |
|
|
4i | 99 |
| 10 |
|
|
4j | 99 |
| 11 |
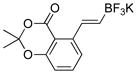
|
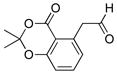
|
4k | 99 |
All reactions were carried out using 1 mmol of aryltrifluoroborate and Oxone (5 mL, 0.2 M in H2O) in 5 mL of acetone for 2 min at rt.
To investigate the stereochemical integrity of the process, the enantiomerically enriched β-trifluoroboratoamide was prepared from the corresponding α,β-unsaturated amide and bis(pinacolato)diboron in a copper-catalyzed process using (R)-(S)-Josiphos as the chiral ligand, according to the procedures previously reported by Yun et al. (Scheme 3).16 With the enantioeriched organotrifluoroborate in hand, this material was subjected to the oxidation conditions with Oxone®. We were pleased to observe that the transformation transpired with complete retention of configuration. The desired alcohol was obtained in 97% yield in only 2 minutes, in a 94 : 6 (R : S) ratio.
SCHEME 3.

In summary, we have successfully developed a general, rapid and efficient method for the oxidation of aryl-, heteroaryl-, alkyl-, and alkenyltrifluoroborates. The reactions were complete in only 2 to 5 minutes at room temperature, affording the desired products in virtually quantitative yield. The use of Oxone® as the oxidant makes the process both economical and environmentally sound. Numerous attendant functional groups were tolerated in this process, and no chromatography was required – a simple aqueous workup followed at most by filtration through a plug of silica gel afforded pure material. The oxidation of enantiomerically enriched secondary alkyltrifluoroborates affords the desired alcohols with complete retention of configuration.
Experimental Section
General Experimental Procedure for the Preparation of Phenols (55 mmol scale). Preparation of Phenol (1a).4
To a 50 mL round bottom flask containing a mixture of potassium phenyltrifluoroborate (10.12 g, 55.0 mmol) and acetone (275 mL, 0.2 M) was added Oxone® (275 mL of a 0.2 M solution in H2O, 1 equiv) in one portion. The reaction was stirred at rt until 11B NMR indicated completion of the reaction (~2 min). To the crude mixture was added H2O (30 mL) and aqueous HCl (0.1 M, 20 mL), and the aqueous layer was extracted with CH2Cl2 (3 x 50 mL). The combined organic layers were dried (Na2SO4), filtered, concentrated, and dried in vacuo. The crude extract was filtered through a small plug of silica topped with charcoal, eluting with CH2Cl2 to afford the desired pure product in 96% yield (5.0 g, 53 mmol) as a light yellow solid, mp 41 – 43 °C (lit. 40 – 42 °C). 1H NMR (500 MHz, CDCl3) δ 7.24 (t, J = 7.5 Hz, 2H), 6.93 (t, J = 7.5 Hz, 1H), 6.83 (d, J = 8.5 Hz, 2H), 4.88 (brs, 1H). 13C NMR (125.8 MHz, CDCl3) δ 155.4, 129.7, 120.8, 115.3.
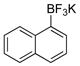
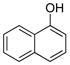
General Experimental Procedure for the Preparation of Phenols (1 mmol scale). Preparation of Naphthalen-1-ol (1b).4
To a 50 mL round bottom flask containing a mixture of potassium naphthalen-1-yltrifluoroborate (0.23 g, 1.0 mmol) and acetone (5 mL, 0.2 M) was added Oxone® (5 mL of a 0.2 M solution in H2O, 1 equiv) in one portion. The reaction was stirred at rt until 11B NMR indicated completion of the reaction (~2 min). To the crude mixture was added H2O (5 mL) and aqueous HCl (0.1 M, 3 mL), and the aqueous layer was extracted with CH2Cl2 (3 x 15 mL). The combined organic layers were dried (Na2SO4), filtered, concentrated, and dried in vacuo. The crude extract was filtered through a small plug of silica topped with charcoal, eluting with CH2Cl2 to afford the desired pure product in 99% yield (0.14 g, 0.99 mmol) as a white solid, mp 91 – 93 °C (lit.3a 92 – 94 °C). 1H NMR (500 MHz, CDCl3) δ 8.17 (m, 1H), 7.81 (m, 1H), 7.50–7.48 (m, 2H), 7.44 (d, J = 8.5 Hz, 1H), 7.30 (t, J = 7.5 Hz, 1H), 6.81 (d, J = 7.5 Hz, 1H), 5.21 (brs, 1H). 13C NMR (125.8 MHz, CDCl3) δ 151.3, 134.8, 127.7, 126.4, 125.8, 125.2, 124.3, 121.5, 120.7, 108.6.
Naphthalen-2-ol (1c).4
The general procedure was employed using potassium naphthalen-2-yltrifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 97% yield (0.14 g, 0.97 mmol) as a white solid, mp 121–123 °C (lit. 122–124 °C). 1H NMR (500 MHz, CDCl3) δ 7.75 (t, J = 8.9 Hz, 2H), 7.67 (d, J = 8.2 Hz, 1H), 7.42 (m, 1H), 7.32 (m, 1H), 7.13 (m, 1H), 7.08 (m, 1H), 5.01 (brs, 1H). 13C NMR (125.8 MHz, CDCl3) δ 134.4, 134.3, 133.1, 130.9, 128.8, 127.9, 126.9, 126.6, 126.0
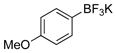
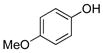
4-Methoxyphenol (1d).4
The general procedure was employed using potassium 4-methoxyphenyltrifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 98% yield (0.12 g, 0.98 mmol) as a white solid, mp 57–58 °C (lit 57–58 °C) . 1H NMR (500 MHz, CDCl3) δ 6.79 – 6.75 (m, 4H), 5.31 (s, 1H), 3.76 (s, 3H). 13C NMR (125.8 MHz, DMSO-d6) δ 161.6, 137.6, 136.0, 114.0, 55.8.
