

Abstract
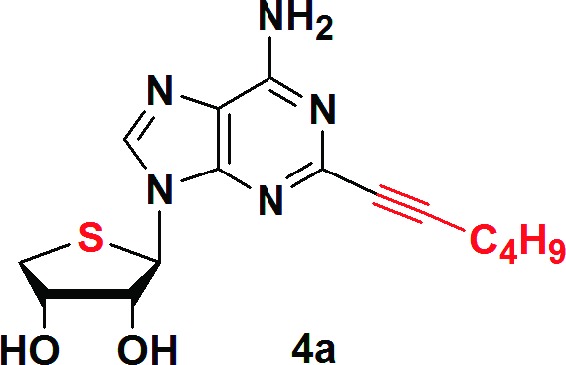
The truncated C2- and C8-substituted 4′-thioadenosine derivatives 4a−d were synthesized from d-mannose, using palladium-catalyzed cross-coupling reactions as key steps. In this study, an A3 adenosine receptor (AR) antagonist, truncated 4′-thioadenosine derivative 3, was successfully converted into a potent A2A AR agonist 4a (Ki = 7.19 ± 0.6 nM) by appending a 2-hexynyl group at the C2-position of a derivative of 3 that was N6-substituted. However, C8-substitution greatly reduced binding affinity at the human A2A AR. All synthesized compounds 4a−d maintained their affinity at the human A3 AR, but 4a was found to be a competitive A3 AR antagonist/A2A AR agonist in cyclic AMP assays. This study indicates that the truncated C2-substituted 4′-thioadenosine derivatives 4a and 4b can serve as novel templates for the development of new A2A AR ligands.
Keywords: A2A adenosine receptor agonists, truncated 2-hexynyl-4′-thioadenosine, palladium-catalyzed cross-coupling reactions, binding mode
The endogenous cytoprotective modulator adenosine (1) exerts its pharmacological effects against hypoxia, ischemia, and inflammation through binding to four subtypes (A1, A2A, A2B, and A3) of adenosine receptors (ARs), members of the G protein-coupled receptor (GPCR) family.1 Among these, activation of A2A AR has been known to play a role in the suppression of immune and inflammatory responses and in vascular responses to adenosine.2−4 It is also highly localized within the central nervous system (CNS), and selective antagonists have become an attractive target for the treatment of Parkinson's disease.5 The A3 AR is the most recently identified subtype and is found in the cardiovascular system, CNS, immune cells, lung, and liver.6,7 The activation of A3 AR is beneficial in models of myocardiac and cerebral ischemia and cancer, while its antagonism is of interest for treating asthma, inflammation, and glaucoma.8
Thio-Cl-IB-MECA (2),9,10 which is bioisosteric with 2-chloro-N6-(3-iodobenzyl)adenosine-5′-N-methyluronamide (Cl-IB-MECA),11 was discovered as a potent and selective A3 AR agonist (Ki = 0.38 nM). This compound showed a potent anticancer activity by inhibiting the Wnt signaling pathway.12 On the basis of rational design, truncated 4′-thioadenosine derivative 3 lacking the 5′-uronamide of 2 essential for the receptor activation was discovered as a potent, selective, and species-independent A3 AR antagonist (Ki = 1.66 nM).13,14 This compound and other related A3 AR antagonists with nucleoside skeletons are expected to be suitable for evaluation in small animal models and for further development as drugs (Figure 1).
Figure 1.

Rationale for the design of A2A AR agonists.
On the basis of the observation that truncation resulting in the 4′-thioadenosine antagonist derivative 3 preserved AR affinity and selectivity, we designed and synthesized the truncated C2- and C8-substituted 4′-thioadenosine derivatives 4a−d as potential new ligands for the A2A AR. This expectation was supported by reports that C2- or C8-substitution sometimes leads to substantial enhancement in the binding affinity or selectivity at the A2A AR or other AR subtype.15−17 C2- or C8-substitution was readily achieved through Sonogashira18 and Suzuki19 cross-coupling reactions. From this study, a C2-alkynyl derivative was found to be potent, mixed A2A AR agonist and A3 AR antagonist, which is an excellent combination for the antiasthmatic activity. Herein, we describe the synthesis and pharmacological activity of novel C2- and C8-substituted 4′-thioadenosine derivatives 4a−d from d-mannose.
