

Abstract
Identifying the genetic basis of complex traits remains an important and challenging problem with the potential to affect a broad range of biological endeavors. A number of statistical methods are available for mapping quantitative trait loci (QTL), but their application to high-throughput phenotypes has been limited as most require user input and interaction. Recently, methods have been developed specifically for expression QTL (eQTL) mapping, but they too are limited in that they do not allow for interactions and QTL of moderate effect. We here propose an automated model-selection-based approach that identifies multiple eQTL in experimental populations, allowing for eQTL of moderate effect and interactions. Output can be used to identify groups of transcripts that are likely coregulated, as demonstrated in a study of diabetes in mouse.
MANY important problems in biology and medicine rely on the accurate identification of the genetic architecture underlying high-throughput phenotypes such as messenger RNA expression. Identifying expression quantitative trait loci (eQTL) and grouping related traits are two primary goals addressed in such endeavors. This manuscript proposes an approach for eQTL mapping and shows how the derived transcript-specific genetic signatures can be used to group transcripts that are likely coregulated.
In the earliest eQTL mapping studies, simple single-QTL mapping methods were repeatedly applied to individual expression traits (Kendziorski and Wang 2006; Williams et al. 2007) and that practice continues today. Certainly powerful and effective methods that provide the flexibility to consider complex genetic models exist (Kao et al. 1999; Sen and Churchill 2001), and they have proven useful in numerous studies. However, the approaches require “fine tuning” (Sen and Churchill 2001) or the choice of thresholds (Kao et al. 1999) to resolve multiple linked QTL and identify interactions for a single trait, and as a result applications to expression data are relatively few.
One of the first methods developed specifically for eQTL mapping was proposed by Storey et al. (2005). In that approach, F-statistics are calculated for each marker and trait, and a primary locus is identified for each trait as the one with a maximal F-statistic. A secondary locus is identified as the one having maximal statistic in a second F-test conditional on the first, with permutations used to estimate the posterior probabilities and thresholds for locus-specific and joint linkage. Zou and Zeng (2009) propose a sequential search for multiple QTL that combines features of Storey's approach with MIM. Both approaches are automated and efficient and therefore useful in eQTL studies. However, the thresholding procedures in identification of primary and secondary loci may exclude potentially important traits affected by moderate and/or interacting QTL.
The methods discussed thus far all consider trait-specific tests or models, whereas some approaches model all traits (Kendziorski et al. 2006; Jia and Xu 2007) or groups of traits (Chun and Keleş 2009) at once. With one model for the data, it is possible to account for multiplicities and estimate false discovery rate across transcripts and markers simultaneously. However, the advantage gained is compromised at the level of interacting loci.
In summary, the state-of-the-art QTL mapping methods are sophisticated and quite capable of identifying complicated genetic architecture, but most require that decisions on the class of models to consider, as well as significance thresholds, be made on a case-by-case basis. This clearly limits applications to studies of high-throughput phenotypes such as expression. Many of the challenges have been met to a great extent by the recently proposed methods designed specifically for eQTL mapping. However, these methods are unable to identify eQTL of small or moderate effect, and they do not allow for automated identification of interactions.
We here propose a new multiple-QTL mapping approach that has the ability to identify both QTL with large effect and those with small or moderate effect as well as interacting QTL. It is automated and efficient and therefore particularly well suited for eQTL studies. Our approach makes use of the results from a single-QTL analysis to reduce the marker search space and thereby reduce the model search space dramatically. The approach is detailed in A multiple-QTL identification approach allowing for interactions.
In addition to the multiple eQTL mapping approach, we propose a clustering method that incorporates eQTL mapping results and trait correlations to identify groups of transcripts that likely share similar biological function. An early consideration of this problem is given in Eisen et al. (1998) in which investigators used hierarchical clustering applied to expression data to identify transcripts with similar function. To date, various clustering algorithms have been proposed in part to address this same goal (for a comprehensive review, see Do and Choi 2007). A particularly powerful and popular approach was proposed by Zhang and Horvath (2005). In their work, they describe a module identification approach that uses hierarchical clustering applied to a biologically meaningful distance derived from pairwise correlations between transcripts. When genetic data including genotypes and a genetic map are available in addition to expression data, ideally mapping information can be incorporated to improve the identification of groups of transcripts that are likely coregulated. To this end, in A model-based clustering method, we detail an approach that extends Zhang and Horvath (2005) to include results from eQTL mapping in the identification of coexpression coregulation (CECR) modules.
MATERIALS AND METHODS
A multiple-QTL identification approach allowing for interactions:
Here, we propose a multiple-QTL mapping approach that has the ability to identify both QTL with large effect and QTL with small or moderate effect as well as interacting QTL. Motivation for our approach is based on the fact that multiple interacting loci induce marginal effects that can be detected by single-QTL mapping methods, as shown for two loci in Lan et al. (2001). Given this, the search space for models with first-order interactions can be dramatically reduced. Instead of considering interactions between all markers, we focus on markers with relatively high LOD scores, even if those LOD scores are not statistically significant.
The multiple-QTL mapping approach uses preselected markers in a stepwise regression to identify main effects and interactions. Details follow for a single phenotype:
Obtain a LOD score profile by applying a single-QTL mapping method, such as interval mapping or Haley–Knott regression.
Preselect markers with relatively high LOD scores. Our approach for doing so is provided in the supporting information, File S1.
