

SUMMARY
The unconventional motor protein, myosin VI, is known to dimerize upon cargo-binding to its C-terminal end. It has been shown that one of its tail domains, called the medial tail domain, is a dimerization region. The domain contains an unusual pattern of alternating charged-residues and a few hydrophobic residues. To reveal the unknown dimerization mechanism of the medial tail domain, we employed molecular dynamics and single-molecule experimental techniques. Both techniques suggest that the formation of electrostatic-based inter-helical salt bridges between oppositely-charged residues is a key dimerization factor. For the dimerization to occur, the two identical helices within the dimer don't bind in a symmetric fashion, but rather with an off-set of about one helical repeat. Calculations of the dimer-dissociation energy find the contribution of hydrophobic residues to the dimerization process to be minor; they also find that the asymmetric homodimer state is energetically favorable over a state of separate helices.
INTRODUCTION
Myosin VI, first identified in Drosophila melanogaster (Kellerman, 1992) and shown schematically in Figure 1, is an actin-based molecular motor in eukaryotic cells. Among 35 myosin superfamily members (Odronitz and Kollmar, 2007), myosin VI occupies a special place since, unlike other myosins, it moves toward the minus (pointed) end of the actin filaments (Wells, et al., 1999). This directionality was revealed to be caused by the lever arm redirection brought about through a unique insert 2 positioned between the converter domain and the IQ motif (Menetrey, et al., 2005; Park, et al., 2007).
Figure 1.
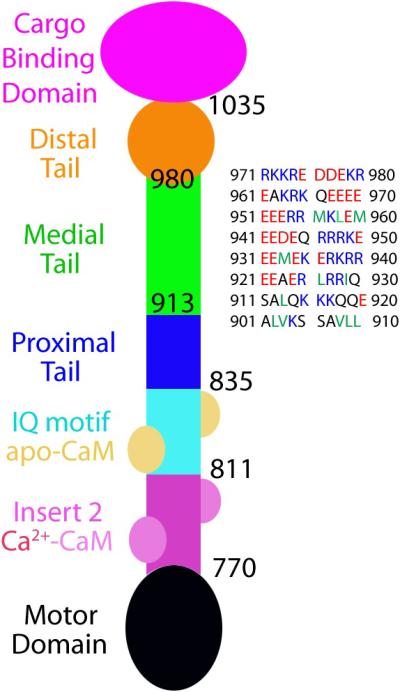
Schematic diagram of myosin VI construct. The N-terminal region starts from a motor domain (black), which is followed by unique insert 2 (pink) and IQ motif (cyan). Insert 2 binds a calmodulin (CaM, light pink) with 4 calcium ions, and the IQ motif binds an apo-CaM (brownish yellow). The proximal tail (PT) domain (blue) consists of a three-helix bundle and serves as the lever-arm extension upon dimerization (Mukherjea, et al., 2009). The medial tail (MT) domain (green) shows a pattern of alternating charged residues with only a small number of hydrophobic residues; it is followed by the distal tail (DT) domain (orange) and the cargo-binding domain (CBD, purple). The sequence of the MT domain is displayed on the right. The color scheme for the displayed sequence is adopted from Spink et al. (Spink, et al., 2008); blue, red, and green colors denote positively- and negatively-charged residues and hydrophobic residues, respectively. The color scheme for the myosin VI diagram is adopted from Phichith et al. (Phichith, et al., 2009).
Like other myosins, myosin VI has a motor domain where ATP (adenosine triphosphate) binds and hydrolysis occurs. The motor domain, which begins at the N-terminus, also contains an actin-binding region and its conformation is tightly regulated by the nucleotide state. As shown in Figure 1, the motor domain is followed by the unique insert 2 and IQ motif, which together form a short (less than 10 nm) lever arm of myosin VI. In addition to the IQ motif, insert 2 also binds calmodulin (CaM) (Bahloul, et al., 2004). The region between the myosin VI C-terminus and the lever arm is known as the tail domain. Following the terminology of Spink et al., the tail domain is divided into four sub-domains: Proximal tail (PT), medial tail (MT), distal tail (DT) domains, and cargo-binding domain (CBD) (Spink, et al., 2008).
Despite its short lever arm, myosin VI can take a large step (30~36 nm) (Nishikawa, et al., 2002; Okten, et al., 2004; Rock, et al., 2001) with a broad step size distribution (Yildiz, et al., 2004). Owing to the conformational rearrangement of the converter domain itself in the pre-powerstroke state (Menetrey, et al., 2007), myosin VI has a large (12~18 nm) powerstroke size (Bryant, et al., 2007; Lister, et al., 2004; Menetrey, et al., 2007; Rock, et al., 2005). Yet, the large average step size is difficult to explain in the context of the short lever arm. Spink et al. (Spink, et al., 2008) and Sivaramakrishnan et al. (Sivaramakrishnan, et al., 2008; Sivaramakrishnan, et al., 2009) suggest that the MT domain consists of a stiff, ~10 nm long, stable single α-helix (SAH) with a 15 nm persistence length and provides myosin VI with the additional extension required for the observed large step. However, recently we demonstrated that the MT domain is actively involved in myosin VI dimerization and does not contribute to the myosin VI step size. Instead, extension needed for the large step size should derive from an unfolding of the three-helix bundle of the PT domain that occurs upon myosin VI dimerization (Mukherjea, et al., 2009). In the present study, we investigate the dimerization properties of the MT domain.
