

Abstract
Aims
Acute ischaemic preconditioning (IPC) induces protection against cardiac ischaemia–reperfusion (IR) via post-translational modification of key proteins. Lysine (Lys) acetylation is an important regulator of protein function, but this type of modification has not been studied in the context of IPC. We investigated Lys acetylation in IPC and its upstream regulation by SIRT1.
Methods and results
Hearts from C57BL/6 mice were Langendorff-perfused and subjected to IPC and IR injury. Mice were exposed to IPC by in vivo coronary artery occlusion. An isolated cardiomyocyte model of IPC was also developed. Lys acetylation was measured by western blotting, and pharmacological modulators of Lys acetylation were tested. More Lys deacetylation was observed in IPC, in the Langendorff, in vivo, and cellular IPC models; this was concurrent with an increase in SIRT1 activity measured by p53 Lys379 deacetylation. IPC was not accompanied by changes in SIRT1 protein level, but evidence was obtained for SIRT1 modification by Small Ubiquitin-like Modifier (SUMOylation) in IPC. Furthermore, the specific SIRT1 inhibitor splitomicin reversed both IPC-mediated Lys deacetylation and IPC-induced cardioprotection. Inhibition of nicotinamide phosphoribosyltransferase (Nampt, an important enzyme which regulates SIRT1 activity by maintaining availability of the substrate NAD+) also blocked both IPC-induced deacetylation and cardioprotection.
Conclusion
Lys deacetylation occurs during IPC and an elevation in SIRT1 activity plays a role in this phenomenon. Inhibition of SIRT1, either directly or by restricting the availability of its substrate NAD+, inhibits IPC. Together these data suggest a role for SIRT1-mediated Lys deacetylation in the mechanism of acute IPC.
Keywords: Lysine acetylation, Ischaemia, Sirtuins, Preconditioning, SUMO
1. Introduction
Cardiac ischaemic preconditioning (IPC) is a mild ischaemic stress which activates signalling pathways, leading to protection against long-term ischaemia and reperfusion (IR) injury.1,2 Two time windows of IPC protection exist: delayed IPC (24–72 h), which is primarily mediated by gene regulation,3 and acute IPC (0–3 h), which is thought to be mediated by protein post-translational modifications (PTMs). Mounting evidence supports roles for protein phosphorylation4 and S-nitrosation5,6 in acute IPC, while another PTM, lysine (Lys) acetylation, is largely un-studied in this context.
Recently protein (de)acetylation has emerged as an important PTM involved in many cell signalling pathways in several compartments.7,8 In the nucleus, class I/II histone deacetylases (HDACs) regulate gene expression via deacetylation of histones and other nuclear proteins.8,9 Interestingly, pharmacologic inhibition of class I/II HDACs is known to confer cardioprotection.10,11
In contrast, the class III HDACs may have an opposite role in protective signalling: these NAD+-dependent HDACs, also known as sirtuins (SIRTs), are thought to play important roles in the regulation of metabolism and apoptosis via deacetylation of cytosolic and mitochondrial enzymes.12,13 SIRT1 in particular initiates several signalling events relevant to cardioprotection, including: activation of endothelial nitric oxide synthase,14 insulin receptor signalling,15 and autophagy.16 In addition SIRT1 activation elicits resistance to oxidative stress17 via regulation of transcription factors and co-activators such as FOXO 1,3,4,18–20 Hif-2α,21 and NF-kB.22
Although SIRT1 has previously been shown to confer protection in various models of cardiovascular oxidative stress,17,23,24 the role of SIRT1 in IPC is unknown. Furthermore, the enzyme nicotinamide phosphoribosyltransferase (Nampt) is known to be an important regulator of SIRT activity by maintaining the availability of its substrate NAD+.25,26 However, the role of Nampt in IPC has also not been investigated. In the current study, we hypothesized that SIRT1 and Nampt play a role in IPC by regulating Lys acetylation of cardiac proteins. This hypothesis was tested using a pharmacological approach coupled with in vitro and in vivo models of IR injury and IPC.
2. Methods
For full methods see Supplementary material online. Male C57BL6 mice (20–25 g) were purchased from Harlan (Indianapolis, IN, USA) and handled in accordance with a protocol approved by the University Committee on Animal Research (UCAR), and in accordance with the NIH Guide for the Care and Use of Laboratory animals (NIH Publication #85-23, 1996). Mice were housed under a 12 h light/dark cycle with food and water available ad libitum.
Mouse hearts were subjected to Langendorff perfusion as previously described.27 Mice were divided into seven groups: (i) IR (n = 6); (ii) Splitomicin (10 µM) + IR (n = 5); (iii) IPC + IR (n = 8); (iv) Splitomicin (10 µM) + IPC + IR (n = 6); (v) FK866 (1 µM) + IR (n = 5); (vi) FK866 (1 µM) + IPC + IR (n = 8); (vii) SRT1720 (1 µM) + IR (n = 5). Splitomicin is a specific SIRT1 inhibitor,28,29 FK866 is a specific Nampt inhibitor,30 and SRT1720 is reported to be a SIRT1 activator.31 Pharmacologic agents were delivered via a port above the aortic perfusion cannula, for 20 min prior to IR. In IPC treatments, agents were delivered 5 min prior to, and during each reperfusion cycle of, IPC. IR injury comprised 25 min index ischaemia plus 60 min reperfusion. IPC comprised 3 × 5 min cycles of ischaemia interspersed with 5 min reperfusion, prior to IR. Left ventricular pressure was monitored throughout by a balloon-linked transducer, and at the end of reperfusion infarct size was measured by 2,3,5-triphenyltetrazolium chloride (TTC) staining, using a modification of a previously described protocol 32 (see Supplementary material online).