Perform stepwise regression to obtain a baseline model, one with main effects only. Candidates for main effects in this step are the preselected markers and relevant covariates (e.g., sex, age).
Perform stepwise regression to obtain the best model with interactions allowed. The potential interactions are between the preselected markers or interactive covariates in the baseline model and all preselected markers.
In steps 3 and 4, a model selection criterion is needed. Many criteria take the form –2 log L + k × c(n), where L is the likelihood on n samples given a genetic model with k parameters. For example, c(n) = 2 is the classical Akaike information criterion (AIC) (Akaike 1974); c(n) = log(n) is the Bayesian information criterion (BIC) (Schwarz 1978). The BIC is used in many studies, but as Broman and Speed (2002) point out, its use can result in QTL models with many extraneous variables. Zou and Zeng (2008) discuss more conservative penalties such as c(n) = 2 log(n) and c(n) = 3 log(n), which we here refer to as BIC(2) and BIC(3), respectively. The recently proposed penalized LOD score (pLOD) criterion (Manichaikul et al. 2009) could also be used.
A model-based clustering method:
In eQTL studies, it is desirable to identify groups of coregulated traits that share similar biological function. Here we propose a clustering approach designed to accomplish this task. It incorporates both trait correlation and evidence of comapping. A measurement to quantify evidence in favor of comapping, as measured by the similarity of estimated mapping models, is introduced in the following.
Similarity between QTL models:
For any pair of models M1, M2, defined by the locations of QTL, 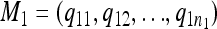 and
and 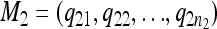 , a similarity measure s should satisfy the following two conditions: (i) s(M1, M2) ∈ [0, 1] and (ii) s(M, M) = 1 for all M.
, a similarity measure s should satisfy the following two conditions: (i) s(M1, M2) ∈ [0, 1] and (ii) s(M, M) = 1 for all M.
Assume, without loss of generality, that n1 ≤ n2. Let φp be a one-to-one mapping from 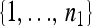 to a subset of
to a subset of 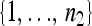 with n1 elements; there are then
with n1 elements; there are then 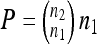 ! possible mappings [that is, φp(i) = φp(j) implies i = j]. We define the model similarity to be
! possible mappings [that is, φp(i) = φp(j) implies i = j]. We define the model similarity to be
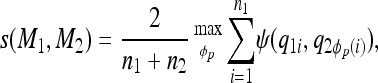 |
where ψ is a measure of similarity between two QTL,
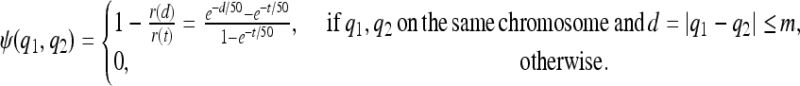 |
m is a parameter set by the user that specifies the genetic distance within which two QTL can be considered similar; t ≥ m is a tuning parameter that quantifies the extent of similarity between two QTL within this distance. As t increases, the similarity between any two QTL within the window increases. Supporting information, Figure S1 is a plot of similarity between QTL vs. distance between QTL in cM when m = 2.5 cM for various tuning parameters. The choice of m is application dependent. When small genomic regions are of interest and dense maps and large sample sizes are available, two QTL that are 1 or 2 cM apart might not be considered similar. In such a case, m would be chosen to be relatively small compared to situations in which larger regions are of interest with fewer markers and samples. Once m is specified, graphs such as that shown in Figure S1 should be used to choose t.
To examine some of the properties of the model similarity defined here, we calculated the similarities among 11 QTL models with 1, 2, and 3 QTL and provide them in Table S1 and Table S2. As shown there, the similarity measure is a function of QTL proximity between models as well as total number of QTL. Consider, for example, the similarity calculated between a model M1 that has a single QTL and a series of nested QTL models M2, M5, and M9, where M2, M5, and M9 each contain a QTL 0.5 cM from the QTL in M1. M5 and M9 also contain one and two additional QTL, respectively. The similarity measure maintains the following ordering: s(M1, M2) > s(M1, M5) > s(M1, M9). This is a desired property since intuitively the similarity between two models should decrease as the number of discrepant loci increases.
Model-based clustering method:
A measurement of the adjacency between two traits that incorporates both correlation and mapping information is defined as
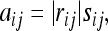 |
where rij is the correlation between traits i and j and
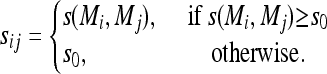 |
Instead of directly using s(Mi, Mj) in the definition of aij, we use sij so that adjacencies are not zero necessarily for pairs of traits whose model similarities equal 0. Such traits may still be related, and in this case we allow for the relationship to be directly assessed by correlation. A choice for s0 is s0 = min {s(Mi, Mj), s(Mi, Mj) ≠ 0}.
As in Zhang and Horvath (2005), we use average linkage hierarchical clustering coupled with the topological overlap matrix (TOM) distance to group traits into modules corresponding to branches of the hierarchical clustering tree (dendrogram). We extend their adjacency measure to the one given in a Model-based clustering method. Since it accommodates both correlation and comapping, we refer to modules constructed using this approach as CECR modules.
Enrichment test:
Given a list of mapping transcripts, it is often of interest to determine whether the transcripts are enriched for any GO (gene ontology) terms in BP (biological process), CC (cellular component), MF (molecular function) categories, or KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways. The hypergeometric test implemented in the R package GOstats was used here for this purpose (R Development Core Team 2009). The hypergeometric calculation tends to result in small P-values when groups with few transcripts are considered and as a result, it has been suggested that one consider only terms with small P-values and a reasonable number of genes (10 or more) (Gentleman 2004). Unless otherwise stated, we report terms with P-value < 0.001, 10 or more genes on the chip, and 5 or more genes in the list annotated with that term.