The most prominent property of the MT domain (residue number 913-980) is that it exhibits a pattern of four positive residues followed by four negative residues, called the ER/K motif, that is repeated, except for a few hydrophobic residues near the N-terminal side of the MT domain (Figure 1). We hypothesized that these hydrophobic residues act as a hydrophobic core, forming a short coiled-coil (Mukherjea, et al., 2009). In the present study, we investigate how far the ER/K motif contributes to the dimerization of the myosin VI MT domain.
For this purpose, we employ molecular dynamics (MD) and experimental single-molecule methods. Dimerization of α-helical proteins has been investigated previously using MD (Henin, et al., 2005; Hsin, et al., 2009), and we adopt a similar treatment in the present investigation.
Our MD simulation results confirm indeed that positive and negative residues across different monomers can interact to form a stable homo-dimer. In order for this to happen, the two identical monomers must assume, however, a spatial offset with respect to each other, such that positive residues on one monomer interact with negative residues on the other. In agreement with the simulation, experimental data support the key roles of Coulomb attraction between positive and negative residues participating in myosin VI dimerization. It is observed, furthermore, that mutations of hydrophobic residues on the MT domain do not abolish myosin VI dimerization, suggesting that Coulomb attraction in the form of salt bridges is the major contributor to dimerization. This conclusion is also supported by calculations of dimerization energies.
RESULTS
Myosin VI MT domain is capable of self-association
It was suggested that the myosin VI MT domain undergoes dimerization both in the case of a truncated 991 construct (residues 1 to 991) (Park, et al., 2006), which does not have a cargo-binding domain, and in the case of a further truncated 940 construct (residues 1 to 940) (Mukherjea, et al., 2009). To discern how the MT domain dimerizes, MD calculations were carried out for two constructs of the MT domain, a full MT domain segment consisting of residues 907 to 980 and a truncated MT domain segment consisting of residues 907 to 940. First, a microsecond simulation was conducted starting with two free MT segments, initially separated by 30 Å (Figure 2A; Figure S1, Movie S1). This simulation was performed in a so-called coarse-grained representation (see Methods), which permits surveying of the microsecond timescale relevant for self-assembly processes (Shih, et al., 2007a; Shih, et al., 2007b). During the simulation, the two MT domain segments were seen to associate after 500 ns starting from their N-terminal ends, with the association reaching the C-terminal ends around 950 ns. To investigate the molecular interactions driving the association, all-atom resolution was recovered from the coarse-grained representation and the system then was subjected to a 50-ns equilibrium MD simulation (Figure 2B; Movie S2). During this simulation, the still separated C-terminal ends were seen to also associate, completing the dimerization as recognized through a rise of buried molecular surface between the two associating segments (Figure 2C).
Figure 2.

Dimerization of the myosin VI MT domain (907 to 980). (A) Spontaneous self-association of the MT segments in a 1-μs RBCG simulation. The MT segments are colored according to residue type (red: negatively charged; blue: positively charged; white: hydrophobic; green: polar). (B) All-atom equilibrium MD of the MT dimer over a 50-ns duration. The dimer is seen to be stable with a significant buried joint molecular surface (black trace in (C)). The red trace in (C) is the buried molecular surface of the hydrophobic residues. The MT dimer contact map is shown in (D), with five observed salt-bridges displayed in the insets. The main inter-helical interactions are seen to lie below the diagonal, indicating a vertical offset between the helices that can be also recognized in (B); the diagonal in the contact map is indicated by a dashed line. See also Supplemental Movies S1, S2, S3 and S4 and Supplemental Figures S1 and S2.
A vertical offset developed between the MT domain segments, with one segment positioned few residues above the other. This offset was identified also in the contact map between the two segments, depicted in Figure 2D, highlighting the key inter-segment interactions. The map shows the region of high molecular interaction to be consistently shifted below the diagonal, the shift corresponding to the vertical offset mentioned. The shift brings the charged residues in one segment in contact with oppositely charged residues of the other segment, permitting thereby formation of salt bridges. Indeed, five salt bridges are seen to form in the modeled MT domain dimer, shown in the inset of Figure 2D.
The dimerization propensity of the truncated MT construct, being 40 amino acids less than the full-length construct and including residues 907 to 940, was tested through the same MD protocol as the full construct. The results are summarized in Figure S2 in Supplemental Information. Spontaneous association of the two MT segments was observed again in a microsecond coarse grained MD simulation (Movie S3). During the subsequent all-atom MD simulation, the two segments remained stable (Movie S4). Formation of three salt bridges was observed and a vertical offset similar to the one in Figure 2D is seen in the contact map (Figure S2).
Buffer with higher ionic strength impedes dimerization of myosin VI
Based on the above simulations, we hypothesized that the electrostatic interactions between the positive and negative residues in each MT domain play an essential role in domain-domain aggregation. Consequently, increasing the ionic strength of the myosin VI buffer should reduce the propensity for dimerization. To test this hypothesis, actin filaments were decorated with high concentration (100 nM) of the truncated myosin VI 940 construct (a gift from Sweeney lab). This tends to hold the monomeric myosins in close proximity. Upon adding ATP, a certain percentage of the myosins begin to walk along the actin filament processively, implying that they are properly dimerized. This percentage of processive myosins was calculated for two different buffer conditions, one with 52 mM ionic strength, the other with 149 mM ionic strength (see Methods). As expected, the buffer with higher ionic strength impeded the dimerization and processivity of the myosin VI 940 construct. While 14.0 ± 3.8 % (average ± s.d.) of the construct was processive with the 52 mM ionic strength buffer, only 3.8 ± 2.9 % of the myosins were processive with the 149 mM one (Figure 3).
