

Abstract
We introduce a simple micro-fluidic device containing an actuated flexible membrane, which allows the viscoelastic characterization of cells in small volumes of suspension by loading them in compression and observing the cell deformation in time. From this experiment, we can determine the characteristic time constant of recovery of the cell. To validate the device, two cell types known to have different cytoskeletal structures, 3T3 fibroblasts and HL60 cells, are tested. They show a substantially different response in the device and can be clearly distinguished on the basis of the measured characteristic recovery time constant. Also, the effect of breaking down the actin network, a main mechanical component of the cytoskeleton, by a treatment with Cytochalasin D, results in a substantial increase of the measured characteristic recovery time constant. Experimental variations in loading force, loading time, and surface treatment of the device also influence the measured characteristic recovery time constant significantly. The device can therefore be used to distinguish between cells with different mechanical structure in a quantitative way, and makes it possible to study changes in the mechanical response due to cell treatments, changes in the cell’s micro-environment, and mechanical loading conditions.
Keywords: Cell characteristic recovery time constant, Micro-fluidic device, Viscoelastic properties, Cell mechanics, Cell deformation
Introduction
The internal structure of most biological cells is governed by the cytoskeleton. This network of protein filaments provides a continuous, dynamic connection between nearly all intracellular structures. The cytoskeleton is involved in regulating cell shape and resistance to deformation. It also plays a role in active processes such as cell division, locomotion and the transport of intracellular particles (Alberts et al. 2008). The cytoskeletal structure determines the mechanical properties of a cell, such as its stiffness and its time-dependent response to mechanical stimuli.
As a consequence, different types of cells having a different cytoskeleton, will have different mechanical properties. Depending on its stage of development a specific type of cell may show a development in stiffness due to the corresponding change in cytoskeleton. Also, many diseases significantly change the stiffness of cells by a rearrangement of the cytoskeletal structure. Known examples are cancer (Lekka et al. 1999; Suresh et al. 2005; Suresh 2007), malaria (Glenister et al. 2002; Suresh et al. 2005), atherosclerosis (Wautier et al. 1999), and cardiac myopathies (Bornemann and Goebel 2001). On the basis of a reliable measurement of mechanical properties of cells, in particular cell stiffness and time-dependent responses, it should therefore be possible to distinguish and select a certain type of cell, cells in particular stages, or isolate affected cells from healthy cells for a variety of diseases.
Several techniques exist for the measurement of cellular mechanical properties. The techniques can be roughly divided into those methods that probe only part of the cell, and those that deform cells globally. The first category includes techniques such as atomic force microscopy (AFM) (Rotsch and Radmacher 2000; Rosenbluth et al. 2006), magnetic twisting cytometry (Maksym et al. 2000; Puig-de-Morales-Marinkovic et al. 2007), cytoindentation (Suresh 2007), and micro-rheology measurements (Wirtz 2009). Although these approaches can provide a quantitative mechanical analysis, a disadvantage is that the response may depend significantly on the precise measurement location, since only part of the cell is probed. As a result, these techniques generally show a large cell-to-cell spread. Techniques in which cells are deformed globally are laser/optical cell stretching (Dao et al. 2003), microplate stretching (Thoumine and Ott 1997), micropipette aspiration (Evans and Yeung 1989; Hochmuth 2000) and cell compression testing (Peeters et al. 2005).
Many of these techniques are performed on a per-cell basis. Therefore, they require single-cell handling which can make them very tedious and slow (several cells per hour). Furthermore, in many methods it is difficult to control the micro-environment around the cells to be tested, which would be desirable for testing the effect of environment on mechanical properties, or for creating physiologically relevant conditions.
Recently, micro-fluidic approaches have been used to characterize the mechanical properties of biological cells. A micro-fluidic device contains sub-millimeter channels and chambers, down to sizes of just a few micrometers, in which liquids and (bio-)molecules can be manipulated. One advantage of micro-fluidics is that it offers opportunities to study mechanical properties of single, non-isolated cells in a controlled micro-environment, such as a capillary-like microenvironment under physiological conditions, for example in fresh blood. An additional advantage of micro-fluidic devices is that high-throughput testing of cells is possible by a proper design of the micro-fluidic channel structure, so that a statistical analysis can be done while the testing time is limited.
Shelby et al. qualitatively monitored the deformability and the shape recovery time of single uninfected and malaria infected red blood cells by flushing through capillary-like channels in such a micro-fluidic chip (Shelby et al. 2003). Rosenbluth et al. developed a micro-fluidic device that resembles the micro-capillary network in vivo, and characterized the properties of leukemic cells (Rosenbluth et al. 2008). Guck et al. combined an optical stretcher and a micro-fluidic channel to study the stiffness of normal and cancerous fibroblasts and epithelial cells (Guck et al. 2005).
