

Abstract
The mevalonate pathway accounts for conversion of acetyl-CoA to isopentenyl 5-diphosphate, the versatile precursor of polyisoprenoid metabolites and natural products. The pathway functions in most eukaryotes, archaea, and some eubacteria. Only recently has much of the functional and structural basis for this metabolism been reported. The biosynthetic acetoacetyl-CoA thiolase and HMG-CoA synthase reactions rely on key amino acids that are different but are situated in active sites that are similar throughout the family of initial condensation enzymes. Both bacterial and animal HMG-CoA reductases have been extensively studied and the contrasts between these proteins and their interactions with statin inhibitors defined. The conversion of mevalonic acid to isopentenyl 5-diphosphate involves three ATP-dependent phosphorylation reactions. While bacterial enzymes responsible for these three reactions share a common protein fold, animal enzymes differ in this respect as the recently reported structure of human phosphomevalonate kinase demonstrates. There are significant contrasts between observations on metabolite inhibition of mevalonate phosphorylation in bacteria and animals. The structural basis for these contrasts has also recently been reported. Alternatives to the phosphomevalonate kinase and mevalonate diphosphate decarboxylase reactions may exist in archaea. Thus, new details regarding isopentenyl diphosphate synthesis from acetyl-CoA continue to emerge.
Keywords: mevalonate pathway, isoprenoid biosynthesis, HMG-CoA, sterol biosynthesis
INTRODUCTION
The mevalonate (MVA1) pathway for biosynthesis of isoprenoids from acetate (Scheme 1) represents the initial steps in a series of enzymatic reactions [1] that have, for decades, been established to account for production of polyisoprenoids (e.g. dolichol) and sterols (e.g. lanosterol, ergosterol, cholesterol) in fungi, plant cytoplasm, animals, most other eukaryotes, archaea and some eubacteria.
Scheme 1.

Mevalonate (MVA) Pathway for Isopentenyl Diphosphate Biosynthesis.
In recent years, much attention has been generated by the elucidation of an alternate pathway (the deoxyxylulose phosphate, methylerithritol phosphate, or nonmevalonate pathway [2]) that is operative in many bacteria, plant chloroplasts, and some eukaryotic parasites. This pathway represents an alternative series of reactions for production of isopentenyl diphosphate.
While much progress continues to be made in work on the deoxyxylulose phosphate pathway, there had been relatively modest effort invested in the application of recombinant DNA methodology and structural investigation to the mevalonate pathway enzymes, with the possible exception of HMG-CoA reductase, the target of the family of statin inhibitors of cholesterol biosynthesis. These enzymes had been isolated and classical enzymological and biochemical characterization had been performed prior to the evolution of experimental tools that have expedited the understanding the molecular basis of enzyme function. These enzymes are significant in animals since several have been identified as potentially useful targets for modulation of polyisoprenoid and sterol biosynthesis. Also, defects of some of these enzymes have been implicated in human inherited disease. These include the cytosolic mevalonate kinase as well as mitochondrial isoforms of biosynthetic acetoacetyl-CoA thiolase and HMG-CoA synthase. The enzymes occur in several gram-positive bacteria, including some that are pathogenic to humans. Genetic work [3] has demonstrated that disruption of genes encoding various enzymes in the mevalonate pathway blocks proliferation of these pathogens. These observations suggest the merit of working toward a more detailed understanding of the mevalonate pathway enzymes.
Much of the work on identification and functional assignments of catalytically important active site residues, high resolution structure determinations, as well as the investigation of the basis for the inhibition or regulation of some of these enzymes has only appeared in recent years. This review will primarily focus on some of these more recent developments. While the characterization of several enzymes is still in progress, the contributions of these recent studies in addressing the molecular basis for isoprenoid biosynthesis by these enzymes are interpreted in the context of the earlier reports to provide an updated understanding of the mevalonate pathway. Reviews of selected enzymes and reactions in this pathway have also appeared [4, 5, 6].
ACETOACETYL-CoA THIOLASE
The thiolase catalyzed Claisen condensation and cleavage reactions [7, 8], whereby an acyl-CoA molecule is extended or shortened (respectively) by a two carbon acetyl module are widely observed in metabolism with the enzyme being found in a wide variety of prokaryotes and eukaryotes. Thiolytic cleavage, catalyzed by 3-ketoacyl-CoA thiolases (EC 2.3.1.16) is perhaps most familiar as a key step in Knoop’s pathway of beta oxidation of fatty acids. In the context of the mevalonate pathway, the biosynthetic (condensation) reaction to form acetoacetyl-CoA from two acetyl-CoA molecules will be emphasized. This biosynthetic reaction is catalyzed by acetoacetyl-CoA thiolase (or acetyl-CoA acetyl transferase), classified by the International Union of Biochemistry in the EC 2.3.1.9 category:
The chemistry of this reaction, depicted in the following scheme (Scheme 2), involves a tranferase step, i.e. formation from substrate acetyl-CoA of a covalent acetyl-S-enzyme and release of CoASH. The subsequent condensation step involves deprotonation of C2 of the second acetyl-CoA substrate and attack of the resulting carbanion on C1 of the acetyl-S-enzyme reaction intermediate to produce the product acetoacetyl-CoA and release free enzyme.
Scheme 2.

Chemical steps in the biosynthetic acetoacetyl-CoA thiolase reaction.
An interesting homologous reaction has been recently reported [9] to involve condensation of acetyl-CoA with malonyl-CoA to form acetoacetyl-CoA, catalyzed by the NphT7 protein from Streptomyces. Such a reaction appears chemically similar to the formation of acetoacetyl-acyl carrier protein in the initial condensation step of bacterial fatty acid biosynthesis. The NphT7 protein is encoded by an open reading frame mapping within a gene cluster proposed to account for other mevalonate pathway enzymes. It will be interesting to learn whether other examples of this variation in more typical acetoacetyl-CoA biosynthesis exist.
Functional observations
Identification of the specific cysteine involved in acetyl-S-enzyme intermediate formation was accomplished for a degradative ketoacyl-CoA thiolase [10]. Soon afterwards, a cytosolic liver acetoacetyl-CoA thiolase was highly purified and characterized [11]. Much of the more recent mechanistic and mutagenesis work on biosynthetic thiolase, reported by the Walsh, Sinskey, and Masamune labs, has employed a recombinant form of the Zoogloea ramigera protein, which supports bacterial polyhydroxybutyrate production. For this protein, Cys-89 is involved in formation of the acetyl-S-enzyme reaction intermediate [12]. The active site base (B:) that activates the second acetyl-CoA substrate molecule for condensation with the reaction intermediate has been identified as Cys-378 [13].
Structural observations
Several reports of structural work from the Wierenga lab have involved the Z. ramigera protein. In particular, results were obtained using crystals that were flash frozen after soaking with acetyl-CoA to produce a crystal containing the acetylated enzyme reaction intermediate and a bound acetyl-CoA molecule [14]. This approach led to structures that confirmed Cys-89 as the site of reaction intermediate formation and also Cys-378 as the base that deprotonates the second acetyl-CoA substrate prior to condensation (Figure 1). Cys-378 is situated within 3.3 Å of C2 of substrate acetyl-CoA, supporting a functional assignment as general base catalyst. The C2 of acetyl-CoA is closely juxtaposted (3.0 Å) to C1 of acetyl-enzyme, as required for an efficient condensation reaction. A positively charged conserved His-348 could interact with the thioester carbonyl of this acetyl-CoA (3.3 Å between His-348 and the C1 carbonyl oxygen) to stabilize the carbanion that is produced after proton abstraction. Such stabilization by a basic amino acid residue of the thioester carbonyl of acyl-CoA metabolites at which negative charge develops during the reaction is a recurring theme in a variety of enzyme catalyzed Claisen condensation/cleavage reactions [15, 16, 17, 18, 19].
Figure 1.

Active site residue triad in Z. ramigera acetoacetyl-CoA thiolase, based on the structural coordinates 1DM3. The structure [14] indicates the acetyl-enzyme intermediate formed at Cys-89 as well as the acetyl-CoA that condenses with the reaction intermediate. His-348 interacts with the C1 carbonyl of bound acetyl–CoA to provide a charge sink that stabilizes the C2 carbanion formed after proton extraction by the general base Cis-378. The carbanion is in close proximity to C1 of the acetyl-enzyme intermediate and supports efficient condensation to form acetoacetyl-CoA and regenerate free enzyme.
The Cys-His-Cys “triad” of thiolase active site residues evokes similar motifs found in the family of “initial condensation enzymes” and may be compared with the Cys-His-Asn triad described for the initial condensing enzyme of bacterial type two fatty acid biosynthesis [20]. Another variation of such a triad is observed for the enzyme that catalyzes the next mevalonate pathway reaction, HMG-CoA synthase.
HMG-CoA SYNTHASE
The biosynthesis of 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) by condensation of acetyl-CoA with acetoacetyl-CoA was demonstrated using a preparation of yeast protein [21]. HMG-CoA synthase (EC 2.3.3.10; formerly EC 4.1.3.5) catalyzes a reaction that is physiologically irreversible but has recently been demonstrated [18] to also support slow catalysis of the cleavage of HMG-CoA:
Work with yeast enzyme [22] supported formation of a covalent reaction intermediate. The purified avian liver enzyme was used to identify cysteine in formation of acetyl-S-enzyme and enzyme-S-HMG-CoA covalent reaction intermediates [23, 24], which are depicted in the following scheme (scheme 3) that outlines the chemistry of the reaction:
Scheme 3.

Chemical steps in biosynthesis of HMG-CoA.
A mitochondrial isoform [25] supports the ketogenic pathway for acetoacetate biosynthesis while a cytosolic isoform [26] participates in the mevalonate pathway for isoprenoid biosynthesis. Several bacteria utilize the mevalonate pathway and the Enterococcus faecalis mvaS protein catalyzes the HMG-CoA synthase reaction [27].
