

Abstract
Previous studies have established the potential of the oligo-acyl-lysyl (OAK) concept in generating simple chemical mimics of host defense peptides (HDPs) with improved antimicrobial properties. We investigated the antibacterial properties of such an OAK, C16(ω7)-KK-C12-Kamide, to obtain a better understanding of the complex mode(s) of action of cationic antibacterial peptides. The average MIC, determined against a multispecies panel of 50 strains, was 6 ± 5 μg/ml. However, although the OAK exerted an essentially dose-dependent bactericidal effect (time-kill curves typically exhibited 99% death within 2 h), marked differences in the killing rates occurred among inter- and intraspecies strains. Mechanistic comparison between equally sensitive and related strains revealed death of one strain to stem from the OAK's capacity to breach the cell membrane permeability barrier, whereas the death of the related strain resulted from the OAK's direct interference with DNA functions in vivo, without detectable membrane damage. These findings therefore support the notion that the antibacterial mechanism of action of a single HDP can vary among inter- and intraspecies strains. In addition, we present data illustrating the differential effects of environmental conditions (pH, ionic strength and temperature), on the OAK's antibacterial properties, and ultimately demonstrate potency enhancement (by orders of magnitude) through selection of optimal incubation conditions. Such attributes might be useful in a variety of antibacterial applications.
The ubiquitous occurrence of host defense peptides (HDPs) and their critical roles as major immunity effectors is well established. Many were found to exhibit activity against a broad spectrum of microorganisms, including viruses, bacteria, fungi, and protozoa (19, 38, 65). Although various details of their mechanism of action are poorly understood, several potential targets have been described. These include the cytoplasmic membrane, cell division, and the synthesis of cell wall and macromolecules (6, 39, 55). As therapeutic agents, HDPs could present various a priori advantages over antibiotics, including molecular simplicity, broad-spectrum activity (27, 45, 59), potentially low levels of induced resistance (41), and concomitant anti-inflammatory activities (11, 25, 36). Nevertheless, as potential drug candidates, HDPs can present a variety of shortcomings that need to be addressed (30, 61). Namely, the antimicrobial activity of many HDPs is significantly reduced in the presence of salts (1, 22, 37) and is susceptible to pH changes (31, 52) and/or to plasma components (40, 45, 49). HDPs can also display poor pharmacokinetic properties and systemic toxicity and are associated with high production costs (5, 23, 34, 64).
Various approaches were proposed to alleviate one or more of these shortcomings, including the use of peptides composed of d-amino acids (2, 56, 62), combinatorial libraries (3, 12), and a wide array of sequence templates and/or minimalistic strategies (8, 9, 13, 28, 59, 60). Other approaches opted for the de novo design of peptide-like constructs that mimic HDPs structure and/or function (29, 47, 57, 58). Oligo-acyl-lysyls (OAKs) are among the simpler HDP-mimic designs wherein the two most important characteristics for activity—hydrophobicity and charge—are represented by tandem repeats of amide-linked fatty acids and lysines. This design was shown to overcome limitations of conventional HDPs with respect to in vivo efficacy and toxicity (43, 44) and, more recently, showed promise toward systemic therapy (33, 53).
In addition to their enhanced potential applications, simple yet robust HDP-mimics might be useful in better understanding various mechanistic aspects of cationic peptide-based antimicrobials. Previous mechanistic studies of OAKs have substantiated the idea that different sequences can exert a number of distinct antibacterial modes of action, as suggested for HDPs. For instance, whereas the sequence C16(ω7)K-β12 exerted a typical rapid bactericidal effect (54), its analog C12(ω7)K-β12 was limited to a bacteriostatic effect over Gram-positive bacteria, mediated by high-affinity but superficial membrane interactions (16, 53). Also, the octamer C12K-7α8 exerted bactericidal activity through cytoplasmic membrane disruption of Gram-negative bacteria (44). However, when compared against the very same bacteria, the hexameric version, C12K-5α8 was unable to disrupt the plasma membrane (48). It was suggested that C12K-5α8 exerts its antibacterial effect after translocating into the cytoplasm, where the OAK inhibits biosynthesis of macromolecule through direct interaction with DNA.
A growing list of HDPs (6, 39) is proposed to exert antibacterial activity through interaction with intracellular targets. Therefore, combined with the OAK findings, the collective data suggest that small differences in charge and/or hydrophobicity might be sufficient for an antibacterial compound to switch from one mechanism to another. However, since the vast majority of mechanistic insights published in the literature were based on investigations that used a single strain of a single bacterial species, this prohibits conclusion on whether a particular compound can be expected to always use a particular mechanism versus using distinct mechanisms on distinct strains. It is also possible that an antibacterial compound might use multiple/combined mechanisms, as sometimes touted in the literature but yet to be convincingly demonstrated experimentally. Here, we attempted to address this hypothesis by investigating the mechanism of action of a representative OAK [C16(ω7)K-β12; the molecular structure is shown in Fig. 1] against a multispecies and multistrain panel of bacteria to determine whether a single antibacterial sequence can use multiple mechanisms of action.
FIG. 1.

Molecular structure of C16(ω7)K-β12. The N terminus is hexadecenoyl-lysyl. The sequence lysyl-aminododecanoyl-lysyl-amide is referred to as a β12 subunit.
MATERIALS AND METHODS
Peptide synthesis.
C16(ω7)K-β12 was synthesized by the solid-phase method (16) by applying the 9-fluorenylmethyloxy carbonyl (Fmoc) active-ester chemistry (Applied Biosystems, model 433A peptide synthesizer) essentially as described previously (44). The crude compounds were purified to chromatographic homogeneity in the range of >95% by using reversed-phase high-performance liquid chromatography with a mass spectrometer (Alliance-ZQ Waters).
Bacteria.
Unless specified otherwise, bacteria were cultured in LB medium (10 g of tryptone/liter, 5 g of yeast extract/liter, 85 mM NaCl [pH 7.4]).
Gram-positive bacteria.
