

Abstract
BACKGROUND AND PURPOSE
Surprisingly high contractile activity was reported for 11-deoxy-16,16-dimethyl prostaglandin E2 (DX-DM PGE2) on pig cerebral artery when used as a selective EP3 receptor agonist. This study investigated the selectivity profile of DX-DM PGE2, focusing on the interaction between its EP3 and TP (thromboxane A2-like) agonist activities.
EXPERIMENTAL APPROACH
Contraction of guinea-pig trachea (EP1 system) and aorta (EP3 and TP systems) was measured in conventional organ baths.
KEY RESULTS
Strong contraction of guinea-pig aorta to sulprostone and 17-phenyl PGE2 (EP3 agonists) was only seen under priming with a second contractile agent such as phenylephrine, histamine or U-46619 (TP agonist). In contrast, DX-DM PGE2 induced strong contraction, which on the basis of treatment with (DG)-3ap (EP3 antagonist) and/or BMS-180291 (TP antagonist) was attributed to self-synergism arising from co-activation of EP3 and TP receptors. EP3/TP self-synergism also accounted for contraction induced by PGF2α and its analogues (+)-cloprostenol and latanoprost-FA. DX-DM PGE2 also showed significant EP1 agonism on guinea-pig trachea as defined by the EP1 antagonists SC-51322, (ONO)-5-methyl-1 and AH-6809, although AH-6809 exhibited poor specificity at concentrations ≥3 µM.
CONCLUSIONS AND IMPLICATIONS
EP3/TP self-synergism, as seen with PGE/PGF analogues in this study, may confound EP3 agonist potency comparisons and the characterization of prostanoid receptor systems. The competitive profile of a TP antagonist may be distorted by variation in the silent/overt contraction profile of the EP3 system in different studies. The relevance of self-synergism to in vivo actions of natural prostanoid receptor agonists is discussed.
Keywords: isolated smooth muscle preparation; contractile self-synergism; prostanoid receptors; 11-deoxy-16,16-dimethyl PGE2; cloprostenol; latanoprost; I-BOP; EP3 receptor antagonist; TP receptor antagonist; BMS-180291; AH-6809
Introduction
Prostaglandin E2 (PGE2) has a broad range of actions including excitation of smooth muscle cells mediated by prostanoid EP1 and EP3 receptors and inhibition mediated by EP2 and EP4 subtypes (see Coleman et al., 1994; receptor nomenclature follows Alexander et al., 2009). Its contractile action on pig isolated cerebral artery has been linked to activation of both EP1 and EP3 receptors operating through the phosphatidyl inositol pathway (Jadhav et al., 2004). We had two major concerns about this study. First, very high concentrations (30–300 µM) of the EP1 antagonist AH-6809 (Coleman et al., 1985; 1987;) were used to identify the EP1 component and also to gauge the selectivity of 11-deoxy-16,16-dimethyl PGE2 (DX-DM PGE2), which was used as a selective EP3 agonist. Second, DX-DM PGE2 was about 50 times more potent and had a higher maximum response than sulprostone, a potent full agonist for the EP3 receptor (Lawrence et al., 1992; Sharif et al., 1998). Given that pig cerebral artery contains a contractile TP receptor system (Wallis and Martin, 2000) and other 11-deoxy PGE and PGF analogues show considerable TP agonism (Jones et al., 1982; Banerjee et al., 1985), the remarkably high activity of DX-DM PGE2 may reflect simultaneous activation of EP3 and TP receptors.
It was decided therefore to examine the profile of DX-DM PGE2 on guinea-pig isolated trachea, which contains a contractile EP1 system (Jones et al., 1982; Coleman and Kennedy, 1985) and guinea-pig aorta, which contains EP3 and TP contractile systems (Jones et al., 1998; 2002;). AH-6809 and two chemically distinct EP1 receptor antagonists, SC-51322 (Hallinan et al., 1994) and (ONO)-5-methyl-1 (Jones et al., 2010) were used to block EP1 receptors. EP3 receptors were blocked by the acyl-sulphonamide (DG)-3ap, which achieves steady-state antagonism faster than its relatives L-798106 and L-826266 (Gallant et al., 2002; Belley et al., 2005; Schlemper et al., 2005) possibly due to its lower lipophilicity (Jones et al., 2010). TP receptors were blocked with BMS-180291, chosen for its high affinity (pA2= 9.8, Zhang et al., 1996).