We have used another micro-fluidic device to characterize the time-dependent mechanical properties of cells. The working principle of our device is sketched in Fig. 1. It consists of two flow reservoirs, separated by a flexible elastomeric membrane. During experiments, the control chamber is filled with water and the cell chamber contains a cell suspension. The control chamber is pressurized, so that the flexible membrane deflects into the cell chamber, touches the cells, and deforms them. This process is observed using an optical microscope. After a sudden release of the pressure, the membrane returns to its original position, and the cells recover, in time, back to their undeformed state. From the observation of this process over time, the global viscoelastic time constant of the cells can be estimated, which is a measure of their viscoelastic mechanical response.
Fig. 1.

The working principle of our device, illustrated by a cross-sectional sketch (not drawn to scale). (a) The non-actuated state. (b) The control chamber is pressurized, and the deflecting membrane deforms the cells present in the cell chamber; cell deformation is monitored by a high-speed camera connected to the optical microscope
As a proof-of-concept of this method, we have measured the characteristic time constants of two cell types known to have different cytoskeletal structures, namely white blood cells from the HL60 neutrophilic cell line, and 3T3 fibroblast cells. Also, we have studied the effect of the variation of several parameters such as surface treatment of the device, disruption of the actin network of the cells using Cytochalasin D, and other experimental parameters. The device proposed by Kim et al. is analogous to our device; they used it to mechanically induce cellular damage in epithelial cells, and for mechanical lysis of cells (Kim et al. 2007).
Materials and methods
The micro-fluidic device
Our micro-fluidic device consists of a polydimethylsiloxane (PDMS) structure bonded to a glass substrate, as shown in Fig. 2. The cell chamber is 100 μm in height and 15 × 5 mm in the lateral directions, and the control chamber is also 100 μm high but has lateral dimensions 10 × 5 mm. To keep the flexible membrane flat in the beginning of the experiment, a relatively thick PDMS membrane is used, i.e. about 500 μm thick.
Fig. 2.

Our PDMS-based device consists of two micro-fluidic chambers separated by a flexible PDMS membrane
The microfluidic device was fabricated from PDMS silicone elastomer using multilayer soft lithography as illustrated in Fig. 3 (Unger 2000). A layer (5 mm thick) was formed by pouring the PDMS prepolymer-crosslinker mixture (Dow Corning Sylgard 184, 10:1 ratio of prepolymer to crosslink agent) onto a mold that contained the (negative) shape of the control chamber (step 1 in Fig. 3). The mold had been fabricated using SU-8 processing on a glass substrate. The PDMS was cured on a hotplate for 30 min at a temperature of 80°C. To make the PDMS membrane, the PDMS prepolymer-crosslinker mixture was poured on a piece of glass (glass A in Fig. 3) and the glass was kept standing at 85° with the horizontal for about 1 min allowing the PDMS to flow, resulting in a 500 μm thick PDMS film that was subsequently cured at 80°C. Both the layers peeled from the mold and the film were treated by a handheld corona treater (Electro-Technic Products, see Haubert et al. 2006) for 15 s to activate the surfaces before bonding the two surfaces together. Then, the PDMS film plus top layer was peeled off from the glass substrate (Step 2, Fig. 3). To fabricate the bottom chamber, another PDMS film was made using the process described earlier, and manually a square area (15 × 5 mm) was cut out from the film and removed from its glass substrate (glass B in Fig. 3). The remaining PDMS was treated by the corona treater for 15 s to bond with the PDMS layer made before (Step 3, Fig. 3). The whole PDMS construction was then peeled off from glass B. Finally, the complete PDMS part was treated by the corona treater and bonded to a clean glass substrate (glass C in Fig. 3), to finally form the bottom cell chamber (Step 4 in Fig. 3).
Fig. 3.

Schematic of the fabrication procedure of the device
To form the fluidic connections, the PDMS layers were punched by 7 G flat needles (BD) and self-made tips were inserted for connection with syringes.
Cell culture and treatments
We used two types of cell for the experiments reported here, namely HL60 cells and 3T3 fibroblast cells. HL60 cells are human promyelocytic leukemia cells. They are predominantly a neutrophilic promyelocyte (precursor). 3T3 cells are from a standard fibroblast cell line. It is known that these cells have different mechanical properties due to a difference in cytoskeletal structure: for adherent 3T3 cells, an elastic modulus of 3–12 kPa was measured with AFM (Rotsch et al. 1999); for HL60, a value of 0.2–1.4 kPa was found using AFM (Rosenbluth et al. 2006).
The HL60 cells were grown in RPMI 1640 medium (32404-014, Invitrogen Ltd, United Kindom) supplemented with 10% fetal bovine serum (FBS) and 1% L-glutamine. The 3T3 cells were cultured in DMEM (BE12-707F, Lonza, Switzerland) supplemented with 10% FBS, 5% Penicillin/Streptomycin (Lonza, Switzerland) and 1% L-glutamine. The 3T3 cells were detached from the culture flasks by trypsin. After centrifugation at 1,000 rpm for 5 min (for both cell types), the old medium was carefully removed and the remaining cell suspension was resuspended with 10 mL of its own fresh medium.