Figure 3.
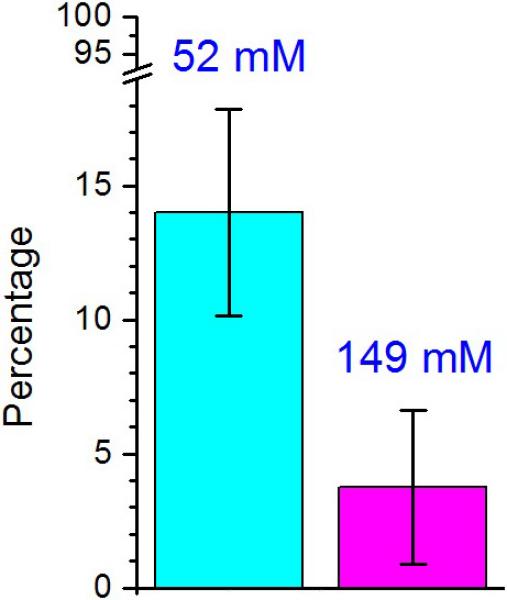
Percentages of dimerized myosins at different ionic strengths. The percentage of processive myosins was measured with 52 mM and 149 mM buffer. 14.0±3.8 % and 3.8±2.9 % (average ± s.d.) of the construct properly dimerized and walked processively for buffers with 52 mM and 149 mM ionic strengths, respectively. Percentages were calculated from six separated measurements. A total of 764 and 1037 myosins were counted for buffers with 52 mM and 149 mM ionic strength, respectively. Error bars represent standard deviation.
Mutations of hydrophobic residues in the medial tail domain do not abolish dimerization
From the simulations and experiments above we are led to believe that the bonding of positive and negative residues on different monomers is a key for the dimerization of myosin VI MT domains. However, a question remains whether the hydrophobic residues of the MT domains also contribute to dimerization. To address this question, we reasoned that disrupting hydrophobic residues in the medial tail should inhibit dimerization if hydrophobic interactions were the predominant dimerization mechanism. Specifically, five glycines were substituted for hydrophobic residues in the 940 construct, namely L909, L913, L926, I929, M933.
Again the actin saturation method (with 50 nM of myosin concentration) was employed. Surprisingly, the mutant 940 construct (a gift from Sweeney lab) showed processive movements after ATP addition, indicating properly dimerized constructs. We then determined the step sizes of the mutated construct by tracking the position of a CY3 dye labeled on the IQ-Calmodulin using the single-molecule FIONA (Fluorescence Imaging with One-Nanometer Accuracy) technique (Yildiz, et al., 2003). The mutant construct showed a typical stair-like trajectory (Figure 4A), and the average step size (± s.d.) was 53.3 ± 19.1 nm for forward steps (Figure 4B). This value is nearly indistinguishable from that of the wild-type (WT) 940 construct of 54.3 ± 19.4 nm, which we have recently reported (Mukherjea, et al., 2009). It is interesting to note that the mutated construct also has a wide step size distribution.
Figure 4.

Insensitivity of processivity to mutations of hydrophobic residues in the medial tail. (A) Example trace of the mutant 940 construct (see text). The graph plots the position of the fluorescent labeled CY3 on one of the IQ-CaMs along actin filament versus time. Calculated step sizes are denoted by numbers in the graph. (B) Measured step size histogram for the mutant 940 construct. The average forward step size is 53.3±19.1 nm (N=143) and the average backward step size is -26.3 nm (N=1). (C) Comparison of the percentage of properly dimerized myosin VI 940 for WT and mutant-type constructs. A fraction of 5.1±3.5 % and 6.6±4.3 % (average ± s.d.) of myosin VI was seen to be processive for mutant- and WT constructs, respectively. Percentages were calculated from nine separate measurements; a total of 1007 and 995 myosins was counted for mutant- and WT, respectively. Ionic strength of buffer was 52 mM for both cases.
After observing no significant difference between the step sizes of wild type and mutant constructs, we proceeded to determine if there is a difference in the efficiency of dimerization. When experiments were performed in the same conditions, the percentages (± s.d.) of processive myosin constructs for the mutant and the WT 940 constructs were 5.1 ± 3.5 % and 6.6 ± 4.3 %, respectively, showing again no significant difference between them.
Dimerization of the mutated construct tested in MD
The experiments above suggest that dimerization is possible between mutated MT segments. To investigate the molecular interactions that contribute to such dimerization, MD simulations were carried out to compare the dimerization of WT and mutant MT domains. The full MT-domain dimer resulting from the all-atom equilibrium MD described above was truncated at residue 940, resulting in a shorter WT dimer, which was then subjected to equilibrium MD for another 30 ns (Movie S5). In a separate all-atom simulation, mutations were introduced into the truncated WT dimer, in which five hydrophobic residues were replaced by glycines, in accordance with the experimental setup, and the mutated dimer was similarly subjected to a 30-ns equilibrium MD (Movie S6). In both all-atom simulations, the dimers remained associated (Figure 5A for the WT construct; Figure 5B for the mutated construct). The buried molecular surface between the MT segments remained high for both cases (Figure 5C). The only discernible difference between WT and mutant is that the contribution to inter-helical interactions from the hydrophobic residues is less in the mutant case (Figure 5C; purple trace: wild type; blue trace: mutant).