Unexpectedly, sulprostone induced minimal contraction of the guinea-pig aorta preparation; there was however powerful synergism with strong contractile agents, including the TP receptor agonist U-46619. The potential for DX-DM PGE2 to self-synergize through co-activation of EP3 and TP receptors therefore became a major focus of the investigation. Synergism profiles were also determined for PGF2α and two of its analogues (+)-cloprostenol and latanoprost-FA, the last being a selective FP receptor agonist (see Jones et al., 2009).
Our experiments have revealed that DX-DM PGE2 potently activates EP3 and TP receptors in guinea-pig aorta, resulting in contractile self-synergism. EP3/TP self-synergism also accounts for the contractile activities of the FP receptor agonists examined. Moreover, variation in the silent/overt contractility of the EP3 receptor system may distort the competitive profile of a TP antagonist owing to the EP3 activity of the TP receptor agonist at high concentration. DX-DM PGE2 is also a moderately potent EP1 receptor agonist. In terms of EP1 receptor antagonism, AH-6809 is much less specific than SC-51322 and (ONO)-5-methyl-1, suppressing the contractile activity of prostanoid and non-prostanoid agonists on trachea and aorta at concentrations of 3 µM and above. These findings have important implications for the characterization of prostanoid receptor systems. The role of self-synergism in the in vivo actions of natural prostanoid receptor agonists is discussed.
Methods
Isolated smooth muscle preparations
All animal care and experimental procedures were in compliance with the UK Animals (Scientific Procedures) Act 1986. Cervical trachea and descending thoracic aorta were dissected from male Dunkin-Hartley guinea-pigs (400–500 g, Harlan, UK) after killing by exposure to CO2. Four contiguous rings, 4 mm in length, were suspended between L-shaped stainless steel wire holders, one of which was attached to a Grass FT03 transducer, in conventional 10 mL tissue baths. The isometric tension signal was relayed to a AD Instruments PowerLab preamplifier-digitizer/Dell computer system. The bathing fluid was Krebs-Henseleit solution (118 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM MgSO4, 1.18 mM KH2PO4, 25 mM NaHCO3, 10 mM glucose) aerated with 95% O2/5% CO2, maintained at 37°C, and containing 1 µM indomethacin to inhibit cyclo-oxygenase activity. Resting tension was adjusted to 1.2 g for both preparations. Endothelium was removed from some aorta preparations by gentle rotation of the vessel ring on a wooden cocktail stick (Jones et al., 1998); this procedure abolished relaxation induced by 3 µM acetylcholine against established contraction to 2 µM phenylephrine, without affecting similar relaxation induced by 3 nM cicaprost (IP receptor agonist).
Experimental protocols
Cumulative addition of compounds was routinely performed. Initial sequences were: trachea: 40 mM K+; 1, 3, 10 nM 17-phenyl PGE2, aorta: 40 mM K+; 1, 5 µM phenylephrine.
Agonist protocols on trachea: 3–4 prostanoid agonist sequences were obtained at 2 h intervals in the presence of 1 µM BMS-180291 (45 min pretreatment). A full concentration–response curve to carbachol (10–1000 nM) was obtained at the end of the experiment.
Antagonist inhibition-curve protocols on trachea and aorta
After establishment of a E70 response to a particular agonist (with priming as necessary), a series of antagonist doses was added. K+ was applied without any correction for increase in ionic strength.
Antagonism pretreatment protocols on aorta
The following sequences were applied: 10 nM 17-phenyl PGE2 under 10 nM U-46619 priming (90 min after set-up); 1, 3, 10 µM phenylephrine (190 min); antagonist (TP, EP3, TP + EP3) or vehicle (225 min) followed by a prostanoid agonist sequence under phenylephrine priming (270 min). Preparations were then washed for 90 min in the presence of the EP3/TP receptor antagonist combination before a full curve to phenylephrine (0.3–30 µM) was obtained. Precise time-matching was essential in each experiment owing to the gradual increase in the maximum responses of strong agonists (typically 50–70% for phenylephrine, histamine and U-46619) throughout the experimental period (6 h); small increases in corresponding pEC50 values also occurred (0.1–0.2 log units).
Data analysis
Contractile responses were measured from the resting level and normalized to the carbachol and phenylephrine maximum responses on trachea and aorta respectively. Log concentration–response curves were fitted by a four-parameter (variable slope) sigmoidal curve with constraint of the lower asymptote to resting tone/priming level/E70 as appropriate (GraphPad Prism software, La Jolla, CA, USA). Theoretical sigmoidal curves were constructed using the same software. pA2 values were calculated by substitution of dose ratios into the Gaddum-Schild equation: pA2= log (dose ratio − 1) – log[antagonist]. Dunnett's multiple comparison test (GraphPad Prism) was applied to data from the antagonist pretreatment experiments (control = vehicle treatment); the significance level was set at P = 0.05. The error associated with a mean value is the s.e.mean.