Functional observations
The condensation reaction proceeds with inversion of stereochemistry to produce the S-isomer of HMG-CoA [28]. Mechanism based inhibitor labeling [29] and protein sequencing [30] approaches have been used in our lab to identify Cys-129 (cytosolic enzyme numbering) as residue involved in formation of the reaction intermediates. Recombinant forms of the avian, human, and bacterial enzymes have become available [31, 32, 17, 33]. Using these tools, the essential nature of Cys-129 was demonstrated and, in contrast to acetoacetyl-CoA thiolase, replacement with serine did not support measurable reactivity [31]. Inhibition of cytosolic human enzyme by the beta lactone containing natural product, hymeglusin (structure shown below), also involves this active site cysteine in formation of a covalent enzyme-inhibitor adduct [32].

This inhibitor has been demonstrated to be a potent inhibitor of hepatic steroidogenesis [34]. Mutagenesis and mechanistic enzymology approaches were used in our lab to implicate a histidine (His-264) in substrate acetoacetyl-CoA binding [35] as well as a glutamate residue (Glu-95) in general acid/base catalysis [36]. The role of Glu-95 was confirmed as the general base in studies which demonstrated that substitutions of this glutamate by alanine or glutamine impair enolization of the acetyl-CoA analog, acetyldithio-CoA [37].
Structural observations
The first three dimensional structural data for HMG-CoA synthase were generated using recombinant bacterial (mvaS) enzymes [17], [18], [33]. The structure of plant enzyme with bound beta lactone inhibitor has been subsequently reported [38]. Only very recently have structures of animal cytosolic and mitochondrial isoforms been published [39]. There is reasonable agreement between structures of prokaryotic and eukaryotic enzymes in terms of confirmation/assignment of catalytic residues and functional roles.
Structural results [17] on the Staphylococcus aureus protein noncovalent complex with acetoacetyl-CoA indicated a thiolase-like fold and confirmed the close juxtaposition of active site cysteine, histidine, and glutamate residues that, as mentioned above, had previously been identified. Work on the E. faecalis enzyme [33] detected a substrate derived covalent adduct between an acetoacetyl moiety and the active site cysteine; this novel species had never been reported for animal enzyme. In a subsequent paper [40], the structure of protein containing substitution of glycine for the conserved alanine that precedes the active site cysteine was reported by Stauffacher and colleagues; a structural rationale for the marked increase in catalytic activity documented for this mutant was provided. Collaborative work between the Harrison and Miziorko labs produced unexpected observations when solutions containing product HMG-CoA and S. aureus enzyme were crystallized and structures determined [18]. Structures not only of enzyme-product complex but also of a complex of acetyl-enzyme reaction intermediate with substrate acetoaceyl-CoA were obtained. Solution experiments under crystallization conditions demonstrated that enzyme dependent cleavage of 14C-HMG-CoA occurs slowly (0.3 per day) and that HMG-CoA is activated to form the E-S-HMG-CoA reaction intermediate. This activation proceeds through a tetrahedral carbon. Reversibility of this activation step accounts for incorporation into reisolated HMG-CoA of two 18O atoms at C5 when the reaction is performed in H2O18 [18]. The structure of the acetyl-S-enzyme complex with acetoacetyl-CoA was particularly noteworthy. These results clearly confirmed (figure 2) not only the important assignment of the active site cysteine but also the function of glutamate, in close proximity (3.0 Å) to C2 of acetyl-S-enzyme, as the general base catalyst. Additionally, close interactions (both 3.1 Å) of the active site histidine with the C1 and C3 oxygens of bound acetoacetyl-CoA were observed. C3 of acetoacetyl-CoA is situated 3.3 Å from C2 of acetyl-enzyme so that, after deprotonation by the general base glutamate, efficient condensation to form enzyme-S-HMG-CoA will occur. Solvent hydrolysis releases product and reforms free enz-SH.
Figure 2.

Active site residue triad in S. aureus HMG-CoA synthase (mvaS), based on the structural coordinates 1XPL. The structure [18] depicts the acetyl-enzyme reaction intermediate formed at Cys-111/129 (bacterial/animal protein numbering). The enzyme’s second substrate, acetoacetyl-CoA is bound with His-233/264 interacting with C1 and C3 oxygens, providing a charge sink to facilitate attack by a carbanion on C3. This carbanion is produced upon abstraction of a proton from C2 of acetyl-enzyme by the general base Glu-79/95.
The structural data have been interpreted in the context of a catalytic Cys-His-Glu triad, which contrasts with the triads proposed for thiolase (Cys-His-Cys; [14]) and other condensing fold enzymes. Comparison of the thiolase triad with the HMG-CoA synthase triad (Figs. 1,2) reveals a contrast in positions of the general base residues, in accordance with the different chemical steps in these reactions and the particular roles for the particular residues that support acetyl-CoA deprotonation in thiolase versus acetyl-S-enzyme deprotonation in HMG-CoA synthase.
HMG-CoA REDUCTASE
In elucidation of the pathway that accounts for incorporation of acetate into isoprenoids and sterols, it was important to account for the observation that mevalonic acid is a good isoprenoid/sterol precursor. The investigation [41] of a yeast HMG-CoA reductase accounted for mevalonate production:
The enzyme (EC 1.1.1.34) is found in eukaryotes, archaebacteria, and some eubacteria. The conversion of the thioesterified HMG-CoA carboxyl to an alcohol represents a two step reduction, accounting for the stoichiometry of NADPH in the reaction. The reaction thus proceeds through the successive reduction steps to first produce bound mevaldyl-CoA, collapse of the thiohemiacetal to release CoASH and form mevaldehyde; the second reduction step then forms product mevalonate (Scheme 4):
Scheme 4.

Chemical steps in the HMG-CoA reductase reaction.
The eukaryotic proteins (class I HMG-CoA reductases) are associated with the endoplasmic reticulum and interact through membrane spanning helices in the N-terminal domain [42]. It follows, therefore, that the catalytic domain follows this membrane anchoring sequence. These class I enzymes are potently inhibited by the class of statin drugs that effectively modulate sterol synthesis and, as a result, have been heavily investigated. There is no homologous sequence for membrane association at the N-terminus in the bacterial HMG-CoA reductases (class II) and a few of these (some in recombinant form) have been isolated as soluble proteins. The Pseudomonas mevalonii enzyme [43] has a degradative function, allowing this microbe to grow on mevalonate as a carbon source. In contrast, the Staphyloccus aureus enzyme [44] has a biosynthetic function and is encoded by a gene within a mevalonate pathway gene cluster.
Functional observations
The soluble P. mevalonii enzyme has been a useful model for mutagenesis and functional investigations; a recombinant soluble hamster catalytic domain has also been employed. These studies in the Rodwell lab aimed at identification of functionally important active site residues [45, 46] and used not only mutagenesis but also employed the reaction intermediate mevaldehyde to study partial reactions catalyzed by wild type and mutant forms of this enzyme. These results implicated active site histidine, aspartate, and glutamate residues prior to the availability of three dimensional structural information.
Structural observations
The structures of various dead-end complexes of P. mevalonii enzyme were elucidated in collaborative work from the Rodwell and Stauffacher labs [47, 48] and an active site lysine was also identified. These results were followed by the Deisenhofer lab’s publication of structures of the soluble catalytic domain of the human enzyme, also liganded to substrates or products [49]. Despite the low overall sequence homology (<20%) and overall protein structure architecture between class I and class II enzymes, there is considerable similarity between these enzymes in the positioning of active site residues important to catalytic function. Residues from two different subunits contribute to an active site. The histidine proposed to function in protonation of product Coenzyme A is appropriately positioned for this role. The aspartate implicated by mutagenesis is located within the active site and involved in a hydrogen bond network with the lysine and the glutamate that have been identified by functional studies (figure 3). While both lysine and glutamate are in close proximity to the HMG-CoA thioester carbonyl that is reduced to form mevalonate, there are different proposals [47, 48] concerning their precise roles in substrate carbonyl polarization and/or the proton transfers that accompany substrate reduction by NADPH.
Figure 3.

Active site residues in the soluble catalytic domain of human HMG-CoA reductase, based on the structural coordinates 1DQA. The structure [49] includes liganded hydroxymethylglutarate and indicates the positions of three residues (Asp-767, Lys-691, and Glu-559) implicated in enzyme function. Both lysine and glutamate would be in close proximity to the thioester carbonyl of HMG-CoA that is subjected to a two reductive steps, resulting in formation of the C5 alcohol of mevalonate. There are different proposals [48, 49] regarding the precise roles of these residues in substrate carbonyl polarization and/or the proton transfers that accompany NADPH reduction.
Efficacy of inhibition by various compounds in the family of statin inhibitors is markedly dependent (e.g. nanomolar versus millimolar inhibitor affinity) on whether class I or class II enzyme is used. The inhibitors are characterized by hydroxymethylglutaryl (HMG)-like moieties linked to an extensive hydrophobic scaffold (e.g. a decalin ring in the case of mevastatin and simvastatin). Complexes of human enzyme catalytic domain with a variety of statins have been crystallized and X-ray structures have been published [50]. The structural results indicated binding of the HMG moiety in the active site pocket where the catalytic glutamate and lysine residues are located. In contrast, the NADPH substrate site is not occupied upon inhibitor binding. The structural results indicating that access to the substrate HMG-CoA is blocked by inhibitor binding are in accord with the observation of competitive inhibition with respect to HMG-CoA [51]. A variety of additional polar interactions with the HMG moiety have also been documented. The hydrophobic portion of the inhibitors is bound in a shallow hydrophobic groove of the human protein. A large number of van der Waals contacts between nonpolar amino acids in this groove and the diverse hydrophobic substituents that are a common feature of the various statins are proposed to represent the dominant contribution to high affinity binding. A structure of lovastatin bound to the class II P. mevalonii enzyme has also been reported [52]. As in the case of the class I enzyme, the structure indicates interactions with residues (e.g. lys, glu) identified in catalysis as well other polar residues in this pocket, with some hydrogen bonds mediated by water molecules. The hydrophobic decalin ring component of the inhibitor blocks closure of the C-terminal flap domain of the protein, which includes the histidine residue that has been implicated in catalysis. Thus, inhibitor binding both blocks the active site and makes correct orientation of active site amino acids impossible. A large difference between class I and class II enzyme interactions with the statin inhibitor is suggested to involve a pocket formed, in part, by the alpha helix of the P. mevalonii enzyme that contains a conserved “THNK motif”. The structural results [51] could expedite potential design of HMG-CoA reductase inhibitors that can discriminate between Class I and Class II enzymes. Additionally, an approach proposed to increase inhibitor affinity [50] involves derivatization of the parent inhibitory compound to incorporate chemical substituents effective in interaction with the NADPH site.