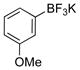
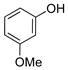
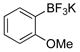
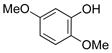
3-Methoxyphenol (1e).4
The general procedure was employed using potassium 3-methoxyphenyltrifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 93% yield (0.11 g, 0.93 mmol) as a light yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.12 (m, 1H), 6.50 (m, 1H), 6.44 – 6.41 (m, 2H), 5.62 (brs, 1H), 3.75 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 160.8, 156.6, 130.2, 107.9, 106.5, 101.6, 55.3.
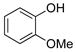
2-Methoxyphenol (1f).4
The general procedure was employed using potassium 2-methoxyphenyltrifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 97% yield (0.12 g, 0.97 mmol) as a light yellow oil. 1H NMR (500 MHz, CDCl3) δ 6.93 (m, 1H), 6.88 – 6.84 (m, 3H), 5.61 (brs, 1H), 3.89 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 146.6, 145.7, 121.5, 120.1, 114.5, 110.7, 55.9.
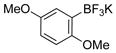
2,4-Dimethoxyphenol (1g).17
The general procedure was employed using potassium 2,4-dimethoxyphenyltrifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 95% yield (0.15 g, 0.95 mmol) as a light yellow oil. 1H NMR (500 MHz, CDCl3) δ 6.75 (d, J = 9.0 Hz, 1H), 6.56 (d, J = 3.0 Hz, 1H), 6.37 (m, 1H), 5.78 (brs, 1H), 3.81 (s, 3H), 3.73 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 154.5, 146.4, 141.0, 111.5, 104.5, 101.8, 56.5, 55.6.
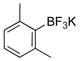
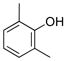
2,6-Dimethylphenol (1h).4
The general procedure was employed using potassium 2,6-dimethyltrifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 94% yield (0.11 g, 0.94 mmol) as a white solid, mp 43–45 °C (lit. 42–43 °C). 1H NMR (500 MHz, CDCl3) δ 6.96 (d, J = 7.5 Hz, 2H), 6.75 (t, J = 7.5 Hz, 1H), 4.61 (brs, 1H), 2.23 (s, 6H). 13C NMR (125.8 MHz, CDCl3) δ 152.1, 128.6, 123.0, 120.2, 15.8.
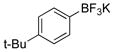
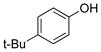
4-tert-Butylphenol (1i).3d
The general procedure was employed using potassium 4-tert-butylphenyltrifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 99% yield (0.15 g, 0.99 mmol) as a white solid, mp 97–99 °C (lit. 96–99 °C). 1H NMR (500 MHz, CDCl3) δ 7.25 (d, J = 6.5 Hz, 2H), 7.17 (d, J = 6.5 Hz, 2H), 4.66 (brs, 1H), 1.29 (s, 9H). 13C NMR (125.8 MHz, CDCl3) δ 153.1, 143.6, 126.4, 114.7, 34.1, 31.5.
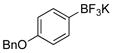
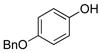
4-(Benzyloxy)phenol (1j).18
The general procedure was employed using potassium 4-(benzyloxy)phenyltrifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 97% yield (0.19 g, 0.97 mmol) as a light yellow solid, mp 117–121 °C (lit. 118 – 122 °C). 1H NMR (500 MHz, CDCl3) δ 7.42–7.36 (m, 4H), 7.32 (m, 1H), 6.85 (d, J = 9 Hz, 2H), 6.75 (d, J = 9 Hz, 2H) 5.00 (s, 2H), 4.46 (brs, 1H). 13C NMR (125.8 MHz, CDCl3) δ 149.6, 137.2, 128.5, 127.9, 127.5, 116.1, 116.0, 70.8.
(Z)-5-(4-Hydroxyphenyl)pent-4-enenitrile (1k)
The general procedure was employed using potassium (Z)-4-(4-cyanobut-1-enyl)phenyltrifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 98% yield (0.17 g, 0.98 mmol) as a white solid. mp 67–69 °C. 1H NMR (500 MHz, CDCl3) δ 7.12 (d, J = 8.5 Hz, 2H), 6.81 (d, J = 8.0 Hz, 2H) 6.52 (d, J = 11.5 Hz, 1H), 5.55 (m, 1H), 5.00 (brs, 1H), 2.69 – 2.65 (m, 2H), 2.44 (t, J = 7.0 Hz, 2H). 13C NMR (125.8 MHz, CDCl3) δ 154.8, 131.6, 130.1, 129.2, 126.0, 119.2, 115.2, 24.4, 17.6. IR (neat) 3325, 2257, 1608, 1514, 1228, 836 cm−1. HRMS (ESI) m/z calcd. for C11H11NONa (M+Na)+ 196.0738, found 196.0741.
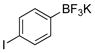
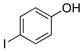
4-Iodophenol (2a).19
The general procedure was employed using potassium 4-iodophenyltrifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 85% yield (0.19 g, 0.85 mmol) as a light yellow solid mp 90–93 °C (lit. 92–95 °C). 1H NMR (500 MHz, CDCl3) δ 7.50 (d, J = 9.0 Hz, 2H), 6.61 (d, J = 9.0 Hz, 2H), 5.00 (brs, 1H). 13C NMR (125.8 MHz, CDCl3) δ 155.2, 138.4, 117.8, 82.8.
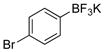
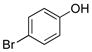
4-Bromophenol (2b).20
The general procedure was employed using potassium 4-bromophenyltrifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 98% yield (0.17 g, 0.98 mmol) as a light yellow solid mp 55–58 °C (lit. 54–64 °C). 1H NMR (500 MHz, CDCl3) δ 7.31 (d, J = 8.5 Hz, 2H), 6.70 (d, J = 9.0 Hz, 2H), 5.64 (brs, 1H). 13C NMR (125.8 MHz, CDCl3) δ 154.4, 132.5, 117.2, 112.9.