DATA SETS CONSIDERED FOR EVALUATION
To assess the proposed methodology we consider many individual traits from the QTL Archive, expression traits collected in a study of diabetes, and simulated data. Details regarding each of these data sources follow.
QTL Archive studies:
The QTL Archive (http://churchill.jax.org/datasets/qtlarchive.shtml) created and maintained by the Jackson Laboratory (Bar Harbor, ME) provides access to raw data and result scripts from various QTL studies using rodent inbred line crosses. There were 31 studies in the QTL Archive as of June 29, 2008. The mapping method described in A multiple-QTL identification approach allowing for interactions was applied to data from the QTL Archive. BIC was used for model selection, with results evaluated and compared using BIC, BIC(2), and BIC(3).
Microarray experiment:
The C57BL/6J (B6) and BTBR mice are two inbred mouse populations maintained at the Jackson Laboratory and often used in studies of type 2 diabetes. When made obese by a leptin mutation, B6 mice are diabetes resistant while the BTBR mice are diabetes susceptible (Clee et al. 2005). In this study, expression profiles were obtained from 499 F2 – ob/ob mice generated from the C57BL/6J (B6) and BTBR founder strains. The profiles probed islet tissue using custom ink-jet microarrays manufactured by Agilent Technologies (Palo Alto, CA). The microarrays consisted of 1,048 control probes and 39,524 noncontrol probes. Mouse islets were homogenized and total RNA extracted using Trizol reagent (Invitrogen, CA) according to the manufacturer's protocol. Total RNA was reverse transcribed and labeled with flurochrome. Labeled complementary RNA (cRNA) from each animal was hybridized against a pool of labeled cRNAs constructed from equal aliquots of RNA from all of the animals. All hybridizations were performed in fluor-reversal for 48 hr in a hybridization chamber, washed, and scanned using a confocal laser scanner. Expressions were quantified on the basis of spot intensity relative to background, adjusted for experimental variation between arrays using the average intensity over multiple channels, and fitted to a previously described error model to determine significance (type I error) (He et al. 2003). Gene expression measures are reported as the ratio of the mean log10 intensity (mlratio). Plasma insulin levels were also measured in each of the 499 mice at approximately 10 weeks of age.
To ameliorate the effect of outliers, we performed a normal score transformation on the basis of ranks. In particular, for a trait (insulin level or expression trait) with measurements on n individuals, let Ri be the rank of the measurement for individual i, and then the transformed measurement for individual i is yi = Φ−1(Ri/(n + 1)), where Φ−1 is the inverse of the standard normal cumulative distribution function. All analyses in this diabetes study are based on the normal scores unless explicitly stated otherwise. Mice were genotyped using the Affymetrix mouse 5K SNP panel (http://www.affymetrix.com); 1,953 SNPs on 19 autosomes reliably segregated for the founders and were used for QTL mapping.
RESULTS
QTL Archive studies:
There were 31 studies in the QTL Archive as of June 29, 2008. To be included in our analysis, a study or trait had to satisfy the following conditions: (1) the data set provided in the QTL Archive had to match the description in the article; (2) the trait to be mapped had to be continuous and suitably handled by the normal model (perhaps following transformation); and (3) the markers closest to the identified QTL had to be given explicitly. This results in 24 traits in 11 studies (Clemens et al. 2000; Farmer et al. 2001; Mähler et al. 2002; Lyons et al. 2003a,b and 2004a,b; Dipetrillo et al. 2004; Ishimori et al. 2004a,b; Korstanje et al. 2004).
Figures S2–S12 and Tables S3–S13 compare the models derived using the proposed approach to those published. As shown in the figures, there is much similarity between models for regions with relatively high LODs. In particular, 67% (63%) of the loci identified in the published models with LODs exceeding 5.0 (4.0) are identified by the proposed approach; 80% (75%) are identified approximately (by markers within 5 cM of the published locus). The published models were also compared to those derived from the proposed approach using standard model selection criteria. Most of the QTL Archive studies derived models using the approach given in Sen and Churchill (2001). As prescribed there, a multiple imputation algorithm is used to fill in missing genotypes. When comparing models derived using the proposed approach to those published, differences due to randomness induced by imputing missing data are not of interest and, as a result, we compare models under two scenarios. The first considers a one-time imputation where each model is evaluated on the same set of imputed data; in the second we impute data 10 times, evaluate each set of imputed data, and report the median BIC. BIC* is the BIC obtained with missing genotypes filled in by a one-time imputation and BIC** is the median BIC.
Table S14 lists BIC* and BIC** corresponding to the published model (superscript 1) and the model identified using the proposed approach (superscript 2). The model complexity, indicated by (No. main effects, No. interactions), and missing genotype proportions are also given. As suggested by Kass and Raftery (1995), we consider two models to be different if their corresponding BICs differ by more than 10 units. Both BIC* and BIC** suggest that the models identified by the proposed approach are comparable to published models when the amount of missing genotype data is small, and they may be advantageous in some cases. In particular, BIC* (BIC**) associated with the proposed approach is comparable (within 10 BIC units) to the BICs derived from published models for 7 of the 16 traits considered when the amount of missing data is less than 35%. For the 9 traits showing significant difference in BICs, the BICs derived from the proposed approach are smaller. However, when the proportion of missing genotype data exceeds 50%, the proposed approach performs rather poorly, showing comparable BICs in some cases and much larger BICs in others.
