

Abstract
Inflammatory bowel disease (IBD), consisting of Crohn's disease and ulcerative colitis, is a source of substantial morbidity and remains difficult to treat. New strategies for beneficial anti-inflammatory therapies would be highly desirable. Apolipoprotein (apo) E has immunomodulatory effects and synthetically derived apoE-mimetic peptides are beneficial in models of sepsis and neuroinflammation. We have reported that the antennapedia-linked apoE-mimetic peptide COG112 inhibits the inflammatory response to the colitis-inducing pathogen Citrobacter rodentium in vitro by inhibiting NF-κB activation. We now determined the effect of COG112 in mouse models of colitis. Using C. rodentium as an infection model, and dextran sulfate sodium (DSS) as an injury model, mice were treated with COG112 by intraperitoneal injection. With C. rodentium, COG112 improved the clinical parameters of survival, body weight, colon weight, and histologic injury. With DSS, COG112 ameliorated the loss of body weight, reduction in colon length, and histologic injury, whether administered concurrently with induction of colitis, during induction plus recovery, or only during the recovery phase of disease. In both colitis models, COG112 inhibited colon tissue inducible nitric-oxide synthase (iNOS), KC, TNF-α, IFN-γ, and IL-17 mRNA expression, and reduced nuclear translocation of NF-κB, as determined by immunoblot and immunofluorescence confocal microscopy. IκB kinase (IKK) activity was also reduced, which is necessary for activation of the canonical NF-κB pathway. Isolated colonic epithelial cells exhibited marked attenuation of expression of iNOS and the CXC chemokines KC and MIP-2. These studies indicate that apoE-mimetic peptides such as COG112 are novel potential therapies for IBD.
Keywords: Cytokine, Immunology, Inflammation, Innate Immunity, Intestine, NF-κB, Nitric-oxide Synthase, Colitis
Introduction
Inflammatory bowel disease (IBD)2 arises from the interplay between luminal bacteria and the colonic mucosa, and comprises two major forms: ulcerative colitis and Crohn's disease (1, 2). Approximately 1.4 million Americans are affected (1, 2). Although the precise mechanisms of the inflammation and immune responses are still being actively investigated, various inflammatory mediators, including chemotactic peptides and pro-inflammatory cytokines have been implicated in the disease process (3). In particular, increased secretion of proinflammatory cytokines is thought to be important for exacerbation of IBD, and a number of therapeutic approaches targeting these factors are available. Administration of anti-TNF-α antibody in mice (4, 5) and in humans (Infliximab) has demonstrated efficacy (6–8), but these treatments are often complicated by multiple side effects and are expensive. In addition, these anti-TNF-α therapies are only effective in about half of patients (9), indicating the need to reduce more than just TNF-α as a target. Therefore, new treatment approaches are still necessary.
Apolipoprotein (apo) E has an important role in cholesterol and lipid metabolism, and has also been shown to alter both innate and adaptive immune responses (10). Notably, mice lacking apoE exhibit increased inflammatory responses and higher mortality following lipopolysaccharide challenge (11), suggesting that apoE has anti-inflammatory effects. The apoE-mimetic peptide COG133 was created from amino acid residues 133–149 located in the receptor binding region of the apoE holoprotein (12). COG133 was shown to inhibit LPS-stimulated TNF-α and NO production in microglial cells in vitro (12), and it suppresses brain and systemic inflammatory responses in LPS-injected mice (13). To enhance transmembrane permeability, COG133 was fused to a protein transduction domain derived from the Drosophila antennapedia protein to create COG112 (14). This molecular fusion has been shown to enhance the bioactivity of COG133, such that there was substantial clinical improvement and protection from inflammation and demyelination injury in the spinal cord of a murine experimental autoimmune encephalomyelitis (EAE) model of multiple sclerosis (14). We have reported that COG112 inhibits the inflammatory response in colonic epithelial cells in vitro to Citrobacter rodentium (15), a bacterium known to cause colitis in mice (16). This was demonstrated to occur by inhibition of nuclear factor κB (NF-κB) activation and resulted in reduced expression of inducible nitric-oxide synthase (iNOS), and the CXC chemokines MIP-2 and KC (15).
NF-κB has been shown to be critically important in chronic inflammatory diseases including IBD (17). Activation of NF-κB leads to the gene expression of inflammatory cytokines and other mediators involved in the pathogenesis of IBD. In many cell types, the most abundant form of NF-κB is the p50/p65 heterodimer, which remains in an inactive state in the cytoplasm (18), forming a ternary complex with the inhibitory protein IκB-α (18). Upon stimulation, IκB-α is rapidly phosphorylated by IκB kinase (IKK), ubiquitinated, and subsequently degraded by proteasomes, allowing translocation of p50/p65 to the nucleus (19). NF-κB-induced cytokine production contributes to the stimulation, activation, and differentiation of immune cells, thus perpetuating inflammation (20, 21). Inhibition of NF-κB activation has been suggested as an anti-inflammatory strategy in IBD (17). Many established drugs are known to mediate anti-inflammatory effects, at least in part, via inhibition of NF-κB activity. We therefore hypothesized that COG112 treatment could have beneficial effects in models of colitis.
C. rodentium is the rodent equivalent of enteropathogenic Escherichia coli (EPEC), which causes diarrhea in humans. C. rodentium colonizes the surface of colonic epithelial cells, resulting in signal transduction events, cytoskeletal rearrangements, formation of attaching and effacing lesions, epithelial hyperplasia, and a strong mucosal Th1 response (22), as well as a Th17 response (23). Murine infection with C. rodentium provides an ideal colitis model with reproducible histological changes similar to human IBD. The main gross pathology associated with C. rodentium infection is diarrhea and thickening of the colon (22). In some cases the inflammation is transmural (16), mimicking Crohn's disease.
Dextran sulfate sodium (DSS) is a heparin-like polysaccharide that has been successfully used to induce both acute and chronic colitis in mice (24). DSS-induced colitis exhibits several characteristics resembling human ulcerative colitis, including weight loss, severe diarrhea, rectal bleeding, ulceration, and loss of epithelium, and leukocyte infiltration (24–26). Although the exact mechanism by which DSS causes colitis is not fully understood, it has been linked to direct cytotoxicity and interference with the normal interaction between intestinal lymphocytes and epithelial cells (24–26).
We now report that in both the C. rodentium and DSS models of colitis, COG112 improves clinical parameters, histological injury, and pro-inflammatory cytokine and chemokine responses. Furthermore, we show that COG112 inhibits NF-κB activation, as indicated by decreased stimulated p65 translocation and IKK activity.
EXPERIMENTAL PROCEDURES
Animals
6-week-old male C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Animals were maintained on a 12 h light/12 h dark cycle under biosafety level 3 conditions. The mice had ad libitum access to a standard diet and water until reaching the desired age of 7 weeks. All procedures using animals were reviewed and approved by the IACUC of Vanderbilt University Medical Center.
ApoE-Mimetic Peptide
The apoE-mimetic peptide COG112 was created by fusing the antennapedia protein transduction domain (RQIKIWFQNRRMKWKKC) to COG133 (LRVRLASHLRKLRKRLL) (14). The resulting sequence of COG112 (acetyl-RQIKIWFQNRRMKWKKCLRVRLASHLRKLRKRLL-amide) was synthesized using standard Fmoc chemistries and purified by NeoMPS, Inc. (San Diego, CA). In all experiments, COG112 was administered by intraperitoneal injection.