In vivo IPC was performed by transient occlusion of the left anterior descending (LAD) coronary artery, in tribromoethanol-anesthetized, intubated mice, as described previously.32 Mice were divided into two groups: (i) Ctrl. (n = 10), (ii) IPC (n = 10). At the end of surgical protocols, tissue was either harvested immediately (n = 5) or mice were allowed to recover for 24 h (n = 5) and then tissue was harvested. Cardiac tissue fractionation was performed by differential centrifugation using established methods.33 The area-at-risk (AAR) was delineated from the area-not-at-risk (ANAR) by visualization of pallor upon transient LAD occlusion, immediately prior to tissue dissection. For delayed IPC in vivo, the suture used to induce IPC (3 × 5 min cycles) was left in place un-occluded for 24 h, thereby allowing transient re-occlusion to ascertain the AAR for dissection. An isolated cardiomyocyte model was also developed (see Supplementary material online).
SIRT1, p53, K379Ac-p53, and global protein acetylation, were all analysed by western blotting34 in a subset of perfused and in vivo hearts (n = 5). For p53 and K379Ac-p53 blots, lysis buffers contained Trichostatin A to inhibit class I/II HDACs. Samples were separated by SDS–PAGE and electro-blotted to nitrocellulose. Membranes were blocked with 5% non-fat dry milk in TBST. Primary antibodies were anti-SIRT1 (Abcam, Cambridge, MA, USA) at 1/1000 dilution in blocking buffer, or anti-K-Ac (Cell Signalling, Danvers, MA, USA) at 1/1000 in blocking buffer. Samples were also run on two-dimensional (2D) gels (pH gradient 3–10), as previously described.32
SIRT1 proteins were immunoprecipitated from the whole heart homogenate using monoclonal anti-SIRT1 antibodies similar to previously published protocols.35 Immunoprecipitated samples or whole extracts (input) were separated by SDS–PAGE as above, and blotted using anti-SIRT1, anti-Phospho Ser/Thr/Tyr (AnaSpec Inc., San Jose, CA, USA), anti-K-Ac, anti-SUMO2/3 (ABGENT, San Diego, CA), or anti-SUMO1 (Invitrogen, Carlsbad, CA, USA) antibodies, all at 1/1000 dilution. All blots were developed with HRP-linked anti-mouse or anti-rabbit secondaries, and enhanced chemiluminescence.
Densitometry was performed on western blots using NIH Image software. For single proteins (e.g. SIRT, p53) density of the appropriate band was determined. For full-length western blots (e.g. global Lys acetylation), total density for all bands in a given molecular weight range was determined. Comparison between two densitometry values was always performed on the same physical western blot. Statistical significance of single parameters between two groups was determined using Student's t-test (paired, where appropriate). Significance between multiple groups was determined using multiple way analysis of variance (ANOVA).
3. Results
Protein acetylation was analysed by western blotting in fractionated cardiac tissue from perfused hearts exposed to control perfusion or acute IPC. Figure 1A shows that acute IPC caused a significant deacetylation of proteins in the cytosolic fraction. In vivo acute IPC elicited similar effects: western blots of IPC-exposed cardiac cytosol revealed marked deacetylation of several proteins in the AAR compared with ANAR (Figure 1B). Densitometry (typical profiles are shown in Supplementary material online, Figure S1C and D) revealed significant differences in acetylation in IPC across a broad molecular weight range, although variability between animals was seen at the level of individual bands. The identity of deacetylated proteins is not known, due to the inherent limitations of applying protein ID to bands cut from 1D gels, hence we chose to run 2D gels for protein ID purposes.
Figure 1.

Protein acetylation during IPC in perfused and in vivo mouse heart. (A) Mouse hearts were subjected to either control perfusion (Ctrl.) or IPC (without subsequent IR injury) followed by immediate tissue fractionation. (B) Mice were subjected to in vivo IPC (without subsequent IR injury). The AAR was separated from the ANAR followed by tissue fractionation. In both panels, protein fractions were separated by SDS–PAGE and K-Ac visualized by western blot. Representative blots for cytosolic fractions are shown, with other cell fractions in Supplementary material online, Figure S1. Molecular weight markers (kDa) are to the left of blots. Arrows highlight differences in protein acetylation between control and IPC groups. Panels below show densitometry in the range 25–100 kDa, normalized to protein across the same molecular weight range (representative Ponceau S stained membranes are in Supplementary material online, Figure S1). Each pair of data points connected by a line represents a single experiment. The ratio of K-Ac/Protein was calculated and presented for each individual experiment. *P < 0.05 (paired Student's t-test) between control and IPC groups, n ≥ 5.
Analysis of other cell fractions (homogenate, nucleus, mitochondria) on 1D gels did not reveal changes in Lys acetylation in IPC (see Supplementary material online, Figure S1). Analysis of 2D gels both recapitulated the 1D gel result for cytosol, and also revealed small changes in Lys acetylation in mitochondria in IPC (see Supplementary material online, Figure S2). Whereas mitochondrial acetylation changed in both directions in IPC, cytosolic acetylation changes were unidirectional (decreased only) and were more robust (i.e. visible on a 1D gel alone). These characteristics prompted us to focus the current study on the origin of IPC-induced changes in cytosolic acetylation, with acetylation in other fractions being the subject of future and ongoing studies.
One potential cause of the decreased density on K-Ac western blots in IPC cytosol could be translocation of acetylated proteins between the cytosol and other cell fractions. Such translocation events are known to occur in IPC.36–40 However, 2D gels (see Supplementary material online, Figure S2) showed that spots which were lost from cytosol in IPC did not appear in mitochondria, and vice versa, suggesting that translocation between these fractions unlikely accounts for the observed changes in K-Ac western blots (see also data w.r.t. splitomicin, below). Furthermore, 2D gels on homogenates revealed several changes in Lys acetylation in IPC (see Supplementary material online, Figure S3), which clearly cannot be due to translocation, since the material analysed contains all cell fractions. Notably, not all deacetylation events seen in cell fractions were visible on the homogenate 2D gels, likely due to the limited loading capacity of IPG strips, which excludes less soluble proteins. This highlights the importance of cell fractionation and 2D gels, to identify low abundance (de)acetylated proteins in more detail.