MEVALONATE KINASE
Mevalonate kinase (MK; MVK; ATP:mevalonate 5-phosphotransferase; EC 2.7.1.36) catalyzes transfer of ATP’s γ-phosphoryl to the C5 hydroxyl oxygen of mevalonic acid, resulting in formation of mevalonate 5-phosphate and ADP. The reaction was characterized in yeast [53]; the protein is found in eukaryotes, archaea, and certain eubacteria. The enzyme was highly purified from porcine liver and was demonstrated to catalyze a sequential reaction with mevalonate substrate binding first and MgADP product released last [54]:
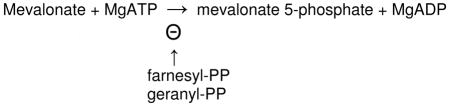
The enzyme is subject to potent feedback inhibition by geranyl diphoshophate and farnesyl diphosphate, downstream intermediates in the polyisoprenoid biosynthetic pathway [55]. High affinity (10−8 M) feedback inhibition has also been observed using a recombinant human protein and contrasted with lower potency inhibition (10−5 M) of the Staphylococcus aureus enzyme [56]. Recently, there has also been a report that mevalonate 5-diphosphate is a potent (submicromolar) inhibitor of the Streptococcus pneumoniae enzyme [57]; this metabolite has not been observed to be effective in inhibition of the Staphylococcus enzyme [56]. Inherited human mevalonate kinase (MVK) mutations are correlated with two diseases, accounting for mevalonic aciduria (MIM6103770) and Hyper-IgD syndrome (MIM260920).
Functional observations
Cloning of the cDNA encoding the rat [58] and human proteins [59] was a prelude to large scale production of recombinant forms of these enzymes [60], [61]. Subsequent mutagenesis work established Ala-334 as the basis for an inherited enzyme deficiency [62]. Other mutagenesis experiments on rat and human MVK were conducted in our lab prior to the availability of any high resolution 3-D structures for the protein. The results implicated conserved Lys-13, Ser-146, Glu-193, and Asp-204 as functionally important active site residues [60, 61, 63]. Ser-146 is the second of two tandem serines that map within a glycine rich ATP consensus sequence. Subsequent work indicated the importance of His-20 in rat MVK [64] and Arg-196 in Methanococcus jannaschii MVK [65].
Structural observations
The X-ray structure of unliganded M. jannaschii MVK [66] indicated the anticipated fold for an enzyme in the family of galactokinase/homoserine kinase/mevalonate kinase/phosphomevalonate (GHMP) kinase proteins. A monomer was observed in the crystal instead of the dimeric form typically reported for MVKs in solution. Based on the active site residues that had been previously identified and the features of the substrate binding sites observed for other GHMP kinase proteins, a model was constructed (a syn conformation of bound ATP was presumed) and offered to explain MVK reaction chemistry. The structure of dimeric rat MVK liganded to Mg-ATP was also elucidated [67]. ATP is bound to rat MVK in an anti conformation. Glu-193 and Ser-146 were confirmed to coordinate to bound cation ligand. Lys-13 interacts with the γ-phosphoryl group of ATP and makes a salt bridge with Asp-204. Thus, Asp-204 is reasonably positioned to support the predicted catalysis of phosphoryl transfer and modeling of mevalonate into the vacant pocket for this substrate produces a ternary complex orientation that is compatible with such a function for Asp-204. The following view (figure 4) of Mg-ATP bound to rat MVK illustrates the orientation of active site residues.
Figure 4.
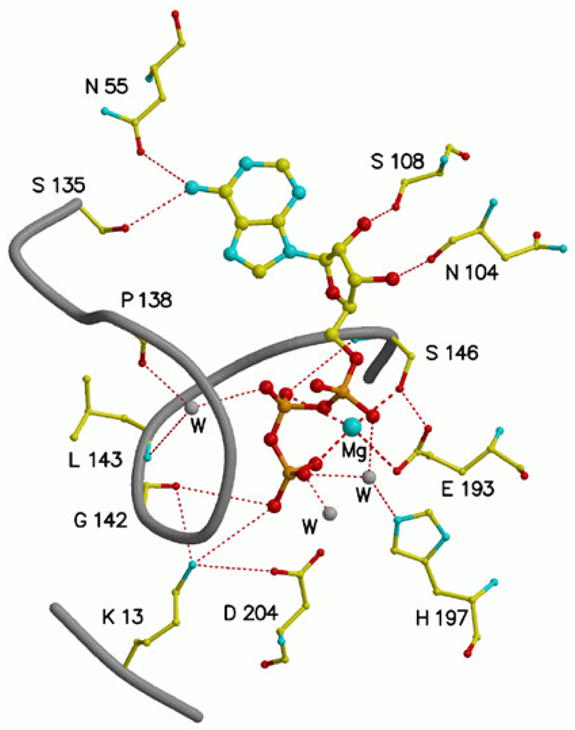
The MgATP binding site in rat mevalonate kinase [67], based on the structural coordinates 1KVK. ATP is bound in an anti conformation. Bound magnesium is coordinated to Glu-193 and Ser-146 as well as the beta and gamma phosphoryls of ATP. The catalytic residue Asp-204 is positioned to support transfer of the ATP gamma phosphoryl to an acceptor substrate. Conserved Lys-13 interacts with both Asp-204 and the gamma phosphoryl of substrate ATP. Dashed lines indicate coordination to magnesium; dotted lines indicate hydrogen bonds.
Structural information has also become available for enzyme with bound metabolites that negatively regulate MVK activity. The structure of a Streptococcus pneumoniae MVK binary complex with mevalonate 5-diphosphate has been elucidated [68]. The bound metabolite was originally characterized as an allosteric inhibitor of this bacterial enzyme [57]. It is situated in the catalytic cleft of the crystallized protein in such a way that it interacts with aspartate, serine, and lysine residues that are homologous to those demonstrated to interact with bound MgATP in rat MVK [67]. Additionally, mevalonate diphosphate interacts with a threonine previously shown to influence the Km for mevalonate [63]. Thus, the structural results [68] are interpreted to suggest that mevalonate diphosphate interacts with MVK as a partial bisubstrate analog, with its pyrophosphoryl moiety mimicking a linkage between mevalonate and the ATP phosphoryl donor.
Structural characterization of a feedback inhibitor bound to animal MVK relied on the observation [56] that farnesyl thiodiphosphate (FSPP), a thio analog of farnesyl diphosphate (FPP), is as effective a competitive inhibitor with respect to ATP as the naturally occurring metabolite. The original observation that crystallization of human MVK in the presence of FPP led only to the structure of an enzyme-pyrophosphate complex prompted the use of rat MVK with FSPP, which is expected to be more resistant to hydrolysis.
Crystallization of enzyme-FSPP solutions in the presence of Mg2+ produced a structure [69] (figure 5) of rat MVK bound to farnesyl thiophosphate (FSP), indicating either some disorder of the β phosphoryl group or some hydrolysis of inhibitor during crystal growth. Nonetheless, enzyme occupancy was adequate for detection of electron density for the entire FSP molecule. The inhibitor binds in the ATP site with its thiophosphoryl situated in the position observed for the β phosphoryl group of ATP. Asp-204 and Ser-146 function as cation ligands and K13 is expected to interact with the beta phosphoryl of FSPP (or FPP). The FSP polyisoprenoid chain overlaps the site at which the adenosine moiety of ATP binds. FSP’s last 10 carbons (C6-C15; two isoprenyl units) interact with nonpolar residues (e.g. Leu-53, Val-56, I-196). Previously, a 1000-fold inflation in inhibitor constants for FPP and FSPP had been observed upon comparison of human and Staphylococcus MVKs [56]. Comparison of the binding surface measured for FSP/FSPP binding to animal MVK and modeled for Streptococcus MVK reveals the basis for this differential affinity. Animal MVK contains a pocket that embeds inhibitor atoms C6-C15 (figure 6). In contrast, the C15 chain of the feedback inhibitor is largely solvent exposed in the model for the bacterial MVK.
Figure 5.
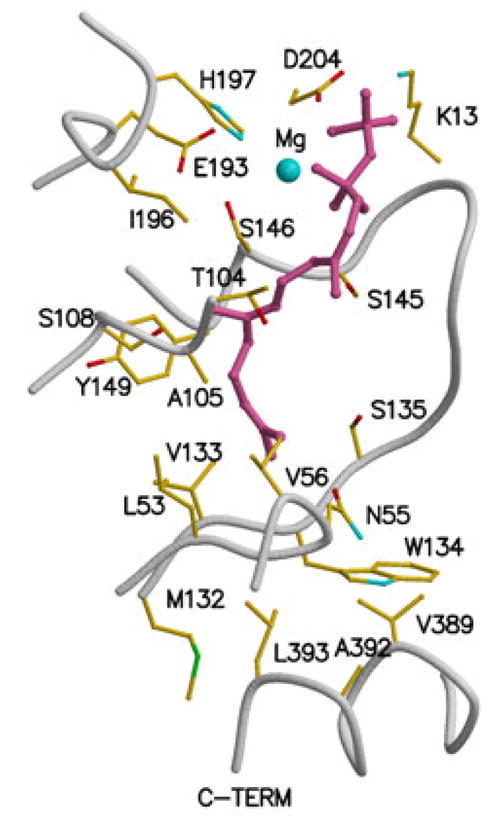
Feedback inhibitor binding to rat mevalonate kinase, based on the structural coordinates 2R42. Residue numbering is based on human mevalonate kinase. Inhibitor ligand electron density is fit using farnesyl thiodiphosphate (FSPP), although due to disorder or hydrolysis the beta phosphoryl is not well defined. The inhibitor binds in the ATP site [69] with its alpha phosphoryl group situated where the beta phosphoryl group of ATP would be located. Asp-204 and Ser-146 function as ligands to cation. Lys-13 would be expected to interact with the beta phosphoryl of inhibitor. The last 10 carbons of the farnesyl moiety interact with a number of nonpolar residues (e.g. Leu-53, Val-56, Val-133, Ile-196).