Induction of C. rodentium Colitis
Mice were orally inoculated with wild-type C. rodentium as described previously (16, 22, 27). Briefly, bacteria were grown overnight in Luria broth and mice were infected by oral gavage with 0.1 ml of broth containing 1 × 108 CFU of C. rodentium. Control mice received sterile broth. At the selected end point, the animals were sacrificed and the colons were removed, cleaned, and Swiss-rolled for histology. In addition, two proximal and two distal 2 mm2 samples were reserved, such that one from each region was combined, snap-frozen, and used for RNA and protein analysis.
Induction of DSS Colitis
DSS (MW 36,000–50,000, MP Biomedical, Solon, OH) was added to the drinking water of mice (28), for varying durations as indicated under “Results.” The animals were allowed free access to the DSS-containing water during the experiment. Mice were then sacrificed and the colons were removed, cleaned, and Swiss-rolled for histology with proximal and distal samples used for RNA and protein analysis as above.
Survival and Body Weight Measurement
To assess the effects of C. rodentium infection or DSS treatment on the mice, survival and changes in body weight of the animals were monitored daily over the course of colitis development. Mice were monitored throughout the experiment and any that showed extreme distress, became moribund, or lost more than 20% of initial body weight were euthanized.
Assessment of Histological Score
Formalin-fixed and paraffin-embedded sections of Swiss-rolled colons were stained with hematoxylin and eosin and examined in a blinded manner by pathologists (M. B. P. for C. rodentium and M. K. W. for DSS colitis). For C. rodentium-induced colitis acute (neutrophilic) and chronic (lymphocytic) inflammation, extent of inflammation, and epithelial damage were each scored on 0–3 scale (16). The aggregate histologic injury score was the sum of acute and chronic inflammation multiplied by the extent of inflammation, plus the epithelial injury (0–21). For DSS-induced colitis, tissues were scored on a 0–40 scale based on the parameters of inflammation severity (0–3), extent (0–3), and crypt damage (0–4) each multiplied by the percent involvement (1 = 0–25%; 2 = 25–50%; 3 = 50–75%; and 4 = 75–100%) as described previously (29, 30).
Isolation of Colonic Epithelial Cells
Colonic epithelial cells were isolated by a dissociation and dispersion method as described previously (31). Briefly, colons were removed, cut open longitudinally, cleaned, and cut in 2–5 mm pieces and then incubated in DTT and EDTA. Epithelial cells were detached by vigorous shaking and filtered with 70-μm nylon mesh. Epithelial cell were enriched using magnetic beads and E-cadherin antibody.
mRNA Analysis
RNA was extracted from tissue samples using the RNeasy Mini kit from Qiagen (Valencia, CA). 1 μg of RNA was reverse-transcribed using an iScript cDNA synthesis kit (Bio-Rad). Each PCR reaction was performed with 2 μl of cDNA and 2× iQTM SYBR Green Supermix (Bio-Rad). Primers for iNOS, KC, IFN-γ, TNF-α, and β-actin were used as described (15, 16). For IL-17, the primer sequences were: sense, 5′-GCTCCAGAAGGCCCTCAGA-3′ and antisense, 5′-CTTTCCCTCCGCATTGACA-3′. The thermal cycling conditions and the method used to calculate relative expression have been described previously (32–34).
Western Blot Analysis
For p65, nuclear and cytosolic extracts were prepared using an extraction kit from Pierce/Thermo Scientific. Rabbit polyclonal antibody to a synthetic peptide corresponding to amino acids near the C-terminal domain of human p65 (Rel A) from EMD Biosciences (Gibbstown, NJ) was used at a dilution of 1:1000 (15). Rabbit polyclonal fibrillarin antibody from Santa Cruz Biotechnology, Inc (Santa Cruz, CA) was used at a dilution of 1:1000. All proteins were detected by chemiluminescence, as described previously (15, 34).
Immunofluorescence Staining
Sections from the paraffin-embedded Swiss-rolls were deparaffinized, treated with citrate buffer for antigen retrieval, and blocked in 5% goat serum for 1 h at room temperature. Slides were incubated at 4 °C for 16 h with primary antibody to p65, described above, at 1:1000 dilution, followed by incubation with Alexa Fluor 488-labeled goat anti-rabbit secondary antibody (BD Biosciences, San Jose, CA) for 2 h at room temperature. Sections were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) for nuclear staining. Slides were dried and mounted, using Vectashield mounting medium (Vector Laboratories, Inc. Burlingame, CA). Slides were visualized using an OLYMPUS FLUOVIEW FV1000 confocal microscope (32, 35).
IκB Kinase (IKK) Activity Assay
The K-LISA IKK-β screening kit (Calbiochem/EMD Biosciences) was used to measure IKK kinase activity in cytosolic extracts as described (15). Lysates were incubated in glutathione-coated wells with a glutathione S-transferase (GST)-tagged IκB-α fusion polypeptide substrate. GST-IκB-α substrate phosphorylated by IKK was detected using horseradish peroxidase-conjugated anti-phospho-IκB-α antibody. Kinase activity was determined from the optical density using a standard curve generated using recombinant IKK-β according to the manufacturer's instructions.
Statistical Analysis
Quantitative data are shown as the mean ± S.E. Comparisons between multiple groups were made by using analysis of variance with the Student-Newman-Keuls posthoc multiple comparisons test. When comparisons between only 2 groups were made, Student's t test was performed. The log-rank (Mantel-Cox) test was used for the survival analysis in the C. rodentium model.
RESULTS
COG112 Reduces C. rodentium-induced Colitis in C57BL/6J Mice
In our previous study, we observed that COG112 inhibited the inflammatory response in C. rodentium-stimulated colonic epithelial cells (15). Therefore to elucidate the anti-inflammatory role of COG112 in vivo we began with the murine model of C. rodentium-induced colitis. The selected COG112 dosing schedule, 1 mg/kg daily intraperitoneal was chosen because this dose of COG112 was shown to have beneficial effects in a murine model of experimental autoimmune encephalomyelitis (14). COG112 had a marked effect on the survival of C. rodentium-infected mice (Fig. 1A). When mice were infected with C. rodentium, mortality was first observed at day 4 post-inoculation and peaked at day 9, with a 24% incidence. COG112 treatment significantly improved survival, such that only a single death was observed at day 9 (4% mortality; p = 0.021; Fig. 1A).
FIGURE 1.