The Gram-positive bacteria examined were as follows: Staphylococcus aureus, ATCC strains 25923, 29213, and 43300; methicillin-sensitive S. aureus (MSSA), clinically isolated strains (CI) 15877, 15668, 15886, and 20745; methicillin-resistant S. aureus (MRSA), CI strains 15903, 15819, 15852, 15918, and 17314; Staphylococcus xylosus ATCC 29971; Staphylococcus epidermidis ATCC 12228; Listeria seeligeri ATCC 35967; Listeria grayi ATCC 19120; Listeria ivanovii ATCC 19119; Listeria innocua ATCC 33090; Listeria welshimeri ATCC 35897; Bacillus polymyxa ATCC 842; Bacillus cereus ATCC 11778; and Enterococcus faecalis ATCC strains 29212 and 51299. Listeria monocytogenes Li2, ATCC 19115, was grown overnight in BHI medium (Bacto brain heart infusion; Difco Labs, France) also containing 85 mM NaCl at pH 7.
Gram-negative bacteria.
The Gram-negative bacteria examined were as follows: Escherichia coli, ATCC strains 35218, 25922, and 43894 and CI strains 16223, 16287, 16147, 16229, 16348, 16327, 16329, 14213, and 16302; Pseudomonas aeruginosa, ATCC strains 9027 and 27853 and CI strains 13216, 11816, 11662, 12777, and 8732; Salmonella enterica serovar Typhimurium ATCC strains 14028 and 7308; Klebsiella pneumonia, CI strains 1287, 1286, and 1331; Proteus mirabilis, CI strain 1285; and Stenotrophomonas maltophilia, CI strains 748 and 749.
Growth inhibition assay.
The MICs were determined by microdilution assay in sterilized 96-well plates (final volume, 200 μl) as follows. Bacteria were grown overnight in medium (LB or BHI) and diluted 10,000-fold in growth medium. A 100-μl portion of culture medium containing bacteria (2 × 105 to 4 × 105 CFU/ml) was added to 100 μl of medium containing C16(ω7)K-β12 in serial 2-fold dilutions. Inhibition of proliferation was determined by measuring the optical density at 620 nm (OD620) after incubation overnight at 37°C. The term “standard growth conditions” refers to growing bacteria in LB medium containing 85 mM NaCl (pH 7) at 37°C. To determine the MIC under nonstandard conditions, bacteria and OAK were preconditioned for 15 min prior to transferring them to the specified incubation conditions.
Kinetic studies.
Bactericidal kinetics were assessed as follows: bacteria were grown overnight in medium (LB or BHI) and diluted 100-fold in growth medium. A 100-μl portion of growth medium containing bacteria was added to 900 μl of the medium containing C16(ω7)K-β12 at final concentrations equal to four times the MIC value, followed by incubation at 37°C under shaking. At the specified time points aliquots were diluted (serial 10-fold dilutions in saline) and plated on LB or BHI agar for the CFU count, using the drop-plate method (three 20-μl drops onto agar plates). CFU were counted after an overnight incubation at 37°C. The reported results are from two independent experiments. To assess the effect of temperature variations, bacteria were incubated for 15 min at the specified temperatures (i.e., 4, 25, and 48°C) prior to the experiment. For pH and salt variations, the culture medium was brought to the desired pH by adding NaOH or HCl (1 N) or to the desired saline concentration by adding NaCl. For all tested conditions, the OAK concentration was 10 μg/ml.
Cytoplasmic membrane permeability assays.
The OAK effect on bacterial membrane integrity was determined by measurement of a non-membrane-permeabilizing fluorophore propidium iodide (PI) from the BacLight kit by Molecular Probes, Inc. (Eugene, OR). PI exerts its fluorescence after incorporation to DNA. To assess the translocation of PI from the medium to the cytoplasm, mid-logarithmic-phase E. coli ATCC 35218 and S. aureus ATCC 29213 cells (OD600 = 0.05) were washed in phosphate-buffered saline (PBS; pH 7.4) and incubated with OAK samples at 4× the MIC value for different time intervals. At the designated time points bacteria were centrifuged and resuspended in PBS containing PI (30 μM). After 15 min of incubation, the fluorescence was monitored (excitation of 485 nm and emission of 645 nm). The results shown here are from three independent experiments.
A cytoplasmic membrane depolarization assay was performed with S. aureus (ATCC 29213) and E. coli (ATCC 35218 and CI 16327) strains as follows. S. aureus samples in mid-logarithmic phase were washed and resuspended in 5 mM HEPES buffer. E. coli strains were resuspended in5 mM HEPES buffer containing 2 mM EDTA. These cell suspensions were incubated with diSC3-5 (0.4 mM), and quenching was allowed to occur at room temperature for 60 min. KCl (100 mM) was added to equilibrate the cytoplasmic and external K concentrations. OAK (four multiples of the MIC value) was added to the bacterial suspensions, and changes in fluorescence were continuously recorded (using excitation and emission wavelengths at 622 and 670 nm, respectively).
Inhibition of bacterial biosynthesis.
Macromolecular synthesis and bacterial killing assays were performed as described previously (48).
Gel electrophoresis assay.
C16(ω7)K-β12 was incubated at different concentrations with pUC19 (200 ng) for 30 min in a final volume of 15 μl at room temperature. The plasmid and marker (DNA Ladder Mix) were applied on 1% agarose gel containing ethidium bromide. Electrophoresis was carried at 80 V for 1 h.
Intracellular localization using DNA binding assay.
The assay was performed with E. coli K-12 containing pUC19 plasmid as described previously (48). Mid-logarithmic-phase bacterial cultures (grown on LB containing ampicillin) were washed in PBS (pH 7.4) and resuspended in the same buffer. The OAK at 4× the MIC value was incubated with bacterial cells (106 CFU/ml) for 30 min. After this preincubation step, the bacteria were thoroughly washed (four wash-spin cycles to prevent the carryover of unbound OAK). Pellets from the last wash were submitted to plasmid purification procedure using a miniprep plasmid DNA purification kit (Qiagen, Inc.). The plasmid was then incubated for 1 h at 37°C with DNase that opens the plasmid in one point (XbaI) according the manufacturer's procedure (New England Biolabs, Inc.). The plasmids and marker (λ-HindIII) were run in 1% agarose gel for 40 min. Control experiments performed to assess the OAK binding to DNA were carried out as described above, but instead of incubating OAK with bacteria OAK was incubated with 150 ng of purified pUC19 (extracted from E. coli K-12 and purified with plasmid DNA purification kit after 30 min of incubation with the OAK). The reported results are from three independent experiments.