Figure 6.
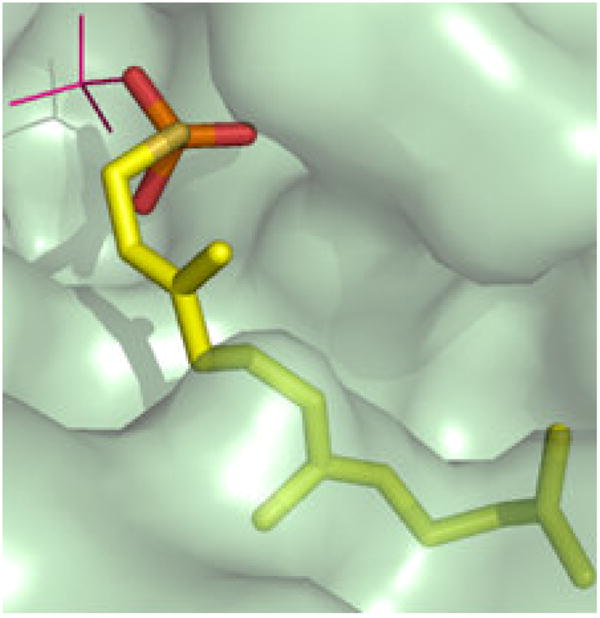
A solvent accessible surface representation of the feedback inhibitor (farnesylSP/farnesylSPP) binding site of mammalian mevalonate kinase, based on the structural coordinates 2R42. The animal MVK embeds farnesyl atoms C6-15 in a pocket to which the side chains of I196, T104, L53, N54, and I56 contribute [69]. This observation explains the high affinity (10−8 M; [56]) observed for feedback inhibition, which is competitive with respect to ATP. The image is adapted from Fu et al., Biochemistry 47 (2008) 3715–3724 [69], with permission of the American Chemical Society.
PHOSPHOMEVALONATE KINASE
Phosphomevalonate kinase (PMK; EC 2.7.4.2) catalyzes the reversible reaction of mevalonate 5-phosphate and ATP to form mevalonate-5-diphosphate and ADP:
Activity of this enzyme was demonstrated in pig liver [70] and the pig liver enzyme has subsequently been isolated and more extensively characterized [71]. Phosphomevalonate kinase is found in eukyaryotes and some eubacteria. The amino acid sequences for animal and low homology invertebrate PMK proteins are not orthologous to those for PMK in plants, fungi, and bacteria [72]. Thus, the proteins that catalyze the enzymatic reaction differ widely, depending on their source. Animal and invertebrate PMK proteins exhibit the fold typical of the nucleoside monophosphate (NMP) kinase family while the other PMK proteins are members of the GHMP kinase family. The tissue-isolated pig enzyme is reported to catalyze an ordered sequential bi-bi reaction with mevalonate 5-phosphate assigned as the first substrate bound and ADP as the last product released [73]. A recombinant form of Streptococcus pneumoniae has been characterized [74] and reported to catalyze a random sequential bi-bi reaction. A recombinant form of Enterococcus faecalis PMK has also been isolated and characterized [75]. The sequence of human PMK has been deduced [76]. The protein encoding DNA was used to produce a GST-PMK fusion construct and this recombinant fusion protein was used to test the site of feedback inhibition in the mevalonate pathway [77]. The recombinant human protein has also been expressed and isolated in an N-terminal His-tagged form. This enzyme has been used for mutagenesis studies on active site functional residues [78] as well as for solution structure experiments.
Functional observations
The N-terminus of the protein harbors a variation on the P-loop motif associated with ATP binding. Work in our lab with the recombinant human protein indicated that Lys-22, as well as Arg-18, have a substantial influence on catalysis in accordance with their location in a P-loop [78]. Evaluation of the importance of other conserved basic residues [79] implicated Arg-110 as making a large contribution to catalysis, Arg-111 and Arg-84 as influencing mevalonate 5-phosphate binding, and Arg-141 as affecting ATP binding. Using the recombinant Streptococcus pneumoniae enzyme, mutations in a series of proposed solvent accessible residues have been characterized [80] and interpreted in the context of a structural model of protein with MgATP and phosphomevalonate ligands. Mutation of Asp-105 mainly results in a kcat effect, while mutations at Lys-9 and Ser-291 have considerable affects on the Km for phosphomevalonate. An enormous effect (17,500 fold) reflected as inflation of the Km for phosphomevalonate is reported upon mutation of ala-293, which is harbored in a glycine rich substrate recognition loop.
Structural observations
Uniformly 15N-labeled recombinant human PMK protein has also been used for NMR measurements on substrate induced structural changes [81]. NMR estimates of equilibrium binding constants were in reasonable accord with affinity estimates from kinetic characterization studies. Dynamics results were interpreted in the context of regions that may influence domain closure, which is well precedented in NMP kinase fold proteins. In contrast with the observation of comparable ATP or ADP dependent conformational changes, the conservatively substituted analog AMPPNP1 induces few chemical shift perturbations, complicating evaluation of the effect of active site occupancy by the gamma phosphoryl moiety of an adenine nucleotide. Based on observed chemical shift perturbations, binding of either mevalonate 5-phosphate or ATP induces conformational changes in human PMK. These biophysical observations contrast with expectations for an ordered sequential mechanism, based of steady state kinetic studies on pig PMK [73]. The possibility of random substrate/product binding to human PMK does, however, agree with the observation of product inhibition of human PMK by mevalonate 5-diphosphate that is competitive with respect to substrate mevalonate 5-phosphate [79]. The preparation of a series of 15N labeled human PMK proteins containing single arginine substitutions (e.g. R48M, R111M, R130M, R141M) has facilitated NMR assignments of arginine side chains. Dynamics studies on wild type PMK and these PMK arginine mutants [82] indicate that substrate binding to PMK correlates with a transition from a flexible to a more rigid protein. The observation extends to the arginine side chains, including several that are somewhat remote from the active site. The pervasive rigidification suggested by the results of these dynamics studies seems reasonable in the context of the substantial structural changes documented for NMP kinase proteins as the substrate binding regions move into the proximity of the P-loop domain upon occupancy of phosphoryl donor and/or acceptor binding sites.
Three dimensional structure information has become available from X-ray diffraction experiments on bacterial and human PMK. The structure of unliganded Streptococcus pneumoniae PMK, which exhibits a GHMP kinase fold, was initially reported [83] and subsequently the structure of a ternary MPK-mevalonate phosphate-Mg AMPPNP complex was published [84]. Comparison of apo and ternary complex structures indicates octahedral coordination of Mg that involves Asp-197, Ser-213, mevalonate 5-phosphate, and three solvent waters. An active site water pentamer is noted as interacting extensively with reactive groups of the bound substrate and ATP analog. Lys-9 is observed to interact with the gamma phosphoryl group of bound AMPPNP, an observation similar to that reported for a lysine in a complex of ATP with another GHMP kinase protein, rat mevalonate kinase. Ser-147 is noted to hydrogen bond to the carboxyl group of bound phosphomevalonate.
The structure of an unliganded form of human PMK has recently become available [85]. The results confirm the assignment of the protein as an NMP kinase family member. Consequently, the unliganded structure is expected to reflect an “open” form of the enzyme, i.e. the substrate binding regions or “lids” are not as close in proximity to the P-loop containing core as would be expected in samples containing ligands in the phosphoryl donor and/or acceptor sites. There is, however, a sulfate ion bound in close proximity to the P-loop (figure 7); this is interpreted as a phosphoryl group analog and a marker for the active site. The N-terminal P-loop assignment that was the subject of earlier functional studies [78] was confirmed in the human PMK structure.
Figure 7.

The active site cavity of human phosphomevalonate kinase and the location of conserved amino acid side chains implicated in enzyme function. The figure is based on the structural coordinates 3CH4, as reported by Chang et al. [85]. While the unliganded enzyme exhibits an open active site, the bound sulfate is interpreted as a phosphoryl group marker for the binding site of phosphorylated substrates. Sulfate is located in proximity to the N-terminal P-loop and interacts with the side chain of Arg-141, which has been shown [78] to influence ATP binding. Mutagenesis of Arg-18, Lys-22, Arg-84, Arg-110, and Arg-111 has resulted in observation of significant catalytic or substrate binding effects for these conserved residues.
The bound sulfate ion interacts with the amide nitrogen of P-loop Gly-21 and also the side chain of Arg-141, which has been shown [78] to influence binding of substrate ATP. The conserved residues which exhibit, upon mutagenesis, the largest functional effects on PMK catalytic activity or substrate Km can be mapped in the open cavity that exists between the P-loop containing core and the “lids” expected to harbor binding sites for the phosphoryl donor and acceptor substrates (Fig. 7). More refined functional assignments would be facilitated by the availability of structural information on liganded forms of human PMK.