Materials
Stock solutions (10 mM) of prostanoid ligands and other compounds were prepared in absolute ethanol and water, respectively, unless stated otherwise. Dilutions were prepared with 0.9% NaCl solution (saline); the first dilutions of AH-6809, (DG)-3ap and U-46619 were solubilized with a trace of NaHCO3. Sources of prostanoid ligands: Allergan, USA, cicaprost, (ONO)-5-methyl-1 ({[2-[isobutyl(phenylsulphonyl)amino]-5-(methyl)phenoxy]methyl}benzoic acid; 5-methyl derivative of compound 1 described by Naganawa et al. (2006), 10 mM in DMSO): Biomol International, UK, SC-51322 (8-chlorodibenz[b,f][1,4]oxazepine-10(11H)-carboxylic acid, 2-[3[2-(furanylmethyl)thio]1-oxopropyl]hydrazine, 10 mM in DMSO): Bristol-Myers Squibb, USA; BMS-180291 ([1S-(exo,exo)]-2-[[3-[4-[(pentylamino)carbonyl]-2-oxazolyl]-7-oxabicyclo[2.2.1]hept-2-yl]methyl]-benzenepropanoic acid, Ifetroban): Cayman Chemical, USA; AH-6809 (6-isopropoxy-9-oxoxanthene-2-carboxylic acid), (+)-cloprostenol, I-BOP ([1S-(1a,2b(5Z),3a(1E,3S),4a]-7-[3-(3-hydroxy-4-(4′-iodophenoxy)-1-butenyl)-7-oxabicyclo-[2.2.1]heptan-2-yl]-5-heptenoic acid, 0.2 mM in ethanol), latanoprost-FA (PhXA 85), PGF2α, 17-phenyl-ω-trinor PGE2, U-46619 (15S-hydroxy-11α,9α-epoxymethano-prosta-5Z,13E-dienoic acid): Target Molecules, UK; (DG)-3ap (1-(3-methoxybenzyl)-3a-methyl-[3,3a,4,5,6-hexahydroindol-2-one-7-acrylic acid, 3,4-difluorobenzenesulphonamide, compound 3ap described by O'Connell et al. (2009), 10 mM in DMSO). Sources of other compounds: Fluka Chemical, Switzerland: phenylephrine hydrochloride, carbachol chloride; Sigma-Aldrich, USA: acetylcholine chloride, indomethacin (20 mM in ethanol).
Results
Selective block of EP1 and EP3 receptors
Concentrations of antagonists used to selectively block EP1 and EP3 receptors in subsequent experiments were determined by cumulative inhibition of an established E70 response (inhibition-curve protocol). On guinea-pig trachea, EP1, TP and muscarinic M3 receptor-mediated contractions (typically 2.5–3.5 g) were induced with 17-phenyl PGE2 (3–5 nM), U-46619 (20–30 nM) and carbachol (50–60 nM) respectively. On the guinea-pig aorta, the EP3 response was elicited by 17-phenyl PGE2 (25 nM) under priming with the α1-adrenoceptor agonist phenylephrine (0.3–1.0 µM, cf. Figure 1C). 17-Phenyl PGE2 was preferred to the more potent EP3 agonist sulprostone (3 nM) owing to its faster onset and offset of action (see Jones et al., 2010 for further details). Although 17-phenyl PGE2 responses were enhanced under threshold priming (<5% of phenylephrine maximum), priming to 20–25% afforded less variation in the EP3 maximum response both within and between experiments, and also allowed the effects of prostanoid antagonists on the priming response to be accurately assessed (see later). Higher phenylephrine priming (40–50%) often resulted in fading of EP3 responses greater than E60. The TP receptor antagonist BMS-180291 (1 µM), which had no effect on established 17-phenyl PGE2 responses on trachea (not shown) and aorta (Figure 1C), was present throughout the EP1 and EP3 inhibition-curve protocols.
Figure 1.