d-Mannose was converted to the glycosyl donor 5 according to our previously published procedure.13,14 The glycosyl donor 5 was condensed with 2-amino-6-chloropurine in the presence of TMSOTf as a Lewis acid to give the β-anomer 6 (30%) as a single stereoisomer (Scheme 1). The anomeric assignment was easily accomplished in a 1H NMR experiment that showed a nuclear Overhauser effect between H-8 and 3′-H. Treatment of 2-amino-6-chloro derivative 6 with isoamyl nitrite, iodine, and methylene iodide in the presence of CuI afforded the 2-iodo-6-chloro derivative 7, which was converted to the 2-iodo-6-amino derivative 8 upon treatment with methanolic ammonia. A Sonogashira18 coupling reaction of 8 with 1-hexyne in the presence of bis(triphenylphosphine)palladium dichloride yielded the 2-hexynyl derivative 9. Finally, removal of the isopropylidene of 9 with 1 N HCl produced the final 2-hexynyl-4′-thioadenosine derivative 4a. Suzuki19 coupling reaction of the 2-iodo derivative 8 with (E)-1-catecholboranylhexene,20 prepared by treating with 1-hexyne and catecholborane, in the presence of tetrakis(triphenylphosphine)palladium(0) afforded the 2-hexenyl derivative 10. Removal of the acetonide of 10 with 1 N HCl gave the 2-hexenyl-4′-thioadenosine derivative 4b.
Scheme 1. Synthesis of the 2-Substituted Derivatives 4a and 4b.

Reagents and conditions: (a) Silylated 2-amino-6-chloropurine, TMSOTf, DCE, room temperature to 80 °C, 3 h. (b) CuI, isoamyl nitrite, I2, CH2I2, THF, 110 °C, 45 min. (c) NH3/MeOH, 80 °C, 2 h. (d) 1-Hexyne, CuI, TEA, DMF, bis(triphenylphosphine)palladium dichloride, room temperature, 3 h. (e) 1 N HCl, THF, room temperature, 15 h. (f) (E)-1-catecholboranylhexene, then tetrakis(triphenylphosphine)palladium(0), Na2CO3, DMF, H2O, 90 °C, 15 h.
Using a strategy similar to Scheme 1, 8-substituted adenosine derivatives 4c and 4d were synthesized from the glycosyl donor 5 (Scheme 2). Condensation of 5 with 8-bromoadenine21 under Lewis acid conditions afforded the 8-bromo derivative 11. Coupling of 11 with 1-hexyne under Sonogashira conditions gave 8-hexynyl derivative 12, which was treated with 1 N HCl to yield the final 8-hexynyl-4′-thioadenosine derivative 4c.
Scheme 2. Synthesis of the 8-Substituted Derivatives 4c and 4d.

Reagents and conditions: (a) Silylated 8-bromoadenine, TMSOTf, DCE, room temperature to 90 °C, 2 h. (b) 1-Hexyne, CuI, TEA, DMF, bis(triphenylphosphine) palladium dichloride, room temperature, 3 h. (c) 1 N HCl, THF, room temperature, 15 h. (d) (E)-1-catecholboranylhexene, then tetrakis(triphenylphosphine) palladium(0), Na2CO3, DMF, H2O, 90 °C, 15 h.
The 8-bromo derivative 11 was condensed with (E)-1-catecholboranylhexene20 under Suzuki conditions19 to give the 2-hexenyl derivative 13. Removal of the isopropylidene group of 13 under acidic conditions afforded the final 8-hexenyl-4′-thio adenosine derivative 4d.
Binding assays were carried out using standard radioligands and membrane preparations from Chinese hamster ovary (CHO) cells stably expressing the human (h) A1 or A3 AR or human embryonic kidney cells (HEK-293) expressing the hA2A AR.22−25 Unlike the parent N6-substituted compound 3 that only weakly bound to the A2A AR, C2-substituted variations of compound 3 led to a dramatic increase in the binding affinity (Ki = 7.19 ± 0.6 nM for 4a and 72.0 ± 19.1 nM for 4b) at the hA2A AR, while maintaining high binding affinity (Ki = 11.8 ± 1.3 nM for 4a and 13.2 ± 0.8 nM for 4b) at the hA3 AR (Table 1).