MEVALONATE DIPHOSPHATE DECARBOXYLASE
Mevalonate diphosphate decarboxylase (EC 4.1.1.33; various abbreviations appear in the literature: MDD, MVD, MPD, DPM-DC) catalyzes the ATP dependent decarboxylation of mevalonate 5-diphosphate (MVAPP) to form isopentenyl 5-diphosphate [86], as indicated in the equation below:
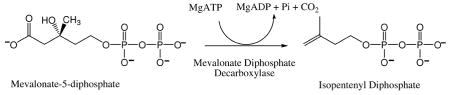
This reaction is essential to the mevalonate pathway of polyisoprenoid and sterol synthesis [87]. Activity has been measured in animals [88], plants [89], and yeast [86]. Genetic complementation has implied activity in the Staphylococcus aureus and Trypanosoma brucei [90] proteins. Highly purified active enzyme has been prepared from avian [88], porcine [91], and rat [92] tissue and in recombinant form from bacteria expressing the yeast [93], human [94], [77], [95]], Trypanosoma brucei, and Staphylococcus aureus [90] proteins. Characterization of the tissue isolated protein implied the presence of an arginine that influences activity of the avian enzyme [96] and documented the selectivity for divalent cation [97]. The avian enzyme was also used to demonstrate that the transient phosphoryl transfer to the C3 oxygen proceeds with inversion of stereochemistry [98] and, thus, no covalent E-P intermediate forms.
Enzyme inhibitors
Early observations that metabolite analogs could block polyisoprenoid/sterol biosynthesis suggested that the MDD reaction is involved and prompted synthesis and evaluation of a variety of MDD inhibitors. The use of 6-fluoromevalonic acid in tissue extracts [99] implicated MDD as the target for a downstream metabolite (6-fluoro-MVAPP) that blocked sterol production. In a survey of several fluorinated mevalonic acid derivatives, Reardon and Abeles [100] showed with purified MDD that 6-fluoro-MVAPP was the potent inhibitor that accounted for the block in sterol biosynthesis. These studies were extended [101] to indicate that a 3-phospho-6-fluoro-MVAPP analog of the normal reaction intermediate could be trapped as an MDD-bound species. In addition, a nitrogen substituted transition state analog (N-methyl-N-carboxymethyl-2-pyrophosphoethanolamine) was synthesized and characterized [101]; results suggested that a positively charged nitrogen atom mimicked a reaction intermediate with carbocation character. This carbocation proposal was supported by synthesis of N-diphosphoglycolyl proline and the compound’s potent inhibition of MDD was attributed to its positively charged nitrogen atom [102]. On the basis of these various studies, a relatively detailed mechanism for the MDD reaction could be proposed well in advance of the availability of recombinant enzymes that could be used to investigate how MDD active site amino acids might support the predicted chemistry.
Voynova et al. [95] used recombinant human MDD to confirm the high affinity inhibition (competitive with respect to MVAPP) of enzyme by both 6-fluoro-MVAPP and diphosphoglycolylproline. Qiu and Li [103] proposed that 2-fluoromevalonate diphosphate could irreversibly inactivate MDD in a time dependent fashion but a subsequent report [104] indicated that 2-fluoro- and 2-difluoro-MVAPP are reversible competitive inhibitors with respect to MVAPP. Recently, a series of mevalonate analogs were prepared and tested as substrates for Streptococcus pneumoniae MVK, PMK, and MDD [105]. Results suggested that this bacterial MDD is very tolerant of replacement of the 6-methyl group with small substituents. Resistance of MDD to compounds designed to covalently modify and inactivate the enzyme has prompted the hypothesis that decarboxylation and phosphate elimination of a 3-phospho-MVAPP intermediate are concerted rather than dissociative processes. The hypothesis [105] included the prediction that little carbocation character develops in the reaction intermediate.
Functional observations
Mutagenesis of yeast MDD and characterization of the mutant proteins by kinetic and biophysical methods [93, 106] initially focused on the Lys-18/Asp-305 pair of conserved residues that are homologous to a similar pair implicated in rat MK function. Replacement of the Asp-305 side chain carboxyl (D305N; D305A) was observed to result in large (103–105 fold) decreases in catalytic activity. Extension of this approach to alanine substitution of a series of conserved serine residues suggested that Ser-121 had a large influence on catalysis (>104 fold effect decrease upon mutation to alanine). The selective influence of this second of two tandem conserved serines in an ATP-consensus motif had previously been observed for rat MK [63]. The results also implicated Ser-153 in a possible interaction with ATP and Ser-155 in MVAPP binding. The Ser-155 mutant exhibited inflated Km and Ki values for both MVAPP and diphosphoglycolylproline. In work on human MDD, Voynova et al. [95] demonstrated that conservative mutation of Arg-161 (R161Q) decreased catalytic activity by 1000-fold. Such an observation is in agreement with the earlier implication of an active site arginine by protein modification methods [96]. Mutation of Asn-17 (N17A) results in inflated Km and Ki estimates for MVAPP, diphosphoglycolyl proline, and 6-fluoro-MVAPP, suggesting that this residue participates in interactions that affect binding of the phosphoryl acceptor substrate. Mutation of recombinant rat MDD [104] resulted in observation of no detectable activity upon mutation of Lys-23 (K23A) or Arg-162 (R162A).
Structural observations
A molecular docking and simulation-base approach [107] has recently been reported; the study used structural information from the initial X-ray structure for MDD (yeast enzyme; [108]). While no structure of liganded MDD has yet been elucidated, this computational study modeled a ternary complex of enzyme with ATP and phosphoryl acceptor. The conclusions suggest that a series of conserved residues implicated by functional studies are situated in the active site. In particular, a water molecule is predicted to mediate interaction of a catalytic aspartate with the C3 hydroxyl of MVAPP. The critical serine in the glycine rich consensus ATP motif is predicted to interact with MVAPP.
Structures of Trypanosoma brucei and Staphylococcus aureus MDD proteins have been empirically determined [90] by Hunter’s lab. The proteins are unliganded except for a sulfate anion. In the high resolution structure of the T. brucei protein, a Lys-18/Asp-293 salt bridge is detected; this is analogous to the observation for rat mevalonate kinase. ATP and MVAPP were positioned into the protein structure to produce a ternary complex model, which predicts an Arg-77 interaction with the beta phosphate of MVAPP. Also, interaction of Tyr-19 and Arg-149 with the C1 carboxyl of MVAPP is proposed.
The structure of human MDD has been elucidated [95]. Protein in the crystal is also unliganded except for a sulfate/phosphate anion (from the crystallization buffer); the anion is bound to Lys-26 and Arg-78. The Z-dock algorithm (http://zdock.bu.edu) was used to model a binary complex of human MDD with mevalonate diphosphate. The beta phosphate of modeled substrate is within 1 Å of the Lys-26, Arg-78 bound anion (a putative phosphoryl mimic); this observation suggests that the binary complex model is reasonable. Overlay of the binary ATP-MK structure allows the positioning of ATP into a MDD-ATP-MVAPP ternary model without any steric conflicts, supporting the positioning of the docked ligands. This model (Figure 8) predicts juxtapositioning of the Arg-161 guanidinium group within 3.5 Å of the C1 carboxyl of MVAPP; Asn-17 has been observed to form a hydrogen bond with Arg-161. These interactions are in agreement with functional/mutagenesis results for these residues. The carboxyl side chain of Asp-305 is situated within 4 Å of the substrate’s C3 hydroxyl; this prediction is in accord with a catalytic function for Asp-305. Ser-127’s positioning near ATP’s phosphoryl groups is also appropriate for the predicted active site role of this residue; the homologous serine in rat mevalonate kinase interacts with ATP and divalent cation.
Figure 8.
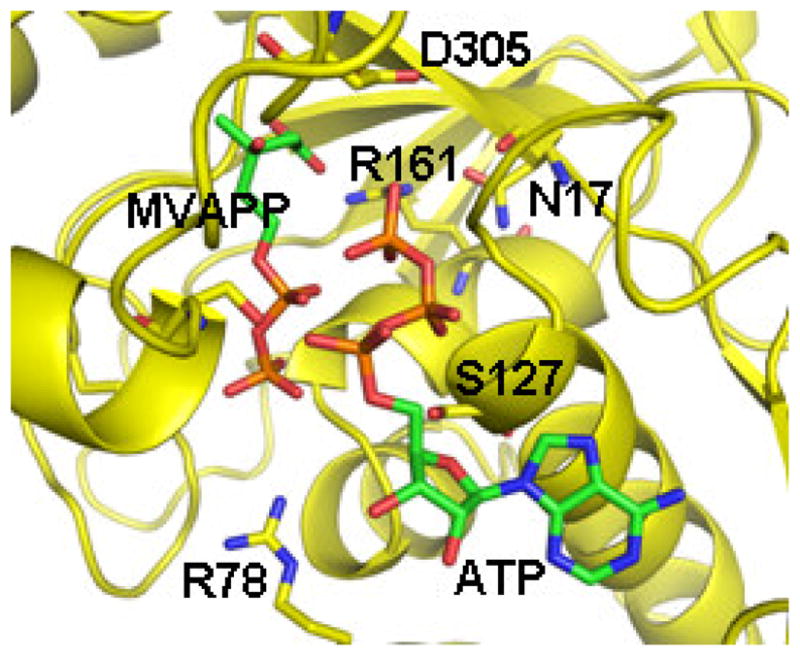
A model of the ternary complex of human mevalonate diphosphate decarboxylase with ATP and mevalonate 5-diphosphate. A binary complex model was generated using the Z-dock algorithm (http://zdock.bu.edu) and coordinates of mevalonate diphosphate (from 2OI2) and the human enzyme (3D4J). The position of ATP is based on an overlay of a structure of the mevalonate kinase-ATP complex (1KVK) on the human MDD structure [95]. The predicted position of S127 agrees with the previous suggestion of its interaction with the phosphoryl chain of ATP [106]. The juxtapositioning of ATP’s gamma phosphoryl group with respect to MVAPP’s C3 oxygen is in accord with production of a 3-phosphoMVAPP reaction intermediate and supports the docking position of MVAPP in the binary complex model. The model predicts juxtapositioning Arg-161 and the C1 carboxyl of MVAPP. Asn-17 has been observed to hydrogen bond to Arg-161. The side chain of Asp-305 is close to the substrate’s C3 hydroxyl. Such interactions are in accord with functional roles for these conserved residues, as suggested by characterization of mutant MDD enzymes. The image has been adapted, with permission (Elsevier) from Voynova et al. Arch Biochem Biophys 480 (2008) 58–67.