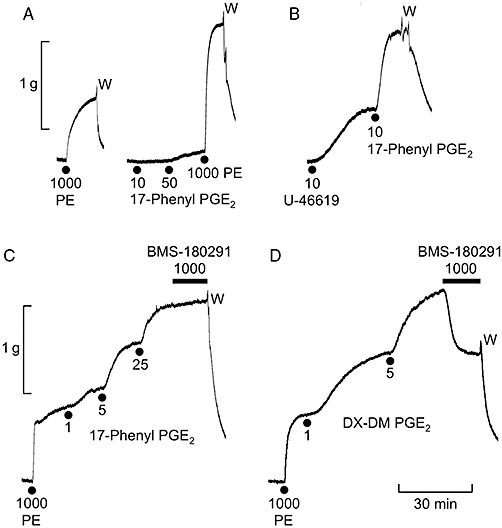
Experimental records illustrating properties of the EP3 receptor contractile system in guinea-pig isolated aorta. (A) Weak contraction/enhancement of the response to phenylephrine (PE) induced by the EP3 standard agonist 17-phenyl PGE2. (B) Enhanced response to 17-phenyl PGE2 under priming with the TP receptor agonist U-46619. (C) Enhanced responses to 17-phenyl PGE2 under phenylephrine priming and nil effect of the TP antagonist BMS-180291 on the established contraction. (D) Partial inhibition of phenylephrine-primed DX-DM PGE2 contraction by BMS-180291. W = wash. Concentrations in nM.
High EP1 receptor selectivity was shown by SC-51322 (pIC50= 7.26 ± 0.10, n = 4), with <10% inhibition of EP3, TP and M3 receptor agonism at 10 µM (Figure 2). The EP1 receptor antagonism of (ONO)-5-methyl-1 was slow to reach steady state and cumulative concentrations of 5 and 15 nM only were applied; it was about nine times more potent than SC-51322 (pIC50∼8.2). At 1–3 µM, it significantly inhibited EP3 and TP responses, while M3 agonism was essentially unaffected. AH-6809 was the least potent (pIC50= 6.78 ± 0.03 on trachea) and least selective EP1 antagonist. At 3 and 10 µM, it significantly inhibited TP and M3 receptor-mediated contraction on trachea, and also EP3, TP, α1-adrenoceptor and histamine H1 receptor agonsim on aorta (inset in Figure 2C, only 10 µM data shown); inhibitions at 1 µM AH-6909 were all minimal. SC-51322, (ONO)-5-methyl-1 and AH-6809 concentrations of 1.0, 0.1 and 1.0 µM, respectively, were considered suitable for selective EP1 receptor blockade
Figure 2.
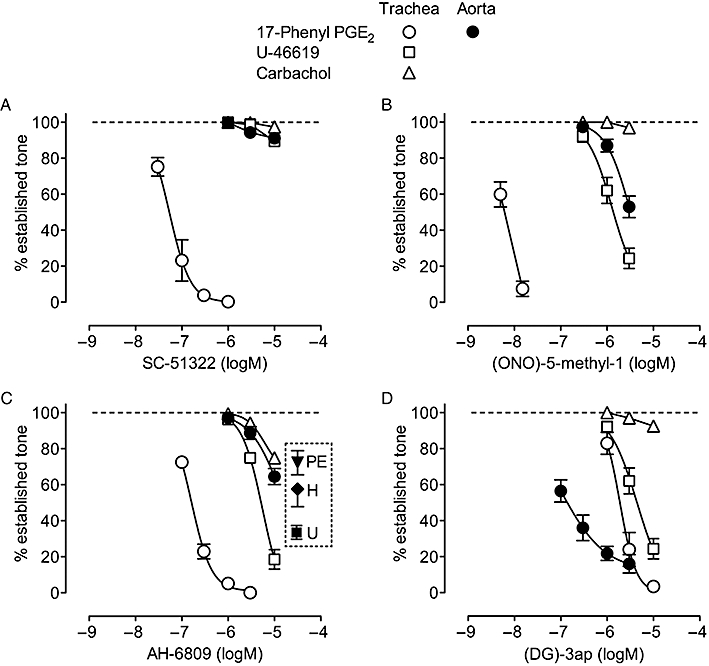
Selectivity of EP receptor antagonists: cumulative inhibition curves for (A) SC-51322 (B) (ONO)-5-methyl-1 (C) AH-6809 and (D) (DG)-3ap on guinea-pig trachea and aorta. Contraction was established with 17-phenyl PGE2, U-46619 and carbachol on trachea and 17-phenyl PGE2 primed with phenylephrine (net EP3 response = 100%) on aorta. The inset in panel C shows the effect of 10 µM AH-6809 against phenylephrine (2 µM, PE), histamine (1.5 µM, H) and U-46619 (20–30 nM, U) on guinea-pig aorta; inhibition with 1 µM AH-6809 was <5% in each case. BMS-180291 (1 µM) was present for all 17-phenyl PGE2 tests. Error bars indicate s.e.mean (n = 4). Carbachol activates M3 receptors in guinea-pig trachea (Morrison and Vanhoutte, 1992).