Figure 5.
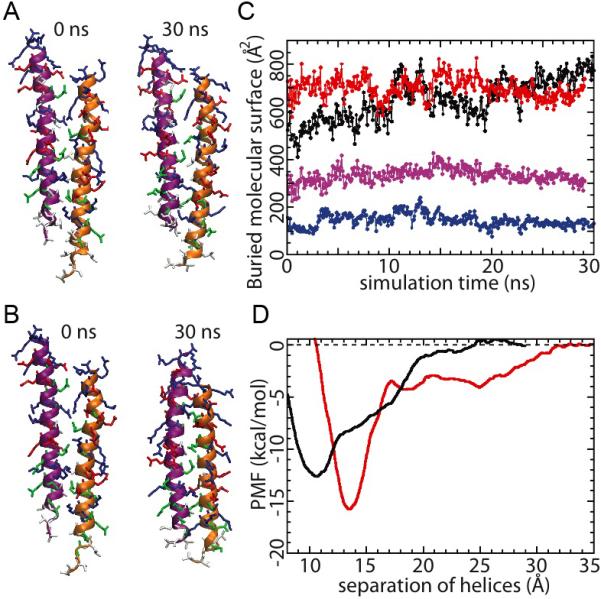
Equilibrium MD of truncated (907-940) MT segment dimers. Both WT (A) and mutant (B) dimers remained stable over the simulation. (C) Evolution of buried molecular surface area between the dimerized MT segments (red: WT dimer; black: mutated dimer; purple: buried molecular surface area from the hydrophobic residues of the WT dimer; blue: buried molecular surface area from the hydrophobic residues of the mutated dimer). (D) Potential of mean force characterizing the dimerization of the MT segments (red: WT; black: mutated). See also Supplemental Movies S5 and S6.
To quantitatively address the dimerization propensity of both WT and mutant constructs, free energy calculations were performed on both dimers to provide an estimate of the dimer disassociation energy (Figure 5D; red trace: wild type; black trace: mutant). The dimerization free energy, estimated from the plot of the potential of mean force (PMF) as a function of the helix-helix distance (Figure 5D), was calculated to be 15.8 ± 0.4 kcal/mol (1.67 ± 0.04 kcal/mol per helical turn) for the WT construct, and 12.6 ± 0.2 kcal/mol (1.33 ± 0.02 kcal/mol per helical turn) for the mutant construct. The result suggests that the WT construct has slightly higher dimerization strength, albeit by just a few kcal/mol. This is consistent with experiment where we found no significant difference between the ability to dimerize for the WT and mutant myosin VI. The dimerization free energies measured here compare reasonably well to that of the GCN4 leucine zipper (1.68 kcal/mol per helical turn) (Gebhardt, et al., 2010), which implies that the values for WT and mutant myosin VI's are neither particularly large nor particularly small. From Figure 5D, one can also discern that the mutated dimer prefers closer helix-helix packing, as the local energy minimum occurs at a smaller helix separation, possibly due to the smaller side-chain size of the glycines.
DISCUSSION
The average step size of myosin is related to the number of calmodulins (CaMs) and/or CaM-like light chains of the protein, and it had been shown that engineered myosin V with smaller number of IQ-domains has a shorter average step size than native myosin V has (Sakamoto, et al., 2005). Myosin VI challenged this relationship, since its step size is comparable to that of myosin V despite having only two CaM binding sites (Bahloul, et al., 2004) compared to six for myosin V. Spink et al. proposed then a model in which the MT domain, a region with a stable single α-helix (SAH), allows myosin VI to take such a large step, dimerization taking place in the cargo-binding domain (CBD) (Spink, et al., 2008). However, based on experiments with truncated 940 constructs (Mukherjea, et al., 2009) along with 1050 and 991 constructs (Park, et al., 2006), all of which do not possess a CBD, we showed that the MT domain is actually a dimerization region. Furthermore, we showed that the three-helix bundle unfolding upon myosin dimerization is the key contributor to myosin VI's large step size (Mukherjea, et al., 2009). The question naturally arises what interactions between the two MT domains give rise to dimerization.