The role of sirtuins in protein deacetylation,13 coupled with recent interest in SIRT1 protection against oxidative stress,17 encouraged us to examine whether SIRT1 may play a role in the deacetylation in IPC. Consistent with increased SIRT1 activity in IPC, the SIRT1 substrate K379 of p5341–43 was deacetylated by acute IPC, in both perfused heart and in vivo systems (Figure 2A).
Figure 2.

p53 acetylation and SIRT1 protein level during IPC in perfused and in vivo mouse heart. (A) Homogenates from perfused hearts subjected to control perfusion (Ctrl.) or IPC were western blotted for p53 lysine 379 acetylation (left upper panel) and total p53 (left lower panel). Homogenates from AAR and ANAR of hearts exposed to IPC in vivo were also blotted. The ratio of Ac p53/p53 was calculated by densitometry and is shown below the blots (mean ± SEM, n ≥ 3). *P < 0.05 (paired Student's t-test) between control and IPC. (B) Homogenates from perfused hearts were obtained as described above, and western blotted for SIRT1 (upper panel) or actin (lower panel). Where indicated, the proteasome inhibitor MG132 was present during sample preparation. The ratio of SIRT1/actin was calculated by densitometry and is shown below the blots (mean ± SEM, n ≥ 4). (C) Mice were subjected to IPC in vivo, and heart tissue harvested from the AAR or ANAR either immediately (i.e. acute IPC), or 24 h later (i.e. delayed IPC). Heart homogenate was western blotted for SIRT1 (upper panel) or actin (lower panel). The ratio of SIRT1/actin was calculated by densitometry and is shown below the blots (mean ± SEM, n ≥ 4). This SIRT1 antibody yields two bands on a western blot, and the literature consensus is that the upper band is SIRT1 whereas the lower is of unknown origin.66 Loading controls (representative Ponceau S stained membranes) for all blots are shown in Supplementary material online, Figure S5.
The mechanism of increased SIRT1 activity in IPC was next examined. Figure 2B shows that SIRT1 protein did not change in acute IPC in perfused hearts. Furthermore, the proteasome inhibitor MG-13244 did not affect SIRT1 levels, indicating no role for proteasomal degradation in regulating SIRT1 in this system. To investigate whether SIRT1 may be regulated by delayed IPC, SIRT1 protein level was also analysed in in vivo mouse models of both acute and delayed IPC. Figure 2C shows that in both cases SIRT1 protein level was not altered. Furthermore, no alterations in SIRT1 level were observed when IPC-exposed heart was fractionated into nuclear and cytosolic fractions (see Supplementary material online, Figure S4). Thus, increases in SIRT1 protein do not appear to underlie increased SIRT1 activity in IPC.
Although protein levels of SIRT1 were not altered in IPC, SIRT1 activity may still be regulated by PTM. Previously, SIRT1 has been shown to be phosphorylated45 and SUMOylated,46 and notably SUMOylation is enhanced in neuronal IPC.47 As shown in Figure 3, immunoprecipitation of SIRT1 followed by western blotting found no evidence for modification of SIRT1 by phosphorylation, acetylation, or SUMO2/3. However, SIRT1 did appear to be modified by SUMO1 during IPC in the perfused heart (Figure 3A) and in vivo (Figure 3B).
Figure 3.

PTMs of SIRT1 during IPC. (A) SIRT1 was immunoprecipitated from homogenates of perfused hearts subjected to control perfusion (Ctrl.) or IPC, without subsequent IR injury. In each blot, lanes 1 and 2 show the input to the immunoprecipitation (i.e. the tissue lysate), lanes 3 and 4 show the immunoprecipitated pellet, and lanes 5 and 6 show an experiment in which immunoprecipitation was performed without the primary (SIRT1) antibody. Panels (top to bottom) show western blots for SIRT1, phospho-Ser/Thr/Tyr, K-Ac, SUMO2/3, and SUMO1. (B) SIRT1 was immunoprecipitated from the AAR or ANAR of hearts subjected to IPC in vivo. Western blots for SIRT1 (upper panel) and SUMO1 (lower panel) are shown. Assignment of lanes 1–6 is as detailed for (A). In both panels, numbers to the left are molecular weight markers (kDa). Blots are representative of at least two independent immunoprecipitation experiments in the in vitro or in vivo condition. The ratio of SUMO1/Sirt1 was calculated and is shown below the blots (mean ± standard deviation). *P < 0.05 (Student's t-test) between control and IPC. Loading controls (representative Ponceau S stained membranes) for all blots in are shown in Supplementary material online, Figure S6.
The observation of an increase in SIRT1 activity, concurrent with Lys deacetylation in IPC (Figures 1 and 2, see Supplementary material online, Figures S2 and S3), led us to hypothesize that the former may be responsible for the latter. To test this, Lys acetylation was examined in cytosol from hearts subjected to various perfusion protocols, with infusion of the specific SIRT1 inhibitor splitomicin. Figure 4 shows that splitomicin treatment inhibited IPC-induced cytosolic Lys deacetylation, but did not significantly affect (de)acetylation in control hearts. These data suggest that a splitomicin-sensitive target (presumably SIRT1) is responsible for Lys deacetylation in IPC, but does not regulate acetylation under control conditions. The ability of splitomicin to inhibit IPC-induced Lys acetylation is also consistent with the notion that observed changes in acetylation are not caused by protein translocation during IPC (vide supra). Splitomicin is a popular inhibitor of protein deacetylation, but has no reported effect on protein localization.
Figure 4.