As shown in Figure 1, the same result holds generally when BIC(2) and BIC(3) are used. Figure 1 (left) shows the adjusted BIC difference between the two models for each trait as
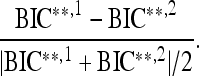 |
Figure 1.—

Adjusted BIC difference for QTL Archive studies. Positive (negative) absolute differences equal to or exceeding 10 units are highlighted in blue (red); absolute differences smaller than 10 units are highlighted in green.
Similar plots are shown for BIC(2) (middle) and BIC(3) (right). The traits are ordered (top to bottom) by the proportion of missing genotypes (least to most), as in Table S14. Detailed numerical results for the BIC(2) and BIC(3) evaluations are given in Table S15 and Table S16, respectively.
The results here demonstrate that the models derived using the proposed automated approach largely overlap those found with other methods for regions with relatively high LODs, and most often they show improvement as assessed by the BIC, BIC(2), and BIC(3) when the amount of missing genotype data is relatively small.
Diabetes study:
This study considers an F2 intercross between B57BL/6 (B6) and BTBR mice to study type 2 diabetes. When made obese, B6 mice are resistant to diabetes, whereas BTBR are severely diabetic.
Identification of eQTL and comparison of methods:
To identify eQTL and further reveal the genetic architecture underlying expression traits in islet tissue, we applied two mapping approaches, a single-QTL mapping approach, and the multiple-QTL mapping approach detailed in A multiple-QTL identification approach allowing for interactions.
The single-QTL mapping approach used here is Haley–Knott regression (Haley and Knott (1992), implemented in R/qtl (Broman et al. 2003). LOD score profiles were obtained at a 2-cM resolution for each trait. For both insulin and the expression traits, sex was included as a main effect and an interactor. A cluster analysis of the 499 mice based on their expression profiles in islet indicated that not only sex but also the date on which the chips were run had effects on the expression measurements. Therefore, for expression traits, date was also included as a main effect. On each chromosome, the locus with maximum LOD score is claimed as a QTL if the LOD score is greater or equal to 5.0, which controls a genome-wide type-I error rate at 0.05.
The proposed approach was applied to each trait by first selecting markers with relatively high LOD scores from the Haley–Knott regression profiles. The variable search space was reduced dramatically since the numbers of potential marker effects retained from 1,953 markers ranged from 46 to 83. Two stepwise regressions were then performed for model selection, as described in A multiple-QTL identification approach allowing for interactions, using pLOD as the model selection criterion (Manichaikul et al. 2009). As in the single-QTL mapping analysis, sex was included as a main effect and a potential interactor for both insulin and the expression traits. For the expression traits, date was also included as a main effect. Table 1 summarizes the complexity of the models for expression traits in islet. In particular, 20,798 (52.62%) out of the 39,524 transcripts mapped to at least one QTL. Among the 20,798 mapping transcripts, 2 or more QTL were identified for 40.44% of the transcripts.
TABLE 1.
The number of transcripts having 1,…, 7 and more than 7 main effects
| No. of QTL | 1 | 2 | 3 | 4 | 5 | 6 | 7 | >7 | Total |
|---|---|---|---|---|---|---|---|---|---|
| No. of transcripts | 12387 | 4877 | 1960 | 849 | 407 | 184 | 84 | 50 | 20798 |
| Percentage 1 | 31.34 | 12.34 | 4.96 | 2.15 | 1.03 | 0.47 | 0.21 | 0.13 | 52.62 |
| Percentage 2 | 59.56 | 23.45 | 9.42 | 4.08 | 1.96 | 0.88 | 0.4 | 0.24 | 100 |
The percentages are given as percentages of the 39,524 transcripts (percentage 1) and the 20,798 mapping transcripts (percentage 2).
Although it is well known that a multiple-QTL mapping analysis is often advantageous over single-QTL mapping, a comparison is helpful to determine the particular advantages of the proposed approach. As expected, more eQTL are identified overall using the proposed approach (Figure 2, top). What is perhaps less expected is that the increase is almost entirely due to the identification of additional trans-acting eQTL (eQTL located outside a 5-cM window centered at the physical location of the expression transcript; see the Figure 2, bottom). In particular, the proposed approach identifies >92% of the cis-acting QTL (eQTL located within a 5-cM window centered on the physical location of the expression transcript) identified by the single-QTL mapping approach along with a few others. It also identifies >80% of the trans-acting QTL identified by a single-QTL analysis and identifies 50% more trans-acting QTL for most chromosomes. Table S17 provides the total counts in detail.
Figure 2.—

Top: The total number of QTL (cis-QTL + trans-QTL) identified by the single- and multiple-QTL mapping approaches. Bottom: Comparison between the number of cis-QTL identified by the multiple-QTL mapping approach and that identified using single-QTL mapping (open bars), and similarly for trans-QTL (shaded bars).
A closer look considers the number of QTL within 5-cM windows. Table 2 lists the number of transcripts mapped by each method for several of the hottest windows. Notably, on chromosome 17, the hottest window (one with the most mapping transcripts) from the proposed approach is centered at 17 cM while the hottest window from the single-QTL analysis is centered at 8.4 cM. Interestingly, the transcripts mapped to 17 cM through the single-QTL analysis did not enrich for any GO BP terms, while those mapped by the proposed approach enriched for GO BP terms mitosis, M phase of mitotic cell cycle, M phase, and cell-cycle phase with P-value < 0.001. Our group has recently detailed evidence for the role of islet cell-cycle transcripts in diabetes (Keller et al. 2008).