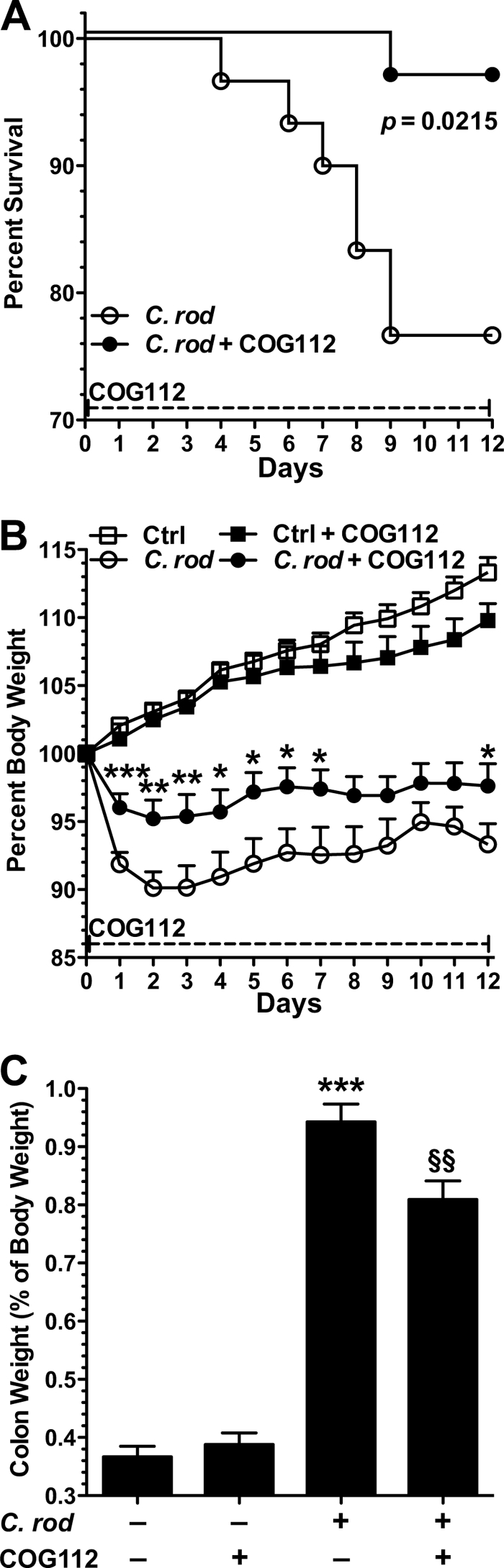
COG112 improves clinical parameters in C. rodentium-induced colitis in mice. 7-week-old C57BL/6J mice were orally inoculated with 1 × 108 CFU of C. rodentium or broth at day 0. COG112 (1 mg/kg) or PBS was injected intraperitoneal each day. A, animal mortality is presented as % survival, p = 0.0215. B, body weights of the mice were measured daily and are presented as a percentage of their initial weight. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus C. rodentium-infected mice. C, upon sacrifice on day 12, colons were removed, cleaned, and weighed. Colon weights are presented as % of mouse body weight on the day of sacrifice. ***, p < 0.001 versus control; §§, p < 0.01 versus C. rodentium-infected. In A–C, n = 11 for control, n = 9 for control + COG112, n = 30 for C. rodentium infected and for C. rodentium + COG112.
Decreasing body weight is an indicator of colitis, and we found that C. rodentium infection induced a rapid body weight loss of 9.9 ± 1.2% (90.1 ± 1.2% of initial body weight) that peaked at day 2, and continued through day 12 (Fig. 1B). This weight loss was significantly attenuated by COG112 administration (39.2 ± 10.2% improvement at day 2; Fig. 1B). COG112 also reduced C. rodentium-induced thickening of the colon, measured as changes in colon weight (Fig. 1C). At the time of sacrifice there was a 2.6 ± 0.1-fold (Fig. 1C) increase in colon weight compared with uninfected mice that was reduced by 23.2 ± 5.5% with COG112 (p < 0.01). C. rodentium infection caused significant colon inflammation and increased histologic injury (Fig. 2, A and B; p < 0.001). COG112 treatment of these mice led to a significant improvement in histological injury score (p < 0.05; Fig. 2A). It must be noted that the histologic improvement with COG112 was likely underestimated because early mortality precluded obtaining histologic samples in the 7 of 30 mice that died prematurely with C. rodentium alone (versus only 1 of 30 in the COG112-treated group) and these mice would be expected to have had very high injury scores. Hematoxylin and eosin-stained sections revealed negligible inflammation in control mice while C. rodentium-infected mice exhibited characteristic infiltration of inflammatory cells (neutrophils and lymphocytes) into the mucosa and submucosa. Treatment of infected mice with COG112 reduced mucosal and submucosal immune cells, and enhanced restitution of epithelial gland architecture, with reversal of mucin depletion (Fig. 2B).
FIGURE 2.

COG112 improves histologic injury in C. rodentium-induced colitis. Colons were Swiss-rolled and stained with hematoxylin and eosin and then scored for histologic injury. A, histologic injury scores, as described under “Experimental Procedures.” ***, p < 0.001 versus controls; §, p < 0.05 versus C. rodentium-infected; n = 11 for control and n = 9 for control + COG112, n = 23 for C. rodentium-infected, and n = 29 for C. rodentium + COG112. B, representative photomicrographs of paraffin-embedded, hematoxylin and eosin-stained sections of colon.
COG112 Reduces DSS-induced Colitis in C57BL/6J Mice
To determine whether the beneficial effects of COG112 in colitis may be generalized, we utilized a second model of colitis in which mice were administered DSS in their drinking water. Mice began losing body weight after 5 days of treatment with DSS and the loss reached 4.7 ± 0.8% (95.3 ± 0.8% of starting body weight) by day 6 and 8.9 ± 0.8% (91.1 ± 0.8% of starting body weight) by day 7 (Fig. 3A). In contrast, the body weight loss of mice receiving daily injections of COG112 (1 mg/kg) beginning on day 0 was only 1.3 ± 0.5% (98.7 ± 0.5% of starting body weight) and 4.4 ± 0.7% (95.6 ± 0.7% of starting body weight) at day 6 and 7 respectively, a significant improvement. In the DSS model of colitis, colon length is a macroscopic indicator of disease severity. Colon length in DSS-treated mice was significantly reduced compared with controls (p < 0.001, Fig. 3B) while COG112 treatment of colitic mice led to a significant improvement in colon length (p < 0.01). We also tested a higher dose of COG112 (10 mg/kg) and monitored changes in body weight. As above, we observed an improvement in body weight at day 6 and 7 of DSS treatment (p < 0.05), but the effect was no greater than seen with the 1 mg/kg dose of COG112 (supplemental Fig. S1). Also, when we tested the effect of COG112 (1 mg/kg) in mice treated with a lower dose of DSS (3%) there was a similar degree of improvement in body weight (supplemental Fig. S2) as observed with the 4% DSS model.
FIGURE 3.

COG112 improves clinical parameters and histologic injury in DSS-induced acute colitis in mice. Mice were given 4% DSS in the drinking water for 7 days. COG112 (1 mg/kg) or PBS was injected daily. A, percent body weight of mice with or without DSS and 1 mg/kg COG112 for 7 days. Body weights were measured every day and are presented as percent of initial weight. *, p < 0.05; ***, p < 0.001 versus DSS-treated only. B, lengths of freshly removed colons were measured from rectum to ileocecal junction on day 7. ***, p < 0.001 versus control, §§, p < 0.01 versus DSS-treated alone. In A-B, n = 11 for control, n = 9 for control + COG112, n = 30 for DSS-treated, and n = 30 for DSS + COG112. C, histolologic injury score of mice given DSS ± COG112 for 7 days. Colons were Swiss-rolled and stained with hematoxylin and eosin and then scored for histologic injury, as described under “Experimental Procedures.” **, p < 0.01 versus DSS alone. D, representative photomicrographs of paraffin-embedded, hematoxylin and eosin-stained sections of colon.
To assess the effect of COG112 on the recovery phase of DSS colitis, mice were given 4% DSS for 7 days, and were then switched to water for an additional 9 days (Fig. 4). In these studies, COG112 was injected daily throughout the experiment. Maximal weight loss was observed at day 11. Both DSS- and DSS + COG112-treated mice began regaining weight at day 12, but the recovery was greater in COG112-treated mice. We observed that DSS + COG112-treated mice actually reached their initial body weight at day 15, and were 103.7 ± 2.3% of initial body weight at day 16, while the DSS-treated mice never reached their initial body weight (Fig. 4A). We again found that there was a blunting of the DSS-induced effects on colon length with COG112 treatment (Fig. 4B).
FIGURE 4.

COG112 improves DSS-induced colitis during the recovery phase. Mice were given DSS in the drinking water from day 0 to day 7 and then switched to normal drinking water after day 7 for recovery. COG112 or PBS was injected every day starting from day 0. A, change in body weight during the course of the experiment, n = 5 for control and control + COG112, n = 20 for DSS treatment, and n = 10 for DSS + COG112. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus DSS-treated only. B, lengths of colons were measured on day 16. ***, p < 0.001; versus control and §, p < 0.05 versus DSS-treated only. C, histological injury score on 0–40 scale. *, p < 0.05 versus DSS-treated only. D, representative photomicrographs of paraffin-embedded, hematoxylin and eosin-stained sections of colon.