Effect of SIRT1 and Nampt inhibition on lysine acetylation in IPC. Cytosolic fractions from hearts subjected to control perfusion (Ctrl.) or IPC, in the absence or presence of splitomicin (Sp, 10 µM) or FK-866 (FK, 1 µM) were western blotted for K-Ac. Representative blot is shown, numbers to the left are molecular weight markers (kDa). Densitometry was performed in the range 25–100 kDa, normalized to protein across the same molecular weight range (representative Ponceau S stained membrane is shown in Supplementary material online, Figure S7). The ratio of K-Ac/Protein was calculated and is shown below the blots (mean ± SEM, n ≥ 5). *P < 0.05 (ANOVA) between the indicated group and all other groups.
An isolated cardiomyocyte model of IPC was also developed, and yielded similar results to those seen in intact hearts; IPC led to deacetylation of cytosolic proteins, in a manner that was inhibited by splitomicin (see Supplementary material online, Figure S8). This suggests that the IPC-induced, splitomicin-sensitive deacetylation event seen in whole hearts originates at the level of cardiomyocytes, and not another cell type (e.g. endothelium). This result is consistent with a previous observation that the SIRT1 protein level is up-regulated in isolated cardiomyocytes by exposure to hypoxia 48.
Recently, Nampt has been identified as an important regulator of SIRT1 activity, by maintaining the availability of its substrate NAD+.25,26 Thus, we tested the effect of the specific Nampt inhibitor FK-866 on Lys acetylation in IPC. Figure 4 shows that, similar to splitomicin, FK-866 reversed IPC-mediated deacetylation of cytosolic proteins, while having no effect on acetylation under control conditions.
The effects of splitomicin and FK-866 on IPC-induced deacetylation prompted an investigation into the role of SIRT1 in the mechanism of IPC-mediated cardioprotection. As shown in Figure 5 and Supplementary material online, Figure S10, treatment of perfused hearts with splitomicin or FK-866 mitigated the protective effects of IPC both at the level of functional recovery (rate-pressure product) and infarct size. Furthermore, neither splitomicin nor FK-866 enhanced IR injury (in fact FK-866 was even slightly cardioprotective), suggesting that these compounds did not impact IPC by inducing secondary pathology. We also confirmed these results in isolated cardiomyocytes; Supplementary material online, Figure S8 shows that IPC protection against IR was prevented by splitomicin. Together these data support a role for Nampt/SIRT1-mediated Lys deacetylation in endogenous cardioprotection by acute IPC.
Figure 5.
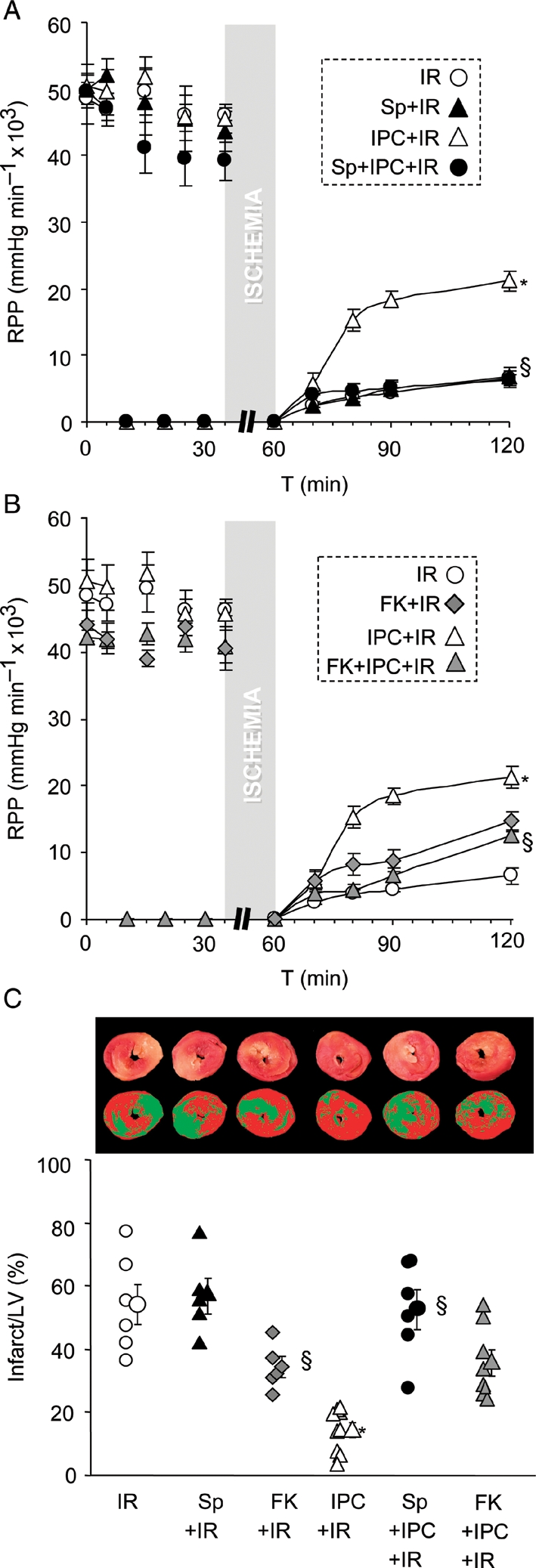
Effect of SIRT1 and Nampt inhibition on IPC-induced cardioprotection. (A/B) Perfused hearts were subjected to IR injury, with or without IPC, and with or without infusion of Sp or FK (see Methods). Left-ventricular (LV) contractile function was monitored and graphs show rate pressure product (RPP). For clarity, experimental groups with Sp and FK treatments are shown in two separate panels; although IR alone and IPC + IR groups were the same in each panel. Mean ± SEM, n ≥ 5. *P < 0.05 vs. IR alone, §P < 0.05 vs. IPC + IR (ANOVA). (C) Following IR protocols, hearts were stained with TTC and infarct size measured as % of LV area. Upper images show representative TTC stained hearts, with image masks used to calculate infarct size below (see Supplementary material online). In the graph, infarct is quantified, with individual data points for each condition shown on the left and means ± SEM on the right (n ≥ 5). *P < 0.05 vs. IR alone, §P < 0.05 vs. IPC + IR (ANOVA).