In some of the experiments, the cells were pre-treated by Cytochalasin D (CytoD). This is known to disrupt actin, which forms one of the components of the cytoskeleton of cells (Goddettes and Frieden 1986). Hence, this intentional disruption of the cell structure enables to study its influence on the mechanical properties, in particular recovery time, in our measurements. Both the HL60 and the 3T3 cells were exposed to 4 μM CytoD (Sigma) for 1 h. After that, the cells were removed from the flask and centrifuged at 1,000 rpm for 5 min. The medium was carefully removed and the cells were resuspended in 10 mL of their own fresh medium.
The change in cytoskeletal structure by the CytoD treatment was assessed by staining actin filaments with phalloidin and subsequent imaging with fluorescent microscopy. Briefly, 3T3 cells were seeded and grown for 2 days on coverslips coated with 0.1% gelatin. HL60 suspensions (around 2 × 104 cells) were spun down on slides in a cytocentrifuge for 10 min at 500 rpm (Rotofix 32 A, Hettich, Tuttlingen, Germany). After fixation in formalin, both 3T3 and HL60 cells were incubated for 10 min in 0.5% Triton-X-100 to improve antibody penetration. Non-specific binding was avoided by incubation in 1% horse serum (HS) for 20 min. Then, samples were stained in dark for 1 h with phalloidin fluorescein isothiocyanate (1:200, Sigma, Netherlands) in order to visualize filamentous actin and were counterstained with DAPI (Sigma, Netherlands) in order to visualize nuclei. It should be noted that, contrary to the experimental condition in the micro-fluidic device, the 3T3 cells were allowed to attach before staining. The cytoskeletal structure in the staining experiment might then not be fully representative of the experimental situation, however it does show the effect of CytoD on the actin filaments.
Figure 4 shows the fluorescent images obtained for both cells, with and without treatment by CytoD. It is clear that after CytoD treatment, in 3T3 cells the long actin filament bundles have been disrupted and short, not-cross-linked aggregates of F-actin are present. For HL60 cells, the effect is less evident but still a strong disintegration of actin cytoskeletal structure can be observed.
Fig. 4.

Fluorescence images of 3T3 (a–c), CytoD treated 3T3 (d–f), HL60 (g–i), CytoD treated HL60 (j–l) stained with phalloidin (green, showing actin) and DAPI (blue, showing nuclei). The first column visualizes both the actin (green) and the nuclei (blue), whereas the second and third columns show respectively the nuclei and the actin only
The figure also shows, by comparison of (c) and (i), that the two cell types have a different cytoskeletal structure. Stationary tightly adherent cells, such as 3T3 fibroblast, are characterized by thick and well structured stress-fiber bundles across the cell (Mitchison and Cramer 1996), whereas such structures cannot be found in more motile cells, such as HL60, where actin fibers are primarily concentrated on the cortical shell area (Jeon 2002). This difference can be seen in Fig. 4.
Measurement procedure
The cell chamber was treated by Pluronic F127 (Sigma Aldrich Pluronic® F-127, Zwijndrecht, Netherlands) which acts to prevent cell adhesion to the PDMS (Wang et al. 2005); a 1.0% (w/w) F127 solution in water was left in the device for 3 days. Prior to the experiment, the chamber was flushed with phosphate buffered saline (PBS). The cells were kept at 37°C in an incubator before use. At the start of the experiment, the control chamber was filled with water and connected to a syringe pump. In the cell chamber, that was kept open to atmospheric pressure during the experiments, a cell solution (around 6 × 106 cells/mL) was entered. To start the membrane deflection, the syringe pump was switched on at 4 μL/min, injecting water into to the control chamber, and the PDMS membrane was deformed slowly by the resulting pressure increase in the control camber. At sufficient membrane deflection, the cells present in the cell chamber were touched by the membrane and subsequently deformed between the bottom wall and the membrane. The deformation of the cells, which was visible as a change in the observed projected surface area, was observed by a microscope equipped with a camera. The pump was stopped when a particular cell compression ratio was reached, typically after 1 min. After a specific holding time during which the cell deformation was kept constant, the pressure was suddenly removed from the system and the PDMS membrane unloaded in less than a second. The cell started to recover back to its original shape and this process was recorded.
All the cell recovery movies were recorded using an optical microscope (Leica) and a CCD camera. The movies were captured at 200× magnification at a frame rate of 15 Hz and split by MATLAB to one frame per second. The projected surface area of the cell was analyzed by Cellprofiler (http://www.cellprofiler.org). All results discussed in the next section are based on 5 or 6 individual measurements on different cells.