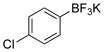
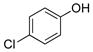
4-Chlorophenol (2c).4
The general procedure was employed using potassium 4-chlorophenyltrifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 99% yield (0.13 g, 0.99 mmol) as a white solid, mp 43–46 °C (lit. 44–45 °C) 1H NMR (500 MHz, CDCl3) δ 7.18 (d, J = 9.0 Hz, 2H), 6.75 (d, J = 8.5 Hz, 2H), 5.52 (brs, 1H). 13C NMR (125.8 MHz, CDCl3) δ 153.9, 129.5, 125.7, 116.7.
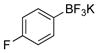
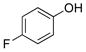
4-Fluorophenol (2d).4
The general procedure was employed using potassium 4-fluorophenyltrifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 99% yield (0.11 g, 0.99 mmol) as a white solid, mp 45–47 °C (lit. 46–47 °C). 1H NMR (500 MHz, CDCl3) δ 6.95 – 6.90 (m, 2H), 6.79 – 6.74 (m, 2H), 4.88 (brs, 1H). 13C NMR (125.8 MHz, CDCl3) δ 157.2 (d, J = 235.9 Hz), 151.4, 116.2 (d, J = 10.0 Hz, 2C), 116.0 (d, J = 22.9, 2C). 19F NMR (470.8 MHz, CDCl3) δ −124.3.
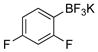
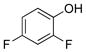
2,4-Difluorophenol (2e).5f
The general procedure was employed using potassium 2,4-difluorophenyltrifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 97% yield (0.13 g, 0.97 mmol) as a light yellow oil. 1H NMR (500 MHz, CDCl3) δ 6.95 (m, 1H), 6.85 (m, 1H), 6.76 (m, 1H), 5.75 (brs, 1H). 13C NMR (125.8 MHz, CDCl3) δ 155.9 (dd, J = 240.9, 10.3 Hz), 150.4 (dd, J = 240.1, 12.2 Hz), 139.9 (dd, J = 14.1, 3.5 Hz), 117.3 (d, J = 6.4 Hz), 111.7 – 110.6 (m), 104.5 – 103.3 (m).19F NMR (470.8 MHz, CDCl3) δ −121.0, −135.7.
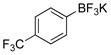
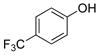
4-(Trifluoromethyl)phenol (2f).21
The general procedure was employed using potassium 4-(trifluoromethyl)phenyltrifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 98% yield (0.16 g, 0.98 mmol) as a light yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.50 (d, J = 8.5 Hz, 2H), 6.90 (d, J = 9 Hz, 2H) 5.45 (brs, 1H). 13C NMR (125.8 MHz, CDCl3) δ 158.1, 127.2 (q, J = 3.8 Hz), 125.4, 123.7 – 122.7 (m), 115.4. 19F NMR (470.8 MHz, CDCl3) δ −61.5.
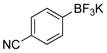
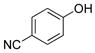
4-Hydroxybenzonitrile (2g).5f
The general procedure was employed using potassium 4-cyanophenyltrifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 98% yield (0.12 g, 0.98 mmol) as a light yellow solid mp 107–109 °C (lit. 108–109 °C). 1H NMR (500 MHz, CDCl3) δ 7.57 (d, J = 9.0 Hz, 2H), 6.94 (d, J = 8.5 Hz, 2H), (brs, 1H). 13C NMR (125.8 MHz, CDCl3) δ 160.0, 134.3, 119.2, 116.4, 103.3.
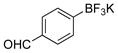
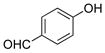
4-Hydroxybenzaldehyde (2h).4
The general procedure was employed using potassium 4-formylphenyltrifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 99% yield (0.12 g, 0.99 mmol) as a white solid, mp 114–117 °C (lit. 114–116 °C). 1H NMR (500 MHz, CDCl3) δ 9.87 (s, 1H), 7.82 (d, J = 8.5 Hz, 2H), 6.97 (d, J = 9.0 Hz, 2H), 6.73 (brs, 1H). 13C NMR (125.8 MHz, CDCl3) δ 191.1, 161.4, 132.4, 129.9, 115.9.
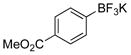
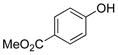
Methyl 4-Hydroxybenzoate (2i).22
The general procedure was employed using potassium 4-(methoxycarbonyl)phenyltrifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 99% yield (0.15 g, 0.99 mmol) as a white solid, mp 121–123 °C (lit. 127–129 °C). 1H NMR (500 MHz, acetone-d6) δ 9.12 (brs, 1H), 7.89 (d, J = 9.0 Hz, 2H), 6.92 (d, J = 9.0 Hz, 2H), 3.82 (s, 3H). 13C NMR (125.8 MHz, acetone-d6) δ 167.8, 163.4, 133.2, 123.3, 116.8, 52.6.
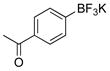
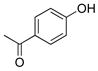
1-(4-Hydroxyphenyl)ethanone (2j).20
The general procedure was employed using potassium 4-acetylphenyltrifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 99% yield (0.13 g, 0.99 mmol) as a white solid , mp 105–107 °C (lit. 108–109 °C). 1H NMR (500 MHz, CDCl3) δ 8.24 (brs, 1H), 7.92 (d, J = 9.0 Hz, 2H), 6.97 (d, J = 9.0 Hz, 2H), 2.59 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 199.0, 161.6, 131.3, 129.4, 115.6, 26.3.
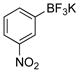
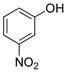
3-Nitrophenol (2k).4
The general procedure was employed using potassium 3-nitrophenyltrifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 98% yield (0.14 g, 0.98 mmol) as a light yellow solid mp 96–99 °C (lit. 98–99 °C). 1H NMR (500 MHz, CDCl3) δ 7.82 (m, 1H), 7.71 (t, J = 2.0 Hz, 1H), 7.41 (t, J = 8.0 Hz, 1H), 7.19 (m, 1H), 5.61 (brs, 1H). 13C NMR (125.8 MHz, CDCl3) δ 156.3, 130.3, 122.0, 115.9, 110.6.