RESULTS
Broad-spectrum antibacterial activity.
Antibacterial activity of C16(ω7)K-β12 was previously assessed against a limited number of bacteria (four strains), suggesting a potential broad-spectrum activity (54). Thus, the first aim of the present study was to verify this postulate by assessing its activity against a multispecies panel, including 50 bacterial strains. Table 1 summarizes the outcome in terms of MICs. Although Gram-positive species were often somewhat more sensitive than Gram-negative species, C16(ω7)K-β12 was significantly active against all of the strains tested, displaying an average MIC of 6 ± 5 μg/ml, where the growth of 90% of the strains tested was fully inhibited at 10 μg/ml or less (MIC90 = 10 μg/ml). For comparison, conventional antibiotics, such as penicillin G and ciprofloxacin, assessed under identical conditions against four representative multidrug-resistant clinical isolates—S. aureus strains 15903 and 15852 and E. coli strains 16329 and 16327—yielded MIC values that ranged from 16 to >512 μg/ml (data not shown). These results established C16(ω7)K-β12 as a potent broad-spectrum antibacterial compound.
TABLE 1.
Antibacterial activity of C16(ω7)K-β12 in a panel of 50 strains
| Bacterium | No. of strains tested | MIC range (μg/ml) |
|---|---|---|
| Gram-positive | ||
| Listeria spp. | 5 | 1.2-2.5 |
| Staphylococcus spp. | 14 | 1.2-5 |
| B. cereus | 1 | 2.5 |
| E. faecalis | 2 | 5-10 |
| B. polymyxa | 1 | 20 |
| Gram-negative | ||
| P. aeruginosa | 7 | 2.5-10 |
| E. coli | 12 | 5-20 |
| S. enterica serovar Typhimurium | 2 | 5-10 |
| K. pneumoniae | 3 | 10-20 |
| P. mirabilis | 1 | 10 |
| S. maltophilia | 2 | 10 |
Environmental conditions differentially affect antibacterial activity.
Table 2 summarizes both the MIC values and the bactericidal rates (time required for reducing viability by 99% at 10 μg of OAK/ml) obtained under different incubation conditions, as assessed against two pathogenic species—Gram-positive Listeria monocytogenes (ATCC 19115) and Gram-negative Escherichia coli O157:H7 (ATCC 43894)—selected due to their ability to grow under a wide range of environmental conditions. Generally, although some extreme incubation conditions have affected both OAK-treated and OAK-untreated bacteria, significant differences could be observed nevertheless. Namely, the data obtained with Listeria showed that the OAK's activity was slightly reduced or improved, respectively, at an extremely acidic or alkaline pH (by 2-fold each), whereas the mode of action evolved from growth inhibition at low pH to a gradually fast bactericidal mode at higher pH values. Extremely high salt concentrations (up to 1,030 mM NaCl) had no effect on the MIC value, although they slowed down the bactericidal rates. Low temperatures hardly affected the MIC value (a 2-fold reduction was noted at between 37 and 4°C), whereas a higher temperature (48°C) significantly enhanced potency, reducing the MIC from 5 to 1.2 μg/ml and the bactericidal kinetics from 15 to 5 min.
TABLE 2.
Effects of incubation conditions on MIC and on bactericidal kinetics of C16(ω7)K-β12a
 |
Shaded rows represent the standard incubation conditions. GI, Growth inhibition effect defined as reduction of CFU count by < 1 log unit after 4 h exposure to OAK.
That is, the optimal incubation conditions: pH 8.5, 85 mM NaCl, and 48°C.
Interestingly, activity against E. coli revealed a substantially different profile with respect to pH and salt: the changes in pH did not alter the MIC value, but the bactericidal effect was inhibited at low pH, whereas salt did increase the MIC values while inhibiting the killing mode. Only the effect of temperature was relatively comparable to that observed with Listeria in that, low temperatures did not alter the MIC value and the high temperature enhanced potency by 4-fold. Also, as observed with Listeria, the bactericidal rates of E. coli increased with increasing temperatures.
Remarkably, when tested under the combined optimal conditions (i.e., 85 mM NaCl [pH 8.5] and 48°C), the OAK displayed 250-fold enhanced potency against L. monocytogenes (the MIC was reduced from 5 to 0.02 μg/ml). Similarly, the MIC against E. coli was reduced by 62-fold (i.e., from 5 to 0.08 μM). This increase in potency was also reflected in terms of the bactericidal rates; an effective reduction of 99% of the bacterial population was observed after 5 and 15 min, respectively. As shown in Fig. 2, altering the pH values and salt concentrations did not significantly modify the OAK's light scattering at the relevant concentration range, suggesting that the observed functional properties were not extensively biased by self-assembly phenomena (54). Nevertheless, the discrepancies observed upon pH and NaCl variations were uncanny, raising again the possibility that the OAK acted on Listeria and E. coli by distinct mechanisms.
FIG. 2.
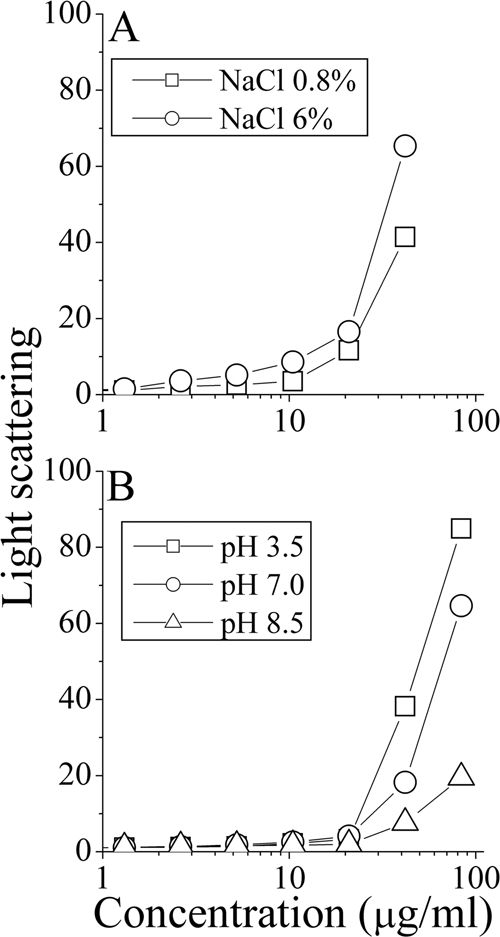
Self-assembly of C16(ω7)K-β12 in solution. Shown are representative dose dependence plots of the light-scattering intensities obtained for PBS solutions of C16(ω7)K-β12 at the specified salt concentrations (A) and pH values (B).