A range of experiments was conducted, including the following variations:
Cell type: HL60; 3T3
Cell treatment: no particular treatment; treatment with Cytochalasin-D (see above)
Surface treatment device: no treatment; three days treatment with Pluronic F127
Cell compression ratio at maximum load, defined as the projected undeformed cell area divided by the area at maximum compression: 0.3; 0.5; 0.7
Holding time at maximum compression: 5 min, 30 min
The conditions in bold are the reference conditions.
Data analysis
Several mechanical models, with varying level of complexity, have been used to describe the mechanical properties of cells, see Lim et al. 2006 for an overview. The cell deformation images we obtain are analyzed by assuming that the mechanical properties of the cells can be described as a homogeneous linear viscoelastic solid model (LVS). This was proposed by Schmid-Schonbein et al. 1981 to study the deformation of human leukocytes undergoing micropipette aspiration, and later applied by others to other cell types, see e.g. Lim et al. 2006. The model includes an elastic element (modulus G2) parallel to a Maxwell element, in which another elastic element (modulus G1) and a viscous element (viscosity η1) are combined. Mathematically, the model is represented by the equation:
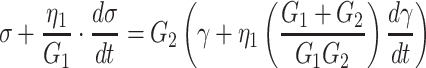 |
1 |
In a recovery experiment, in which a stress is applied and suddenly removed, just as in our device, this model shows a recovery to the original strain according to the following expression:
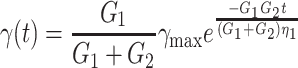 |
2a |
in which γmax is the maximum strain just before starting the recovery at t = 0. The characteristic time constant of recovery can thus be expressed by:
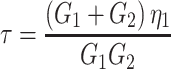 |
2b |
To apply Eqs. 2a and 2b we assume that the cell is spherical with radius R 0 in the undeformed state, and will take an ellipsoidal shape during recovery, as sketched in Fig. 5. Also, we assume that the cell volume remains constant during the recovery process. The z-direction is perpendicular to the deflecting membrane, i.e. the direction in which the cell is loaded. During recovery, the length R z lengthens, while R x and R y shorten equally, all approaching the value R 0 in the end. The quantity we observe during the experiment is the projected cell area given by:
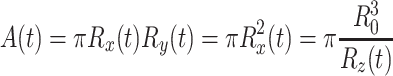 |
3 |
The area strain is defined by:
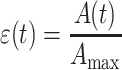 |
4 |
in which A max is the maximum projected area at maximum deformation, just before unloading. The linear strain γ is defined as:
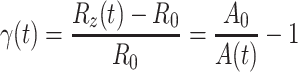 |
5 |
in which (3) was used, and A 0 = πR 0 2. Note that γ is negative in our experiments, since the cell is compressed. The combination of Eqs. 4 and 5 yields:
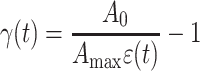 |
6 |
Fig. 5.

The cell deformation process showing only the upper half of the cell. (a) Before deformation; (b) during deformation. The z-axis is oriented perpendicularly to the moving membrane
We assume now that Eqs. 2a and 2b applies to the linear strain of Eq. 6. Effectively, we thereby assume that the deformation process is uniaxial, which is only approximately true. Substitution of Eq. 6 into Eqs. 2a and 2b results in:
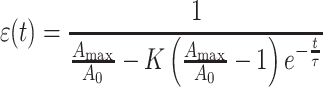 |
7 |
in which
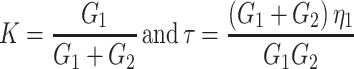 |
8 |
where τ is now called the characteristic recovery time constant of the cell.
Equation 7 was fitted to the measured recovery curves using MATLAB to obtain K and the characteristic recovery time constant τ. The fit was done for each individual cell recovery curve separately, and subsequently the fit results were averaged for all cells (typically 5 or 6 different cells) for each condition or population. To assess the goodness of fit, the percent relative standard deviation (%RSD) was computed. To assess the significance of the differences in fit parameters found, Student’s t-test were carried out and p-values were calculated. For p-values smaller than 0.01, the differences were considered to be statistically significant.
Results and discussion
Figure 6 shows snapshots from a movie of the recovery process of both an HL60 and a 3T3 cell. Also, the corresponding recovery curves and the fitted function according to Eq. 7 are depicted. In these experiments, the reference experimental conditions were used, i.e. the cell compression ratio at maximum load was 0.5, the holding time at maximum compression was 5 min, the cells were not treated with CytoD, and the device was treated with F127. Both cell types gradually recover to their undeformed cell compression ratio value of 0.5, and both cells seem to undergo a slight instantaneous elastic response at t = 0. These characteristics are consistent with the linear viscoelastic solid model described in the previous section. It takes typically 100 s or more for the cells to recover completely. Clearly, the 3T3 fibroblast has a longer recovery time than the HL60 cells.
Fig. 6.