Histological scoring of inflammation revealed that COG112 significantly improved the injury score of DSS-treated mice in both acute (Fig. 3C) and recovery (Fig. 4C) experiments. DSS colitis is characterized by infiltration of immune cells in the mucosa and submucosa, epithelial ulceration, and crypt damage. Hematoxylin and eosin-stained sections of the distal colon of control mice showed no inflammation (data not shown) while colons of DSS-treated mice exhibited severe inflammatory infiltrates (neutrophils and lymphocytes), crypt loss, and ulceration, at both the acute time point (Fig. 3D) and after the recovery period (Fig. 4D). In contrast, DSS-treated mice receiving COG112 displayed reduced inflammatory infiltrates and less crypt loss and ulceration (Figs. 3D and 4D).
Having observed that COG112 improved clinical parameters and histology in both the acute and the recovery phase while given throughout the experiments, we sought to determine the effect of COG112 if given after onset of the disease as a treatment. Mice were administered DSS for 5 days, at which time they were switched to water for an additional 7 days (12 day model). In these experiments COG112 was injected only during the recovery phase from day 6 to day 12. Maximal weight loss was observed at day 8 when DSS-treated mice reached 82.8 ± 1.6% of initial weight, and recovery was enhanced in the mice treated with COG112 (Fig. 5A). At the time of sacrifice the DSS-treated group had a body weight of 91.2 ± 1.4% of initial weight, whereas the DSS + COG112 group had a body weight of 98.3 ± 2.5% (p < 0.05). Colon length was also improved in these treatment experiments (p < 0.05, Fig. 5B). In parallel, with COG112 there was an improvement in histologic injury scores (Fig. 5C) with enhanced restitution of the epithelium readily apparent (Fig. 5D).
FIGURE 5.

COG112 improves DSS-induced colitis when given after the onset of disease. Mice were given 4% DSS in their drinking water for 5 days. COG112 or PBS was injected intraperitoneal daily only during the 7-day recovery phase. A, body weight data. *, p < 0.05; ***, p < 0.001 versus DSS-treated only. n = 5 for control, n = 5 for control + COG112, n = 15 for DSS-treated and n = 10 for DSS + COG112. B, colon lengths. ***, p < 0.001 versus control, §, p < 0.05 versus DSS-treated. C, histolologic injury score of mice given DSS for 5 days followed by 7 days ± COG112 during the recovery phase. D, representative photomicrographs of hematoxylin and eosin-stained sections of colon.
COG112 Inhibits iNOS and Cytokine mRNA Expression in C. rodentium- and DSS-induced Colitis
iNOS has been shown to play a role in the pathophysiology of experimental colitis (16, 25). Previously, we reported that iNOS is up-regulated in colonic tissues infected with C. rodentium and that iNOS−/− mice exhibited less severe colitis in this model (16). In our current experiments, iNOS mRNA was increased 572.3 ± 100.5-fold in C. rodentium-infected colon tissues compared with controls (p < 0.001; Fig. 6A). When C. rodentium-infected mice were treated with COG112, iNOS mRNA levels were down-regulated by 33.6 ± 9.1% (p < 0.05). We also measured mRNA levels of KC (CXCL1), because this CXC-chemokine has been reported to be up-regulated in C. rodentium-induced colitis (27, 36). KC was induced by 68.1 ± 10.1-fold (Fig. 6B) in C. rodentium-infected colon tissues. Treatment with COG112 led to a significant down-regulation of this gene by 39.6 ± 9.5% (p < 0.05). TNF-α, a marker of inflammation, was induced 12.6 ± 3.4-fold by C. rodentium infection (p < 0.05) and was reduced by 70.8 ± 4.3% (p < 0.01) with COG112 treatment (Fig. 6C). When we investigated the modulation of the Th1 cytokine IFN-γ and the Th17 cytokine IL-17 by C. rodentium infection and COG112 treatment, we found that both followed similar patterns to the other genes investigated (Fig. 6, D and E). IFN-γ was increased 566.2 ± 194.5-fold, (p < 0.001) and down-regulated by 66.9 ± 8.6% (p < 0.01) with COG112, and IL-17 was increased 61.8 ± 14.3-fold, (p < 0.01) and reduced by 45.5 ± 15.6% (p < 0.05) with COG112 treatment. Similar results were observed in colon tissues from DSS-treated mice. DSS treatment increased iNOS mRNA levels by 100 ± 34.1-fold (p < 0.001), which were reduced by 76.9 ± 10.5% with COG112 (p < 0.001; Fig. 6F). DSS treatment also stimulated increased expression of KC (37.7 ± 15.4-fold, p < 0.001, Fig. 6G), TNF-α (7.8 ± 1.1- fold, p < 0.01, Fig. 6H), IFN-γ (40.3 ± 6.0-fold, p < 0.01, Fig. 6I), and IL-17 (15.6 ± 3.1-fold, p < 0.01, Fig. 6J), and these inflammatory genes were down-regulated by 79.9 ± 4.1%, 60.1 ± 7.8%, 74.7 ± 7.9% and 52.3 ± 2.1%, respectively, with COG112 treatment (Fig. 6, G–J).
FIGURE 6.

Effect of COG112 on mRNA expression of iNOS, KC, and cytokines in C. rodentium- and DSS-induced colitis tissues. RNA was isolated from the tissues and mRNA levels were assessed by real-time PCR using SYBR green with standardization to β-actin levels in tissues from the C. rodentium (A–E) and DSS (F–J) models. **, p < 0.01; ***, p < 0.001 versus controls; §, p < 0.05; §§, p < 0.01; §§§, p < 0.001 versus C. rodentium-infected or DSS-treated; n = 3–6 for controls and control + COG112, and n = 7–10 for C. rodentium, DSS, C. rodentium + COG112, and DSS + COG112. In A–E, control mice (Ctrl) received gavage with LB broth, and in F–J, control mice received normal drinking water.
COG112 Inhibits iNOS and Chemokine mRNA Expression in Colonic Epithelial Cells from DSS-treated Mice
After 7 days of DSS ± COG112 treatment, colonic epithelial cells were isolated by the dissociation and dispersion method and selected using E-cadherin antibody (supplemental Fig. S3). By real-time PCR, there was a 60.5 ± 5.9-fold increase in iNOS mRNA expression in colon cells isolated from DSS-treated mice, which was decreased by 69.4 ± 3.9% in cells from DSS + COG112-treated mice (p < 0.001, Fig. 7A). We also analyzed expression of the chemokines MIP-2 (CXCL2) and KC, and both were up-regulated in epithelial cells isolated from DSS-treated mice and were markedly decreased by 95.7 ± 3.1% and 92.6 ± 5.3% by COG112 treatment, respectively (p < 0.001, Fig. 7, B and C).
FIGURE 7.