Bactericidal rates are strain dependent.
Figure 3 A to D depicts the dose-dependent time-kill curves under standard conditions of four bacteria representing Gram-positive (S. aureus and E. faecalis) and Gram-negative (P. aeruginosa and E. coli) species, while Fig. 3E and F compare the time-kill curves of four different strains of S. aureus and E. coli, respectively, at a single concentration representing four multiples of the MIC value. Clearly, the OAK exerted a dose-dependent bactericidal effect in all cases. However, significant inter- and intraspecies rate variations were observed. For instance, among the four S. aureus strains, C16(ω7)K-β12 reduced the CFU count by 6 log units within 15 to 30 min in two strains, whereas, in the remaining two strains, the rapid reduction in CFU count (∼2 log units) observed initially significantly slowed down thereafter. Such a phenomenon and its possible mechanism were recently reported with another OAK sequence (33).
FIG. 3.

Bactericidal properties of C16(ω7)K-β12. Panels A to D show the dose-dependent bactericidal kinetics as assessed against E. faecalis ATCC 29212 (A), S. aureus ATCC 29213 (B), P. aeruginosa ATCC 9027 (C), and E. coli ATCC 35218 (D). Symbols (A to D): ○, untreated control; □, 1× the MIC; ▵, 2× the MIC; ✳;, 4× the MIC. Panels E and F show the bactericidal kinetics against four representative S. aureus (E) and E. coli (F) strains, at a single concentration representing 4× the MIC. Symbols in panel E: ○, CI 15903; □, CI 17314; ▿, CI 15668; ▵, ATCC 29213. Symbols in panel F: ○, ATCC 35218; □, ATCC 43894; ▿, CI 16327; ▵, CI 16329. The dashed and solid lines in panels E and F represent untreated and OAK-treated bacteria, respectively. Bacteria were sampled at the specified time periods, subjected to serial 10-fold dilutions, and plated on LA agar for CFU counting after overnight incubation at 37°C.
Evocative rate differences were also observed among different E. coli strains, although among all species tested, E. coli displayed a particularly odd behavior in that the initial killing rate was repeatedly and predominantly slow (e.g., during the first 30 min), even among strains displaying relatively fast kinetics. This was true for the four strains shown in Fig. 3F, as well as for three additional strains that we tested (data not shown). Thus, combined with the observations described above, these kinetic discrepancies also insinuated the possible occurrence of multiple mechanisms of action and suggested E. coli species as ideal candidates for verifying this notion. Therefore, we selected two equally sensitive E. coli strains (E. coli 16327 and 35218, herein referred to as Ec1 and Ec2, respectively) displaying identical MIC value but differential killing curves (Fig. 3F) and compared them to two S. aureus strains (ATCC 29213 and 17314, referred to as Sa1 and Sa2, respectively).
Comparing the mechanism of action against related bacterial strains.
The potentially sensitive dye diSC3-5 (66) is often used as an indicator for membrane depolarization resulting from disruption of the plasma-membrane integrity (15, 35, 66). We compared the OAK's effect on the plasma membranes of strains Ec1, Ec2, Sa1, and Sa2 and simultaneously monitored viability by CFU counts performed on aliquots drawn from the diSC3-5 assay. As shown in Fig. 4 A, upon exposure to OAK (4× the MIC), an immediate burst of fluorescence (increase in dye release) occurred in strain Ec1; this is believed to reflect membrane depolarization (66). The addition of another dose of C16(ω7)K-β12 or of an HDP (dermaseptin, known for its rapid disruption of the cytoplasmic membrane) ∼10 min after the first OAK treatment had little effect, indicating that the initial OAK treatment was responsible for near-total membrane depolarization. A CFU count performed concomitantly under the specific experimental conditions of the diSC3-5 assay (i.e., in the presence of EDTA) showed a clear correlation in that the OAK induced concomitantly rapid reduction in bacterial viability, thus supporting the notion that the bactericidal mechanism of C16(ω7)K-β12 over this E. coli strain stemmed from damaging its cytoplasmic membrane.
FIG. 4.

Aptitude of C16(ω7)K-β12 (4× the MIC) to disrupt the cytoplasmic membrane. Panels A to C show the simultaneous determination of bacterial viability and membrane depolarization using E. coli CI 16327 (A), E. coli ATCC 35218 (B), and S. aureus ATCC 29213 (C). Symbols in panels A to C: ○, untreated control; □, OAK-treated bacteria. Black traces show online monitoring of the fluorescence increase representing release of the potential-sensitive dye diSC3-5 in OAK-treated bacteria. Arrows point to the times of addition of the OAK. The second arrow in panel A points to the time for dermaseptin addition (4× the MIC). Panel D compares the PI uptake kinetics by OAK-treated E. coli ATCC 35218 (○) and S. aureus ATCC 29213 (▪).
In sharp contrast, these same conditions failed to induce depolarization in strain Ec2, and the CFU count did not detect lower viability than with the untreated control, albeit some growth inhibition seems to have taken place (Fig. 4B). Figure 4C shows the rapid depolarization and death of strain Sa1, while strain Sa2 showed a similar outcome (data not shown). To validate the data collected from the diSC3-5 assay, we compared the uptake kinetics of the normally excluded dye, propidium iodide (PI). As shown in Fig. 4D, strain Ec2 revealed no detectable evidence for PI uptake, unlike strain Sa1, whose exposure to the OAK led to rapid PI uptake concomitantly with membrane depolarization (shown in Fig. 4C). Combined with the observed lack of depolarization (Fig. 4B), the lack of PI uptake by strain Ec2 provided further support to the view that C16(ω7)K-β12 did not damage the cytoplasmic membrane of strain Ec2, as opposed to the staphylococcal strain. It should be noted that the OAK concentration used (4× the MIC) does not allow one to rigorously conclude that observations reflect a mechanism of killing versus a postdeath event.