Table 1. Binding Affinities of Known A3 AR Antagonist 3 and Truncated 2- and 8-Substituted 4′-Thioadenosine Derivatives 4a−d at Three Subtypes of hARs.
| affinitya |
|||
|---|---|---|---|
| compounds | hA1 (% inhibition) | hA2A (% inhibition) | hA3 (Ki, nM) |
| 3 | 37.9 | 17.7 | 1.66 ± 0.90 |
| 4a | 38.9 ± 9.9 | 97.2 ± 4.1 | 11.8 ± 1.3 |
| 4b | 16.2 ± 8.4 | 95.9 ± 8.7 | 13.2 ± 0.8 |
| 4c | 49.3 ± 4.9 | 46.5 ± 4.3 | 20.0 ± 4.0 |
| 4d | 3.7 ± 2.9 | 22.8 ± 6.4 | 259 ± 10 |
All binding experiments were performed using adherent mammalian cells stably transfected with cDNA encoding the appropriate hAR (A1 AR and A3 AR in CHO cells and A2A AR in HEK-293 cells). Binding was carried out using 1 nM [3H]CCPA, 10 nM [3H]CGS21680, or 0.5 nM [125I]I-AB-MECA as radioligands for A1, A2A, and A3 ARs, respectively. Values are expressed as means ± SEMs, n = 3−4 (outliers eliminated) and normalized against a nonspecific binder, 5′-N-ethylcarboxamidoadenosine (NECA, 10 μM). Values expressed as a percentage refer to the percent inhibition of specific radioligand binding at 10 μM, with nonspecific binding defined using 10 μM NECA.
These results indicate that bulky hydrophobic pockets exist in the binding sites of A2A AR and A3 AR, allowing the C2-substituent to form favorable hydrophobic interactions. The 2-alkynyl derivative 4a showed a better binding affinity than the 2-alkenyl derivative 4b. Interestingly, C8-substitution on 3 abolished the binding affinity at the hA2A AR, but the binding affinity at the hA3 AR was maintained although decreased. These findings suggest that a bulky hydrophobic group at position 8 could be tolerated at the binding site of the hA3 but not hA2A AR. The ability to enhance affinity at the A2A AR in the truncated series by extending an unsaturated carbon chain at the 2-position implies a mode of receptor binding in common with the riboside series.15−17 All compounds showed very weak binding affinity at the hA1 AR.
Compounds 4a and 4b were found to be potent and full antagonists in a cyclic AMP functional assay at the hA3 AR. In this assay, 4a dose dependently shifted the concentration−response curve for agonist Cl-IB-MECA to the right as an antagonist, corresponding to a KB value of 1.69 nM calculated by Schild analysis (Figure 2). This is consistent with previous studies in which truncated N6-substituted 4′-thioadenosine derivatives have generally displayed A3 AR antagonist activity.13,14 However, in a cyclic AMP functional assay at the hA2A AR expressed in CHO cells, compound 4a behaved as a full agonist as compared to the standard 2-[p-(2-carboxyethyl)phenyl-ethylamino]-5′-N-ethylcarboxamidoadenosine (CGS21680) and displayed an EC50 of 12 nM. At the hA2B AR expressed in CHO cells, 4a was a weak partial agonist in cyclic AMP accumulation (EC50 ∼10 μM). The finding that compound 4a is both a potent and a selective agonist at the hA2A AR and a competitive antagonist at the hA3 AR is similar to the pharmacological profile of a more heavily 2,5′-substituted adenosine derivative,26 which inhibited both formation of reactive oxygen species and eosinophils degranulation for the antiasthmatic activity.
Figure 2.

Parallel right shifts induced by compound 4a on the concentration−response curve of a full agonist in the inhibition of cyclic AMP production at the hA3 AR expressed in CHO cells (A), the corresponding Schild plot (B), and the activity of 4a as a full agonist at the hA2A AR expressed in CHO cells, as compared to CGS21680 (C).