Dibenzo[b,d]furan-4-ol (3a)
The general procedure was employed using potassium dibenzo[b,d]furan-4-yltrifluoroborate, and the reaction was complete in 5 min. The desired pure product was obtained in 97% yield (0.18 g, 0.97 mmol) as a white solid, mp 98–100 °C. 1H NMR (500 MHz, CDCl3) δ 7.93 (d, J = 7.7 Hz, 1H), 7.57 (d, J = 8.3 Hz, 1H), 7.51 (d, J = 7.7 Hz, 1H), 7.46 (t, J = 7.8 Hz, 1H), 7.35 (t, J = 7.5 Hz, 1H), 7.23 (m, 1H), 7.02 (d, J = 7.9 Hz, 1H), 5.39 (s, 1H). 13C NMR (125.8 MHz, CDCl3) δ 156.1, 144.0, 141.1, 127.3, 125.7, 124.6, 123.7, 123.0, 121.0, 113.6, 112.8, 111.8. IR (neat) 3277, 1603, 1437, 1249, 744 cm−1. HRMS (ESI) m/z calcd. for C12H7O2 (M-H)− 183.0446, found 183.0454.
Dibenzo[b,d]thiophen-4-ol (3b)
The general procedure was employed using potassium dibenzo[b,d]thiophen-4-yltrifluoroborate, and the reaction was complete in 5 min. The desired pure product was obtained in 97% yield (0.18 g, 0.97 mmol) as a white solid. mp 158–160 °C. 1H NMR (500 MHz, CDCl3) δ 8.13 (m, 1H), 7.89 (m, 1H), 7.78 (d, J = 7.9 Hz, 1H), 7.50 – 7.42 (m, 2H), 7.34 (t, J = 7.8 Hz, 1H), 6.88 (d, J = 7.7 Hz, 1H), 5.35 (s, 1H). 13C NMR (125.8 MHz, CDCl3) δ 150.5, 139.6, 137.9, 135.9, 126.9, 126.5, 125.7, 124.4, 123.1, 122.0, 114.5, 111.7. IR (neat) 3230, 1569, 1444, 1253, 744 cm−1. HRMS (ESI) m/z calcd. for C12H7OS (M-H)− 199.0218, found 199.0222.
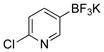
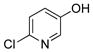
6-Chloropyridin-3-ol (3c)
The general procedure was employed using potassium 6-chloropyridin-3-yltrifluoroborate, and the reaction was complete in 5 min. The desired pure product was obtained in 91% yield (0.12 g, 0.97 mmol) as a white solid, mp 155–157 °C. 1H NMR (500 MHz, acetone-d6) δ 9.04 (brs, 1H), 7.99 (d, J = 2.9 Hz, 1H), 7.40 – 7.14 (m, 2H). 13C NMR (125.8 MHz, acetone-d6) δ 154.9, 142.5, 138.9, 127.6, 126.1. IR (neat) 3004, 1573, 1464, 1278, 1228, 826 cm−1. HRMS (ESI) m/z calcd. for C5H3NOCl (M-H)− 127.9903, found 127.9902.
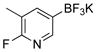
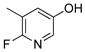
6-Fluoro-5-methylpyridin-3-ol (3d)
The general procedure was employed using potassium 6-chloropyridin-3-yltrifluoroborate, and the reaction was complete in 5 min. The desired pure product was obtained in 94% yield (0.12 g, 0.94 mmol) as a white solid. mp 120–122 °C. 1H NMR (500 MHz, CDCl3) δ 7.69 (brs, 1H), 7.61 (s, 1H), 7.22 (m, 1H), 2.25 (s, 3H).13C NMR (125.8 MHz, CDCl3) δ 157.1, 155.2, 151.1, 129.8 (dd, J = 44.4, 9.5 Hz), 120.7 (d, J = 34.1 Hz), 14.4. 19F NMR (470.8 MHz, CDCl3) δ −83.9. IR (neat) 3048, 1471, 1232, 770 cm−1. HRMS (ESI) m/z calcd. for C6H7NOF (M+H)+ 128.0512, found 128.0510.
Benzo[b]thiophen-2(3H)-one (3e).23
The general procedure was employed using potassium benzothiophen-2-yltrifluoroborate, and the reaction was complete in 5 min. The desired pure product was obtained in 99% yield (0.15 g, 0.99 mmol) as a light yellow oil. 1H NMR (500 MHz, acetone-d6) δ 7.44 (m, 1H), 7.38 (d, J = 7.5 Hz, 1H), 7.33 (m, 1H), 7.25 (m, 1H), 4.09 (s, 2H). 13C NMR (125.8 MHz, acetone-d6) δ 203.7, 138.3, 134.7, 129.8, 127.7, 126.7, 124.5, 48.4.
Benzofuran-2(3H)-one (3f).24
The general procedure was employed using potassium benzofuran-2-yltrifluoroborate, and the reaction was complete in 5 min. The desired pure product was obtained in 97% yield (0.13 g, 0.97 mmol) as a light yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.32 – 7.24 (m, 2H), 7.13 (m, 1H), 7.08 (d, J = 8.1 Hz, 1H), 3.71 (s, 2H). 13C NMR (125.8 MHz, CDCl3) δ 174.0, 154.6, 128.8, 124.5, 123.9, 123.0, 110.6, 32.8.