4. Discussion
The main findings of this investigation are: (i) cytosolic protein deacetylation occurs in IPC; (ii) SIRT1 activity is increased in IPC, but SIRT1 protein levels do not change; (iii) SIRT1 SUMOylation occurs in IPC; (vi) Inhibition of either SIRT1 or Nampt inhibits both IPC-mediated deacetylation and IPC cardioprotection. Together these data suggest that during IPC, deacetylation of cardiomyocyte cytosolic proteins is likely mediated by SIRT1, and this process may play a role in the mechanism of IPC signalling.
Additional studies in our laboratory showed that infusion of the SIRT1 activator molecule SRT1720 alone did not elicit cardioprotection (see Supplementary material online, Figure S9), suggesting that SIRT1 may be necessary but not sufficient for IPC cardioprotection. However, the recent discovery that SRT1720 and related molecules may not be specific SIRT1 activators49 somewhat clouds interpretation of these data. Whereas resveratrol is known to elicit cardioprotection,50,51 the role of SIRTs in this process is controversial in light of recent reports that resveratrol is not a direct SIRT1 activator.52 The sirtuin field has recently been plagued by such reports of reagent non-specificity, including criticism of a widely used fluorescent SIRT1 activity assay.49,53 The assay utilized herein (p53 K379 deacetylation) is specific for SIRT1,41–43 but overall, these findings highlight the need for better tools to investigate this intriguing family of proteins.
Several proteins implicated in cardioprotective signalling have been identified as substrates for SIRT1-mediated deacetylation, including: eNOS,14 IRS-1,15 PARP-1,54 autophagy,16 and HIF-2α.21 The role of each of these proteins in mediating the downstream effects of SIRT1 in this system remains to be elucidated. In addition to its deacetylase activity, SIRT1 may have other effects in IPC related to NAD+ consumption. For example, the depletion of NAD+ by SIRTs may restrict availability of this co-factor for other enzymes important in IR injury and cardioprotection, including: PARP-1,55 ALDH2,56 and mitochondrial complex I (Cx I).5 The observation that inhibiting Nampt gave similar effects to inhibiting SIRT1 (Figures 4 and 5), must be tempered with the caveat that Nampt may also regulate NAD+ availability for these other enzymes. Interestingly, it has also been shown that AMP-dependent protein kinase, which is activated during IPC,57 can up-regulate SIRT1 activity by modulating NAD+ levels.58 Overall, it appears reasonable to suggest that these diverse NAD+/NADH-dependent enzymes may cross-talk during stress conditions such as IR injury and IPC.
The current study focused on SIRT1, based on both direct evidence for SIRT1 activation in IPC (Figure 2A) and the effects of a specific SIRT1 inhibitor (Figures 4 and 5, see Supplementary material online, Figure S8). Although the regulation of protein (de)acetylation by other enzymes in IPC has not been investigated, our observation that the Nampt inhibitor FK-866 blocked IPC-mediated deacetylation suggests that this deacetylation is mediated by NAD+-dependent enzymes, most likely the SIRTs. In contrast, the class I/II HDACs are NAD+ independent, and inhibitors of these enzymes are cardioprotective,10,11 implying a deleterious role in IR injury. Given the changes in mitochondrial protein acetylation observed in IPC (see Supplementary material online, Figure S2), an intriguing candidate for further investigation is SIRT3. SIRT3 is known to regulate Cx I,59 which is both required for IPC,32 and is emerging as an important regulatory enzyme in the context of IR injury.60 Clearly the development of tools such as inhibitors and specific assays for investigating SIRT3 function will aid in elucidating its role in IPC.
Corollary to investigating the downstream effects of SIRT1, we also investigated its upstream regulation. In contrast to a previous study in porcine myocardium which reported elevation in SIRT1 protein levels in acute IPC,48 we did not observe any change in SIRT1 protein in either acute or delayed IPC, either in perfused hearts or in vivo. Thus, it appears that regulation of SIRT1 protein levels in IPC may be species-specific. We did, however, observe that SIRT1 becomes modified by SUMO-1 in IPC, suggesting SUMOylation as a potential upstream mechanism regulating SIRT1 activity in this setting. Notably SUMOylation is known to be up-regulated by IPC in neurons,47 although this has not been studied in cardiac IPC. One additional possibility is that SIRT1 may be subject to oxidative PTM, as has recently been reported.61–63
Furthermore, it has been shown that SIRT1 can translocate to the nucleus under stress conditions such as cardiomyopathy or myocardial infarction, where it may regulate adaptive protein synthesis.64 However, our results (see Supplementary material online, Figure S4) show that IPC did not trigger such translocation, suggesting that the effects of SIRT1 in IPC are mainly cytosolic.
Although our data show that several proteins were (de)acetylated during IPC, the opposing pathway of acetylation also remains poorly studied in the context of stress. The recent discovery that the acetyl-CoA required for protein acetylation is primarily derived from ATP-citrate lyase65 provides a link between the metabolic state of the cell and the regulation of stress signalling.
In summary, herein we demonstrated that IPC is accompanied by cytosolic protein deacetylation, and that inhibition of the protein deacetylase SIRT1 blocks this process, and blocks IPC. The molecular targets of SIRT1 in the cardiomyocyte which mediate this effect, the role of other SIRTs, and the upstream signalling pathways that regulate SIRT1 will be fertile ground for future studies.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was funded by NIH grant HL071158. P.S.B. discloses a consulting agreement with Galleon Pharmaceuticals Inc., Horsham, PA, USA. I.R. is funded by NIH grants HL085613 and HL097751.