The net EP3 response of guinea-pig aorta induced by 25 nM 17-phenyl PGE2 was not completely inhibited by (DG)-3ap (pIC50∼7.0, Figure 2D), possibly due to residual synergism maintained by α1 agonism (priming) alone (Jones et al., 2010). Inhibition of EP1 and TP responses by (DG)-3ap on trachea was significant (pIC50= 5.73 ± 0.08 and 5.35 ± 0.09, respectively), while suppression of M3-receptor responses was minimal. A (DG)-3ap concentration of 1 µM was chosen for selective EP3 receptor blockade. In the presence of 1 µM (DG)-3ap, established contraction of the aorta to a 20-fold higher concentration of 17-phenyl PGE2 (500 nM) was unaffected by addition of the three EP1 receptor antagonists at the chosen concentrations (n = 3, data not shown).
Agonist profiles of PGE and PGF analogues on guinea-pig trachea and aorta
In terms of EP1 receptor agonism on guinea-pig trachea, 17-phenyl PGE2 (standard agonist), DX-DM PGE2 and (+)-cloprostenol exhibited parallel log concentration–response curves with pEC50 values of 8.79 ± 0.08 (n = 8), 6.81 ± 0.13 (n = 4) and 5.85 ± 0.12 (n = 4) respectively (1 µM BMS-180291 present). The corresponding equi-effective molar ratios are 1.0, 95 and 870. Established EC70 responses to DX-DM PGE2 and (+)-cloprostenol were abolished by the chosen concentrations of the three EP1 receptor antagonists (all n = 3).
In relation to EP3 receptor agonism on guinea-pig aorta, DX-DM PGE2, PGF2α (+)-cloprostenol and latanoprost-FA differed radically from 17-phenyl PGE2 (and sulprostone) in inducing strong contraction under resting tone; pEC50 values were 8.25 ± 0.17, 6.58 ± 0.07, 6.55 ± 0.05 and 5.25 ± 0.15 respectively (n = 4). Moreover, 1 µM BMS-180291 partially inhibited their phenylephrine-primed E70 responses, typically by 25–50% (see Figure 1D for DX-DM PGE2). Contraction induced by 17-phenyl PGE2 was markedly enhanced by priming with U-46619 (5–10 nM, Figure 1B). It thus appeared that DX-DM PGE2 and the PGF analogues might activate both EP3 and TP receptors resulting in a pronounced contractile synergism. Enhancement of EP3 receptor agonism was also seen under priming with histamine (1 µM, H1 agonist) or K+ (20–25 mM) (data not shown). Removal of the aortic endothelium by gentle abrasion did not significantly alter the agonist profiles of 17-phenyl PGE2 and DX-DM PGE2 shown in Figure 1 (data not shown).
Effect of EP3/TP receptor antagonist pretreatment on primed responses to PGE and PGF analogues on guinea-pig aorta
It was considered useful to obtain separate EP3 and TP receptor concentration–response profiles for DX-DM PGE2 and the three PGF analogues on guinea-pig aorta under the same degree of priming. Preparations were therefore pretreated with vehicle, 1 µM (DG)-3ap, 1 µM BMS-180291 and a combination of the antagonists for 45 min before prostanoid agonist was applied cumulatively under phenylephrine priming (Figure 3). Statistical analysis using Dunnett's multiple comparison test showed that, for each prostanoid agonist examined, the initial TP and EP3 receptor agonist sensitivities of the four groups of aorta preparations were uniform: no significant differences (P > 0.05) between responses to 10 nM U-46619 and between incremental (primed) responses to 10 nM 17-phenyl PGE2 (cf. Figure 1B). Using the same analysis, phenylephrine priming responses in the presence of the antagonist treatments were not significantly different to the vehicle control (P > 0.05). A log concentration–response curve for 17-phenyl PGE2 (0.5–312.5 nM) in the presence of 1 µM BMS-180291 obtained in contemporaneous experiments (n = 10) is also shown in Figure 3B; fitting parameters (mean, 95% CI) were: priming level = 21.5% (18–25), maximum = 78% (73.5–82.5), pEC50= 8.42 (8.28–8.55), nH= 0.77 (0.71–0.84).