The SAH was first experimentally identified for myosin X among myosin family members (Knight, et al., 2005), and recently, several more charged single α-helices (CSAH), particularly the ER/K motifs, have been identified (Sivaramakrishnan, et al., 2008; Suveges, et al., 2009), with the MT domain of myosin VI being a prominent example. Although the properties of the ER/K motif have been investigated by various groups (Lyu, et al., 1992; Sivaramakrishnan, et al., 2008; Sivaramakrishnan, et al., 2009; Suveges, et al., 2009), no detailed study on the interaction between two ER/K motifs in myosin VI had been reported so far. In our previous myosin VI publications, we simply speculated that a few hydrophobic residues in the MT domain form a short coiled-coil structure (Mukherjea, et al., 2009; Phichith, et al., 2009). Indeed, the PAIRCOIL algorithm (Berger, et al., 1995) predicts a strong coiled-coil tendency in the MT domain based on its sequence (Rock, et al., 2005). However, it was known that PAIRCOIL could misrepresent CSAHs (Suveges, et al., 2009). The MD simulations performed in the present study suggest that electrostatic interactions through several inter-helical salt bridges, made possible by a small vertical offset (~10 Å) between the MT helices, are a key contributor to the potential dimerization of MT segments. It is then understandable why our prior circular dichroism (CD) data of an artificially dimerized PT-MT construct (834-991-GCN4) did not find a significant amount of coiled coil, but rather demonstrated a high level of α-helical content even after dimerization of the construct (Mukherjea, et al., 2009). This earlier circular dichroism observation is compatible with the results of our present simulation and experiments, since by interacting via electrostatic salt bridges, two SAHs would actually maintain their α-helical structure (without forming a large segment of coiled-coil along the entire MT domain). We do note, however, that formation of a short coiled-coil from the few MT domain hydrophobic residues, not giving a discernable change in the CD spectra, is still possible.
The experimental observation of lower dimerization propensity with higher ionic strength in the buffer reported here further implicates electrostatic interaction mediated by salt bridges to play a role in MT domain dimerization. If formation of salt-bridges were not the dimerization mechanism, we would not have been able to observe such a significant dimerization dependence on ionic strength of buffer. In a buffer with higher ionic strength, interstitial ions weaken inter-helical interactions between two MT domains (and perhaps also intra-helical interactions that are responsible for the formation of SAH).
In this study we successfully combined MD methods with single-molecule experiments to help answer questions about myosin VI dimerization. While the MD methods applied suggest how the myosin VI MT domains undergo dimerization, we do not claim that the all-atom model of the MT domain dimer revealed in the present study to be a unique dimerization conformation. However, it seems very likely that the dimerized MT domains exhibit a slight vertical offset as observed computationally, which enables electrostatic interactions via salt bridges between the two helices. However, the formation of salt bridges is possibly non-specific, i.e., there might be multiple feasible conformations involving different sets of amino acid forming salt bridges for a given myosin VI MT dimer. It is also conceivable that the vertical offset seen in this study might provide the PT domain with some shearing force which enables the PT domain unfolding. Clearly, more studies are necessary to test this conjecture.
Additional evidence for a key role of salt bridges in the dimerization of MT domains further supports our findings. If hydrophobic interactions were the only contributor to dimerization, then substitution of five hydrophobic residues in the truncated 940 construct (1-940) into five (small size) glycines should significantly lower the dimerization propensity of the construct. However, single-molecule experiments showed that the mutated 940 construct actually does not show a significant decrease in terms of probability of dimerization compared to that of the WT 940 construct (5.1 % vs. 6.6 %) (Figure 4C). It should be noted, though, that we do not rule out the participation of hydrophobic interactions in the dimerization of MT helices, but it is unclear how much the hydrophobic residues contribute to the dimerization of the MT segments, given the close values (5.1% vs. 6.6%). Certainly, the experiments with the mutant construct reveal that hydrophobic interactions (if any) are not the dominant dimerization mechanism and strongly suggest that there must be something else controlling dimerization. Combining all experimental and simulation data, we are led to believe that the key role is played by inter-helical salt bridges. It is interesting to note that salt bridges play various roles – contributing not only to dimerization mechanism of myosin VI, but also to motor function in the rigor to post-rigor transition of various myosins (Tehver and Thirumalai, 2010).
The MD simulations appear to support the experimental observation and show that a mutant MT dimer remains structurally stable much like the WT counterpart. Calculations on the dimerization free energy of WT and mutant dimers confirm that both constructs require significant amount of energy to disassociate; however, the dimerization of the mutated construct is possibly weaker than that of the WT construct by a few kcal/mol, which could affect myosin VI's intramolecular strain modulation.
Recently, two groups showed that cargo-binding mediates dimerization of myosin VI (Phichith, et al., 2009; Yu, et al., 2009), implying that cargo-binding plays a role in bringing two monomers close enough for association. According to our previous model, cargo-binding initiated dimerization at the distal-end of the tail domain brings about the dimerization of the more proximal parts via a short coiled-coil formation in the hydrophobic amino acid region of the MT domain; then the three-helix bundle of the PT domain unfolds and provides a large portion of the step size (Mukherjea, et al., 2009; Phichith, et al., 2009). Our present study modifies this model by suggesting that the internal dimerization of the MT domain is held together via the formation of salt bridges alone or in combination with a short coiled-coil (Figure 6).
Figure 6.

Present model of the dimerized full-length myosin VI. The color scheme is adopted from Figure 1, except for the motor domain, which is shown in dark grey. MT domain (green) is the key dimerization region; the unfolded three-helix PT domain (blue) provides myosin VI with sufficient step length for the two motor domains (dark grey) to span a 36 nm step distance on the actin filament. There is a slight (~10 Å) offset in the relative placement of the two MT domain segments.
Several questions remain to be answered. For example, what is the functional rational for myosin VI to adopt the stated dimerization strategy in a cellular environment? Are the few hydrophobic residues in the MT domain actually a part of the PT domain, providing even additional length to its step size, or do they indeed form a short coiled-coil? All of the above questions await more experimental and theoretical work.
EXPERIMENTAL PROCEDURES
In this section the experimental and computational protocols are summarized. Further information can be found in the references cited and in Supplemental Information.