To investigate the binding mode, we performed a study of docking the C2- and C8-substituted 4′-thioadenosine derivatives 4a−d in the hA2A AR X-ray crystallographic structure (PDB code: 3EML)27 using GOLD software,28 considering the flexibility of the binding site residues. As shown in Figure 3, the C2-substituted 4′-thioadenosine derivatives 4a and 4b, whose binding affinities are in the nanomolar range, occupied the binding site very well. Their bulky and rigid C2-substituents oriented toward the extracellular region, forming hydrophobic interactions. The adenine moieties appeared to form three H-bonds with Glu169 and Asn253, and the ring systems were in π−π stacking with Phe168. Also, the thio-sugar rings were located deep inside the binding pocket, and the 3′-OH groups were able to donate a H-bond to Ser277. On the basis of this result, the C8-substituents were expected to be oriented in a hydrophobic pocket deep inside the binding site, which was not occupied by 4a and 4b in Figure 3. However, the C8-substitued derivatives 4c and 4d showed various binding modes (data not shown). In addition to the expected binding mode, the C8-substituents alternately pointed toward the extracellular region through a rotation of the bond between the adenine and the thio-sugar rings. It might be due to spatial restriction of the long and rigid C8-substituents in the hydrophobic pocket inside the binding site, which would cause the nucleosides to lose some H-bonding and/or π−π stacking interactions that were shown for the C2-substituted derivatives. These results might explain why the A2A AR affinities of 4c and 4d were reduced.
Figure 3.

Predicted binding modes of (A) 4a and (B) 4b docked in the hA2A AR crystal structure. The key interacting residues are marked and displayed in capped-stick, except Phe168 in ball-and-stick, with carbon atoms in white. The ligands are depicted as ball-and-stick with carbon atoms in magenta (4a) and purple (4b). Hydrogen bonds are shown in yellow dashed lines. The van der Waals surfaces of the ligands were generated by MOLCAD and colored by hydrogen-bonding property (red, H-bond donating regions; blue, H-bond accepting regions). The fast Connolly surface of the protein is Z-clipped, and nonpolar hydrogens are undisplayed for clarity.
In conclusion, we synthesized the truncated C2- and C8-substituted 4′-thioadenosine derivatives 4a−d, starting from d-mannose, using palladium-catalyzed cross-coupling reactions as key steps. Although the antagonist activity of various truncated 4′-thionucleosides at the A3 AR was well explored previously, this is the first characterization of the functional activity of such derivatives at the A2A AR. From this study, we successfully identified potent and sterically compact A2A AR agonists, 4a and 4b, by placing extended hydrophobic 2-hexynyl or 2-hexenyl groups on truncated and N6-unsubstituted 4′-thioadenosine derivatives. This observation was supported by molecular modeling that placed the chain at the 2-position in a hydrophobic region of the A2A AR. However, C8-substitution greatly reduced binding affinity at the hA2A AR. Thus, the absence of a 5′-uronamide of typical A2A AR agonists2−4 or the native −CH2OH of adenosine did not preclude potent binding and full activation of the hA2A AR. This suggested a major difference between the A2A AR and the A3 AR in the pathway of receptor activation. All synthesized compounds 4a−d maintained their binding affinity at the human A3 AR, and as for the 2-H or 2-Cl analogues that were N6-substituted, competitive A3 AR antagonism was demonstrated. This study establishes that the truncated C2-substituted 4′-thioadenosine derivatives 4a and 4b can serve as a novel template for the development of new A2A AR ligands, although they still may interact at the A3 AR. This mixed activity as A2A AR agonist/A3 AR antagonist might also be advantageous in disease models such as asthma.26 Thorough elucidation of the structure−activity relationship of this series is in progress in our laboratory.
Supporting Information Available
Complete experimental procedures and characterization data and 1H and 13C NMR copies of 4a−d. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by the Basic Science Research grant (NRF-2008-314-E00304), the National Core Research Center grant (R15-2006-020), the World Class University grant (R31-2008-000-10010-0) from National Research Foundation (NRF) (Korea), and the Intramural Research Program of NIDDK, NIH (Bethesda, MD).