5-Bromobenzo[b]thiophen-2(3H)-one (3g)
The general procedure was employed using potassium 5-bromobenzo[b]thiophen-2-yltrifluoroborate, and the reaction was complete in 5 min. The desired pure product was obtained in 94% yield (0.21 g, 0.94 mmol) as a light yellow solid. mp 117–120 °C. 1H NMR (500 MHz, CDCl3) δ 7.49 – 7.37 (m, 2H), 7.21 (d, J = 8.9 Hz, 1H), 3.97 (s, 2H). 13C NMR (125.8 MHz, CDCl3) δ 201.5, 136.1, 133.9, 131.4, 127.9, 124.3, 119.7, 47.1. FT- IR (neat) 1711, 1017, 819 cm−1. HRMS (ESI) m/z calcd. for C8H4OSBr (M-H)− 226.9166, found 226.9158.
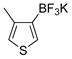
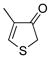
4-Methylthiophen-2(3H)-one (3h)
The general procedure was employed using potassium 4-methylthiophen-2-yltrifluoroborate, and the reaction was complete in 5 min. The desired pure product was obtained in 89% yield (0.10 g, 0.89 mmol) as a light yellow oil. 1H NMR (500 MHz, CDCl3) δ 6.09 (m, 1H), 3.97 (s, 2H), 2.21 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 199.7, 167.7, 129.0, 40.8, 18.9. FT- IR (neat) 2918, 1676, 1096, 653 cm−1. HRMS (ESI) m/z calcd. for C5H7OS (M+H)+ 115.0218, found 115.0215.
Furan-2(3H)-one (3i).25
The general procedure was employed using potassium thiophen-3-yltrifluoroborate, and the reaction was complete in 5 min. The desired pure product was obtained in 98% yield (0.10 g, 0.98 mmol) as a light yellow oil. 1H NMR (500 MHz, CDCl3) δ 6.77 (m, 1H), 5.55 (m, 1H), 3.14 (s, 2H). 13C NMR (125.8 MHz, CDCl3) δ 176.5, 143.8, 105.4, 32.3.
4-Hydroxybutyl Benzoate (4a).26
The general procedure was employed using potassium 4-(benzoyloxy)butyltrifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 99% yield (0.19 g, 0.99 mmol) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 8.04 – 8.02 (m, 2H), 7.53 (m, 1H), 7.44 – 7.40 (m, 2H), 4.35 (t, J = 6.5 Hz, 2H), 3.70 (t, J = 6.5 Hz, 2H), 2.60, (brs, 1H), 1.88 – 1.83 (m, 2H), 1.74 – 1.69 (m, 2H). 13C NMR (125.8 MHz, CDCl3) δ 166.7, 132.8, 130.2, 129.4, 128.3, 64.8, 62.0, 29.1, 25.1.
6-Hydroxyhexyl Benzoate (4b).27
The general procedure was employed using potassium 7-(benzoyloxy)hexyltrifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 99% yield (0.23 g, 0.99 mmol) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 8.05 – 8.03 (m, 2H), 7.53 (m, 1H), 7.44 – 7.41 (m, 2H), 4.31 (t, J = 6.5 Hz, 2H), 3.63 (t, J = 6.5 Hz, 2H), 2.38, (brs, 1H), 1.80 – 1.75 (m, 2H), 1.62 – 1.56 (m, 2H), 1.48 – 1.43 (m, 4H). 13C NMR (125.8 MHz, CDCl3) δ 166.6, 132.7, 130.2, 129.4, 128.2, 64.8, 62.4, 32.4, 28.5, 25.7, 25.3.
3-Bromopropan-1-ol (4c).28
The general procedure was employed using potassium 3-bromopropyltrifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 98% yield (0.14 g, 0.98 mmol) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 3.78 (t, J = 6.0 Hz, 2H), 3.54 (t, J = 6.5 Hz, 2H), 2.69 (brs, 1H), 2.11 – 2.07 (m, 2H). 13C NMR (125.8 MHz, CDCl3) δ 60.1, 34.9, 30.3.
Cyclopentanol (4d).29
The general procedure was employed using potassium cyclopentyltrifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 99% yield (0.09 g, 0.99 mmol) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 4.33 (m, 1H), 1.78 – 1.76 (m, 4H), 1.59 (brs, 1H), 1.58 – 1.56 (m, 4H). 13C NMR (125.8 MHz, CDCl3) δ 74.0, 35.5, 23.2.
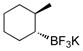
2-Methylcyclohexanol (4e).30
The general procedure was employed using potassium 2-methylcyclohexyltrifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 96% yield (0.11 g, 0.96 mmol) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 3.11 (m, 1H), 1.95 – 1.93 (m, 1H), 1.75 – 1.59 (m, 4H), 1.36 – 1.18 (m, 4H), 1.01 (d, J = 6.5 Hz, 3H). 13C NMR (125.8 MHz, CDCl3) δ 76.5, 40.2, 35.5, 33.6, 25.7, 25.2, 18.5.
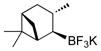
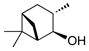
3,6,6-Trimethylbicyclo[3.1.1]heptan-2-ol (4f).31
The general procedure was employed using potassium 3,6,6-trimethylbicyclo[3.1.1]heptan-2-yltrifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 98% yield (0.15 g, 0.98 mmol) as a white solid, mp 49 – 52 ºC (lit. 51–53 ºC). 1H NMR (500 MHz, CDCl3) δ 4.05 (m, 1H), 2.64 (brs, 1H), 2.50 (m, 1H), 2.37 (m, 1H), 2.00 – 1.90 (m, 2H), 1.79 (m, 1H), 1.73 (m, 1H), 1.21 (s, 3H), 1.13 (d, J = 7.4 Hz, 3H), 1.06 (d, J = 9.7 Hz, 1H), 0.92 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 71.3, 47.7, 47.4, 41.67, 38.8, 38.1, 34.2, 27.6, 23.6, 20.6. IR (neat) 3260, 2905, 1469, 1043, 1006, 923 cm−1. HRMS (ESI) m/z calcd. for C10H17 (M-OH)+ 137.1330, found 137.1326.