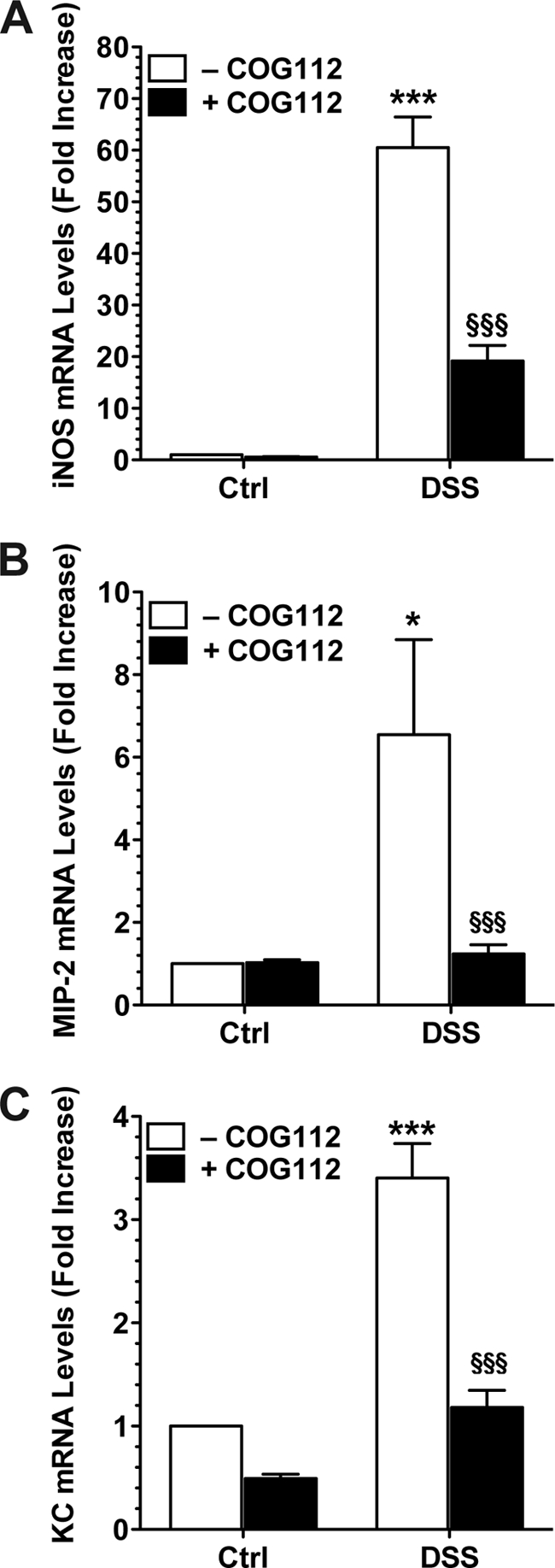
Effect of COG112 on mRNA expression of iNOS and chemokines in colonic epithelial cells isolated from DSS-treated colons. Mice received 4% DSS or water alone (Ctrl) for 7 days. Mice were injected daily with COG112 (1 mg/kg) or PBS vehicle. Colons were harvested, and epithelial cells were isolated and positively selected with antibody to E-cadherin using magnetic beads (supplemental Fig. S3). mRNA was extracted and real-time PCR was performed for iNOS (A), MIP-2 (B), and KC (C) standardized to β-actin using SYBR green. *, p < 0.05; **, p < 0.01 versus control; §§, p < 0.01 versus DSS alone. n = 4 mice per group.
COG112 Reduces NF-κB Activation in Both C. rodentium- and DSS-Induced Colitis
C. rodentium colitis is known to involve activation of NF-κB in both macrophages and epithelial cells (27, 37). NF-κB activation has also been shown to be important in DSS colitis (38, 39). Using anti-p65 antibody, we observed nuclear translocation of p65 in colon tissues from both C. rodentium and DSS colitis, which appeared to occur in both epithelial and immune cells (Fig. 8 and supplemental Figs. S4 and S5). In mice treated with COG112, much less nuclear p65 staining was observed (Fig. 8) in both colitis models, with less overall p65 staining (supplemental Figs. S4 and S5). When nuclear protein was isolated from colonic tissues and immunoblotted for p65, we noted increased p65 protein in C. rodentium- and DSS-treated mice compared with controls, that was reduced when mice were administered COG112 (Fig. 9, A and B). Nuclear translocation of p65 requires phosphorylation of the inhibitory protein IκB-α and its dissociation from p65 in the cytoplasm. We therefore assessed IKK activity in the cytoplasmic fractions of colonic tissue extracts. As shown in Fig. 9, C and D, C. rodentium- and DSS-induced colitis tissues exhibited significantly more IKK activity compared with controls (p < 0.001), and this kinase activity was reduced with COG112 treatment by 91.5 ± 4.0% (p < 0.001) in C. rodentium colitis and 85.9 ± 12.3% (p < 0.01) in DSS-treated mice.
FIGURE 8.

Nuclear translocation of NF-κB in C. rodentium- and DSS-induced colitis is reduced by COG112. C. rodentium tissues were obtained on day 12 post-inoculation. DSS tissues were obtained after 7 days of DSS treatment. Formalin-fixed, paraffin-embedded sections (5 μm) of Swiss-rolled tissues were stained for p65 detected with Alexa Fluor 488-conjugated secondary antibody (green, 1:1000) after antigen retrieval. Nuclei were visualized with DAPI (blue, 1:10,000 dilution). Images were captured by confocal microscopy and 600× merged images are shown. Scale bars, 20 μm in full size images and 10 μm in zoom insets.
FIGURE 9.

COG112 inhibits nuclear accumulation of NF-κB and IKK activity in C. rodentium- and DSS-induced colitis. A and B, representative Western blots of p65 (65 kDa) and fibrillarin (36 kDa) as nuclear loading control; 20 μg of nuclear protein/lane was used. C and D, ELISA assay for IKK activity was performed in the cytosolic fractions of tissues. ***, p < 0.001 versus control; §§, p < 0.01, §§§, p < 0.001 versus C. rodentium-infected or DSS-treated.
DISCUSSION
The objective of this study was to determine whether the apoE-mimetic peptide COG112 could lessen disease in models of colitis and thus could have potential as a therapy in human IBD. We previously reported that COG112 inhibits the inflammatory response to C. rodentium in colonic epithelial cells in vitro by preventing NF-κB activation (15). To evaluate the effect of COG112 in colitis in vivo, we used two well-established mouse models. Here we report that COG112 has anti-inflammatory properties and improves clinical disease parameters in C. rodentium-infected and DSS-treated mice.
We have chosen the dose and route of administration of COG112 on the basis of prior reports (14), in which the i.p route and the 1 mg/kg dose was the most effective. Pharmacokinetic studies have shown that significantly higher amounts of COG112 accumulated in the blood when administered intraperitoneal versus i.v (11). COG112 can be detected in blood as early as 10 min. after intraperitoneal administration (13, 40). It should be noted that we also performed pilot studies with daily subcutaneous injection of COG112 (1 mg/kg) in the 4% DSS model, and observed improvement in clinical parameters, but these effects were less marked than with intraperitoneal administration at the same dose (data not shown).
C. rodentium, like EPEC and enterohemorrhagic E. coli (EHEC), attaches to the luminal surface of the intestinal epithelium and provides an excellent in vivo model for infectious colitis, including how the host recognizes pathogens in the intestinal lumen. Infection with C. rodentium is characterized by a transient colonization of the large intestine, crypt hyperplasia, mucosal thickening, and diarrhea (41). C. rodentium may cause focal erosions on the surface of the colon and induces a Th1 response, with expression of IL-12, TNF-α, and IFN-γ (42), as well as a Th17 response (23, 43). COG112 significantly ameliorated the clinical changes induced by C. rodentium infection. We demonstrated improvement in survival of C. rodentium-infected mice. It has been speculated that C. rodentium-infected mice die due to overwhelming sepsis (44). The enhanced survival with COG112 that we demonstrated may have resulted from decreased transmural inflammation and enhanced integrity of the epithelium that we observed on histologic assessment, as well as the diminished expression of proinflammatory cytokines and iNOS, both of which contribute to the hemodynamic alterations of sepsis (45, 46). We observed a modest decrease in colonic colonization with C. rodentium in mice treated with COG112 (C. rodentium alone, 4.3 × 107 ± 2.9 × 107 CFU/g; C. rodentium + COG112, 1.7 × 107 ± 8.0 × 106 CFU/g, p = 0.46); although this could account for some of the effect, this small difference is unlikely to fully explain the enhanced survival.