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Olah M. E.; Stiles G. L. The role of receptor structure in determining adenosine receptor activity. Pharmacol. Ther. 2000, 85, 55–75. [DOI] [PubMed] [Google Scholar]
- Fredholm B. B.; Cunha R. A.; Svenningsson P. Pharmacology of adenosine receptors and therapeutic applications. Curr. Top. Med. Chem. 2002, 3, 413–426. [DOI] [PubMed] [Google Scholar]
- Sitkovsky M. V.; Lukashev D.; Apasov S.; Kojima H.; Koshiba M.; Cladwell C.; Ohta A.; Thiel M. Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A2A receptors. Annu. Rev. Immunol. 2004, 22, 657–682. [DOI] [PubMed] [Google Scholar]
- Lappas C. M.; Sullivan G. W.; Linden J. Adenosine A2A agonists in development for the treatment of inflammation. Expert Opin. Invest. Drugs 2005, 14, 797–806. [DOI] [PubMed] [Google Scholar]
- Svenningsson P.; Hall H.; Sedvall G.; Fredholm B. B. Distribution of adenosine receptors in the postmortem human brain: An extended autoradiographic study. Synapse 1997, 27, 322–335. [DOI] [PubMed] [Google Scholar]
- Linden J. Cloned adenosine A3 receptors: Pharmacological properties, species differences and receptor functions. Trends Pharmacol. Sci. 1994, 15, 298–306. [DOI] [PubMed] [Google Scholar]
- Baraldi P. G.; Cacciari B.; Romagnoli R.; Merighi S.; Varani K.; Borea P. A.; Spalluto G. A3 adenosine receptor ligands: History and perspectives. Med. Res. Rev. 2000, 20, 103–128. [DOI] [PubMed] [Google Scholar]
- Jacobson K. A.; Gao Z.-G. Adenosine receptors as therapeutic targets. Nature Rev. Drug Discovery 2006, 5, 247–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong L. S.; Jin D. Z.; Kim H. O.; Shin D. H.; Moon H. R.; Gunaga P.; Chun M. W.; Kim Y.-C.; Melman N.; Gao Z.-G.; Jacobson K. A. N6-Substituted d-4′-thioadenosine-5′-methyluronamides: Potent and selective agonists at the human A3 adenosine receptor. J. Med. Chem. 2003, 46, 3775–3777. [DOI] [PubMed] [Google Scholar]
- Jeong L. S.; Lee H. W.; Jacobson K. A.; Kim H. O.; Shin D. H.; Lee J. A.; Gao Z.-G.; Lu C.; Duong H. T.; Gunaga P.; Lee S. K.; Jin D. Z.; Chun M. W.; Moon H. R. Structure-activity relationships of 2-chloro-N6-substituted-4′-thioadenosine-5′-uronamides as highly potent and selective agonists at the human A3 adenosine receptor. J. Med. Chem. 2006, 49, 273–281. [DOI] [PubMed] [Google Scholar]
- Kim H. O.; Ji X.-d.; Siddiqi S. M.; Olah M. E.; Stiles G. L.; Jacobson K. A. 2-Substitution of N6-benzyladenosine-5′-uronamides enhances selectivity for A3 adenosine receptors. J. Med. Chem. 1994, 37, 3614–3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E. J.; Min H. Y.; Chung H. J.; Park E. J.; Shin D. H.; Jeong L. S.; Lee S. K. A novel adenosine analog, thio-Cl-IB-MECA, induces G0/G1 cell cycle arrest and apoptosis in human promyelocytic leukemia HL-60 cells. Biochem. Pharmacol. 2005, 70, 918–924. [DOI] [PubMed] [Google Scholar]
- Jeong L. S.; Choe S. A.; Gunaga P.; Kim H. O.; Lee H. W.; Lee S. K.; Tosh D. K.; Patel A.; Palaniappan K. K.; Gao Z.-G.; Jacobson K. A.; Moon H. R. Discovery of a new nucleoside template for human A3 adenosine receptor ligands: D-4′-thioadenosine derivatives without 4′-hydroxymethyl group as highly potent and selective antagonists. J. Med. Chem. 2007, 50, 3159–3162. [DOI] [PubMed] [Google Scholar]