TABLE 2.
The number of transcripts mapping in 5-cM windows identified by the single and multiple-QTL analysis (n.mapping.s and n.mapping.m, respectively)
| Chr | Pos (cM) | n.mapping.s | n.mapping.m |
|---|---|---|---|
| 6 | 108.1 | 1978 | 2373 |
| 2 | 96.0 | 1578 | 1678 |
| 2 | 73.0 | 959 | 1592 |
| 2 | 89.2 | 796 | 984 |
| 7 | 4.0 | 495 | 890 |
| 12 | 8.0 | 402 | 843 |
| 17 | 8.4 | 621 | 778 |
Position of the window center is shown in centimorgans.
Table 3 shows the results from an enrichment test applied to transcripts mapping to the window on chromosome 6; m.count and s.count are the numbers of genes annotated with the term among the list from the proposed approach and from a single-QTL analysis, respectively. For the 29 terms listed, m.count ≥ s.count, and for 20 terms, m.P-value < s.P-value, suggesting generally stronger enrichment results for transcripts identified using the proposed approach.
TABLE 3.
Results from an enrichment test applied to transcripts mapping to the 5-cM window centered at 108.1 cM on chromosome 6
| Set | Term | Size | s.count | s.P-value | m.count | m.P-value |
|---|---|---|---|---|---|---|
| GOBP | Multicellular organismal process | 3774 | 236 | 9.56e-07 | 292 | 5.07e-08 |
| GOBP | Multicellular organismal development | 2087 | 137 | 5.04e-05 | 166 | 2.86e-05 |
| GOBP | Cell adhesion | 585 | 44 | 0.0021 | 59 | 4.42e-05 |
| GOBP | Biological adhesion | 585 | 44 | 0.0021 | 59 | 4.42e-05 |
| GOBP | Phosphate transport | 76 | 8 | 0.029009 | 14 | 0.000135 |
| GOBP | Organ development | 1319 | 89 | 0.000554 | 108 | 0.000317 |
| GOBP | Tube development | 190 | 22 | 0.000118 | 24 | 0.000384 |
| GOBP | System development | 1612 | 103 | 0.001324 | 126 | 0.000651 |
| GOBP | Proteolysis | 605 | 47 | 0.000763 | 55 | 0.001063 |
| GOBP | Tube morphogenesis | 134 | 16 | 0.000692 | 17 | 0.002481 |
| GOBP | Embryonic development | 437 | 37 | 0.000583 | 40 | 0.004356 |
| GOCC | Proteinaceous extracellular matrix | 278 | 30 | 4.54e-05 | 42 | 3.87e-08 |
| GOCC | Extracellular matrix | 282 | 30 | 5.92e-05 | 42 | 5.86e-08 |
| GOCC | Extracellular region | 2483 | 164 | 2.44e-05 | 210 | 1.03e-07 |
| GOCC | Collagen | 37 | 6 | 0.008744 | 13 | 1.27e-07 |
| GOCC | Extracellular matrix part | 93 | 10 | 0.015876 | 20 | 5.82e-07 |
| GOCC | Extracellular region part | 2037 | 133 | 0.000289 | 172 | 2.34e-06 |
| GOCC | Extracellular space | 1919 | 118 | 0.005395 | 156 | 5.94e-05 |
| GOCC | Intrinsic to plasma membrane | 648 | 51 | 0.000605 | 60 | 0.000695 |
| GOMF | Extracellular matrix structural constituent conferring tensile strength | 29 | 6 | 0.00263 | 13 | 4.09e-09 |
| GOMF | Extracellular matrix structural constituent | 59 | 8 | 0.008438 | 16 | 2.99e-07 |
| GOMF | Peptidase activity | 625 | 50 | 0.000599 | 59 | 0.000545 |
| GOMF | Metalloendopeptidase activity | 105 | 16 | 5.87e-05 | 16 | 0.000608 |
| GOMF | Cytokine activity | 214 | 18 | 0.020571 | 26 | 0.000619 |
| GOMF | Transmembrane receptor activity | 1891 | 124 | 0.000615 | 147 | 0.000961 |
| GOMF | Endopeptidase activity | 408 | 37 | 0.000307 | 41 | 0.001155 |
| GOMF | Carbohydrate binding | 259 | 26 | 0.000526 | 28 | 0.002367 |
| KEGG | Cell communication | 125 | 16 | 0.000246 | 23 | 1.72e-06 |
| KEGG | Ecm-receptor interaction | 84 | 11 | 0.001933 | 16 | 4.47e-05 |
Terms with size ≥ 10 and P-value ≤ 0.001 on either list from the single (s) and multiple (m)-QTL analysis are listed.
As discussed earlier, one advantage of our proposed approach is the ability to identify interactions, particularly ones that involve moderate main effects. Among the 20,798 mapping transcripts, sex-by-marker or marker-by-marker interactions were identified for 7,985 (38.39%) transcripts. Among 8,411 transcripts mapping to 2 or more markers, 797 marker-by-marker interactions were identified across 763 transcripts. Figure 3 illustrates the types of interactions identified. The first part of Figure 3 highlights an interaction for which the main effect associated with each interacting term would have been found using the single-QTL approach; the second shows a case for which only one of the QTL would have been found; and the third shows a case in which neither locus is found significant in a single-QTL scan.