To confirm the beneficial effects of COG112, we employed a second model of colitis, which is primarily an injury model. Chemical disruption of the epithelial barrier by DSS triggers crypt damage, ulceration, and neutrophil infiltration (47). This model system has been shown to have similarities with human UC, including weight loss, erosion of the colonic mucosa and shortening of the colon. Notably, COG112 significantly reduced the clinical parameters of body weight loss, shortening of the colon, and histologic damage. Importantly, COG112 had consistently beneficial effects, whether tested during the onset of acute colitis, continued during the recovery period, or used only as a treatment once colitis was established. The latter findings indicate that COG112 could have benefit in treating human IBD during both acute flares of increasing activity, as well as chronic persistent inflammation.
iNOS has been purported to be linked to both protection and exacerbation of colitis (48), but clearly it is an excellent marker of intestinal inflammation (16, 25). In the current study, we identified large increases in iNOS expression in both models of colitis and found that COG112 treatment of the mice significantly reduced iNOS levels. These findings are strongly consistent with our previous report that COG112 inhibited iNOS expression in colonic epithelial cells and macrophages exposed to C. rodentium in vitro (15). Furthermore, we have reported that C. rodentium colitis is improved in iNOS-deficient mice in vivo (16), suggesting that the reduction in iNOS with COG112 is likely contributing to its benefit in this model. The role of iNOS in injury models of colitis is variable; for example, worsening of trinitrobenzene sulfonic acid (TNBS) colitis in mice with deletion of iNOS has been reported (49), while iNOS deletion or use of a chemical inhibitor of iNOS (1400W) has been shown to improve DSS colitis (25). As in the C. rodentium model, we also observed that iNOS levels were reduced with COG112 treatment in the DSS model. However, in both models, whether this reduction in iNOS is a mechanism underlying disease improvement or is only a marker of decreased inflammation could not be directly determined.
Neutrophil accumulation plays an important role in DSS-induced colitis, since the disease can be attenuated when anti-neutrophil serum is administered (50). As part of the amelioration of colon histologic injury in the DSS model, COG112 reduced neutrophil infiltration. Consistent with this, COG112 reduced the increased levels of the neutrophil-attracting chemokines KC and MIP-2 in colonic epithelial cells derived from DSS-treated mice. Similarly, there was a reduction in KC mRNA levels in colitis tissues from either DSS- or C. rodentium-treated mice. These results provide support for COG112 as an anti-inflammatory agent, because chemokine expression is recognized as a key early step in the pathophysiology of inflammatory responses to colonic pathogens and has been directly linked to the pathophysiology of human IBD (29).
COG112 also reduced TNF-α mRNA expression in both our models of colitis. This cytokine is an important mediator of inflammation and it has been reported that treatment with the TNF-α antagonist Etanercept reduces inflammatory lesions in DSS-induced chronic colitis in mice (51). In addition, Infliximab (52–54), which is a chimeric murine-human monoclonal antibody to TNF-α, is effectively used in humans to treat Crohn's disease and ulcerative colitis (6). The down-regulation of TNF-α by COG112 contributes to the concept that it may be a useful treatment for IBD. Because only about half of all patients receiving anti-TNF-α antibody have a sustained benefit, and it has been shown in mouse models, such as DSS colitis, to have only a partial efficacy (4, 5), our findings that COG112 also effectively reduced other proinflammatory mediators, namely chemokines, Th1 and Th17 cytokines, and iNOS, suggests that it may be broadly beneficial in IBD patients.
We also investigated the effect of COG112 on T cell-derived cytokines. IFN-γ produced by Th1 lymphocytes has been shown to be involved in the pathophysiology of IBD (16). In both of our colitis models, IFN-γ mRNA expression was markedly up-regulated. The recently described Th17 subset of lymphocytes may play an important role in the immunopathogenesis of IBD (1). IL-17 is a prototype cytokine product of these cells, and its up-regulated expression in C. rodentium infection has been specifically demonstrated (23). When we measured mRNA levels of IL-17 in DSS- and C. rodentium-induced colitis we observed a significant up-regulation of gene expression. In both colitis models, COG112 inhibited colonic expression of these potent T cell inflammatory cytokines, suggests an additional potential mechanism for the beneficial effects of COG112.
In our previous study, we reported that C. rodentium induced NF-κB activation in vitro was inhibited by COG112 (15). In the current report, we have confirmed this observation using two in vivo models of colitis. We verified the nuclear translocation of p65 by immunoblotting and immunofluorescence in C. rodentium and DSS colitis tissues that was blocked by COG112, and demonstrated that COG112 inhibits upstream IKK activity necessary for NF-κB activation. These data are consistent with our previous demonstration that COG112 inhibits IKK activity by inhibiting formation of the IKK complex in colonic epithelial cells (15). Further, our findings that COG112 effectively inhibits IKK activity, and to the same degree as it inhibits p65 translocation, suggests that COG112 acts predominantly on the canonical NF-κB signaling pathway (55). Activation of NF-κB is believed to be an important step in the development of human IBD and inhibition of NF-κB activation is associated with reduced colonic inflammation (17). Many of the drugs used to treat the disease including corticosteroids, salicylates (56), and sulfasalazine (57) inhibit NF-κB activation. It has also been reported that decoy oligonucleotides mimicking the NF-κB consensus binding sequence attenuate both Th1- and Th2-mediated experimental colitis (58). Resveratrol and piceatannol, naturally occurring substances found in peanuts, grapes and red wines, have also been shown to protect mice from DSS colitis by preventing NF-κB activation (59, 60). The NEMO binding domain, which specifically inhibits NF-κB activation, was shown to ameliorate both DSS and TNBS colitis in mice (61, 62).
Currently, the role of NF-κB in colitis is still unclear; in some models NF-κB inhibition is beneficial, and in others it is deleterious. Tissue-specific deletion of IKK-β in enterocytes and macrophages reduces the development of inflammation-associated cancer (63). However, selective blockade of NF-κB activation in mouse intestinal epithelial cells (NEMOIEC-KO) by ablation of IKK-γ results in severe chronic inflammation, diarrhea, and rectal bleeding (64). In the DSS model, complete deletion of p65 in colonic epithelial cells results in disease exacerbation (65). Another concern is that inhibition of NF-κB promotes development of squamous cell carcinomas in keratinocytes (66), and increases the incidence of liver tumors in a chemical carcinogenesis model (67). These previous findings suggest that complete NF-κB inactivation may have different roles depending on the tissues. Although the literature is complex, our data show that significant, but not complete inhibition of NF-κB by COG112 was associated with reduced Th1/Th17 activation, proinflammatory cytokine and chemokine expression, and clinical and histologic improvement in the two colitis models tested. From a different point of view, it should be noted that COG112 can affect immune cells in addition to epithelial cells (15), which could also explain its in vivo benefit.