2-Phenylethanol (4g).27
The general procedure was employed using potassium phenethyltrifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 99% yield (0.12 g, 0.99 mmol) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.30 – 7.27 (m, 2H), 7.21 – 7.18 (m, 3H), 3.78 (t, J = 6.5 Hz, 2H), 2.81 (t, J = 6.5 Hz, 2H), 2.20 (brs, 1H). 13C NMR (125.8 MHz, CDCl3) δ 138.6, 129.0, 128.5, 126.4, 63.5, 39.1.
Phenylmethanol (4h).32
The general procedure was employed using potassium benzyltrifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 99% yield (0.11 g, 0.99 mmol) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.32 – 7.25 (m, 5H), 4.56 (s, 2H), 2.90 (brs, 1H). 13C NMR (125.8 MHz, CDCl3) δ 140.8, 128.4, 127.4, 126.9, 64.9.
11-Hydroxydodecanal (4i).33
The general procedure was employed using potassium (E)-11-hydroxydodec-1-en-1-yltrifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 99% yield (0.2 g, 0.99 mmol) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 9.76 (t, J = 1.9 Hz, 1H), 3.79 (m, 1H), 2.44 – 2.40 (m, 2H), 1.69 – 1.50 (m, 2H), 1.41 (d, J = 14.2 Hz, 2H), 1.29 (s, 12H), 1.19 (d, J = 6.2 Hz, 3H). 13C NMR (125.8 MHz, CDCl3) δ 68.2, 43.9, 39.3, 29.6, 29.3, 29.1, 25.7, 22.1.
Methyl 6-Oxohexanoate (4j).34
The general procedure was employed using potassium (E)-6-methoxy-6-oxohex-1-en-1-yltrifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 99% yield (0.14 g, 0.99 mmol) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 9.77 (t, J = 1.2 Hz, 1H), 3.67 (s, 3H), 2.55 – 2.25 (m, 4H), 1.67 (m, 4H).13C NMR (125.8 MHz, CDCl3) δ 202.0, 174.0, 173.7, 51.5, 43.5, 33.7, 24.3, 21.5.
2-(2,2-Dimethyl-4-oxo-4H-benzo[d][1,3]dioxin-5-yl)acetaldehyde (4k)
The general procedure was employed using potassium (E)-(2-(2,2-dimethyl-4-oxo-4H-benzo[d][1,3]dioxin-5-yl)vinyl)trifluoroborate, and the reaction was complete in 2 min. The desired pure product was obtained in 99% yield (0.22 g, 0.99 mmol) as a light yellow solid, mp 55 – 57 °C. 1H NMR (500 MHz, CDCl3) δ 9.84 (s, 1H), 7.50 (t, J = 8 Hz, 1H), 6.96 (d, J = 8.3 Hz, 1H), 6.91 (d, J = 7.5 Hz, 1H), 4.22 (s, 2H), 1.75 (s, 6H). 13C NMR (125.8 MHz, CDCl3) δ 198.1, 160.8, 157.1, 137.0, 135.6, 126.4, 117.0, 112.6, 105.7, 49.0, 25.6. IR (neat) 1722, 1292, 1052 cm−1. HRMS (ESI) m/z calcd. for C12H12O4Na (M+Na)+ 243.0633, found 243.0633.
(R)-3-hydroxy-N-(4-methoxyphenyl)butanamide (5).35
The general procedure was employed using 0.1 mmol of potassium (R)-(4-((4-methoxyphenyl)amino)-4-oxobutan-2-yl)trifluoroborate (R : S) (97 : 3), and the reaction was complete in 2 min. The desired pure product was obtained in 97% yield (0.020 g, 0.097 mmol) as a white solid, mp 136–138 °C (lit. 135 °C). 1H NMR (500 MHz, CDCl3) δ 1H NMR (500 MHz, CDCl3) δ 7.75 (brs, 1H), 7.39 (d, J = 9.0 Hz, 2H), 6.85 (d, J = 9.0 Hz, 2H), 4.28 (m, 1H), 3.78 (s, 3H), 3.43 (d, J = 2.9 Hz, 1H), 2.52 – 2.41 (m, 2H), 1.27 (d, J = 6.5 Hz, 3H). 13C NMR (125.8 MHz, CDCl3) δ 170.3, 156.5, 130.5, 122.0, 114.1, 64.9, 55.5, 44.8, 22.9.
Supplementary Material
Acknowledgments
This work was generously supported by the NIGMS (R01 GM086209, GM035249), and a Conselho Nacional de Desenvolvimento Cientítico e Tecnológico (CNPq) Graduate Research Fellowship to L.N.C. We also acknowledge Aldrich, BoroChem, and Frontier Scientific for their donation of the boronic acids and Dr. Deidre Sandrock for providing the enantiomerically enriched organotrifluoroborate sample. Dr. Rakesh Kohli (University of Pennsylvania) is acknowledged for obtaining the HRMS data.