Left, images of the recovery of the cells at various points in time, after sudden release of the pressure at t = 0. (a) 3T3; (b) HL60. Right, the recovery curves of 3T3 (solid blue), and HL60 (dashed red), showing the change in time of the area ratio defined in Eq. 4. In these measurements, the reference measurement conditions were used. Each marker represents one data point; the data points of multiple cells are shown. The lines represent fits according to Eq. 7
Fitting Eq. 7 to the measurement data enables us to quantify the characteristic recovery time constants of the cells, given by Eq. 8. The average over 6 measurements (per cell type) is depicted in Fig. 7. The error bars represent the standard deviation on the basis of these 6 measurements. There is a significant difference between the characteristic recovery time constant of HL60 (26 s) and that of 3T3 (55 s). Hence, we can distinguish one type of cell from the other by measuring its characteristic recovery time constant. The measured values for K are 0.96 ± 0.03 and 0.95 ± 0.03 for HL60 and 3T3, respectively. This difference is not statistically significant.
Fig. 7.

Average characteristic recovery time constants for the HL60 and the 3T3 cells under reference conditions
The difference in recovery response times correlates with the entirely different cytoskeletal structure of the two cell types. This difference in cell structure could be seen in Fig. 4. Here, we should note that the 3T3 cells were adherent to the substrate in the staining experiments of Fig. 4, whereas they are in suspension in the recovery experiments. In the suspended condition, a less organized structure may be present due to the lack of tension forces at the focal adhesion points; nevertheless, we expect the actin structure still to be different in 3T3 compared to HL60 cells in the experiments.
The effect of the maximum cell compression ratio A0/Amax maintained during the holding period is shown in Fig. 8; all other measurement conditions were equal to the reference situation in these experiments. The corresponding characteristic recovery time constants are summarized in Fig. 9. The characteristic recovery time constant depends on the maximum cell compression ratio. This proves that, under these measurement conditions, the linear viscoelastic model does not hold strictly, since for this model the characteristic recovery time constant is a constitutive parameter that should be independent of the deformation or loading conditions. The reason is that the deformation itself causes changes in the cells’ cytoskeletal structure such that the effective cell properties change. Larger deformations (i.e. smaller cell compression ratios) result in longer recovery times. For the 3T3 cells recovery times are longer than for the HL60 cells over the entire range of cell compression ratios tested. The fitted values of K for 3T3 are 0.97 ± 0.02, 0.95 ± 0.03, and 0.91 ± 0.04 respectively for the maximum cell compression ratios of 0.6, 0.5 and 0.3, respectively. For HL60, these values are 0.98 ± 0.02, 0.99 ± 0.02, and 0.96 ± 0.03 for the cell compression ratios of 0.5, 0.4 and 0.3
Fig. 8.

Cell recovery curves measured for different maximum cell compression ratios. (a) 3T3 cells, for A0/Amax = 0.6 (blue, N = 6,%RSD = 4.8), 0.5 (black, N = 6,%RSD = 5.8) and 0.3 (red, N = 6,%RSD = 4.7). (b) HL60 cells for A0/Amax = 0.5 (black, N = 6,%RSD = 5.3), 0.4 (green, N = 7,%RSD = 5.2) and 0.3 (red, N = 6,%RSD = 5.4). N is the number of cells tested for each condition. Each marker represents one data point; the data points of multiple cells are shown. The lines represent fits according to Eq. 7
Fig. 9.

The characteristic recovery time constant of the HL60 and 3T3 cells compared, as a function of the maximum cell compression ratio A0/Amax. The p-values indicate that the differences are statistically significant
The treatment with CytoD has a dramatic effect on the cell properties, as is evidenced by Figs. 10 and 11. The characteristic recovery time constant is greatly increased, especially for the HL60 cells. As we have seen in Fig. 4, the CytoD treatment disrupts the actin network in the cells, and therefore it can be expected that the structural coherence of the cell is at least partly lost. The elastic component in response to deformation becomes therefore more insignificant due to the treatment, which will result in longer characteristic recovery time constants (see also Eq. 8 that shows that smaller elastic moduli in the LVS model result in a larger characteristic recovery time constant τ), which is what we observe. For the reference loading conditions, τ increases from 55 s to 110 s for 3T3 cells (i.e. a 100% increase), and from 26 s to 150 s for the HL60 cells (i.e. a 470% increase), see Fig. 11 compared with Fig. 9. Particularly the latter change is substantial. The values for K for the cytoD treated 3T3 cells are 0.95 ± 0.02 and 0.93 ± 0.034 respectively for the area ratios 0.7 and 0.5. For the cytoD treated HL60 cells the fitted K is 0.99 ± 0.03 and 0.98 ± 0.04 for the area ratios 0.6 and 0.5.
Fig. 10.