What then induced the bactericidal effect in strain Ec2? To address this question, we assessed the OAK's aptitude to affect bacterial viability through direct interaction with DNA as previously reported for the OAK sequence C12K-5α8 (48). Essentially, the assay exploits the fact that E. coli K-12 contain a plasmid (pUC19), used here as a reporter for OAK interactions with bacterial DNA. To reveal this interaction, bacteria were preincubated with the OAK; pUC19 was then extracted, exposed to the specific DNase, XbaI (selected due to the presence of a single restriction site on pUC19), and run in an agarose gel. It is assumed that OAK-bound DNA will alter the enzyme's binding affinity to its site of action either by the physical occupation of the binding site on the plasmid or by altering its chemical environment in a manner that will prohibit proper XbaI reaction with pUC19. Figure 5 A shows a control experiment where purified pUC19 was incubated with the OAK (30 min, room temperature) at concentrations roughly corresponding to 1× and 2× the MIC and run on agarose gel. The fact that DNA migration was inhibited in a dose-dependent manner provided initial evidence for the OAK's ability, a priori, to interact with bacterial DNA. Figure 5B shows another control experiment where digestion of pUC19 (in the absence of OAK) yielded a single band, whereas direct incubation of pUC19 with OAK (4× the MIC) prior to exposure to XbaI has shifted the band position to that of a lower molecular weight (similarly to the native plasmid). This suggested that the OAK interaction with pUC19 can protect from XbaI digestion. Next, we assessed this property in vivo when E. coli cultures were pretreated with OAK briefly (for 30 min or even less) and washed thoroughly to minimize potential OAK carryover, and then the pUC19 was extracted, exposed to XbaI, and run in a gel. As shown in Fig. 5B, the OAK prohibited the enzymatic activity. Hence, this finding revealed the cytoplasmic localization of the OAK under conditions where the plasma membrane disruption was not detectable (Fig. 4B).
FIG. 5.

Binding of C16(ω7)K-β12 to DNA and the resulting synthesis inhibition. (A) Agarose gel runs of bacterial plasmid pUC19 (200 ng) after incubation with various OAK concentrations (30 min, room temperature). (B) DNA restriction inhibition assay showing (from left to right) agarose gel runs (in duplicates) of pUC19 in its native state and after treatment with the DNase XbaI or when preincubated (30 min) with OAK (4× the MIC) and then treated with XbaI. The rightmost lanes show the gel runs of pUC19 after extraction from OAK-free and OAK-pretreated bacteria (4× the MIC). (C) Bacterial viability (CFU count) and macromolecule synthesis ([3H]thymidine incorporation assay) using E. coli CGSC 5895. Symbols: circles, untreated control; triangles, OAK-treated bacteria (4× the MIC). Open symbols, radioactivity signals; closed symbols, bacterial viability.
Next, we verified that bacteria exposed to the OAK (4× the MIC) were indeed subject to similarly rapid inhibition in carrying out biosynthesis, as assessed by their ability to incorporate radiolabeled thymidine. Figure 5C shows that C16(ω7)K-β12 inhibited the process of thymidine incorporation, although as evident from CFU enumeration of aliquots from the same experiment, reduced viability was reflected only hours later. This finding supports the notion that the consequence of OAK's direct interaction with bacterial DNA was to inhibit biosynthesis, which leads to cell death only hours thereafter (48). The combined data therefore support the view that C16(ω7)K-β12 can affect the viability of strain Ec2 through interference with processes of DNA maintenance and expression despite its apparent inability to afflict damage to the cytoplasmic membrane.
DISCUSSION
Data collected from the present study provided evidence for the aptitude of a broad-spectrum bactericidal OAK to affect bacterial viability by distinct mechanisms. This notion is based on a variety of direct and indirect evidence. (i) When assessed against a variety of bacterial species, C16(ω7)K-β12 exhibited high heterogeneity in killing rates (Fig. 3). Whereas fast time-kill curves are often associated with abrupt membrane disruption (33, 35), slower rates are more compatible with inhibition of vital processes (48). (ii) C16(ω7)K-β12 is not always able to inflict early membrane-damage, as seen by comparing between two E. coli strains that were equally sensitive to the OAK (identical MIC value). This view was corroborated by two dye-based assays (diSC3-5 and PI). (iii) Different experimental evidence supports the view that C16(ω7)K-β12 can induce bacterial death through interaction with DNA under live conditions. For instance, the OAK was directly responsible for protecting bacterial DNA from XbaI action and inhibited biosynthesis very early, suggesting that bacterial death was the consequence of this inhibition. (iv) Various changes in environmental conditions, such as pH and ionic strength, significantly diverged in their effects over OAK's potency, supporting the view that the OAK is involved in interactions with at least two different targets: cytoplasmic membrane and DNA (discussed further below).
Collectively, these findings add up to provide strong support to the view that while, in some cases, bacterial death can stem from the capacity of C16(ω7)K-β12 to breach the cell permeability barrier; in other cases, death stems from direct interference with DNA functions. By extrapolation, it is possible that at least some HDPs will behave similarly, provided that enough strains are tested. MSI-78, for example, has shown similar heterogeneity in bactericidal rates (20). Various studies have reported that a single HDP (42) or peptidomimetic (14) can exert antibacterial activity by different mechanisms at different peptide concentrations, suggesting that at their lowest inhibitory concentrations, HDPs may be less capable of damaging cell membranes, while they maintain their ability to inhibit macromolecular synthesis. In the present study, however, the same OAK concentration exhibited different mechanisms of action. This phenomenon had not been reported before, to our knowledge. A few closely related references are found in the literature. (i) Rotem et al. (48) provide evidence that analogous OAKs (i.e., hexamer and octamer) acted on a single bacterial strain by distinct mechanisms, inhibition of DNA functions or membrane disruption, respectively, whereas here a single OAK is proposed to use, against different bacteria, one of these mechanisms or the other. (ii) According to Epand et al. (14), a peptidomimetic compound acted by a dose-dependent dual mechanism, i.e., at low concentrations (up to 25 μg/ml) the polymer was proposed to inhibit cytoplasmic components, whereas at higher concentrations the polymer prevents entry to the cytoplasm by blockage of transport across the outer membrane. This is clearly not the case with C16(ω7)K-β12. Similarly, Livne et al. (33) reported that interspecies differential bactericidal rates stem from interactions with cell wall specific components leading to a transient rapid bactericidal stage that over time converts to a bacteriostatic effect.