- Jeong L. S.; Pal S.; Choe S. A.; Choi W. J.; Jacobson K. A.; Gao Z.-G.; Klutz A. M.; Hou X.; Kim H. O.; Lee H. W.; Tosh D. K.; Moon H. R. Structure-activity relationships of truncated d- and l-4′-thioadenosine derivatives as species-independent A3 adenosine receptor antagonists. J. Med. Chem. 2008, 51, 6609–6613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda A.; Shinozaki M.; Yamaguchi T.; Homma H.; Nomoto R.; Miyasaka T.; Watanabe Y.; Abiru T. Nucleosides and Nucleotides. 103. 2-Alkynyladenosines: A novel class of selective adenosine A2 receptor agonists with potent antihypertensive effects. J. Med. Chem. 1992, 35, 241–252. [DOI] [PubMed] [Google Scholar]
- Cristalli G.; Volpini R.; Vittori S.; Camaioni E.; Monopoli A.; Conti A.; Dionisotti S.; Zocchi C.; Ongini E. 2-Alkynyl derivatives of adenosine-5′-N-ethyluronamide (NECA): Selective A2 adenosine receptor agonists with potent inhibitory activity on platelet aggregation. J. Med. Chem. 1994, 37, 1720–1726. [DOI] [PubMed] [Google Scholar]
- Lambertucci C.; Costanzi S.; Vittori S.; Volpini R.; Cristalli G. Synthesis and adenosine receptor affinity and potency of 8-alkynyl derivatives of adenosine. Nucleosides, Nucleotides Nucleic Acids 2001, 20, 1153–1157. [DOI] [PubMed] [Google Scholar]
- Chinchilla R.; Nájera C. The Sonogashira reaction: A booming methodology in synthetic organic chemistry. Chem. Rev. 2007, 107, 874–922. [DOI] [PubMed] [Google Scholar]
- Suzuki A. Carbon-carbon bonding made easy. Chem. Commun. 2005, 38, 4759–4763. [DOI] [PubMed] [Google Scholar]
- Miyaura N.; Suzuki A. Palladium-catalyzed Reaction of 1-alkenylboronates with vinylic halides: (1Z,3E)-1-phenyl-1,3-octadiene. Org. Syn. Coll. Vol. 1993, 8, 532–534. [Google Scholar]
- Laxer A.; Major D. T.; Gottlieb H. E.; Fischer B. (15N5)-Labeled Adenine Derivatives: Synthesis and Studies of Tautomerism by 15N NMR Spectroscopy and Theoretical Calculations. J. Org. Chem. 2001, 66, 5463–5481. [DOI] [PubMed] [Google Scholar]
- Perreira M.; Jiang J. K.; Klutz A. M.; Gao Z. G.; Shainberg A.; Lu C.; Thomas C. J.; Jacobson K. A. “Reversine” and Its 2-Substituted Adenine Derivatives as Potent and Selective A3 Adenosine Receptor Antagonists. J. Med. Chem. 2005, 48, 4910–4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis M. F.; Schulz R.; Hutchison A. J.; Do E.; Sills M. A.; Williams M. [3H]CGS 21680, a selective A2 adenosine receptor agonist directly labels A2 receptors in rat brain. J. Pharmacol. Exp. Ther. 1989, 251, 888–893. [PubMed] [Google Scholar]
- Olah M. E.; Gallo-Rodriguez C.; Jacobson K. A.; Stiles G. L. 125I-4-aminobenzyl-5'-N-methylcarboxamidoadenosine, a high affinity radioligand for the rat A3 adenosine receptor. Mol. Pharmacol. 1994, 45, 978–982. [PMC free article] [PubMed] [Google Scholar]
- Nordstedt C.; Fredholm B. B. A modification of a protein-binding method for rapid quantification of cAMP in cell-culture supernatants and body fluid. Anal. Biochem. 1990, 189, 231–234. [DOI] [PubMed] [Google Scholar]
- Bevan N.; Butchers P. R.; Cousins R.; Coates J.; Edgar E. V.; Morrison V.; Sheehan M. J.; Reeves J.; Wilson D. J. Pharmacological characterisation and inhibitory effects of (2R,3R,4S,5R)-2-(6-amino-2-{[(1S)-2-hydroxy-1-(phenylmethyl)ethyl]amino}-9H-purin-9-yl)-5-(2-ethyl-2H-tetrazol-5-yl)tetra hydro-3,4-furandiol, a novel ligand that demonstrates both adenosine A2A receptor agonist and adenosine A3 receptor antagonist activity. Eur. J. Pharmacol. 2007, 564, 219–225. [DOI] [PubMed] [Google Scholar]
- Jaakola V. P.; Griffith M. T.; Hanson M. A.; Cherezov V.; Chien E. Y. T.; Lane J. R.; IJzerman A. P.; Stevens R. C. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 2008, 322, 1211–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOLD, version 4.1.2; Cambridge Crystallographic Data Centre: Cambridge, United Kingdom, 2009. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.