Footnotes
Supporting Information Available: Copies of 1H, 13C, and 19F spectra for all compounds prepared by the method described. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Rappoport Z. The Chemistry of Phenols. Wiley-VCH; Weinheim: 2003. [Google Scholar]; (b) Tyman JHP. Synthetic and Natural Phenols. Elsevier; New York: 1996. [Google Scholar]
- 2.(a) Fyfe CA. In: The Chemistry of the Hydroxyl Group. Patai S, editor. Vol. 1. Wiley-Interscience; New York: 1971. [Google Scholar]; (b) Hoarau C, Pettus TRR. Synlett. 2003:127. doi: 10.1055/s-2003-36234. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hanson P, Jones JR, Taylor AB, Walton PH, Timms AW. J Chem Soc,Perkin Trans. 2002;2:1135. [Google Scholar]; (d) George T, Mabon R, Sweeney G, Sweeney JB, Tavassoli AJ. Chem Soc, Perkin Trans. 2000;1:2529. and references therein. [Google Scholar]
- 3.(a) Schulz T, Torborg C, Schaffner B, Huang J, Zapf A, Kadyrov R, Borner A, Beller M. Angew Chem Int Ed. 2009;48:918. doi: 10.1002/anie.200804898. [DOI] [PubMed] [Google Scholar]; (b) Tlili A, Xia N, Monnier F, Taillefer M. Angew Chem Int Ed. 2009;48:8725. doi: 10.1002/anie.200903639. [DOI] [PubMed] [Google Scholar]; (c) Zhao D, Wu N, Zhang S, Xi P, Su X, Lan J, You J. Angew Chem Int Ed. 2009;48:8729. doi: 10.1002/anie.200903923. [DOI] [PubMed] [Google Scholar]; (d) Anderson KW, Ikawa T, Tundel RE, Buchwald SL. J Am Chem Soc. 2006;128:10694. doi: 10.1021/ja0639719. [DOI] [PubMed] [Google Scholar]
- 4.Xu J, Wang X, Shao C, Su D, Cheng G, Hu Y. Org Lett. 2010;12:1964. doi: 10.1021/ol1003884. [DOI] [PubMed] [Google Scholar]
- 5.(a) Prakash GKS, Chacko S, Panja C, Thomas TE, Gurung L, Rasul G, Mathew T, Olah GA. Adv Synth Catal. 2009;351:1567. [Google Scholar]; (b) Kianmehr E, Yahyaee M, Tabatabai K. Tetrahedron Lett. 2007;48:2713. [Google Scholar]; (c) Clay JM, Vedejs E. J Am Chem Soc. 2005;127 doi: 10.1021/ja043743j. [DOI] [PMC free article] [PubMed]; (d) Maleczka RE, Shi F, Holmes D, Smith MR., III J Am Chem Soc. 2003;125:7792. doi: 10.1021/ja0349857. [DOI] [PubMed] [Google Scholar]; (e) Benjamin R, Travis BR, Ciaramitaro BP, Borhan B. Eur J Org Chem. 2002:3429. [Google Scholar]; (f) Simon J, Salzbrunn S, Prakash GKS, Petasis NA, Olah GA. J Org Chem. 2001;66:633. doi: 10.1021/jo0015873. [DOI] [PubMed] [Google Scholar]; (g) Webb KS, Levy D. Tetrahedron Lett. 1995;36:5117. [Google Scholar]
- 6.For reviews, see: Darses S, Genêt JP. Chem Rev. 2008;108:288. doi: 10.1021/cr0509758.Doucet H. Eur J Org Chem. 2008:2013.Molander GA, Ellis NM. Acc Chem Res. 2007;40:275. doi: 10.1021/ar050199q.Stefani HA, Cella R, Vieira AS. Tetrahedron. 2007;63:3623.Molander GA, Figueroa R. Aldrichim Acta. 2005;38:49.Darses S, Genêt JP. Eur J Org Chem. 2003:4313.
- 7.For selected examples, see: Molander GA, Raushel J, Ellis NM. J Org Chem. 2010;75:4304. doi: 10.1021/jo1004058.Molander GA, Canturk B, Kennedy LE. J Org Chem. 2009;74:973. doi: 10.1021/jo802590b.Molander GA, Febo-Ayala W, Ortega-Guerra M. J Org Chem. 2008;73:6000. doi: 10.1021/jo800760f.Molander GA, Cooper DJ. J Org Chem. 2008;73:3885. doi: 10.1021/jo800383e.Molander GA, Ellis NM. J Org Chem. 2006;71:7491. doi: 10.1021/jo061324u.Molander GA, Felix LA. J Org Chem. 2005;70:3950. doi: 10.1021/jo050286w.
- 8.For selected examples see: Molander GA, Cavalcanti LN, Canturk B, Pan PS, Kennedy LE. J Org Chem. 2009;74:7364. doi: 10.1021/jo901441u.Kabalka GW, Mereddy AR. Tetrahedron Lett. 2004;45:343.Kabalka GW, Mereddy AR. Organometallics. 2004;23:4519.
- 9.Gillis EP, Burke MD. J Am Chem Soc. 2007;129:6716. doi: 10.1021/ja0716204. [DOI] [PubMed] [Google Scholar]
- 10.(a) Ochiai M, Miyamoto K, Yokota Y, Suefuji T, Shiro M. Angew Chem Int Ed. 2005;44:75. doi: 10.1002/anie.200461375. [DOI] [PubMed] [Google Scholar]; (b) Sorin G, Mallorquin RM, Contie Y, Baralle A, Malacria M, Goddard JP, Fensterbank L. Angew Chem, Int Ed. 2010;49:8721. doi: 10.1002/anie.201004513. [DOI] [PubMed] [Google Scholar]
- 11.Catalog price: U$ 7.00/mol
- 12.(a) Dreher SD, Lim SE, Sandrock DL, Molander GA. J Org Chem. 2009;74:3626. doi: 10.1021/jo900152n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gerbino DC, Mandolesi SD, Schmalz HG, Podesta JC. Eur J Org Chem. 2009:3964. [Google Scholar]; (c) Molander GA, Sandrock DL. J Am Chem Soc. 2008;130:15792. doi: 10.1021/ja807076d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Dreher SD, Dormer PG, Sandrock DL, Molander GA. J Am Chem Soc. 2008;130:9257. doi: 10.1021/ja8031423. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Molander GA, Petrillo DE. J Am Chem Soc. 2006;128:9634. doi: 10.1021/ja062974i. [DOI] [PubMed] [Google Scholar]; (f) Molander GA, Figueroa R. J Org Chem. 2006;71:6135. doi: 10.1021/jo060863w. [DOI] [PubMed] [Google Scholar]; (g) Smoum R, Rubinstein A, Srebnik M. Org Biomol Chem. 2005;3:941. doi: 10.1039/b415957h. [DOI] [PubMed] [Google Scholar]; (h) Yuen AKL, Hutton CA. Tetrahedron Lett. 2005;46:7899. [Google Scholar]; (i) Navarre L, Darses S, Genet JP. Eur J Org Chem. 2004:69. [Google Scholar]; (j) Molander GA, Biolatto B. J Org Chem. 2003;68:4302. doi: 10.1021/jo0342368. [DOI] [PubMed] [Google Scholar]; (k) Molander GA, Biolatto B. Org Lett. 2002;4:1867. doi: 10.1021/ol025845p. [DOI] [PubMed] [Google Scholar]; (l) Molander GA, Ito T. Org Lett. 2001;3:393. doi: 10.1021/ol006896u. [DOI] [PubMed] [Google Scholar]; (m) Vedejs E, Chapman RW, Fields SC, Lin S, Schrimpf MR. J Org Chem. 1995;60:3020. [Google Scholar]
- 13.For selected examples see: Zhu W, Ford WT. J Org Chem. 1991;56:7022.Adam W, Hadjiarapoglou L, Smerz A. Chem Ber. 1991;124:227.Corey PF, Ward FE. J Org Chem. 1986;51:1925.Bloch R, Abecassis J, Hassan D. J Org Chem. 1985;50:1544.Jeyaraman R, Murray RW. J Am Chem Soc. 1984;106:2462. doi: 10.1021/ja00269a069.Curci R, Fiorentino M, Troisi L, Edwards JO, Pater RH. J Org Chem. 1980;45:4758.