Figure 3.
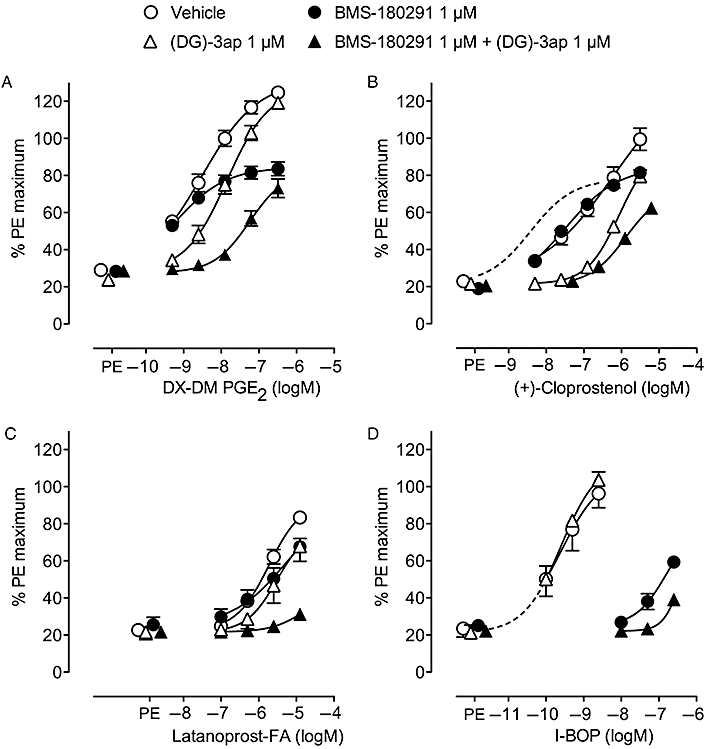
Log concentration–response curves for prostanoid agonists on guinea-pig aorta under phenylephrine (PE) priming in the presence of vehicle, BMS-180291, (DG)-3ap and a combination of BMS-180291 and (DG)-3ap: (A) DX-DM PGE2 (B) (+)-cloprostenol (C) latanoprost-FA and (D) I-BOP. Responses were normalized to the maximum response to phenylephrine on each preparation in the presence of BMS-180291 + (DG)-3ap. Error bars indicate s.e.mean (n = 4). The broken line in B is the fitted curve for 17-phenyl PGE2 in the presence of 1 µM BMS-180291 obtained in contemporaneous experiments (mean priming = 21.5%, n = 10). The broken line in D is an extrapolation of the I-BOP/1 µM (DG)-3ap curve.
Under TP receptor blockade, maximum responses to DX-DM PGE2, PGF2a (data not shown) and (+)-cloprostenol approach 80% of the phenylephrine maximum, similar to 17-phenyl PGE2 and indicative of activation of EP3 receptors. This is supported by the rightward shifts caused by 1 µM (DG)-3ap (in the presence of BMS-180291); corresponding pA2 values are 7.96 ± 0.07, 7.55 ± 0.03 and 8.15 ± 0.03, which are similar to the value of 7.92 ± 0.07 obtained with 17-phenyl PGE2 as agonist (Schild plot, Jones et al., 2010). A similar analysis was not possible for latanoprost-FA owing to its low potency. Nevertheless, there was a clear suppression of its contractile action by (DG)-3ap in the presence of BMS-180291. Under EP3 receptor blockade, BMS-180291 treatment revealed TP receptor agonism for each analogue. Contraction induced by the potent TP receptor agonist I-BOP (Sessa et al., 1990; Matsuda et al., 1994) was particularly slow and it was only possible to challenge with three mid-range doses; responses to the two lower doses may not have reached steady state. (DG)-3ap did not block I-BOP-induced contraction, attesting to its selectivity in this situation. It is likely that the response to the highest concentration of I-BOP (250 nM) in the presence of BMS-180291 + (DG)-3ap contains a TP component owing to I-BOP overcoming the BMS-180291 blockade: dose ratio for EP3+ TP versus EP3 treatment at 35% phenylephrine maximum level is ∼5900; dose ratio calculated for 1 µM BMS-180291 using pA2 of 9.98 obtained in a concurrent study is 9550 (Schild plot, U-46619 as agonist, Jones et al., 2010).