The combined data seem to support the notion that cationic peptide-based antibacterials might in principle have the capacity to affect bacterial viability through a range of mechanisms, suggesting therefore the futility in assigning a mode of action to a particular peptide, as a rule. Future studies will have to face a remarkable challenge in better defining the molecular basis of this phenomenon, a task likely to be more suitable for molecules that are simpler than typical HDPs.
The present study also illustrated the effects of incubation conditions on antibacterial potency. The OAK maintained significant growth inhibitory activity under a variety of extreme incubation conditions while other conditions have radically enhanced potency. From the mechanistic viewpoint, the manner by which different incubation conditions have affected the OAK's activity was both revealing and quite complex for interpretation. While further studies are clearly required to fully comprehend the molecular basis for these observations, these findings seem nonetheless consistent with the occurrence of the stipulated differential mechanisms. Thus, increases in pH values have greatly influenced potency over Listeria, whereas they had a much lower effect on E. coli. This would make sense according to the view of the cytoplasmic localization of the OAK's target in E. coli as opposed to targeting the membrane in Listeria, where the electrostatic interactions are known to play a major role and are therefore expected to affect more severely the membrane-damaging mechanism. For comparison, a similar study performed with the OAK C12K-7α8, which is notorious for its membrane-active properties (48), revealed that changes in pH have equally affected the OAK's potency against both Gram-negative and Gram-positive bacteria (21). Consistent with the dual-mechanism view is the fact that the potency of C16(ω7)K-β12 was inversely affected in a high-ionic-strength milieu, which was expected to influence DNA interaction more severely due to the charge-masking effect of NaCl. Accordingly, an increase in temperature is not expected to differentially affect the OAK's potencies, as observed. In fact, potency was significantly enhanced against both bacterial species. In comparison, the potency of the membrane-active HDP dermaseptin was severely reduced under acidic conditions, high salt concentrations, or low temperatures (52, 63).
In addition to their mechanistic relevance, these data also reveal new means for enhancing antibacterial potency (by up to 250-fold) through proper selection and combination of the most favorable incubation conditions. Although the molecular basis for these observations also warrants further investigation, the findings are likely to contribute to better understanding the antibacterial effects of OAKs and HDPs and the design of new treatment strategies, for instance in topical applications of polymicrobial infections such as in oral mucositis (10) or in diabetic foot ulcers (32).
In addition to their therapeutic potential, HDPs are believed to be useful in a variety of antimicrobial applications, such as in cosmetics or food safety (17, 51). Nisin, for instance, is a widely used bacteriocin (50), including its use to control and/or prevent L. monocytogenes in a variety of foods (7, 17). Nisin, however, has several disadvantages, including low solubility over the physiological pH range, activity restricted to Gram-positive bacteria, and known resistant strains (4). New approaches that combine bacteriocins and heat treatments, pulse fields, or other chemicals have shown improved activity, although they might imply a higher production cost (17). There is thus a need for new approaches that would properly address these problems. Both E. coli and Listeria have highly efficient mechanisms of global stress resistance, which contribute to their low infectious dose and tolerance to stress factors, including acidic pH (4, 46), allowing these pathogens to overcome food preservatives and safety barriers and thereby pose potential risks to human health (18, 26). In this respect, our data point to potential uses of OAKs in food safety since the OAK properties were studied under conditions relevant to food preservation (24).
Acknowledgments
This research was supported by the Israel Science Foundation (grant 283/08) and the Elyahu Pen Foundation.
Footnotes
Published ahead of print on 15 November 2010.
REFERENCES
- 1.Bals, R., et al. 1998. Human beta-defensin 2 is a salt-sensitive peptide antibiotic expressed in human lung. J. Clin. Invest. 102:874-880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bessalle, R., A. Kapitkovsky, A. Gorea, I. Shalit, and M. Fridkin. 1990. All-d-magainin: chirality, antimicrobial activity and proteolytic resistance. FEBS Lett. 274:151-155. [DOI] [PubMed] [Google Scholar]
- 3.Blondelle, S., and K. Lohner. 2000. Combinatorial libraries: a tool to design antimicrobial and antifungal peptide analogues having lytic specificities for structure-activity relationship studies. Biopolymers 55:74-87. [DOI] [PubMed] [Google Scholar]
- 4.Bonnet, M., M. M. Rafi, M. L. Chikindas, and T. J. Montville. 2006. Bioenergetic mechanism for nisin resistance, induced by the acid tolerance response of Listeria monocytogenes. Appl. Environ. Microbiol. 72:2556-2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bradshaw, J. 2003. Cationic antimicrobial peptides: issues for potential clinical use. BioDrugs 17:233-240. [DOI] [PubMed] [Google Scholar]
- 6.Brogden, K. A. 2005. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 3:238-250. [DOI] [PubMed] [Google Scholar]