Recovery curves measured for cells untreated or treated with cytochalasin D. (a) 3T3 cells: untreated, reference conditions (black), treated with CytoD, A0/Amax = 0.5 (green, N = 6,%RSD = 4.8), treated with CytoD, A0/Amax = 0.7 (blue, N = 5,%RSD = 3.0). (b) HL60 cells: untreated, reference conditions (black), treated with CytoD, A0/Amax = 0.5 (green, N = 6,%RSD = 4.8), treated with CytoD, A0/Amax = 0.6 (blue, N = 6,%RSD = 3.9). N is the number of cells tested for each condition. Each marker represents one data point; the data points of multiple cells are shown. The lines represent fits according to Eq. 7
Fig. 11.

The characteristic recovery time constant of both cell types after treatment with Cytochalasin D, as a function of the maximum cell compression ratio. A comparison with Fig. 9 shows that the treatment results in a large increase of characteristic recovery time constant
Cellular shape, elasticity and contraction are to a large extent determined by the actin structure of the cytoskeleton. Therefore, the structural organization of actin fibers determines the mechanical properties of the cell. For this reason, disruption of actin by the CytoD treatment results in a change of cytoskeletal organization and thus a change in the mechanical response (Charras and Horton 2002; Olins et al. 2000; Rotsch and Radmacher 2000; Wakatsuki et al. 2000). In previous studies, it has been shown that actin microfilament disrupting drugs such as CytoD or latrunculin B, diminished cell elasticity (Lekka et al. 1999) and caused a dramatic reduction of cell stiffness (Glenister et al. 2002). Rosenbluth et al. found that the transit time of HL60 cells flowing through narrow micro-channels was decreased substantially by a CytoD treatment (Rosenbluth et al. 2008). The characteristic recovery time constant increase we observe is in line with this. This is due to the fact that actin filaments provide and support contractile stresses generated within the cell. It has been postulated that actin filaments are anchored to the plasma membranes by barbed end capping proteins and for this reason they provide tensile forces in the cell (Wautier et al. 1999). CytoD destabilizes these site of anchorage, disrupts the fibers and consequently reduces the elasticity of the cell and the ability to maintain cell shape. It might be postulated that other cytoskeletal structures, such as microtubules and the cell membrane, then take substantial part in the cell recovery.
Figure 12 shows the effect of an increase of the holding time at which the maximum load is maintained from 5 min to 30 min. The change is dramatic: for the 3T3 cells the characteristic recovery time constant increases to 340 s, i.e. a 7-fold increase. The characteristic recovery time constant increase for the HL60 cells is less, but seems still substantial however the large p-value (p > 0,01) indicates that the change is not statistically significant. The most probable explanation for the influence of the holding time is that the cell structure is rearranged during the holding period. The compression load acting on the cell influences the continuous polymerization and depolymerization processes that happen in the cytoskeleton, thereby substantially changing the cell’s mechanical structure and properties, i.e. biological remodeling takes place.
Fig. 12.

The influence of the holding time on the characteristic recovery time constant. (a) The characteristic recovery time constant of 3T3 cells with a maximum compression ratio around 0.5 for a holding time of 30 min (grey, N = 4) and 5 min (white, N = 6). (b) The characteristic recovery time constant of HL60 cells with a maximum compression ratio around 0.35 when with a holding time of 30 min (grey, N = 4) and 5 min (white, N = 6). N is the number of cells tested for each condition. The difference is not statistically significant for the HL60 cells
To illustrate the influence of the interaction of the cells with the surface, Fig. 13 shows the effect of the Pluronic F127 treatment of the device on the characteristic recovery time constant. Both 3T3 and HL60 cells have a larger characteristic recovery time constant for the untreated surfaces, although the change is not statistically significant for the HL60 cells. It is known that the Pluronic treatment reduces the cell-surface adhesion (Wang et al. 2005). Hence, the interaction between the cells and the surface, in particular adhesion, is larger for the untreated than for the treated surfaces. It is clear from Fig. 13 that the additional interaction can slow down the cell recovery quite substantially, in particular for the 3T3 cells.
Fig. 13.

The effect of device surface treatment on the characteristic recovery time constant. (a) The characteristic recovery time constant of 3T3 for an untreated surface (grey, N = 4) and of the F127 treated surface (white, N = 6), both for a maximum compression ratio of 0.3. (b) The characteristic recovery time constant of HL60 when for an untreated surface (grey, N = 5) and in the F127 treated device (white, N = 6), both for a maximum compression ratio of 0.35. N is the number of cells tested for each condition. Only for the 3T3 cells, the difference is statistically significant
Finally, Table 1 compares our values of the parameters K and τ, with those derived from the LVS parameters G2 G1 and η1 published by other authors for various cell types and using different methods. The value of K does not vary much (between 0.35 and 0.96), at least compared to the value of the characteristic recovery time constant that ranges over orders of magnitude (from 0.3 to 116) for the various cells and methods. Our value for the 3T3 fibroblast is comparable to that measured with microplate stretching by Thoumine and Ott for fibroblasts.