- 14.(a) Elguero J, Marzin C, Katritzky AR, Linda P. The Tautomerism of Heterocyclic Compounds. Academic Press; New York: 1976. [Google Scholar]; (b) Capon B, Kwok FC. Tetrahedron Lett. 1986;27:3275. [Google Scholar]
- 15.For selected examples see: Greehalgh RP. Synlett. 1992:235.Itahara T. Chem Lett. 1991;57:1591.Kettani AE, Bernadou J, Meunier B. J Org Chem. 1989;54:3213.Murray RW, Jeyaraman R. J Org Chem. 1985;50:2847.Evans TL, Grade MM. Synth Commun. 1986;16:1207.Trost BM, Curran DP. Tetrahedron Lett. 1981;22:1287.
- 16.Chea H, Sim HS, Yun J. Adv Synth Catal. 2009;351:855. [Google Scholar]
- 17.Gonzalez RR, Gambarotti C, Liguori L, Bjorsvik HR. J Org Chem. 2006;71:1703. doi: 10.1021/jo0522512. [DOI] [PubMed] [Google Scholar]
- 18.Ohkubo M, Mochizuki S, Sano T, Kawaguchi Y, Okamoto S. Org Lett. 2007;9:773. doi: 10.1021/ol062963u. [DOI] [PubMed] [Google Scholar]
- 19.Zhou CY, Li J, Peddibhotla S, Romo D. Org Lett. 2010;12:2104. doi: 10.1021/ol100587j. [DOI] [PubMed] [Google Scholar]
- 20.Magano J, Chen MH, Clark JD, Nussbaumer T. J Org Chem. 2006;71:7103. doi: 10.1021/jo0611059. [DOI] [PubMed] [Google Scholar]
- 21.Sergeev AG, Schulz T, Torborg C, Spannenberg A, Neumann H, Beller M. Angew Chem Int Ed. 2009;48:7595. doi: 10.1002/anie.200902148. [DOI] [PubMed] [Google Scholar]
- 22.Sivakumar S, Reddy MLP, Cowleyb AH, Vasudevanb KV. Dalton Trans. 2010;39:776. doi: 10.1039/b917256d. [DOI] [PubMed] [Google Scholar]
- 23.Chen S, Zhao X, Chen J, Chen J, Kuznetsova L, Wong SS, Ojima I. Bioconjugate Chem. 2010;21:979. doi: 10.1021/bc9005656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim MJ, Kim JJ, Won JE, Kang S-E, Park SE, Jung KJ, Lee SG, Yoon YJ. Bull Korean Chem Soc. 2008;29:2247. [Google Scholar]
- 25.Yanai H, Takahashi A, Taguchi T. Tetrahedron Lett. 2007;48:2993. [Google Scholar]
- 26.Ochiai M, Miyamoto K, Yokota Y, Suefuji T, Shiro M. Angew Chem Int Ed. 2005;44:75. doi: 10.1002/anie.200461375. [DOI] [PubMed] [Google Scholar]
- 27.Saburi H, Tanaka S, Kitamura M. Angew Chem Int Ed. 2005;44:1730. doi: 10.1002/anie.200462513. [DOI] [PubMed] [Google Scholar]
- 28.Shah STA, Singh S, Guiry PJ. J Org Chem. 2009;74:2179. doi: 10.1021/jo802494t. [DOI] [PubMed] [Google Scholar]
- 29.Abrahm RJ, Koniotou R, Sancassan F. J Chem Soc, Perkin Trans. 2002;2:2025. [Google Scholar]
- 30.Brown HC, Weissman SA, Perumal PT, Dhokte UP. J Org Chem. 1990;55:1217. [Google Scholar]
- 31.Brown HC, Gupta AK. J Organomet Chem. 1988;341:73. [Google Scholar]
- 32.Jagdale AR, Paraskar AS, Sudalai A. Synthesis. 2009:660. [Google Scholar]
- 33.Li X-Q, Zhang C. Synthesis. 2009:1163. [Google Scholar]
- 34.Hickmann V, Alcarazo M, Furstner A. J Am Chem Soc. 2010;132:11042. doi: 10.1021/ja104796a. [DOI] [PubMed] [Google Scholar]
- 35.Chea H, Sim HS, Yun J. Adv Synth Catal. 2009;351:855. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.