Acknowledgements
We thank Keith Nehrke and Jun-Ichi Abe (Rochester) for stimulating discussions regarding these studies, and Samuel Caito (Rochester) for technical assistance.
Conflict of interest: none declared.
References
- 1.Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. doi:10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 3.Rizvi A, Tang XL, Qiu Y, Xuan YT, Takano H, Jadoon AK, et al. Increased protein synthesis is necessary for the development of late preconditioning against myocardial stunning. Am J Physiol. 1999;277:H874–H884. doi: 10.1152/ajpheart.1999.277.3.H874. [DOI] [PubMed] [Google Scholar]
- 4.Hausenloy DJ, Tsang A, Mocanu MM, Yellon DM. Ischemic preconditioning protects by activating prosurvival kinases at reperfusion. Am J Physiol. 2005;288:H971–H976. doi: 10.1152/ajpheart.00374.2004. [DOI] [PubMed] [Google Scholar]
- 5.Nadtochiy SM, Burwell LS, Brookes PS. Cardioprotection and mitochondrial S-nitrosation: effects of S-nitroso-2-mercaptopropionyl glycine (SNO-MPG) in cardiac ischemia-reperfusion injury. J Mol Cell Cardiol. 2007;42:812–825. doi: 10.1016/j.yjmcc.2007.01.010. doi:10.1016/j.yjmcc.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res. 2007;101:1155–1163. doi: 10.1161/CIRCRESAHA.107.155879. doi:10.1161/CIRCRESAHA.107.155879. [DOI] [PubMed] [Google Scholar]
- 7.Lu Z, Scott I, Webster BR, Sack MN. The emerging characterization of lysine residue deacetylation on the modulation of mitochondrial function and cardiovascular biology. Circ Res. 2009;105:830–841. doi: 10.1161/CIRCRESAHA.109.204974. doi:10.1161/CIRCRESAHA.109.204974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spange S, Wagner T, Heinzel T, Kramer OH. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol. 2009;41:185–198. doi: 10.1016/j.biocel.2008.08.027. doi:10.1016/j.biocel.2008.08.027. [DOI] [PubMed] [Google Scholar]
- 9.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. doi:10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suzuki YJ, Nagase H, Day RM, Das DK. GATA-4 regulation of myocardial survival in the preconditioned heart. J Mol Cell Cardiol. 2004;37:1195–1203. doi: 10.1016/j.yjmcc.2004.09.009. doi:10.1016/j.yjmcc.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 11.Zhao TC, Cheng G, Zhang LX, Tseng YT, Padbury JF. Inhibition of histone deacetylases triggers pharmacologic preconditioning effects against myocardial ischemic injury. Cardiovasc Res. 2007;76:473–481. doi: 10.1016/j.cardiores.2007.08.010. doi:10.1016/j.cardiores.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 12.Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587–591. doi: 10.1038/nature08197. doi:10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu J, Auwerx J. Protein deacetylation by SIRT1: an emerging key post-translational modification in metabolic regulation. Pharmacol Res. 2009;62:35–41. doi: 10.1016/j.phrs.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mattagajasingh I, Kim CS, Naqvi A, Yamamori T, Hoffman TA, Jung SB, et al. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 2007;104:14855–14860. doi: 10.1073/pnas.0704329104. doi:10.1073/pnas.0704329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang J. The direct involvement of SirT1 in insulin-induced insulin receptor substrate-2 tyrosine phosphorylation. J Biol Chem. 2007;282:34356–34364. doi: 10.1074/jbc.M706644200. doi:10.1074/jbc.M706644200. [DOI] [PubMed] [Google Scholar]
- 16.Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci USA. 2008;105:3374–3379. doi: 10.1073/pnas.0712145105. doi:10.1073/pnas.0712145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alcendor RR, Gao S, Zhai P, Zablocki D, Holle E, Yu X, et al. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res. 2007;100:1512–1521. doi: 10.1161/01.RES.0000267723.65696.4a. doi:10.1161/01.RES.0000267723.65696.4a. [DOI] [PubMed] [Google Scholar]
- 18.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. doi:10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 19.van der HA, Tertoolen LG, de Vries-Smits LM, Frye RA, Medema RH, Burgering BM. FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1) J Biol Chem. 2004;279:28873–28879. doi: 10.1074/jbc.M401138200. doi:10.1074/jbc.M401138200. [DOI] [PubMed] [Google Scholar]
- 20.Yang Y, Hou H, Haller EM, Nicosia SV, Bai W. Suppression of FOXO1 activity by FHL2 through SIRT1-mediated deacetylation. EMBO J. 2005;24:1021–1032. doi: 10.1038/sj.emboj.7600570. doi:10.1038/sj.emboj.7600570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dioum EM, Chen R, Alexander MS, Zhang Q, Hogg RT, Gerard RD, et al. Regulation of hypoxia-inducible factor 2alpha signaling by the stress-responsive deacetylase sirtuin 1. Science. 2009;324:1289–1293. doi: 10.1126/science.1169956. doi:10.1126/science.1169956. [DOI] [PubMed] [Google Scholar]
- 22.Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369–2380. doi: 10.1038/sj.emboj.7600244. doi:10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Danz ED, Skramsted J, Henry N, Bennett JA, Keller RS. Resveratrol prevents doxorubicin cardiotoxicity through mitochondrial stabilization and the Sirt1 pathway. Free Radic Biol Med. 2009;46:1589–1597. doi: 10.1016/j.freeradbiomed.2009.03.011. doi:10.1016/j.freeradbiomed.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 24.Pillai JB, Isbatan A, Imai S, Gupta MP. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2alpha deacetylase activity. J Biol Chem. 2005;280:43121–43130. doi: 10.1074/jbc.M506162200. doi:10.1074/jbc.M506162200. [DOI] [PubMed] [Google Scholar]