Other unliganded MDD structures (Streptococcus pneumoniae (2GS8) and Legionalla pneumophila (3LTO) proteins) have been generated and deposited in a public database but not published in refereed literature. Information from liganded MDD structures would clearly expedite more detailed functional assignments.
The scheme above (Scheme 5) integrates available information and hypotheses concerning the reaction chemistry and the participation of conserved MDD residues. Asp-305 is shown juxtaposed to MVAPP’s C3 hydroxyl; this is consistent with its observed large contribution to catalytic efficiency as well as positional homology with the proposed general base catalyst in mevalonate kinase. Similar observations on the functional contribution to MDD catalysis and homology with a serine implicated in the mevalonate kinase reaction suggest that Ser-127 interacts with ATP to orient the phosphoryl chain for productive transfer of ATP’s gamma phosphoryl group to MVAPP. Arg-161 has been established to hydrogen bond to Asn-17. The large contribution of Arg-161 to catalysis is compatible with its depicted interaction with the MVAPP carboxyl group. Recent structural studies on PEP carboxykinase [109] provide precedent for the proposed function of Arg-161. The scheme depicts four chemical species that are proposed to participate in the reaction: 1) substrate MVAPP; 2) a transient 3-phosphomevalonate 5-diphosphate intermediate (in which the C3 alcohol is transformed to an improved leaving group needed for formation of the next intermediate; 3) a transient carbocation intermediate, which provides an electron sink to drive decarboxylation; 4) the reaction product isopentenyl diphosphate. The inhibition studies of Dhe-Paganon et al. [101] and Vlattas et al. [102] on N-derivatived substrate analogs have been interpreted to support participation of the positively charged carbocation intermediate in the MDD reaction.
Scheme 5.

Mevalonate diphosphate decarboxylase reaction chemistry and possible contributions of active site residues.
VARIATIONS ON THE MEVALONATE PATHWAY
Isoprenoids represent a major component of archael lipids. Not only do they contribute to the usual isoprenoid derived metabolites like dolichol, but ether linked long chain (e.g. C20) polyisoprenoids are abundant in membranes of archaeal cells. As genome sequences for these organisms became available, it seemed likely that, like most eukaryotes, they utilized the mevalonate pathway for isoprenoid biosynthesis. However, in most archaeal genomes, it was unclear whether genes encoding phosphomevalonate kinase (PMK) and mevalonate diphosphate decarboxylase (MDD) proteins could be assigned [72]. This may have been anticipated given the observation of nonorthologous PMK proteins upon comparison of the enzyme from higher eukaryotes with PMKs from fungi, plants, or bacteria. For MDD, where homology between the animal and bacterial enzymes is more obvious, failure to detect the protein in most archaea is more surprising.
The observation of a protein encoded by Methanocaldococcus jannaschii that catalyzed an ATP dependent phosphorylation of isopentenyl 5-phosphate to produce isopentenyl 5-diphosphate (IPP) [110] seemed to provide an explanation for the lack of detection of a PMK in this organism. The heterologously expressed protein was purified and partially characterized. The ATP dependent phosphorylation could be detected using isopentenyl 5-phosphate as acceptor substrate but not with mevalonate or mevalonate 5-phosphate acceptors. On this basis, the protein was assigned as an isopentenyl phosphate kinase (IPK). While the absence of a more traditional pathway in M. jannaschii for synthesis of isopentenyl 5-diphosphate was not excluded, these observations did prompt the hypothesis of an alternative route to IPP biosynthesis:
The alternative route requires decarboxylation of mevalonate 5-phosphate to produce the substrate for IPK, which would catalyze formation of IPP.
Recently, there have been several reports that confirm the function of an isopentenyl 5-phosphate kinase. Recombinant forms of IPK from Methanothermobacter thermautotrophicus and from Thermoplasma acidophilum have been produced and characterized [111]. These IPK enzymes catalyze reversible interconversions of substrates and products and exhibit maximum activity at 70° C. A sequential mechanism was indicated on the basis of kinetic results. Sequence alignments and pH/rate profiles have been interpreted to suggest that a conserved histidine may be functional within the active site. These proteins were demonstrated to utilize various C4 and C5 monophosphates as substrates, with good activity supported by dimethylallyl phosphate, n-butyl phosphate, and 3-butenyl phosphate. While isopentenyl 5-phosphate was the optimal substrate of several compounds that were tested, these enzymes’ ability to utilize various phosphorylated compounds does raise a question about whether their major physiological role involves support of the proposed mevalonate metabolism. Future work may allow a more definitive answer to this question.
These demonstrations of isopentenyl kinase enzymes have been further supported by the elucidation of X-ray structures for substrate/product complexes of these enzymes from several organisms. Work from Poulter’s lab [112] led to structures of the ADP-liganded Methanothermobacter thermautotrophicus IPK as well as the Thermoplasma acidophilum IPK liganded to either isopentenyl phosphate and ATP or IPP and ADP. The structures establish these IPKs as members of the large amino acid kinase (AAK) family of proteins. The liganded enzyme structures confirm the presence of histidine and lysine active site residues. The active site lysine exhibits multiple interactions with the phosphoryl groups of donor and acceptor substrates to support an in-line phosphoryl transfer. Structures of Methanocaldococcus jannaschii IPK have also been elucidated [113]. While no bound adenine nucleotide has been observed, the structures contain bound isopentenyl phosphate, isopentenyl diphosphate (IPP), or IPPβS. A bound sulfate anion is proposed to indicate the position of an adenine nucleotide phosphoryl. A conserved His-60 has been mutated to produce H60A and H60N proteins that are not observed to support reaction catalysis. The H60Q mutant exhibits a ~ 40-fold decrease in catalytic efficiency as well as modest inflation of Km values for ATP and isopentenyl phosphate substrates. Mutations of a hydrophobic substrate pocket have resulted in “chain length” mutants with ability to phosphorylate the C15 substrate, farnesyl phosphate.
No detailed characterization of a protein with mevalonate phosphate decarboxylase activity (the initial enzyme required for the proposed variation in the mevalonate pathway; Scheme 6) has been reported. However, the collected observations on archaeal IPK proteins and the possibility of IPK homologs in organisms for which all of the traditional mevalonate pathway proteins are also predicted raise intriguing questions about the potential complexity of mevalonate metabolism. These issues suggest that future investigation of this area of biosynthesis will lead to interesting developments.
Scheme 6.

Proposed alternative reactions in biosynthesis of isopentenyl diphosphate.
Acknowledgments
Parts of the work performed in my laboratory were supported by NIH DK21491 or DK53766. The author acknowledges the important contributions of structural collaborators Prof. Jung-Ja P. Kim and Prof. David H.T. Harrison and their lab members. Major contributors to my laboratory’s work on mevalonate pathway enzymes include Ila Misra, Timothy Herdendorf, Prof. Natalia Voynova, Kelly Chun, Christine Behnke, and Chang-Zeng Wang. The author greatly appreciates the generosity of the many colleagues who provided the DNA samples or plasmids used for preparation of recombinant forms of some of the mevalonate pathway enzymes investigated in my lab.