Figure 3.—



Interaction plots and single-QTL LOD profiles for three expression traits. The LOD score for Cdk5rap1 (A) at rs4223605 (M1.chr2 at 156.7 Mb) is 42.37 and at rs13481837 (M2.chr13 at 60.1 Mb) is 14.6; the LOD score for Alkbh6 (B) at rs4226520 (M1.chr7 at 29.5 Mb) is 12.66 and at rs13479518 (M2.chr7 at 127.9 Mb) is 0.64; and the LOD score for Syn3 (C) at rs13476918 (M1.chr2 at 171.3 Mb) is 4.43 and at rs6365385 (M2.chr11 at 46.2 Mb) is 3.81.
Insulin-based coexpression coregulation (CECR) module:
When eQTL colocalize with QTL of a clinical trait, one can hypothesize that a close relationship exists, such as sharing a regulator (Ferrara et al. 2008). The construction of CECR modules has the potential to identify groups of traits that are likely coregulated, since both the correlation in expression along with mapping information is used. To illustrate, we consider the relationship between insulin and selected expression traits. First, the locations to which insulin maps were identified, where evidence of mapping was quantified, using the proposed approach. The model identified for insulin includes 7 QTL and two interactions, one between sex and marker rs3700924 (chromosome 17 at 8.4 cM) and the other between sex and marker rs13476801 (chromosome 2 at 91.7 cM). The 2,854 transcripts comapping with insulin were then identified as those with at least one locus in common. The pairwise similarities among the 2,855 traits (insulin and the comapping transcripts) were calculated using m = 2.5 cM and t = 5 cM, and CECR modules were constructed. Figure 4 shows the resulting modules and the mapping patterns for the traits on the seven chromosomes harboring insulin's QTL. Columns are a series of 5-cM nonoverlapping bins along the seven chromosomes and each row represents a trait. The much thicker top row highlights the model for insulin, with rows following the top row organized into CECR modules indicated by the colors on the far left. The (i, j)th entry is colored (not white) if the ith transcript maps to the jth genomic location as assessed by the proposed approach. The color used represents the single-QTL LOD score with LOD scores >5 shown in black. The top row indicates that insulin maps to seven locations using the proposed approach, with two identified by single-QTL mapping.
Figure 4.—

The mapping patterns for insulin and the 2854 comapping transcripts. Columns are a series of 5-cM nonoverlapping bins across seven chromosomes harboring locations to which insulin maps. Shown are 2855 rows. The first represents insulin and is extra thick so that the locations to which insulin maps can be easily seen. There are 2854 rows following, one for each transcript, with row ordering determined by the CECR module construction. The color bar at the left represents the CECR modules. Insulin is in the turquoise module. Bins containing QTL are colored (not white) with the color representing the magnitude of the LOD score obtained from a single-QTL analysis (LODs > 5 are colored black). In the rare event that a bin contains more than one QTL, the color corresponds to the maximum LOD score.
Enrichment tests were performed to see whether the transcripts in the CECR modules are enriched for any biologically meaningful GO terms or KEGG pathways. The results are listed in Table S18. Insulin is in the turquoise module (a module with 540 transcripts), which enriches for innate immune response, a response known to be connected with insulin and diabetes (Fernández-Real and Pickup 2008). In contrast, the 540 transcripts most correlated with insulin are enriched only for wound healing, adult behavior, regulation of body fluid levels, and response to virus, none of which is particularly striking. From Figure 4, we see that most transcripts in the turquoise module have QTL near insulin's QTL, rs13483664, at 36.8 cM (51 Mb) on chromosome 19. SorCS1 is one, located on chromosome 19 between 50 and 51 Mb. In particular, the QTL model for SorCS1 involves rs13483664 and an interaction between sex and rs13483664. Clee et al. (2006) have shown evidence suggesting that this gene has broad relevance to the development of type 2 diabetes.
DISCUSSION
Many important problems in biology and medicine rely on the accurate identification of QTL contributing to variation in quantitative traits. A number of powerful statistical methods for mapping QTL have proven useful in traditional mapping studies in which one or a few quantitative traits are surveyed. Typically, when thousands of phenotypes are available, as in an eQTL mapping study, single-QTL mapping methods are repeatedly applied to individual expression traits, effectively sacrificing the identification of refined genetic architecture for efficiency. We here propose an efficient and automated eQTL mapping approach that in part addresses this limitation, accommodating QTL of small or moderate effect as well as interactions. The output is used to identify CECR modules, groups of transcripts that are likely coregulated. In practice the approach could and likely should be applied following adjustment for latent variables or population structure such as in Leek and Storey (2007) and Kang et al. (2008). The effects of doing so were not studied here.
The eQTL mapping approach consists of two-stage model selection over a reduced marker search space supported by the fact that interacting QTL induce effects that are detectable marginally. This motivates a first step of identifying markers with relatively large LOD scores following a single QTL scan. Model selection is performed over the selected markers to determine a baseline model of main effects. A second model selection considers possible interactions. As noted, any one of several model selection criteria and procedures for identifying large LODs could be used. The approach results in the identification of a single model per transcript, which specifies the QTL affecting that transcript along with their actions and interactions. The model selection methods considered here have a number of advantages, but they do not target error-rate control and as a result no statements can be made regarding false discovery rates, for example, either within or across transcripts.