Molecular dynamics protocols
To investigate the self-association of the myosin VI MT constructs, the molecular dynamics method was employed using a so-called coarse-grained representation that allows exploring the simulated system on a micro-second timescale, the timescale relevant for the spontaneous assembly of small protein aggregates (Shih, et al., 2007a; Shih, et al., 2007b). The representation used in the present case is the residue-based coarse-graining method (RBCG) (Shih, et al., 2006). All-atom resolution can be recovered from an RBCG system (Shih, et al., 2009) to fully preserve the secondary structure of the MT segments, a flexible fitting method (Trabuco, et al., 2008; Trabuco, et al., 2009; Wells, et al., 2007) was employed, described in Supplemental Information. Equilibrium MD at all-atom resolution can then be performed using standard protocols, reviewed in Phillips et al., 2005 (Phillips, et al., 2005), with the simulation parameters given in Supplemental Information. Calculations of the association free energy were carried out using the adaptive biasing method (Darve and Pohorille, 2001; Henin, et al., 2005; Hénin and Chipot, 2004; Rodriguez-Gomez, et al., 2004). A listing of all simulations performed is given in Table S1 in Supplemental Information.
Dimerization initiation and single-molecule analysis
Myosin VI monomer construct was induced to dimerize as previously explained (Mukherjea, et al., 2009). The following procedures were applied: F-actin (one biotinylated G-actin with six WT G-actins) was immobilized onto the glass surface of a custom-made flow chamber through BSA-biotin (1.6 mg/ml, Sigma Aldrich) and neutravidin (0.8 mg/ml, Pierce) linkages, then 5 mg/ml casein was incubated to block non-specific protein binding for 5~6 minutes. Protein construct with high concentration (100 nM for experiments with 52 mM and 149 mM ionic strengths; 50 nM for experiments of wild- and mutant-type comparison) was incubated to induce its dimerization. Introduction of imaging buffer including ATP (20 μM for step size measurement; 40 μM for the calculation of percentage of processive movement), 5~10 minutes after adding high concentration of myosins, initiated processive movements of the properly dimerized myosin construct. In addition to ATP, the imaging buffer contained 2.5 mM PCA (protocatechuic acid) / 50 nM PCD (protocatechuate-3,4-dioxygenase) as an oxygen scavenging system (Aitken, et al., 2008), 20 % trolox (we saturated trolox in the buffer at physiological pH, then filtered, and 20 μL was used for a total of 100 μL imaging buffer), and surface blocking proteins (0.8 mg/ml BSA, 0.4 mg/ml casein). The pH of experimental buffer was ~7.1 and excess CaMs (0.09 mg/ml) were included in the buffer. The only difference between buffers (20 mM HEPES, 2 mM MgCl2, 2 mM EGTA, 1 mM DTT, KCl) with 52 mM and 149 mM ionic strengths was the concentration of KCl (25 mM for 52 mM ionic strength, 122 mM for 149 mM ionic strength). CY3 labeled on myosin construct was excited by 532 nm laser (CrystaLaser) and emission signals were collected by an EMCCD camera (Andor technology). The center coordinates of the diffraction-limited CY3 signal were calculated using the FIONA (Fluorescence Imaging with One-Nanometer Accuracy) technique (Yildiz, et al., 2003); myosin steps were identified by t-Test coded for by IDL (ITT visual information solutions). For the calculation of the percentage of properly dimerized myosins, myosins showing movements longer than 3 pixels (around 0.3 μm) after adding ATP were considered as properly dimerized processive myosins. The percentage was calculated from the number of moving spots, which satisfy this threshold, divided by the number of total spots in the field-of-view (around 30 × 30 pixels) of interest.
Supplementary Material
ACKNOWLEDGEMENTS
We thank H. Lee Sweeney, Monalisa Mukherjea, and Daniel Safer for their generous gifts for wild- and mutant-type myosin VI 940 constructs (funded by RO1-DC-009100), which were used for experiments in Figures 3 and 4; Anne Houdusse for helpful discussion and for providing us with coordinate information before its release on the PDB. This work is supported by the following grants: NSF PHY0822613 (to P.R.S. and K.S.), NIH GM068625 (to P.R.S.), NIH P41-RR005969 and NIH GM073655 (to K.S.). Computer time was provided via Large Resources Allocation Committee grant MCA93S02. Molecular images were rendered with VMD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Aitken CE, Marshall RA, Puglisi JD. An oxygen scavenging system for improvement of dye stability in single-molecule fluorescence experiments. Biophys. J. 2008;94:1826–1835. doi: 10.1529/biophysj.107.117689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahloul A, Chevreux G, Wells AL, Martin D, Nolt J, Yang Z, Chen LQ, Potier N, Van Dorsselaer A, Rosenfeld S, Houdusse A, Sweeney HL. The unique insert in myosin VI is a structural calcium-calmodulin binding site. Proc. Natl. Acad. Sci. U. S. A. 2004;101:4787–4792. doi: 10.1073/pnas.0306892101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger B, Wilson DB, Wolf E, Tonchev T, Milla M, Kim PS. Predicting coiled coils by use of pairwise residue correlations. Proc. Natl. Acad. Sci. U. S. A. 1995;92:8259–8263. doi: 10.1073/pnas.92.18.8259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant Z, Altman D, Spudich JA. The power stroke of myosin VI and the basis of reverse directionality. Proc. Natl. Acad. Sci. U. S. A. 2007;104:772–777. doi: 10.1073/pnas.0610144104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darve E, Pohorille A. Calculating free energies using average force. J. Chem. Phys. 2001;115:9169–9183. [Google Scholar]