- 7.Cotter, P. D., C. Hill, and R. P. Ross. 2005. Bacteriocins: developing innate immunity for food. Nat. Rev. Microbiol. 3:777-788. [DOI] [PubMed] [Google Scholar]
- 8.Dartois, V., et al. 2005. Systemic antibacterial activity of novel synthetic cyclic peptides. Antimicrob. Agents Chemother. 49:3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deslouches, B., et al. 2005. Activity of the de novo engineered antimicrobial peptide WLBU2 against Pseudomonas aeruginosa in human serum and whole blood: implications for systemic applications. Antimicrob. Agents Chemother. 49:3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Donnelly, J., L. Bellm, J. Epstein, S. Sonis, and R. Symonds. 2003. Antimicrobial therapy to prevent or treat oral mucositis. Lancet Infect. Dis. 3:405-412. [DOI] [PubMed] [Google Scholar]
- 11.Easton, D., A. Nijnik, M. Mayer, and R. Hancock. 2009. Potential of immunomodulatory host defense peptides as novel anti-infectives. Trends Biotechnol. 27:582-590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eichler, J., and R. Houghten. 1995. Generation and utilization of synthetic combinatorial libraries. Mol. Med. Today 1:174-180. [DOI] [PubMed] [Google Scholar]
- 13.Epand, R., et al. 2003. Direct comparison of membrane interactions of model peptides composed of only Leu and Lys residues. Biopolymers 71:2-16. [DOI] [PubMed] [Google Scholar]
- 14.Epand, R. F., et al. 2008. Dual mechanism of bacterial lethality for a cationic sequence-random copolymer that mimics host-defense antimicrobial peptides. J. Mol. Biol. 379:38-50. [DOI] [PubMed] [Google Scholar]
- 15.Epand, R. F., H. Sarig, A. Mor, and R. M. Epand. 2009. Cell-wall interactions and the selective bacteriostatic activity of a miniature oligo-acyl-lysyl. Biophys. J. 97:2250-2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fields, G. B., and R. L. Noble. 1990. Solid phase peptide synthesis utilizing 9-fluorenylmethoxycarbonyl amino acids. Int. J. Peptide Protein Res. 35:161-214. [DOI] [PubMed] [Google Scholar]
- 17.Galvez, A., H. Abriouel, R. L. Lopez, and N. Ben Omar. 2007. Bacteriocin-based strategies for food biopreservation. Int. J. Food Microbiol. 120:51-70. [DOI] [PubMed] [Google Scholar]
- 18.Gandhi, M., and M. L. Chikindas. 2007. Listeria: a food-borne pathogen that knows how to survive. Int. J. Food Microbiol. 113:1-15. [DOI] [PubMed] [Google Scholar]
- 19.Ganz, T. 1999. Defensins and host defense. Science 286:420-421. [DOI] [PubMed] [Google Scholar]
- 20.Ge, Y., et al. 1999. In vitro antibacterial properties of pexiganan, an analog of magainin. Antimicrob. Agents Chemother. 43:782-788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldfeder, Y., F. Zaknoon, and A. Mor. Experimental conditions that enhance potency of an antibacterial oligo-acyl-lysyl. Antimicrob. Agents Chemother. 54:2590-2595. [DOI] [PMC free article] [PubMed]
- 22.Goldman, M. J., et al. 1997. Human beta-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell 88:553-560. [DOI] [PubMed] [Google Scholar]
- 23.Gordon, Y., E. Romanowski, and A. McDermott. 2005. A review of antimicrobial peptides and their therapeutic potential as anti-infective drugs. Curr. Eye Res. 30:505-515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gould, G. W. 2000. Preservation: past, present, and future. Br. Med. Bull. 56:84-96. [DOI] [PubMed] [Google Scholar]
- 25.Guarna, M. M., R. Coulson, and E. Rubinchik. 2006. Anti-inflammatory activity of cationic peptides: application to the treatment of acne vulgaris. FEMS Microbiol. Lett. 257:1-6. [DOI] [PubMed] [Google Scholar]
- 26.Hamon, M., H. Bierne, and P. Cossart. 2006. Listeria monocytogenes: a multifaceted model. Nat. Rev. Microbiol. 4:423-434. [DOI] [PubMed] [Google Scholar]
- 27.Jenssen, H., P. Hammill, and R. E. W. Hancock. 2006. Peptide antimicrobial agents. Clin. Microbiol. Rev. 19:491-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jing, W., H. Hunter, J. Hagel, and H. Vogel. 2003. The structure of the antimicrobial peptide Ac-RRWWRF-NH2 bound to micelles and its interactions with phospholipid bilayers. J. Peptide Res. 61:219-229. [DOI] [PubMed] [Google Scholar]
- 29.Lai, X. Z., et al. 2008. Ceragenins: cholic acid-based mimics of antimicrobial peptides. Acc. Chem. Res. 41:1233-1240. [DOI] [PubMed] [Google Scholar]
- 30.Latham, P. W. 1999. Therapeutic peptides revisited. Nat. Biotechnol. 17:755-757. [DOI] [PubMed] [Google Scholar]
- 31.Lee, I. H., Y. Cho, and R. I. Lehrer. 1997. Effects of pH and salinity on the antimicrobial properties of clavanins. Infect. Immun. 65:2898-2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lipsky, B., K. Holroyd, and M. Zasloff. 2008. Topical versus systemic antimicrobial therapy for treating mildly infected diabetic foot ulcers: a randomized, controlled, double blinded, multicenter trial of pexiganan cream. Clin. Infect. Dis. 47:1537-1545. [DOI] [PubMed] [Google Scholar]
- 33.Livne, L., et al. 2009. Design and characterization of a broad-spectrum bactericidal acyl-lysyl oligomer. Chem. Biol. 16:1250-1258. [DOI] [PubMed] [Google Scholar]
- 34.Marr, A. K., W. J. Gooderham, and R. E. Hancock. 2006. Antibacterial peptides for therapeutic use: obstacles and realistic outlook. Curr. Opin. Pharmacol. 6:468-472. [DOI] [PubMed] [Google Scholar]
- 35.Marynka, K., S. Rotem, I. Portnaya, U. Cogan, and A. Mor. 2007. In vitro discriminative antipseudomonal properties resulting from acyl substitution of N-terminal sequence of dermaseptin s4 derivatives. Chem. Biol. 14:75-85. [DOI] [PubMed] [Google Scholar]