For easier appreciation, the data in Figure 3 have been presented as EP3 and TP receptor activation ranges (Figure 4). Each range corresponds to E3– E97 (obtained by extrapolation as necessary); the range for an agonist with nH of 1.0 covers 3.0 log units. Estimation of EP3 activation ranges (∼4.0 log units, nH∼0.75) is straightforward because of the comprehensive block of TP receptors by BMS-180291. The TP receptor activity ranges for I-BOP and U-46619 cover ∼3.5 log units (nH∼0.87), but are less well-defined for the other prostanoid agonists given the weaker block produced by (DG)-3ap, and this applies particularly to (+)-cloprostenol. The EC50 values for the prostanoid agonists acting alone have also been included.
Figure 4.
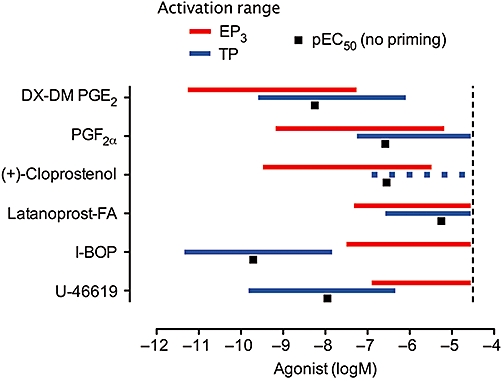
EP3 and TP receptor activation ranges for prostanoid agonists under phenylephrine priming on guinea-pig aorta. Each horizontal bar represents the concentration range corresponding to E3– E97; the broken bar for (+)-cloprostenol indicates difficulty in estimation, because of interference from the other receptor activity. The black squares indicate pEC50 values in the absence of priming; the I-BOP data point derives from Jones et al. (2010) in which steady-state responses were obtained by applying single concentrations of I-BOP to multiple preparations. The vertical line represents a nominal upper limit of 30 µM.
Further investigation of the antagonist profile of BMS-180291 on guinea-pig aorta
Using a conventional Schild protocol, Zhang et al. (1996) obtained a pA2 value of 9.8 for BMS-180291 versus U-46619 (pEC50∼8.0) on guinea-pig aorta, but failed to produce dose ratios in excess of ∼20. This was not the case in the current study, where pretreatment with 300 nM BMS-180291 for 60 min essentially abolished the response to 2.5 µM U-46619 (Figure 5). Furthermore, under phenylephrine priming, 30 nM BMS-180291 afforded a U-46619 dose ratio of ∼295 (pA2= 9.99). BMS-180291 at 300 nM and 3 µM produced larger rightward shifts, but these were less than expected for competitive antagonism according to the Schild equation (see broken curves in Figure 5). The residual response to 2.5 µM U-46619 in the presence of 3 µM BMS-180291 was abolished following addition of 1 µM (DG)-3ap (data not shown), indicating activation of EP3 receptors by U-46619. High sensitivity to 17-phenyl PGE2 was maintained in the presence of 3 µM BMS-180291 (pEC50= 8.33 ± 0.03, n = 4, Figure 5).
Figure 5.
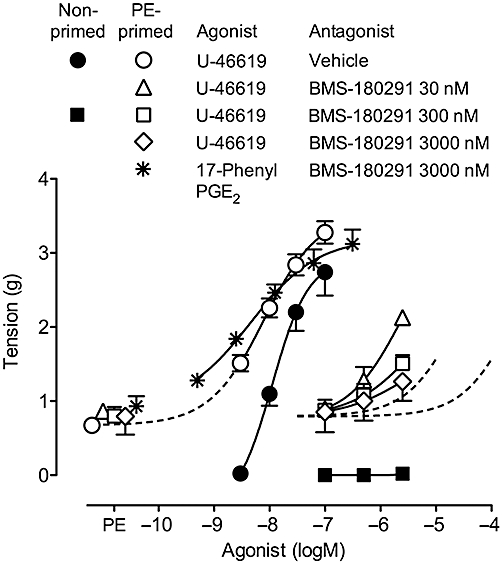
Guinea-pig aorta: effect of the TP receptor antagonist BMS-180291 on log concentration–response curves for U-46619 with and without phenylephrine (PE) priming. The broken line curves on the right are predicted for 300 and 3000 nM BMS-180291 assuming a pA2 of 9.98 (taken from Jones et al., 2010) and nH of 0.87 for U-46619. The curve for PE-primed 17-phenyl PGE2 in the presence of 3000 nM BMS-180291 is also shown. Error bars indicate s.e.mean (n = 4).