Table 1.
Reported parameters for the homogeneous linear viscoelastic solid, compared to our results under reference conditions
| Cell type | K [-] | τ [s] | Method | Reference |
|---|---|---|---|---|
| Myoblasts | 0.48 | 0.3 | Cell compression | Peeters et al. 2005 |
| Fibroblasts | 0.35 | 39 | Microplate stretching | Thoumine and Ott 1997 |
| Human leukocytes | 0.73 | 0.65 | Micropipette aspiration | Schmid-Schönbein et al. 1981 |
| Endothelial cells | 0.67 | 116 | Micropipette aspiration | Sato et al. 1990 |
| 3T3 fibroblasts | 0.95 | 55 | Microfluidic membrane device | The present work |
| HL60 neutrophilic promyelocytes | 0.96 | 26 | Microfluidic membrane device | The present work |
However, one should be very careful in directly comparing these numbers. To a large extent, the variation in τ-values is due to the differences in the structure of the different cell types, but it is also due to differences in loading devices, loading protocols, operating conditions, and degree of cellular attachment. For example, the experiments by Thoumine and Ott and by Sato et al. were done by applying large strains during long timescales, whereas the measurements by Peeters et al. and Schmid-Schönbein et al. were performed at smaller strains and short timescales. Indeed, our own results show that experimental conditions such as loading time and surface conditions can have a large effect on the measured parameters. Strictly, the wide variation of τ implies that the LVS model is not a suitable quantitative constitutive model for this wide range of conditions, although it can still be used to describe our experiments qualitatively.
Conclusion
Our micro-fluidic device, based on the actuation of a flexible membrane, allows the characterization of the viscoelastic properties of cells in small volumes of suspension by loading them in compression and observing the cell deformation in time. From this experiment, we can determine the characteristic time constant of recovery of cells.
As a first proof-of-concept, we characterized two different cell types known to have a different cytoskeletal structure, 3T3 fibroblasts and HL60 neutrophilic promyelocytes. Indeed, these showed a substantially and significantly different response in the device and could be clearly distinguished on the basis of the calculated characteristic recovery time constant. Also, the effect of breaking down the actin network, the main mechanical component of the cytoskeleton, by a treatment with Cytochalasin D, resulted in a dramatic increase of the measured characteristic recovery time constant. Experimental variations in loading force, loading time, and surface treatment of the device also influenced the measured characteristic recovery time constant significantly.
The device can be used to distinguish between cells with different mechanical structure, and makes it possible to study changes in the mechanical response due to cell treatments, changes in the cell’s micro-environment, and mechanical loading conditions. This allows, for example, a systematic study of the effect of biological remodeling of the cell. Also, (adherent) cells may be grown in the device itself before testing. The method may be further developed to aid in the selection of a certain type of cell, cells in particular stages, or in isolating affected cells from healthy cells for a variety of diseases. In conclusion, the main characteristics and advantages of our device that distinguish the method from existing approaches, are:
Whole-cell mechanical properties are probed
The device is simple and easy to use
Only small volumes of cell suspension are needed
The microenvironment of the cells can be controlled by changing the working fluids and temperature, and by surface treatments
The loading conditions can be varied in a flexible way
The device allows, in principle, to characterize multiple cells simultaneously in the central area of the device where uniform loading conditions exist, if the cells have approximately the same size.