- 25.Hsu CP, Oka S, Shao D, Hariharan N, Sadoshima J. Nicotinamide phosphoribosyltransferase regulates cell survival through NAD+ synthesis in cardiac myocytes. Circ Res. 2009;105:481–491. doi: 10.1161/CIRCRESAHA.109.203703. doi:10.1161/CIRCRESAHA.109.203703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem. 2004;279:50754–50763. doi: 10.1074/jbc.M408388200. doi:10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- 27.Prime TA, Blaikie FH, Evans C, Nadtochiy SM, James AM, Dahm CC, et al. A mitochondria-targeted S-nitrosothiol modulates respiration, nitrosates thiols, and protects against ischemia-reperfusion injury. Proc Natl Acad Sci USA. 2009;106:10764–10769. doi: 10.1073/pnas.0903250106. doi:10.1073/pnas.0903250106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bedalov A, Gatbonton T, Irvine WP, Gottschling DE, Simon JA. Identification of a small molecule inhibitor of Sir2p. Proc Natl Acad Sci USA. 2001;98:15113–15118. doi: 10.1073/pnas.261574398. doi:10.1073/pnas.261574398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hirao M, Posakony J, Nelson M, Hruby H, Jung M, Simon JA, et al. Identification of selective inhibitors of NAD+-dependent deacetylases using phenotypic screens in yeast. J Biol Chem. 2003;278:52773–52782. doi: 10.1074/jbc.M308966200. doi:10.1074/jbc.M308966200. [DOI] [PubMed] [Google Scholar]
- 30.Hasmann M, Schemainda I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 2003;63:7436–7442. [PubMed] [Google Scholar]
- 31.Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712–716. doi: 10.1038/nature06261. doi:10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nadtochiy SM, Burwell LS, Ingraham CA, Spencer CM, Friedman AE, Pinkert CA, et al. In vivo cardioprotection by S-nitroso-2-mercaptopropionyl glycine. J Mol Cell Cardiol. 2009;46:960–968. doi: 10.1016/j.yjmcc.2009.01.012. doi:10.1016/j.yjmcc.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cox B, Emili A. Tissue subcellular fractionation and protein extraction for use in mass-spectrometry-based proteomics. Nat Protoc. 2006;1:1872–1878. doi: 10.1038/nprot.2006.273. doi:10.1038/nprot.2006.273. [DOI] [PubMed] [Google Scholar]
- 34.Tompkins AJ, Burwell LS, Digerness SB, Zaragoza C, Holman WL, Brookes PS. Mitochondrial dysfunction in cardiac ischemia-reperfusion injury: ROS from complex I, without inhibition. Biochim Biophys Acta. 2006;1762:223–231. doi: 10.1016/j.bbadis.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 35.Nadtochiy SM, Tompkins AJ, Brookes PS. Different mechanisms of mitochondrial proton leak in ischaemia/reperfusion injury and preconditioning: implications for pathology and cardioprotection. Biochem J. 2006;395:611–618. doi: 10.1042/BJ20051927. doi:10.1042/BJ20051927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garlid KD, Costa AD, Quinlan CL, Pierre SV, Dos SP. Cardioprotective signaling to mitochondria. J Mol Cell Cardiol. 2009;46:858–866. doi: 10.1016/j.yjmcc.2008.11.019. doi:10.1016/j.yjmcc.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Costa AD, Garlid KD, West IC, Lincoln TM, Downey JM, Cohen MV, et al. Protein kinase G transmits the cardioprotective signal from cytosol to mitochondria. Circ Res. 2005;97:329–336. doi: 10.1161/01.RES.0000178451.08719.5b. doi:10.1161/01.RES.0000178451.08719.5b. [DOI] [PubMed] [Google Scholar]
- 38.Eaton P, Awad WI, Miller JI, Hearse DJ, Shattock MJ. Ischemic preconditioning: a potential role for constitutive low molecular weight stress protein translocation and phosphorylation? J Mol Cell Cardiol. 2000;32:961–971. doi: 10.1006/jmcc.2000.1136. doi:10.1006/jmcc.2000.1136. [DOI] [PubMed] [Google Scholar]
- 39.Malik G, Gorbounov N, Das S, Gurusamy N, Otani H, Maulik N, et al. Ischemic preconditioning triggers nuclear translocation of thioredoxin and its interaction with Ref-1 potentiating a survival signal through the PI-3-kinase-Akt pathway. Antioxid Redox Signal. 2006;8:2101–2109. doi: 10.1089/ars.2006.8.2101. doi:10.1089/ars.2006.8.2101. [DOI] [PubMed] [Google Scholar]
- 40.Pyle WG, Chen Y, Hofmann PA. Cardioprotection through a PKC-dependent decrease in myofilament ATPase. Am J Physiol. 2003;285:H1220–H1228. doi: 10.1152/ajpheart.00076.2003. [DOI] [PubMed] [Google Scholar]
- 41.Langley E, Pearson M, Faretta M, Bauer UM, Frye RA, Minucci S, et al. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 2002;21:2383–2396. doi: 10.1093/emboj/21.10.2383. doi:10.1093/emboj/21.10.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Solomon JM, Pasupuleti R, Xu L, McDonagh T, Curtis R, DiStefano PS, et al. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol Cell Biol. 2006;26:28–38. doi: 10.1128/MCB.26.1.28-38.2006. doi:10.1128/MCB.26.1.28-38.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vaziri H, Dessain SK, Ng EE, Imai SI, Frye RA, Pandita TK, et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. doi:10.1016/S0092-8674(01)00527-X. [DOI] [PubMed] [Google Scholar]
- 44.Luss H, Schmitz W, Neumann J. A proteasome inhibitor confers cardioprotection. Cardiovasc Res. 2002;54:140–151. doi: 10.1016/s0008-6363(02)00232-8. doi:10.1016/S0008-6363(02)00232-8. [DOI] [PubMed] [Google Scholar]