In the analysis of individual traits from the QTL Archive, the BIC was used for model selection since many of the published models utilize the BIC to some extent; models were evaluated using the BIC as well as BIC(2) and BIC(3). The results demonstrate that the proposed approach identifies models that largely overlap those found with other methods for loci with large LOD scores, and in most cases they show improvement when there is not a large amount of missing genotype data (<35%). Here, improvement is assessed by decreased BIC, BIC(2), and BIC(3), and of course in practice it is impossible to know which models better approximate reality. When consideration of one or a few phenotypes is of interest, as in the QTL Archive studies, clearly multiple approaches and lines of evidence should be considered during model development and selection. However, when high-throughput phenotypes prohibit such a careful and comprehensive evaluation, the automated approach proposed here can be useful.
As demonstrated in the diabetes case study, the output from the proposed approach can be used to identify groups of transcripts (or transcripts and clinical traits) that are likely coregulated. The so-called CECR module construction extends the definition of module initially proposed by Zhang and Horvath (2005) to accommodate trait-specific mapping information, in particular through the specification of a function that defines model similarity. A specific form of model similarity was considered here, but could be modified through the choice of different tuning parameters and/or a different functional form. An investigation of different similarity measures should prove useful in guiding future applications of CECR module construction as well as related efforts that require a measure of distance between two models. In this work, CECR module construction was used to identify groups of transcripts correlated and comapping with insulin. The groups generally show stronger enrichment for biological functions, suggesting improvement over using correlation measures alone.
Acknowledgments
This work was supported in part by the National Institute of General Medical Sciences (NIGMS) 76274, the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) 66369, NIDDK 58037, and NIGMS training grant 74904.
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.110.122796/DC1.
References
- Akaike, H., 1974. A new look at statistical model identification. IEEE Trans. Automatic Control AC-19 716–723. [Google Scholar]
- Broman, K. W., and T. P. Speed, 2002. A model selection approach for the identification of quantitative trait loci in experimental crosses. J. R. Statist. Soc. B 64 641–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman, K. W., H. Wu, S. Sen and G. A. Churchill, 2003. R/qtl: QLT mapping in experimental crosses. Bioinformatics 19 889–890. [DOI] [PubMed] [Google Scholar]
- Chun, H., and S. Keleş, 2009. Expression quantitative trait loci mapping with multivariate sparse partial least squares regression. Genetics 182 79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clee, S. M., S. T. Nadler and A. D. Attie, 2005. Genetic and genomic studies in the BTBR ob/ob model of type 2 diabetes. Am. J. Ther. 12 491–498. [DOI] [PubMed] [Google Scholar]
- Clee, S. M., B. S. Yandell, K. M. Schueler, M. E. Rabaglia, O. C. Richards et al., 2006. Positional cloning of a type 2 diabetes quanitative trait locus. Nat. Genet. 38 688–693. [DOI] [PubMed] [Google Scholar]
- Clemens, K. E., G. Churchill, N. Bhatt, K. Richardson and F. P. Noonan, 2000. Genetic control of susceptibility to UV-induced immunosuppression by interacting quantitative trait loci. Genes Immun. 1(4): 251–259. [DOI] [PubMed] [Google Scholar]
- DiPetrillo, K., S.-W. Tsaih, S. Sheehan, C. Johns, P. Kelmenson et al., 2004. Genetic analysis of blood pressure in C3H/HeJ and SWR/J mice. Physiol. Genomics 17(2): 215–220. [DOI] [PubMed] [Google Scholar]
- Do, J. H., and D. K. Choi, 2007. Clustering approaches to identifying gene expression patterns from DNA microarray data. Mol. Cells 25(2): 279–288. [PubMed] [Google Scholar]
- Eisen, M. B., P. T. Spellman, P. O. Brown and D. Botstein, 1998. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 95 14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer, M. A., J. P. Sundberg, I. J. Bristol, G. A. Churchill, R. Li et al., 2001. A major quantitative trait locus on chromosome 3 controls colitis severity in IL-10-deficient mice. Proc. Natl. Acad. Sci. USA 98(24): 13820–13825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Real, J. M., and J. C. Pickup, 2008. Innate immunity, insulin resistance and type 2 diabetes. Trends Endocrinol. Metab. 19(1): 10–16. [DOI] [PubMed] [Google Scholar]
- Ferrara, C. T., P. Wang, E. C. Neto, R. D. Stevens, J. R. Bain et al., 2008. Genetic networks of liver metabolism revealed by integration of metabolic and transcriptional profiling. PLoS Genet. 4(3): e1000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman, R., 2004. Using GO for statistical analyses. In Compstat 2004 Proceedings in Computational Statistics. Physica Verlag, Heidelberg, Germany.