In the future, the device can be extended with a pressure sensor and/or a measurement of the PDMS membrane deflection, which would allow the quantitative determination of elastic moduli, though at the expense of an increased complexity,
Acknowledgement
This work is part of the Brainbridge Project, financially supported by Philips Research.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts, P. Walter, Mol. Biol. Cell, 5th edn, (Garland Science 2008)
- Bornemann A, Goebel HH. Brain Pathol. 2001;11:206. doi: 10.1111/j.1750-3639.2001.tb00393.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charras GT, Horton MA. Biophys. J. 2002;82:2970. doi: 10.1016/S0006-3495(02)75638-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao M, Lim CT, Suresh S. J. Mech. Phys. Solids. 2003;15:11. [Google Scholar]
- Evans E, Yeung A. Biophys. J. 1989;56:151. doi: 10.1016/S0006-3495(89)82660-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenister FK, Coppel RL, Cowman AF, Mohandas N, Cooke BM. Blood. 2002;99:1060. doi: 10.1182/blood.V99.3.1060. [DOI] [PubMed] [Google Scholar]
- Goddettes DW, Frieden C. J. Biol. Chem. 1986;261:15974. [PubMed] [Google Scholar]
- Guck J, Schinkinger S, Lincoln B, Wottawah F, Ebert S, Romeyke M, Lenz D, Erickson HM, Ananthakrishnan R, Mitchell D, Kas J, Ulvick S, Bilby C. Biophys. J. 2005;88:3689. doi: 10.1529/biophysj.104.045476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubert K, Drier T, Beebe D. Lab Chip. 2006;6:1548. doi: 10.1039/b610567j. [DOI] [PubMed] [Google Scholar]
- Hochmuth RM. J. Biomech. 2000;33:15. doi: 10.1016/S0021-9290(99)00175-X. [DOI] [PubMed] [Google Scholar]
- K.W. Jeon (ed.), International Review of Cytology, Volume 216, A survey of cell biology (Academic Press, 2002)
- Kim YC, Kang JH, Park S-J, Yoon E-S, Park J-K. Sens. Actuators B. 2007;128:108. doi: 10.1016/j.snb.2007.05.050. [DOI] [Google Scholar]
- Lekka M, Laidler P, Gil D, Lekki J, Stachura Z, Hrynkiewicz AZ. Eur Biophys. J. 1999;28:312. doi: 10.1007/s002490050213. [DOI] [PubMed] [Google Scholar]
- Lim CT, Zhou EH, Quek ST. J. Biomech. 2006;39:195. doi: 10.1016/j.jbiomech.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Maksym GN, Fabry B, Butler JP, Navajas D, Tschumperlin DJ, Laporte JD, Fredberg JJ. J. Appl. Physiol. 2000;89:1619. doi: 10.1152/jappl.2000.89.4.1619. [DOI] [PubMed] [Google Scholar]
- Mitchison TJ, Cramer LP. Cell. 1996;84:371. doi: 10.1016/S0092-8674(00)81281-7. [DOI] [PubMed] [Google Scholar]
- Olins AL, Hermann H, Lichter P, Olins DE. Exp. Cell Res. 2000;254:130. doi: 10.1006/excr.1999.4727. [DOI] [PubMed] [Google Scholar]
- Peeters EAG, Oomens CWJ, Bouten CVC, Bader DL, Baaijens FPT. J. Biomech. Eng. 2005;127:237. doi: 10.1115/1.1865198. [DOI] [PubMed] [Google Scholar]
- Puig-de-Morales-Marinkovic M, Turner K, Butler JP, Fredberg JJ, Suresh S. Am. J. Physiol., Cell Physiol. 2007;293:597. doi: 10.1152/ajpcell.00562.2006. [DOI] [PubMed] [Google Scholar]
- Rosenbluth MJ, Lam WA, Fletcher DA. Biophys. J. 2006;90:2994. doi: 10.1529/biophysj.105.067496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbluth MJ, Lam WA, Fletcher DA. Lab Chip. 2008;8:1062. doi: 10.1039/b802931h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotsch C, Radmacher M. Biophys. J. 2000;78:520. doi: 10.1016/S0006-3495(00)76614-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotsch C, Jacobson K, Radmacher M. Proc. Nat. Acad. Sci. 1999;96:921. doi: 10.1073/pnas.96.3.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- M. Sato, D.P. Theret, L.T. Wheeler, N. Ohshima, R.M. Nerem, Trans. ASME J. Biomech. Eng. 112, 263 (1990) [DOI] [PubMed]
- Schmid-Schonbein GW, Sung KL, Tozeren H, Skalak R, Chien S. Biophys. J. 1981;36:243. doi: 10.1016/S0006-3495(81)84726-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelby JP, White J, Ganesan K, Rathod P, Chiu D. Proc. Nat. Acad. Sci. 2003;100:14618. doi: 10.1073/pnas.2433968100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suresh S. Acta Biomaterialia. 2007;3:413. doi: 10.1016/j.actbio.2007.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suresh S, Spatz J, Mills JP, Micoulet A, Dao M, Lim CT, Beil M, Seufferlein T. Acta Biomaterialia. 2005;1:15. doi: 10.1016/j.actbio.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Thoumine O, Ott A. J. Cell Sci. 1997;110:2109. doi: 10.1242/jcs.110.17.2109. [DOI] [PubMed] [Google Scholar]
- Unger MA. Science. 2000;288:113. doi: 10.1126/science.288.5463.113. [DOI] [PubMed] [Google Scholar]
- Wakatsuki T, Schwab B, Thompson NC, Elson EL. J. Cell Sci. 2000;114:1025. doi: 10.1242/jcs.114.5.1025. [DOI] [PubMed] [Google Scholar]
- Wang Y, Wang T, Su Y, Peng F, Wu H, Jiang Z. Langmuir. 2005;21:11856. doi: 10.1021/la052052d. [DOI] [PubMed] [Google Scholar]
- Wautier JL, Schmid-Schonbein GW, Nash GB. Clin. Hemorheol. Microcirc. 1999;21:7. [PubMed] [Google Scholar]
- Wirtz D. Annual Rev. Biophys. 2009;38:301. doi: 10.1146/annurev.biophys.050708.133724. [DOI] [PubMed] [Google Scholar]