- 45.Sasaki T, Maier B, Koclega KD, Chruszcz M, Gluba W, Stukenberg PT, et al. Phosphorylation regulates SIRT1 function. PLoS One. 2008;3:e4020. doi: 10.1371/journal.pone.0004020. doi:10.1371/journal.pone.0004020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang Y, Fu W, Chen J, Olashaw N, Zhang X, Nicosia SV, et al. SIRT1 sumoylation regulates its deacetylase activity and cellular response to genotoxic stress. Nat Cell Biol. 2007;9:1253–1262. doi: 10.1038/ncb1645. doi:10.1038/ncb1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ja LY, Castri P, Bembry J, Maric D, Auh S, Hallenbeck JM. SUMOylation participates in induction of ischemic tolerance. J Neurochem. 2009;109:257–267. doi: 10.1111/j.1471-4159.2009.05957.x. doi:10.1111/j.1471-4159.2009.05957.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rane S, He M, Sayed D, Vashistha H, Malhotra A, Sadoshima J, et al. Downregulation of miR-199a derepresses hypoxia-inducible factor-1alpha and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ Res. 2009;104:879–886. doi: 10.1161/CIRCRESAHA.108.193102. doi:10.1161/CIRCRESAHA.108.193102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pacholec M, Chrunyk BA, Cunningham D, Flynn D, Griffith DA, Griffor M, et al. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J Biol Chem. 2010;285:8340–8351. doi: 10.1074/jbc.M109.088682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Das S, Das DK. Resveratrol: a therapeutic promise for cardiovascular diseases. Recent Pat Cardiovasc Drug Discov. 2007;2:133–138. doi: 10.2174/157489007780832560. doi:10.2174/157489007780832560. [DOI] [PubMed] [Google Scholar]
- 51.Penumathsa SV, Maulik N. Resveratrol: a promising agent in promoting cardioprotection against coronary heart disease. Can J Physiol Pharmacol. 2009;87:275–286. doi: 10.1139/Y09-013. doi:10.1139/Y09-013. [DOI] [PubMed] [Google Scholar]
- 52.Beher D, Wu J, Cumine S, Kim KW, Lu SC, Atangan L, et al. Resveratrol is not a direct activator of SIRT1 enzyme activity. Chem Biol Drug Des. 2009;74:619–624. doi: 10.1111/j.1747-0285.2009.00901.x. [DOI] [PubMed] [Google Scholar]
- 53.Borra MT, Langer MR, Slama JT, Denu JM. Substrate specificity and kinetic mechanism of the Sir2 family of NAD+-dependent histone/protein deacetylases. Biochemistry. 2004;43:9877–9887. doi: 10.1021/bi049592e. doi:10.1021/bi049592e. [DOI] [PubMed] [Google Scholar]
- 54.Rajamohan SB, Pillai VB, Gupta M, Sundaresan NR, Birukov KG, Samant S, et al. SIRT1 promotes cell survival under stress by deacetylation-dependent deactivation of poly(ADP-ribose) polymerase 1. Mol Cell Biol. 2009;29:4116–4129. doi: 10.1128/MCB.00121-09. doi:10.1128/MCB.00121-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liaudet L, Yang Z, Al-Affar EB, Szabo C. Myocardial ischemic preconditioning in rodents is dependent on poly (ADP-ribose) synthetase. Mol Med. 2001;7:406–417. [PMC free article] [PubMed] [Google Scholar]
- 56.Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;321:1493–1495. doi: 10.1126/science.1158554. doi:10.1126/science.1158554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nishino Y, Miura T, Miki T, Sakamoto J, Nakamura Y, Ikeda Y, et al. Ischemic preconditioning activates AMPK in a PKC-dependent manner and induces GLUT4 up-regulation in the late phase of cardioprotection. Cardiovasc Res. 2004;61:610–619. doi: 10.1016/j.cardiores.2003.10.022. doi:10.1016/j.cardiores.2003.10.022. [DOI] [PubMed] [Google Scholar]
- 58.Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, et al. AMPK regulates energy expenditure by modulating NAD + metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. doi:10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, et al. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci USA. 2008;105:14447–14452. doi: 10.1073/pnas.0803790105. doi:10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Burwell LS, Nadtochiy SM, Brookes PS. Cardioprotection by metabolic shut-down and gradual wake-up. J Mol Cell Cardiol. 2009;46:804–810. doi: 10.1016/j.yjmcc.2009.02.026. doi:10.1016/j.yjmcc.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Caito S, Rajendrasozhan S, Cook S, Chung S, Yao H, Friedman AE, et al. SIRT1 is a redox-sensitive deacetylase that is post-translationally modified by oxidants and carbonyl stress. FASEB J. 2010;24:3145–3159. doi: 10.1096/fj.09-151308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rajendrasozhan S, Yang SR, Kinnula VL, Rahman I. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;177:861–870. doi: 10.1164/rccm.200708-1269OC. doi:10.1164/rccm.200708-1269OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zee RS, Yoo CB, Pimentel DR, Perlman DH, Burgoyne JR, Hou X, et al. Redox regulation of sirtuin-1 is mediated by S-glutathiolation. Antioxid Redox Signal. 2010;13:1023–1032. doi: 10.1089/ars.2010.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tanno M, Kuno A, Yano T, Miura T, Hisahara S, Ishikawa S, et al. Induction of manganese superoxide dismutase by nuclear translocation and activation of SIRT1 promotes cell survival in chronic heart failure. J Biol Chem. 2010;285:8375–8382. doi: 10.1074/jbc.M109.090266. doi:10.1074/jbc.M109.090266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. doi:10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang SR, Wright J, Bauter M, Seweryniak K, Kode A, Rahman I. Sirtuin regulates cigarette smoke-induced proinflammatory mediator release via RelA/p65 NF-kappaB in macrophages in vitro and in rat lungs in vivo implications for chronic inflammation and aging. Am J Physiol. 2007;292:L567–L576. doi: 10.1152/ajplung.00308.2006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.