- Gebhardt JC, Bornschlogl T, Rief M. Full distance-resolved folding energy landscape of one single protein molecule. Proc. Natl. Acad. Sci. U. S. A. 2010;107:2013–2018. doi: 10.1073/pnas.0909854107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hénin J, Chipot C. Overcoming free energy barriers using unconstrained molecular dynamics simulations. J. Chem. Phys. 2004;121:2904–2914. doi: 10.1063/1.1773132. [DOI] [PubMed] [Google Scholar]
- Henin J, Pohorille A, Chipot C. Insights into the Recognition and Association of Transmembrane [alpha]-Helices. The Free Energy of [alpha]-Helix Dimerization in Glycophorin A. J. Am. Chem. Soc. 2005;127:8478–8484. doi: 10.1021/ja050581y. [DOI] [PubMed] [Google Scholar]
- Hsin J, Chipot C, Schulten K. A glycophorin A-like framework for the dimerization of photosynthetic core complexes. J. Am. Chem. Soc. 2009;131:17096–17098. doi: 10.1021/ja905903n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellerman KA. An unconventional myosin heavy chain gene from Drosophila melanogaster. J. Cell Biol. 1992;119:823–834. doi: 10.1083/jcb.119.4.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight PJ, Thirumurugan K, Xu Y, Wang F, Kalverda AP, Stafford WF, 3rd, Sellers JR, Peckham M. The predicted coiled-coil domain of myosin 10 forms a novel elongated domain that lengthens the head. J. Biol. Chem. 2005;280:34702–34708. doi: 10.1074/jbc.M504887200. [DOI] [PubMed] [Google Scholar]
- Lister I, Schmitz S, Walker M, Trinick J, Buss F, Veigel C, Kendrick-Jones J. A monomeric myosin VI with a large working stroke. EMBO J. 2004;23:1729–1738. doi: 10.1038/sj.emboj.7600180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu PC, Gans PJ, Kallenbach NR. Energetic contribution of solvent-exposed ion pairs to alpha-helix structure. J. Mol. Biol. 1992;223:343–350. doi: 10.1016/0022-2836(92)90735-3. [DOI] [PubMed] [Google Scholar]
- Menetrey J, Bahloul A, Wells AL, Yengo CM, Morris CA, Sweeney HL, Houdusse A. The structure of the myosin VI motor reveals the mechanism of directionality reversal. Nature. 2005;435:779–785. doi: 10.1038/nature03592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menetrey J, Llinas P, Mukherjea M, Sweeney HL, Houdusse A. The structural basis for the large powerstroke of myosin VI. Cell. 2007;131:300–308. doi: 10.1016/j.cell.2007.08.027. [DOI] [PubMed] [Google Scholar]
- Mukherjea M, Llinas P, Kim H, Travaglia M, Safer D, Menetrey J, Franzini-Armstrong C, Selvin PR, Houdusse A, Sweeney HL. Myosin VI dimerization triggers an unfolding of a three-helix bundle in order to extend its reach. Mol. Cell. 2009;35:305–315. doi: 10.1016/j.molcel.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa S, Homma K, Komori Y, Iwaki M, Wazawa T, Hikikoshi Iwane A, Saito J, Ikebe R, Katayama E, Yanagida T, Ikebe M. Class VI myosin moves processively along actin filaments backward with large steps. Biochem. Biophys. Res. Commun. 2002;290:311–317. doi: 10.1006/bbrc.2001.6142. [DOI] [PubMed] [Google Scholar]
- Odronitz F, Kollmar M. Drawing the tree of eukaryotic life based on the analysis of 2,269 manually annotated myosins from 328 species. Genome Biol. 2007;8:R196. doi: 10.1186/gb-2007-8-9-r196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okten Z, Churchman LS, Rock RS, Spudich JA. Myosin VI walks hand-over-hand along actin. Nat. Struct. Mol. Biol. 2004;11:884–887. doi: 10.1038/nsmb815. [DOI] [PubMed] [Google Scholar]
- Park H, Li A, Chen LQ, Houdusse A, Selvin PR, Sweeney HL. The unique insert at the end of the myosin VI motor is the sole determinant of directionality. Proc. Natl. Acad. Sci. U. S. A. 2007;104:778–783. doi: 10.1073/pnas.0610066104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Ramamurthy B, Travaglia M, Safer D, Chen LQ, Franzini-Armstrong C, Selvin PR, Sweeney HL. Full-length myosin VI dimerizes and moves processively along actin filaments upon monomer clustering. Mol. Cell. 2006;21:331–336. doi: 10.1016/j.molcel.2005.12.015. [DOI] [PubMed] [Google Scholar]