- 36.McInturff, J. E., et al. 2005. Granulysin-derived peptides demonstrate antimicrobial and anti-inflammatory effects against Propionibacterium acnes. J. Invest. Dermatol. 125:256-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Minahk, C. J., and R. D. Morero. 2003. Inhibition of enterocin CRL35 antibiotic activity by mono- and divalent ions. Lett. Appl. Microbiol. 37:374-379. [DOI] [PubMed] [Google Scholar]
- 38.Mor, A. 2009. Multifunctional host defense peptides: antiparasitic activities. FEBS J. 276:6474-6482. [DOI] [PubMed] [Google Scholar]
- 39.Nicolas, P. 2009. Multifunctional host defense peptides: intracellular-targeting antimicrobial peptides. FEBS J. 276:6483-6496. [DOI] [PubMed] [Google Scholar]
- 40.Oh, J. E., S. Y. Hong, and K. H. Lee. 1999. Design, synthesis and characterization of antimicrobial pseudopeptides corresponding to membrane-active peptide. J. Peptide Res. 54:129-136. [DOI] [PubMed] [Google Scholar]
- 41.Perron, G. G., M. Zasloff, and G. Bell. 2006. Experimental evolution of resistance to an antimicrobial peptide. Proc. R. Soc. Lond. B Biol. Sci. 273:251-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Podda, E., et al. 2006. Dual mode of action of Bac7, a proline-rich antibacterial peptide. Biochim. Biophys. Acta 1760:1732-1740. [DOI] [PubMed] [Google Scholar]
- 43.Radzishevsky, I. S., et al. 2008. Structure-activity relationships of antibacterial acyl-lysine oligomers. Chem. Biol. 15:354-362. [DOI] [PubMed] [Google Scholar]
- 44.Radzishevsky, I. S., et al. 2007. Improved antimicrobial peptides based on acyl-lysine oligomers. Nat. Biotechnol. 25:657-659. [DOI] [PubMed] [Google Scholar]
- 45.Radzishevsky, I. S., et al. 2005. Effects of acyl versus aminoacyl conjugation on the properties of antimicrobial peptides. Antimicrob. Agents Chemother. 49:2412-2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Richard, H., and J. W. Foster. 2004. Escherichia coli glutamate- and arginine-dependent acid resistance systems increase internal pH and reverse transmembrane potential. J. Bacteriol. 186:6032-6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rotem, S., and A. Mor. 2009. Antimicrobial peptide mimics for improved therapeutic properties. Biochim. Biophys. Acta 1788:1582-1592. [DOI] [PubMed] [Google Scholar]
- 48.Rotem, S., et al. 2008. Analogous oligo-acyl-lysines with distinct antibacterial mechanisms. FASEB J. 22:2652-2661. [DOI] [PubMed] [Google Scholar]
- 49.Rozek, A., J. P. Powers, C. L. Friedrich, and R. E. Hancock. 2003. Structure-based design of an indolicidin peptide analogue with increased protease stability. Biochemistry 42:14130-14138. [DOI] [PubMed] [Google Scholar]
- 50.Rulis, A. M. 2009. Agency response letter. GRAS notice no. GRN 000065. Food and Drug Administration, Washington, DC.
- 51.Rydlo, T., J. Miltz, and A. Mor. 2006. Eukaryotic antimicrobial peptides: promises and premises in food safety. J. Food Sci. 71:125. [Google Scholar]
- 52.Rydlo, T., S. Rotem, and A. Mor. 2006. Antibacterial properties of dermaseptin S4 derivatives under extreme incubation conditions. Antimicrob. Agents Chemother. 50:490-497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sarig, H., et al. 2010. A miniature mimic of host defense peptides with systemic antibacterial efficacy. FASEB J. [Epub ahead of print.] [DOI] [PMC free article] [PubMed]
- 54.Sarig, H., S. Rotem, L. Ziserman, D. Danino, and A. Mor. 2008. Impact of self-assembly properties on antibacterial activity of short acyl-lysine oligomers. Antimicrob. Agents Chemother. 52:4308-4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shai, Y. 2002. Mode of action of membrane active antimicrobial peptides. Biopolymers 66:236-248. [DOI] [PubMed] [Google Scholar]
- 56.Shai, Y., and Z. Oren. 1996. Diastereomers of cytolysins, a novel class of potent antibacterial peptides. J. Biol. Chem. 271:7305. [DOI] [PubMed] [Google Scholar]
- 57.Som, A., S. Vemparala, I. Ivanov, and G. N. Tew. 2008. Synthetic mimics of antimicrobial peptides. Biopolymers 90:83-93. [DOI] [PubMed] [Google Scholar]
- 58.Tew, G. N., R. W. Scott, M. L. Klein, and W. F. Degrado. 2010. De novo design of antimicrobial polymers, foldamers, and small molecules: from discovery to practical applications. Acc. Chem. Res. 43:30-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tossi, A., L. Sandri, and A. Giangaspero. 2000. Amphipathic, alpha-helical antimicrobial peptides. Biopolymers 55:4-30. [DOI] [PubMed] [Google Scholar]
- 60.Tossi, A., C. Tarantino, and D. Romeo. 1997. Design of synthetic antimicrobial peptides based on sequence analogy and amphipathicity. Eur. J. Biochem. 250:549-558. [DOI] [PubMed] [Google Scholar]
- 61.Vaara, M. 2009. New approaches in peptide antibiotics. Curr. Opin. Pharmacol. 9:571-576. [DOI] [PubMed] [Google Scholar]
- 62.Wade, D., et al. 1990. All-d-amino acid-containing channel-forming antibiotic peptides. Proc. Natl. Acad. Sci. U. S. A. 87:4761-4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yaron, S., T. Rydlo, D. Shachar, and A. Mor. 2003. Activity of dermaseptin K4-S4 against food-borne pathogens. Peptides 24:1815-1821. [DOI] [PubMed] [Google Scholar]
- 64.Yeaman, M. R., and N. Y. Yount. 2003. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 55:27-55. [DOI] [PubMed] [Google Scholar]
- 65.Zasloff, M. 2002. Antimicrobial peptides of multicellular organisms. Nature 415:389-395. [DOI] [PubMed] [Google Scholar]
- 66.Zhang, L., P. Dhillon, H. Yan, S. Farmer, and R. E. Hancock. 2000. Interactions of bacterial cationic peptide antibiotics with outer and cytoplasmic membranes of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 44:3317-3321. [DOI] [PMC free article] [PubMed] [Google Scholar]