Footnotes
Abbreviations used are: MVA, mevalonic acid/mevalonate; MVAPP, mevalonate 5-diphosphate; HMG-CoA, 3-hydroxy-3-methylglutaryl-CoA; IPP, isopentenyl diphosphate; FPP, farnesyl diphosphate; FSPP, farnesyl thiodiphosphate; MVK, mevalonate kinase; PMK, phosphomevalonate kinase; MDD, mevalonate diphosphate decarboxylase; IPK, isopentenyl phosphate kinase; GHMP, galactokinase, homoserine kinase, mevalonate kinase, phosphomevalonate kinase; NMP, nucleoside monophosphate, AMPPNP, 5′-adenylyl-beta, gamma-imidodiphosphate.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bloch K. Science. 1965;150:19–28. doi: 10.1126/science.150.3692.19. [DOI] [PubMed] [Google Scholar]
- 2.Eisenreich W, Bacher A, Arigoni D, Rohdich F. Cell Mol Life Sci. 2004;61:1401–1426. doi: 10.1007/s00018-004-3381-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilding EI, Brown JR, Bryant AP, Chalker AF, Holmes DJ, Ingraham KA, Iordanescu S, So CY, Rosenberg M, Gwynn MN. J Bacteriol. 2000;182:4319–4327. doi: 10.1128/jb.182.15.4319-4327.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bochar FJDA, Stauffacher CV, Rodwell VW. Biosynthesis of mevalonic acid from acetyl-CoA. Pergamon Press; New York: 1999. [Google Scholar]
- 5.Hedl M, Tabernero L, Stauffacher CV, Rodwell VW. J Bacteriol. 2004;186:1927–1932. doi: 10.1128/JB.186.7.1927-1932.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haapalainen AM, Merilainen G, Wierenga RK. Trends Biochem Sci. 2006;31:64–71. doi: 10.1016/j.tibs.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 7.Lynen WLF, Wieland O, Rueff L. Angewandte Chemie. Vol. 64. 1952. p. 687. [Google Scholar]
- 8.Gehring LFU. Thiolase. Academic Press; New York: 1972. [Google Scholar]
- 9.Okamura E, Tomita T, Sawa R, Nishiyama M, Kuzuyama T. Proc Natl Acad Sci USA. 2010;107:11265–11270. doi: 10.1073/pnas.1000532107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gehring U, Harris JI. Eur J Biochem. 1970;16:492–498. doi: 10.1111/j.1432-1033.1970.tb01108.x. [DOI] [PubMed] [Google Scholar]
- 11.Clinkenbeard KD, Sugiyama T, Moss J, Reed WD, Lane MD. J Biol Chem. 1973;248:2275–2284. [PubMed] [Google Scholar]
- 12.Thompson S, Mayerl F, Peoples OP, Masamune S, Sinskey AJ, Walsh CT. Biochemistry. 1989;28:5735–5742. doi: 10.1021/bi00440a006. [DOI] [PubMed] [Google Scholar]
- 13.Palmer DE, MAJ, Gamboni R, Williams SF, Peoples OP, Walsh CT, Sinskey AJ, Masamune S. J Biol Chem. 1991;266:8369–8375. [PubMed] [Google Scholar]
- 14.Modis Y, Wierenga RK. J Mol Biol. 2000;297:1171–1182. doi: 10.1006/jmbi.2000.3638. [DOI] [PubMed] [Google Scholar]
- 15.Anstrom DM, Kallio K, Remington SJ. Protein Sci. 2003;12:1822–1832. doi: 10.1110/ps.03174303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fu Z, Runquist JA, Montgomery C, Miziorko HM, Kim JJ. J Biol Chem. 2010;285:26341–26349. doi: 10.1074/jbc.M110.139931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Campobasso N, Patel M, Wilding IE, Kallender H, Rosenberg M, Gwynn MN. J Biol Chem. 2004;279:44883–44888. doi: 10.1074/jbc.M407882200. [DOI] [PubMed] [Google Scholar]
- 18.Theisen MJ, Misra I, Saadat D, Campobasso N, Miziorko HM, Harrison DH. Proc Natl Acad Sci U S A. 2004;101:16442–16447. doi: 10.1073/pnas.0405809101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koon N, Squire CJ, Baker EN. Proc Natl Acad Sci U S A. 2004;101:8295–8300. doi: 10.1073/pnas.0400820101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.White SW, Zheng J, Zhang YM, Rock Annu Rev Biochem. 2005;74:791–831. doi: 10.1146/annurev.biochem.74.082803.133524. [DOI] [PubMed] [Google Scholar]
- 21.Ferguson JJ, Jr, Rudney H. J Biol Chem. 1959;234:1072–1075. [PubMed] [Google Scholar]
- 22.Middleton B, Tubbs PK. Biochem J. 1974;137:15–23. doi: 10.1042/bj1370015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miziorko HM, Clinkenbeard KD, Reed WD, Lane MD. J Biol Chem. 1975;250:5768–5773. [PubMed] [Google Scholar]
- 24.Miziorko HM, Lane MD. Journal of Biological Chemistry. 1977;252:1414–1420. [PubMed] [Google Scholar]
- 25.Reed WD, Clinkenbeard D, Lane MD. J Biol Chem. 1975;250:3117–3123. [PubMed] [Google Scholar]
- 26.Clinkenbeard KD, Reed WD, Mooney RA, Lane MD. J Biol Chem. 1975;250:3108–3116. [PubMed] [Google Scholar]
- 27.Sutherlin A, Hedl M, Sanchez-Neri B, Burgner JW, 2nd, Stauffacher CV, Rodwell VW. J Bacteriol. 2002;184:4065–4070. doi: 10.1128/JB.184.15.4065-4070.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cornforth PG, JW, Messner B, Eggerer H. Eur J Biochem. 1974;42:591–604. doi: 10.1111/j.1432-1033.1974.tb03374.x. [DOI] [PubMed] [Google Scholar]
- 29.Miziorko HM, Behnke CE. Biochemistry. 1985;24:3174–3179. doi: 10.1021/bi00334a015. [DOI] [PubMed] [Google Scholar]
- 30.Miziorko HM, Behnke CE. J Biol Chem. 1985;260:13513–13516. [PubMed] [Google Scholar]
- 31.Misra I, Narasimhan C, Miziorko HM. J Biol Chem. 1993;268:12129–12136. [PubMed] [Google Scholar]
- 32.Rokosz LL, Boulton DA, Butkiewicz EA, Sanyal G, Cueto MA, Lachance PA, Hermes JD. Arch Biochem Biophys. 1994;312:1–13. doi: 10.1006/abbi.1994.1273. [DOI] [PubMed] [Google Scholar]
- 33.Steussy CN, Vartia AA, Burgner JW, 2nd, Sutherlin A, Rodwell VW, Stauffacher CV. Biochemistry. 2005;44:14256–14267. doi: 10.1021/bi051487x. [DOI] [PubMed] [Google Scholar]
- 34.Greenspan MD, Yudkovitz JB, Lo CY, Chen JS, Alberts AW, Hunt VM, Chang MN, Yang SS, Thompson KL, Chiang YC, et al. Proc Natl Acad Sci U S A. 1987;84:7488–7492. doi: 10.1073/pnas.84.21.7488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Misra I, Miziorko HM. Biochemistry. 1996;35:9610–9616. doi: 10.1021/bi9605797. [DOI] [PubMed] [Google Scholar]
- 36.Chun KY, Vinarov DA, Zajicek J, Miziorko HM. J Biol Chem. 2000;275:17946–17953. doi: 10.1074/jbc.M909725199. [DOI] [PubMed] [Google Scholar]
- 37.Wang CZ, Misra I, Miziorko HM. J Biol Chem. 2004;279:40283–40288. doi: 10.1074/jbc.M406566200. [DOI] [PubMed] [Google Scholar]
- 38.Pojer F, Ferrer JL, Richard SB, Nagegowda DA, Chye ML, Bach TJ, Noel JP. Proc Natl Acad Sci U S A. 2006;103:11491–11496. doi: 10.1073/pnas.0604935103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shafqat N, Turnbull A, Zschocke J, Oppermann U, Yue WW. J Mol Biol. 2010;398:497–506. doi: 10.1016/j.jmb.2010.03.034. [DOI] [PubMed] [Google Scholar]
- 40.Steussy CN, Robison AD, Tetrick AM, Knight JT, Rodwell VW, Stauffacher CV, Sutherlin AL. Biochemistry. 2006;45:14407–14414. doi: 10.1021/bi061505q. [DOI] [PubMed] [Google Scholar]
- 41.Durr IF, Rudney H. J Biol Chem. 1960;235:2572–2578. [PubMed] [Google Scholar]
- 42.Liscum L, Finer-Moore J, Stroud RM, Luskey KL, Brown MS, Goldstein JL. J Biol Chem. 1985;260:522–530. [PubMed] [Google Scholar]
- 43.Gill JF, Jr, Beach MJ, Rodwell VW. J Biol Chem. 1985;260:9393–9398. [PubMed] [Google Scholar]
- 44.Wilding EI, Kim DY, Bryant AP, Gwynn MN, Lunsford RD, McDevitt D, Myers JE, Jr, Rosenberg M, Sylvester D, Stauffacher CV, Rodwell VW. J Bacteriol. 2000;182:5147–5152. doi: 10.1128/jb.182.18.5147-5152.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Darnay BG, Wang Y, Rodwell VW. J Biol Chem. 1992;267:15064–15070. [PubMed] [Google Scholar]
- 46.Frimpong K, Rodwell VW. J Biol Chem. 1994;269:11478–11483. [PubMed] [Google Scholar]
- 47.Lawrence CM, Rodwell VW, Stauffacher CV. Science. 1995;268:1758–1762. doi: 10.1126/science.7792601. [DOI] [PubMed] [Google Scholar]
- 48.Tabernero L, Bochar DA, Rodwell VW, Stauffacher CV. Proc Natl Acad Sci U S A. 1999;96:7167–7171. doi: 10.1073/pnas.96.13.7167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Istvan ES, Palnitkar M, Buchanan SK, Deisenhofer J. Embo J. 2000;19:819–830. doi: 10.1093/emboj/19.5.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Istvan ES, Deisenhofer J. Science. 2001;292:1160–1164. doi: 10.1126/science.1059344. [DOI] [PubMed] [Google Scholar]
- 51.Endo A, Kuroda M, Tanzawa K. FEBS Lett. 1976;72:323–326. doi: 10.1016/0014-5793(76)80996-9. [DOI] [PubMed] [Google Scholar]
- 52.Tabernero L, Rodwell VW, Stauffacher CV. J Biol Chem. 2003;278:19933–19938. doi: 10.1074/jbc.M213006200. [DOI] [PubMed] [Google Scholar]
- 53.Tchen TT. J Biol Chem. 1958;233:1100–1103. [PubMed] [Google Scholar]
- 54.Beytia E, Dorsey KJ, Marr J, Cleland WW, Porter JW. J Biol Chem. 1970;245:5450–5458. [PubMed] [Google Scholar]
- 55.Dorsey KJ, Porter JW. J Biol Chem. 1968;243:4667–4670. [PubMed] [Google Scholar]