In summary, the present study provides the first in vivo evidence that an apoE-mimetic peptide, COG112, can improve colitis and that it has specific anti-inflammatory properties. COG112 had beneficial effects on clinical parameters in two distinct models of colitis, and was effective in both the induction phase and in the treatment of established colitis. Furthermore, this agent inhibited the expression of inflammatory cytokines and chemokines, and markedly reduced NF-κB activation in both the C. rodentium and DSS colitis models. Because the drug was well tolerated out to the maximum time point tested of 16 days, it is an excellent candidate for future studies in chronic models of colitis and colitis-associated carcinoma.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants R41DK075161 (to K. T. W. and M. P. V.), R01DK053620, R01AT004821, and 3R01AT004821S1 (to K. T. W.), P01CA028842 (to K. T. W.), F31GM083500 (to N. D. L), T32CA009592 (N. D. L.), T32DK007673 (D. P. B.), the Flow Cytometry Core, and the Cell Imaging Core of the Vanderbilt University Digestive Disease Research Center supported by National Institutes of Health Grant P30DK058404; and a Merit Review Grant from the Office of Medical Research, Dept. of Veterans Affairs (to K. T. W.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S5.
- IBD
- inflammatory bowel disease
- C. rod
- C. rodentium
- DSS
- dextran sulfate sodium
- apoE
- apolipoprotein E
- IKK
- IκB kinase
- IFN
- interferon
- iNOS
- inducible nitric-oxide synthase
- KC
- keratinocyte-derived chemokine
- i.p.
- intraperitoneal
- MIP
- macrophage inflammatory protein
- NO
- nitric oxide
- TNF
- tumor necrosis factor
- DAPI
- 4′,6-diamidino-2-phenylindole.
REFERENCES
- 1. Baumgart D. C., Carding S. R. (2007) Lancet 369, 1627–1640 [DOI] [PubMed] [Google Scholar]
- 2. Baumgart D. C., Sandborn W. J. (2007) Lancet 369, 1641–1657 [DOI] [PubMed] [Google Scholar]
- 3. Loftus C. G., Loftus E. V., Jr., Harmsen W. S., Zinsmeister A. R., Tremaine W. J., Melton L. J., 3rd, Sandborn W. J. (2007) Inflamm. Bowel Dis. 13, 254–261 [DOI] [PubMed] [Google Scholar]
- 4. Kojouharoff G., Hans W., Obermeier F., Männel D. N., Andus T., Schölmerich J., Gross V., Falk W. (1997) Clin. Exp. Immunol. 107, 353–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Murthy S., Cooper H. S., Yoshitake H., Meyer C., Meyer C. J., Murthy N. S. (1999) Aliment. Pharmacol. Ther. 13, 251–260 [DOI] [PubMed] [Google Scholar]
- 6. Present D. H. (1999) Aliment. Pharmacol. Ther. Suppl. 4, 23–28 [DOI] [PubMed] [Google Scholar]
- 7. Mouser J. F., Hyams J. S. (1999) Clin. Ther. 21, 932–942; discussion 931 [DOI] [PubMed] [Google Scholar]
- 8. Present D. H., Rutgeerts P., Targan S., Hanauer S. B., Mayer L., van Hogezand R. A., Podolsky D. K., Sands B. E., Braakman T., DeWoody K. L., Schaible T. F., van Deventer S. J. (1999) N. Engl. J. Med. 340, 1398–1405 [DOI] [PubMed] [Google Scholar]
- 9. Clark M., Colombel J. F., Feagan B. C., Fedorak R. N., Hanauer S. B., Kamm M. A., Mayer L., Regueiro C., Rutgeerts P., Sandborn W. J., Sands B. E., Schreiber S., Targan S., Travis S., Vermeire S. (2007) Gastroenterology 133, 312–339 [DOI] [PubMed] [Google Scholar]
- 10. Laskowitz D. T., Lee D. M., Schmechel D., Staats H. F. (2000) J. Lipid Res. 41, 613–620 [PubMed] [Google Scholar]
- 11. Van Oosten M., Rensen P. C., Van Amersfoort E. S., Van Eck M., Van Dam A. M., Breve J. J., Vogel T., Panet A., Van Berkel T. J., Kuiper J. (2001) J. Biol. Chem. 276, 8820–8824 [DOI] [PubMed] [Google Scholar]
- 12. Laskowitz D. T., Thekdi A. D., Thekdi S. D., Han S. K., Myers J. K., Pizzo S. V., Bennett E. R. (2001) Exp. Neurol. 167, 74–85 [DOI] [PubMed] [Google Scholar]
- 13. Lynch J. R., Tang W., Wang H., Vitek M. P., Bennett E. R., Sullivan P. M., Warner D. S., Laskowitz D. T. (2003) J. Biol. Chem. 278, 48529–48533 [DOI] [PubMed] [Google Scholar]
- 14. Li F. Q., Sempowski G. D., McKenna S. E., Laskowitz D. T., Colton C. A., Vitek M. P. (2006) J. Pharmacol. Exp. Ther. 318, 956–965 [DOI] [PubMed] [Google Scholar]
- 15. Singh K., Chaturvedi R., Asim M., Barry D. P., Lewis N. D., Vitek M. P., Wilson K. T. (2008) J. Biol. Chem. 283, 16752–16761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gobert A. P., Cheng Y., Akhtar M., Mersey B. D., Blumberg D. R., Cross R. K., Chaturvedi R., Drachenberg C. B., Boucher J. L., Hacker A., Casero R. A., Jr., Wilson K. T. (2004) J. Immunol. 173, 2109–2117 [DOI] [PubMed] [Google Scholar]
- 17. Schreiber S., Nikolaus S., Hampe J. (1998) Gut 42, 477–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Baeuerle P. A., Henkel T. (1994) Annu. Rev. Immunol. 12, 141–179 [DOI] [PubMed] [Google Scholar]
- 19. DiDonato J. A., Hayakawa M., Rothwarf D. M., Zandi E., Karin M. (1997) Nature 388, 548–554 [DOI] [PubMed] [Google Scholar]
- 20. Mercurio F., Zhu H., Murray B. W., Shevchenko A., Bennett B. L., Li J., Young D. B., Barbosa M., Mann M., Manning A., Rao A. (1997) Science 278, 860–866 [DOI] [PubMed] [Google Scholar]
- 21. Chiao P. J., Miyamoto S., Verma I. M. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 28–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vallance B. A., Deng W., Knodler L. A., Finlay B. B. (2002) Infect. Immun. 70, 2070–2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mangan P. R., Harrington L. E., O'Quinn D. B., Helms W. S., Bullard D. C., Elson C. O., Hatton R. D., Wahl S. M., Schoeb T. R., Weaver C. T. (2006) Nature 441, 231–234 [DOI] [PubMed] [Google Scholar]
- 24. Okayasu I., Hatakeyama S., Yamada M., Ohkusa T., Inagaki Y., Nakaya R. (1990) Gastroenterology 98, 694–702 [DOI] [PubMed] [Google Scholar]
- 25. Krieglstein C. F., Cerwinka W. H., Laroux F. S., Salter J. W., Russell J. M., Schuermann G., Grisham M. B., Ross C. R., Granger D. N. (2001) J. Exp. Med. 194, 1207–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Krieglstein C. F., Cerwinka W. H., Sprague A. G., Laroux F. S., Grisham M. B., Koteliansky V. E., Senninger N., Granger D. N., de Fougerolles A. R. (2002) J. Clin. Invest. 110, 1773–1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Khan M. A., Ma C., Knodler L. A., Valdez Y., Rosenberger C. M., Deng W., Finlay B. B., Vallance B. A. (2006) Infect. Immun. 74, 2522–2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Theiss A. L., Vijay-Kumar M., Obertone T. S., Jones D. P., Hansen J. M., Gewirtz A. T., Merlin D., Sitaraman S. V. (2009) Gastroenterology 137, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sartor R. B. (2006) Nat. Clin. Pract. Gastroenterol. Hepatol. 3, 390–407 [DOI] [PubMed] [Google Scholar]