- Haley, C. S., and S. A. Knott, 1992. A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity 69 315–324. [DOI] [PubMed] [Google Scholar]
- He, Y. D., H. Dai, E. E. Schadt, G. Cavet, S. W. Edwards et al., 2003. Microarray standard data set and figures of merit for comparing data processing methods and experiment designs. Bioinformatics 19 956–965. [DOI] [PubMed] [Google Scholar]
- Ishimori, N., R. Li, P. M. Kelmenson, R. Korstanje, K. A. Walsh et al., 2004. a Quantitative trait loci analysis for plasma HDL-cholesterol concentrations and atherosclerosis susceptibility between inbred mouse strains C57BL/6J and 129S1/SvImJ. Arterioscler. Thromb. Vasc. Biol. 24 161–166. [DOI] [PubMed] [Google Scholar]
- Ishimori, N., R. Li, P. M. Kelmenson, R. Korstanje, K. A. Walsh et al., 2004. b Quantitative trait loci that determine plasma lipids and obesity in C57BL/6J and 129S1/SvImJ inbred mice. J. Lipid Res. 45 1624–1632. [DOI] [PubMed] [Google Scholar]
- Jia, Z., and S. Xu, 2007. Mapping quantitative trait loci for expression abundance. Genetics 176 611–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, H. M., C. Ye and E. Eskin, 2008. Accurate discovery of expression quantitative trait loci under confounding from spurious and genuine regulatory hotspots. Genetics 180 1909–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao, C. H., Z.-B. Zeng and R. D. Teasdale, 1999. Multiple interval mapping for quantitative trait loci. Genetics 152 1203–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass, R. E., and A. E. Raftery, 1995. Bayes factors. J. Am. Statist. Assoc. 90 773–795. [Google Scholar]
- Keller, M. P., Y. J. Choi, P. Wang, D. B. Davis, M. E. Rabaglia et al., 2008. A gene expression network model of type 2 diabetes links cell cycle regulation in islets with diabetes susceptibility. Genome Res. 18 706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendziorski, C., and P. Wang, 2006. On statistical methods for expression quantitative trait loci mapping. Mamm. Genome 17(6): 509–517. [DOI] [PubMed] [Google Scholar]
- Kendziorski, C., M. Chen, M. Yuan, H. Lan and A. D. Attie, 2006. Statistical methods for expression quantitative trait loci (eQTL) mapping. Biometrics 62 19–27. [DOI] [PubMed] [Google Scholar]
- Korstanje, R., R. Li, T. Howard, P. Kelmenson, J. Marshall et al., 2004. Influence of sex and diet on quantitative trait loci for HDL cholesterol levels in an SM/J by NZB/BlNJ intercross population. J. Lipid Res. 45 881–888. [DOI] [PubMed] [Google Scholar]
- Lan, H., C. M. Kendziorski, L. A. Shepel, J. D. Haag, M. A. Newton et al., 2001. Genetic loci controlling breast cancer susceptibility in the Wistar–Kyoto rat. Genetics 157 331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek, J. T., and J. D. Storey, 2007. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 3(9): 1724–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons, M. A., H. Wittenburg, R. Li, K. A. Walsh, G. A. Churchill et al., 2003. a Quantitative trait loci that determine lipoprotein cholesterol levels in DBA/2J and CAST/Ei inbred mice. J. Lipid Res. 44 953–967. [DOI] [PubMed] [Google Scholar]
- Lyons, M. A., H. Wittenburg, R. Li, K. A. Walsh, M. R. Leonard et al., 2003. b New quantitative trait loci that contribute to cholesterol gallstone formation detected in an intercross of CAST/Ei and 129S1/SvImJ inbred mice. Physiol. Genomics 14 225–239. [DOI] [PubMed] [Google Scholar]
- Lyons, M. A., R. Korstanje, R. Li, K. A. Walsh, G. A. Churchill et al., 2004. a Genetic contributors to lipoprotein cholesterol levels in an intercross of 129S1/SvImJ and RIIIS/J inbred mice. Physiol. Genomics 17 114–121. [DOI] [PubMed] [Google Scholar]
- Lyons, M. A., H. Wittenburg, R. Li, K. A. Walsh, R. Korstanje et al., 2004. b Quantitative trait loci that determine lipoprotein cholesterol levels in an intercross of 129S1/SvImJ and CAST/Ei inbred mice. Physiol. Genomics 17 60–68. [DOI] [PubMed] [Google Scholar]
- Mähler, M., C. Most, S. Schmidtke, J. P. Sundberg, R. Li et al., 2002. Genetics of colitis susceptibility in IL-10-deficient mice: backcross versus F2 results contrasted by principal component analysis. Genomics 80(3): 274–282. [DOI] [PubMed] [Google Scholar]
- Manichaikul, A., J. Y. Moon, S. Sen, B. S. Yandell and K. W. Broman, 2009. A model selection approach for the identification of quantitative trait loci in experimental crosses, allowing epistasis. Genetics 181 1077–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team, 2009. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna.
- Schwarz, G., 1978. Estimating the dimension of a model. Ann. Statist. 6 461–464. [Google Scholar]
- Sen, S., and G. A. Churchill, 2001. A statistical framework for quantitative trait. Genetics 159 371–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey, J. D., J. M. Akey and L. Kruglyak, 2005. Multiple locus linkage analysis of genomewide expression in yeast. PLoS Biol. 3(8): e267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, R. B., E. K. Chan, M. J. Cowley and P. F. Little, 2007. The influence of genetic variation on gene expression. Genome Res. 17 1707–1716. [DOI] [PubMed] [Google Scholar]
- Zhang, B., and S. Horvath, 2005. A general framework for weighted gene co-expression network analysis. Statist. Appl. Genet. Mol. Biol. 4 17. [DOI] [PubMed] [Google Scholar]
- Zou, W., and Z.-B. Zeng, 2008. Statistical methods for mapping multiple QTL. Int. J. Plant Genomics, 286561. [DOI] [PMC free article] [PubMed]
- Zou, W., and Z.-B. Zeng, 2009. Multiple interval mapping for gene expression QTL analysis. Genetica 137 125–134. [DOI] [PubMed] [Google Scholar]