- 56.Voynova NE, Rios SE, Miziorko HM. J Bacteriol. 2004;186:61–67. doi: 10.1128/JB.186.1.61-67.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Andreassi JL, 2nd, Dabovic K, Leyh TS. Biochemistry. 2004;43:16461–16466. doi: 10.1021/bi048075t. [DOI] [PubMed] [Google Scholar]
- 58.Tanaka RD, Lee LY, Schafer BL, Kratunis VJ, Mohler WA, Robinson GW, Mosely ST. Proc Natl Acad Sci. 1990;87:2872–2876. doi: 10.1073/pnas.87.8.2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schafer BL, Bishop RW, Kratunis VJ, Kalinowski SS, Mosely ST, Gibson KM, Tanaka RD. J Biol Chem. 1992;267:13229–13238. [PubMed] [Google Scholar]
- 60.Potter D, Wojnar JM, Narasimhan C, Miziorko HM. J Biol Chem. 1997;273:5741–5746. doi: 10.1074/jbc.272.9.5741. [DOI] [PubMed] [Google Scholar]
- 61.Potter D, Miziorko HM. J Biol Chem. 1997;272:25449–25454. doi: 10.1074/jbc.272.41.25449. [DOI] [PubMed] [Google Scholar]
- 62.Hinson DD, Chambliss KL, Hoffmann GF, Krisans S, Keller RK, Gibson KM. J Biol Chem. 1997;272:26756–26760. doi: 10.1074/jbc.272.42.26756. [DOI] [PubMed] [Google Scholar]
- 63.Cho YK, Rios SE, Kim JJ, Miziorko HM. J Biol Chem. 2001;276:12573–12578. doi: 10.1074/jbc.M011478200. [DOI] [PubMed] [Google Scholar]
- 64.Chu X, Li D. Protein Expr Purif. 2003;32:75–82. doi: 10.1016/S1046-5928(03)00221-3. [DOI] [PubMed] [Google Scholar]
- 65.Chu X, Liu X, Yau M, Leung YC, Li D. Protein Expr Purif. 2003;30:210–218. doi: 10.1016/s1046-5928(03)00101-3. [DOI] [PubMed] [Google Scholar]
- 66.Yang D, Shipman LW, Roessner CA, Scott AI, Sacchettini JC. J Biol Chem. 2002;277:9462–9467. doi: 10.1074/jbc.M110787200. [DOI] [PubMed] [Google Scholar]
- 67.Fu Z, Wang M, Potter D, Miziorko HM, Kim JJ. J Biol Chem. 2002;277:18134–18142. doi: 10.1074/jbc.M200912200. [DOI] [PubMed] [Google Scholar]
- 68.Andreassi JL, 2nd, Bilder PW, Vetting MW, Roderick SL, Leyh TS. Protein Sci. 2007;16:983–989. doi: 10.1110/ps.072755707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fu Z, Voynova NE, Herdendorf TJ, Miziorko HM, Kim JJ. Biochemistry. 2008;47:3715–3724. doi: 10.1021/bi7024386. [DOI] [PubMed] [Google Scholar]
- 70.Hellig H, Popjak G. Journal of Lipid Research. 1961;2:235–243. [Google Scholar]
- 71.Bazaes S, Beytia E, Jabalquinto AM, Solis de Ovando F, Gomez I, Eyzaguirre J. Biochemistry. 1980;19:2300–2304. doi: 10.1021/bi00552a003. [DOI] [PubMed] [Google Scholar]
- 72.Smit A, Mushegian A. Genome Res. 2000;10:1468–1484. doi: 10.1101/gr.145600. [DOI] [PubMed] [Google Scholar]
- 73.Eyzaguirre J, Valdebenito D, Cardemil E. Arch Biochem Biophys. 2006;454:189–196. doi: 10.1016/j.abb.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 74.Pilloff D, Dabovic K, Romanowski MJ, Bonanno JB, Doherty M, Burley SK, Leyh TS. J Biol Chem. 2003;278:4510–4515. doi: 10.1074/jbc.M210551200. [DOI] [PubMed] [Google Scholar]
- 75.Doun SS, Burgner JW, 2nd, Briggs SD, Rodwell VW. Protein Sci. 2005;14:1134–1139. doi: 10.1110/ps.041210405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chambliss KL, Slaughter CA, Schreiner R, Hoffmann GF, Gibson KM. J Biol Chem. 1996;271:17330–17334. doi: 10.1074/jbc.271.29.17330. [DOI] [PubMed] [Google Scholar]
- 77.Hinson DD, Chambliss KL, Toth MJ, Tanaka RD, Gibson KM. J Lipid Res. 1997;38:2216–2223. [PubMed] [Google Scholar]
- 78.Herdendorf TJ, Miziorko HM. Biochemistry. 2006;45:3235–3242. doi: 10.1021/bi052231u. [DOI] [PubMed] [Google Scholar]
- 79.Herdendorf TJ, Miziorko HM. Biochemistry. 2007;46:11780–11788. doi: 10.1021/bi701408t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Andreassi JL, 2nd, Leyh TS. Biochemistry. 2004;43:14594–14601. doi: 10.1021/bi048963o. [DOI] [PubMed] [Google Scholar]
- 81.Olson AL, Yao H, Herdendorf TJ, Miziorko HM, Hannongbua S, Saparpakorn P, Cai S, Sem DS. Proteins. 2009;75:127–138. doi: 10.1002/prot.22228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Olson AL, Cai S, Herdendorf TJ, Miziorko HM, Sem DS. J Am Chem Soc. 2010;132:2102–2103. doi: 10.1021/ja906244j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Romanowski MJ, Bonanno JB, Burley SK. Proteins. 2002;47:568–571. doi: 10.1002/prot.10118. [DOI] [PubMed] [Google Scholar]
- 84.Andreassi JL, 2nd, Vetting MW, Bilder PW, Roderick SL, Leyh TS. Biochemistry. 2009;48:6461–6468. doi: 10.1021/bi900537u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chang Q, Yan XX, Gu SY, Liu JF, Liang DC. Proteins. 2008;73:254–258. doi: 10.1002/prot.22151. [DOI] [PubMed] [Google Scholar]
- 86.Bloch K, Chaykin S, Phillips AH, De Waard A. J Biol Chem. 1959;234:2595–2604. [PubMed] [Google Scholar]
- 87.Berges T, Guyonnet D, Karst F. J Bacteriol. 1997;179:4664–4670. doi: 10.1128/jb.179.15.4664-4670.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Alvear M, Jabalquinto AM, Eyzaguirre J, Cardemil E. Biochemistry. 1982;21:4646–4650. doi: 10.1021/bi00262a020. [DOI] [PubMed] [Google Scholar]
- 89.Skilleter DN, Kekwick RG. Biochem J. 1971;124:407–417. doi: 10.1042/bj1240407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Byres E, Alphey MS, Smith TK, Hunter WN. J Mol Biol. 2007;371:540–553. doi: 10.1016/j.jmb.2007.05.094. [DOI] [PubMed] [Google Scholar]
- 91.Chiew YE, O’Sullivan WJ, Lee CS. Biochim Biophys Acta. 1987;916:271–278. doi: 10.1016/0167-4838(87)90170-1. [DOI] [PubMed] [Google Scholar]
- 92.Michihara SMA, Nara Y, Ikeda K, Yamori Y. Purification and characterization of two mevalonate pyrophosphate decarboxylases from rat liver: a novel molecular species of 37 kDa. 1997;122:647–654. doi: 10.1093/oxfordjournals.jbchem.a021802. [DOI] [PubMed] [Google Scholar]
- 93.Krepkiy D, Miziorko HM. Protein Sci. 2004;13:1875–1881. doi: 10.1110/ps.04725204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Toth MJ, Huwyler L. J Biol Chem. 1996;271:7895–7898. doi: 10.1074/jbc.271.14.7895. [DOI] [PubMed] [Google Scholar]
- 95.Voynova NE, Fu Z, Battaile KP, Herdendorf TJ, Kim JJ, Miziorko HM. Arch Biochem Biophys. 2008;480:58–67. doi: 10.1016/j.abb.2008.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jabalquinto AM, Eyzaguirre J, Cardemil E. Arch Biochem Biophys. 1983;225:338–343. doi: 10.1016/0003-9861(83)90038-3. [DOI] [PubMed] [Google Scholar]
- 97.Jabalquinto AM, Cardemil E. Biochim Biophys Acta. 1987;916:172–178. doi: 10.1016/0167-4838(87)90105-1. [DOI] [PubMed] [Google Scholar]
- 98.Iyengar R, Cardemil E, Frey PA. Biochemistry. 1986;25:4693–4698. doi: 10.1021/bi00364a036. [DOI] [PubMed] [Google Scholar]
- 99.Nave JF, d’Orchymont H, Ducep JB, Piriou F, Jung MJ. Biochem J. 1985;227:247–254. doi: 10.1042/bj2270247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Reardon JE, Abeles RH. Biochemistry. 1987;26:4717–4722. doi: 10.1021/bi00389a018. [DOI] [PubMed] [Google Scholar]
- 101.Dhe-Paganon S, Magrath J, Abeles RH. Biochemistry. 1994;33:13355–13362. doi: 10.1021/bi00249a023. [DOI] [PubMed] [Google Scholar]
- 102.Vlattas I, Dellureficio J, Ku E, Bohacek R, Zhang X. Bioorg Med Chem Lett. 1996;6:2091–2096. [Google Scholar]
- 103.Qiu Y, Li D. Biochim Biophys Acta. 2006;1760:1080–1087. doi: 10.1016/j.bbagen.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 104.Qiu Y, Gao J, Guo F, Qiao Y, Li D. Bioorg Med Chem Lett. 2007;17:6164–6168. doi: 10.1016/j.bmcl.2007.09.033. [DOI] [PubMed] [Google Scholar]
- 105.Lefurgy ST, Rodriguez SB, Park CS, Cahill S, Silverman RB, Leyh TS. J Biol Chem. 2010;285:20654–20663. doi: 10.1074/jbc.M109.098350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Krepkiy DV, Miziorko HM. Biochemistry. 2005;44:2671–2677. doi: 10.1021/bi0484217. [DOI] [PubMed] [Google Scholar]
- 107.Weerasinghe S, Samantha Dassanayake R. J Mol Model. 2010;16:489–498. doi: 10.1007/s00894-009-0561-7. [DOI] [PubMed] [Google Scholar]
- 108.Bonanno JB, Edo C, Eswar N, Pieper U, Romanowski MJ, Ilyin V, Gerchman SE, Kycia H, Studier FW, Sali A, Burley SK. Proc Natl Acad Sci U S A. 2001;98:12896–12901. doi: 10.1073/pnas.181466998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sullivan SM, Holyoak T. Biochemistry. 2007;46:10078–10088. doi: 10.1021/bi701038x. [DOI] [PubMed] [Google Scholar]
- 110.Grochowski LL, Huimin X, White RHJ. Bacteriol. 2006;188:3192–3198. doi: 10.1128/JB.188.9.3192-3198.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chen M, Poulter CD. Biochemistry. 2010;49:207–217. doi: 10.1021/bi9017957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mabanglo MF, Schubert HL, Chen M, Hill CP, Poulter CD. ACS Chem Biol. 2010;5:517–527. doi: 10.1021/cb100032g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dellas N, Noel JP. ACS Chem Biol. 2010;5:589–601. doi: 10.1021/cb1000313. [DOI] [PMC free article] [PubMed] [Google Scholar]