- 30. Williams K. L., Fuller C. R., Dieleman L. A., DaCosta C. M., Haldeman K. M., Sartor R. B., Lund P. K. (2001) Gastroenterology 120, 925–937 [DOI] [PubMed] [Google Scholar]
- 31. Whitehead R. H., VanEeden P. E., Noble M. D., Ataliotis P., Jat P. S. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 587–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chaturvedi R., Cheng Y., Asim M., Bussière F. I., Xu H., Gobert A. P., Hacker A., Casero R. A., Jr., Wilson K. T. (2004) J. Biol. Chem. 279, 40161–40173 [DOI] [PubMed] [Google Scholar]
- 33. Cheng Y., Chaturvedi R., Asim M., Bussière F. I., Scholz A., Xu H., Casero R. A., Jr., Wilson K. T. (2005) J. Biol. Chem. 280, 22492–22496 [DOI] [PubMed] [Google Scholar]
- 34. Chaturvedi R., Asim M., Lewis N. D., Algood H. M., Cover T. L., Kim P. Y., Wilson K. T. (2007) Infect. Immun. 75, 4305–4315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Asim M., Chaturvedi R., Hoge S., Lewis N. D., Singh K., Barry D. P., Algood H. S., de Sablet T., Gobert A. P., Wilson K. T. (2010) J. Biol. Chem. 285, 20343–20357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bergstrom K. S., Guttman J. A., Rumi M., Ma C., Bouzari S., Khan M. A., Gibson D. L., Vogl A. W., Vallance B. A. (2008) Infect. Immun. 76, 796–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang Y., Xiang G. S., Kourouma F., Umar S. (2006) Br. J. Pharmacol. 148, 814–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Murano M., Maemura K., Hirata I., Toshina K., Nishikawa T., Hamamoto N., Sasaki S., Saitoh O., Katsu K. (2000) Clin. Exp. Immunol. 120, 51–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Edelblum K. L., Washington M. K., Koyama T., Robine S., Baccarini M., Polk D. B. (2008) Gastroenterology 135, 539–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lynch J. R., Wang H., Mace B., Leinenweber S., Warner D. S., Bennett E. R., Vitek M. P., McKenna S., Laskowitz D. T. (2005) Exp. Neurol. 192, 109–116 [DOI] [PubMed] [Google Scholar]
- 41. Wiles S., Clare S., Harker J., Huett A., Young D., Dougan G., Frankel G. (2004) Cell. Microbiol. 6, 963–972 [DOI] [PubMed] [Google Scholar]
- 42. Higgins L. M., Frankel G., Douce G., Dougan G., MacDonald T. T. (1999) Infect. Immun. 67, 3031–3039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Harrington L. E., Hatton R. D., Mangan P. R., Turner H., Murphy T. L., Murphy K. M., Weaver C. T. (2005) Nat. Immunol. 6, 1123–1132 [DOI] [PubMed] [Google Scholar]
- 44. Bry L., Brenner M. B. (2004) J. Immunol. 172, 433–441 [DOI] [PubMed] [Google Scholar]
- 45. Chalaris A., Adam N., Sina C., Rosenstiel P., Lehmann-Koch J., Schirmacher P., Hartmann D., Cichy J., Gavrilova O., Schreiber S., Jostock T., Matthews V., Häsler R., Becker C., Neurath M. F., Reiss K., Saftig P., Scheller J., Rose-John S. (2010) J. Exp. Med. 207, 1617–1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. MacMicking J. D., Nathan C., Hom G., Chartrain N., Fletcher D. S., Trumbauer M., Stevens K., Xie Q. W., Sokol K., Hutchinson N. (1995) Cell 81, 641–650 [DOI] [PubMed] [Google Scholar]
- 47. Vowinkel T., Mori M., Krieglstein C. F., Russell J., Saijo F., Bharwani S., Turnage R. H., Davidson W. S., Tso P., Granger D. N., Kalogeris T. J. (2004) J. Clin. Invest. 114, 260–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cross R. K., Wilson K. T. (2003) Inflamm. Bowel Dis. 9, 179–189 [DOI] [PubMed] [Google Scholar]
- 49. McCafferty D. M., Miampamba M., Sihota E., Sharkey K. A., Kubes P. (1999) Gut 45, 864–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Domek M. J., Iwata F., Blackman E. I., Kao J., Baker M., Vidrich A., Leung F. W. (1995) Scand. J. Gastroenterol. 30, 1089–1094 [DOI] [PubMed] [Google Scholar]
- 51. Popivanova B. K., Kitamura K., Wu Y., Kondo T., Kagaya T., Kaneko S., Oshima M., Fujii C., Mukaida N. (2008) J. Clin. Invest. 118, 560–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. D'Haens G. R. (1999) Ital. J. Gastroenterol. Hepatol. 31, 519–520 [PubMed] [Google Scholar]
- 53. Hyams J. S., Markowitz J., Wyllie R. (2000) J. Pediatr. 137, 192–196 [DOI] [PubMed] [Google Scholar]
- 54. Kirman I., Whelan R. L., Nielsen O. H. (2004) Eur. J. Gastroenterol. Hepatol. 16, 639–641 [DOI] [PubMed] [Google Scholar]
- 55. Hacker H., Karin M. (2006) Sci. STKE 13, [DOI] [PubMed] [Google Scholar]
- 56. Kopp E., Ghosh S. (1994) Science 265, 956–959 [DOI] [PubMed] [Google Scholar]
- 57. Wahl C., Liptay S., Adler G., Schmid R. M. (1998) J. Clin. Invest. 101, 1163–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fichtner-Feigl S., Fuss I. J., Preiss J. C., Strober W., Kitani A. (2005) J. Clin. Invest. 115, 3057–3071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kim Y. H., Kwon H. S., Kim D. H., Cho H. J., Lee H. S., Jun J. G., Park J. H., Kim J. K. (2008) Int. Immunopharmacol. 8, 1695–1702 [DOI] [PubMed] [Google Scholar]
- 60. Singh U. P., Singh N. P., Singh B., Hofseth L. J., Price R. L., Nagarkatti M., Nagarkatti P. S. (2010) J. Pharmacol. Exp. Ther. 332, 829–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shibata W., Maeda S., Hikiba Y., Yanai A., Ohmae T., Sakamoto K., Nakagawa H., Ogura K., Omata M. (2007) J. Immunol. 179, 2681–2685 [DOI] [PubMed] [Google Scholar]
- 62. May M. J., D'Acquisto F., Madge L. A., Glöckner J., Pober J. S., Ghosh S. (2000) Science 289, 1550–1554 [DOI] [PubMed] [Google Scholar]
- 63. Greten F. R., Eckmann L., Greten T. F., Park J. M., Li Z. W., Egan L. J., Kagnoff M. F., Karin M. (2004) Cell 118, 285–296 [DOI] [PubMed] [Google Scholar]
- 64. Nenci A., Becker C., Wullaert A., Gareus R., van Loo G., Danese S., Huth M., Nikolaev A., Neufert C., Madison B., Gumucio D., Neurath M. F., Pasparakis M. (2007) Nature 446, 557–561 [DOI] [PubMed] [Google Scholar]
- 65. Steinbrecher K. A., Harmel-Laws E., Sitcheran R., Baldwin A. S. (2008) J. Immunol. 180, 2588–2599 [DOI] [PubMed] [Google Scholar]
- 66. Dajee M., Lazarov M., Zhang J. Y., Cai T., Green C. L., Russell A. J., Marinkovich M. P., Tao S., Lin Q., Kubo Y., Khavari P. A. (2003) Nature 421, 639–643 [DOI] [PubMed] [Google Scholar]
- 67. Maeda S., Chang L., Li Z. W., Luo J. L., Leffert H., Karin M. (2003) Immunity 19, 725–737 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.