- Phichith D, Travaglia M, Yang Z, Liu X, Zong AB, Safer D, Sweeney HL. Cargo binding induces dimerization of myosin VI. Proc. Natl. Acad. Sci. U. S. A. 2009;106:17320–17324. doi: 10.1073/pnas.0909748106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. Journal of Computational Chemistry. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock RS, Ramamurthy B, Dunn AR, Beccafico S, Rami BR, Morris C, Spink BJ, Franzini-Armstrong C, Spudich JA, Sweeney HL. A flexible domain is essential for the large step size and processivity of myosin VI. Mol. Cell. 2005;17:603–609. doi: 10.1016/j.molcel.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Rock RS, Rice SE, Wells AL, Purcell TJ, Spudich JA, Sweeney HL. Myosin VI is a processive motor with a large step size. Proc. Natl. Acad. Sci. U. S. A. 2001;98:13655–13659. doi: 10.1073/pnas.191512398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Gomez D, Darve E, Pohorille A. Assessing the efficiency of free energy calculation methods. J. Chem. Phys. 2004;120:3563–3578. doi: 10.1063/1.1642607. [DOI] [PubMed] [Google Scholar]
- Sakamoto T, Yildez A, Selvin PR, Sellers JR. Step-size is determined by neck length in myosin V. Biochemistry. 2005;44:16203–16210. doi: 10.1021/bi0512086. [DOI] [PubMed] [Google Scholar]
- Shih AY, Arkhipov A, Freddolino PL, Schulten K. Coarse Grained Protein− Lipid Model with Application to Lipoprotein Particles†. J Phys Chem B. 2006;110:3674–3684. doi: 10.1021/jp0550816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AY, Arkhipov A, Freddolino PL, Sligar SG, Schulten K. Assembly of lipids and proteins into lipoprotein particles. Journal of Physical Chemistry B-Condensed Phase. 2007a;111:11095–11104. doi: 10.1021/jp072320b. [DOI] [PubMed] [Google Scholar]
- Shih AY, Freddolino PL, Arkhipov A, Schulten K. Assembly of lipoprotein particles revealed by coarse-grained molecular dynamics simulations. J. Struct. Biol. 2007b;157:579–592. doi: 10.1016/j.jsb.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Shih AY, Sligar SG, Schulten K. Maturation of high-density lipoproteins. Journal of The Royal Society Interface. 2009;6:863–871. doi: 10.1098/rsif.2009.0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivaramakrishnan S, Spink BJ, Sim AY, Doniach S, Spudich JA. Dynamic charge interactions create surprising rigidity in the ER/K alpha-helical protein motif. Proc. Natl. Acad. Sci. U. S. A. 2008;105:13356–13361. doi: 10.1073/pnas.0806256105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivaramakrishnan S, Sung J, Ali M, Doniach S, Flyvbjerg H, Spudich JA. Combining single-molecule optical trapping and small-angle x-ray scattering measurements to compute the persistence length of a protein ER/K alpha-helix. Biophys. J. 2009;97:2993–2999. doi: 10.1016/j.bpj.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spink BJ, Sivaramakrishnan S, Lipfert J, Doniach S, Spudich JA. Long single alpha-helical tail domains bridge the gap between structure and function of myosin VI. Nat. Struct. Mol. Biol. 2008;15:591–597. doi: 10.1038/nsmb.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suveges D, Gaspari Z, Toth G, Nyitray L. Charged single alpha-helix: a versatile protein structural motif. Proteins. 2009;74:905–916. doi: 10.1002/prot.22183. [DOI] [PubMed] [Google Scholar]
- Tehver R, Thirumalai D. Rigor to post-rigor transition in myosin V: Link between the dynamics and the supporting architecture. Structure. 2010;18:471–481. doi: 10.1016/j.str.2010.01.019. [DOI] [PubMed] [Google Scholar]
- Trabuco LG, Villa E, Mitra K, Frank J, Schulten K. Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure. 2008;16:673–683. doi: 10.1016/j.str.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabuco LG, Villa E, Schreiner E, Harrison CB, Schulten K. Molecular dynamics flexible fitting: A practical guide to combine cryo-electron microscopy and X-ray crystallography. Methods. 2009;49:174–180. doi: 10.1016/j.ymeth.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells AL, Lin AW, Chen LQ, Safer D, Cain SM, Hasson T, Carragher BO, Milligan RA, Sweeney HL. Myosin VI is an actin-based motor that moves backwards. Nature. 1999;401:505–508. doi: 10.1038/46835. [DOI] [PubMed] [Google Scholar]
- Wells DB, Abramkina V, Aksimentiev A. Exploring transmembrane transport through α-hemolysin with grid-steered molecular dynamics. J. Chem. Phys. 2007;127:125101. doi: 10.1063/1.2770738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildiz A, Forkey JN, McKinney SA, Ha T, Goldman YE, Selvin PR. Myosin V walks hand-over-hand: single fluorophore imaging with 1.5-nm localization. Science. 2003;300:2061–2065. doi: 10.1126/science.1084398. [DOI] [PubMed] [Google Scholar]
- Yildiz A, Park H, Safer D, Yang Z, Chen LQ, Selvin PR, Sweeney HL. Myosin VI steps via a hand-over-hand mechanism with its lever arm undergoing fluctuations when attached to actin. J. Biol. Chem. 2004;279:37223–37226. doi: 10.1074/jbc.C400252200. [DOI] [PubMed] [Google Scholar]
- Yu C, Feng W, Wei Z, Miyanoiri Y, Wen W, Zhao Y, Zhang M. Myosin VI undergoes cargo-mediated dimerization. Cell. 2009;138:537–548. doi: 10.1016/j.cell.2009.05.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.