Discussion
EP3 receptor-mediated contractility on guinea-pig aorta
EP3 receptor agonism on guinea-pig aorta was characterized by weak overt contraction accompanied by pronounced synergism with a second strong contractile agent such as phenylephrine (selective α1-adrenoceptor agonist), U-46619 (TP receptor agonist) or K+ (membrane-depolarizing agent). We presume that these interactions primarily involve smooth muscle cells as contraction profiles were not affected by endothelium removal. Activation of EP3 receptors located on post-synaptic sympathetic neurones in rat and human blood vessel preparations results in suppression of transmitter release (Molderings et al., 1992; 1994;); it is not known whether an analogous action influenced the contractility profiles found in this study. Low maximum/high synergy agonist profiles have been previously described for 5-HT1B, α2-adrenoceptor and neuropeptide Y Y1 receptor systems in vascular smooth muscle (De La Lande et al., 1966; Edvinsson et al., 1984; Wahlestedt et al., 1985; McGrath et al., 1990); the corresponding agonists are often described as ‘silent’. Yildiz et al. (1998) have reviewed the discrimination of this marked contractile synergism from ‘mutual effect amplification’, in which a small degree of synergism arises when two agonists activate receptors (e.g. α1-adrenoceptor/H1 receptor) utilizing the same transduction system(s) (Leff, 1987; Christ and Jean-Jacques, 1991). They further distinguished inertial (threshold) synergism, whereby ‘a certain amount of stimulus must be delivered in order for the tissue to approach the threshold for contraction’ (Ariens et al., 1960; Stupecky et al., 1986).
In contrast to the current study, one of us previously found that EP3 receptor agonists induced much stronger contraction of guinea-pig aorta (30–60% of the tissue maximum; synergism with phenylephrine or U-46619 was moderate (Jones et al., 1998; 2002;). The animals (Dunkin-Hartley strain) in the two situations came from unrelated breeding colonies. Studies in isolated cell systems show that EP3 receptors exist in several isoforms (Sugimoto and Narumiya, 2007). All EP3 isoforms couple efficiently to Gi (as do 5-HT1B, α2-adrenoceptor and neuropeptide Y Y1 receptor subtypes; see Yildiz et al., 1998), while they differ in their abilities to couple to Gq and Gs; their agonist recognition profiles are similar. Thus it is possible that the different contractility profiles are due to variation in the proportions of EP3 receptor isoforms present and/or the amplifications of their transduction systems. Higher overt contraction could be due to a more prominent Gq (PLC)-driven component of signal transduction. Alternatively, a silent profile could indicate operation of a significant Gs (cAMP)-driven component that suppresses contractile activity. In this context (post-synaptic) α2-adrenoceptor systems in blood vessels from different species and peripheral locations vary in their need for priming to achieve strong contraction (see Blaylock and Wilson, 1995).
EP3/TP receptor self-synergism on guinea-pig aorta
Our results indicate that strong contraction of guinea-pig aorta elicited by DX-DM PGE2 and the PGF analogues may be attributed to co-activation of EP3 and TP receptors resulting in a synergistic response, which we have termed self-synergism. The steep lower section of the log concentration–response curve for non-primed U-46619 on the aorta, which is poorly fitted by a symmetrical sigmoidal curve constrained to the resting tension (Figure 5), hints at inertial constraint of the TP receptor input. Priming with phenylephrine exposes excitation at lower U-46619 concentrations and we propose that this condition affords a truer picture of the TP receptor activation range. Thus for DX-DM PGE2 at 1 nM, significant TP activation is accompanied by at least half maximal activation of the EP3 system (Figure 4). A similar relationship is seen for PGF2α and (+)-cloprostenol, while the EP3 and TP inputs for latanoprost-FA overlap more. In contrast, I-BOP maximally activates the TP system before the EP3 system is brought into play at around 30 nM. In this context, Kiriyama et al. (1997) reported binding Kds of 0.56 and 100 nM for I-BOP on mouse recombinant TP and EP3 receptors, respectively; corresponding values for U-46619 were 67 and >10 000 nM. The potent and selective FP receptor agonist latanoprost-FA (Griffin et al., 1997; Sharif et al., 2002; Chen et al., 2005) showed very low activity on the aorta in the presence of TP and EP3 receptor blockade (Figure 3C), arguing against interference from a FP receptor system.
EP1-selective concentrations of SC-51322, (ONO)-5-methyl-1 and AH-6809 failed to inhibit the modest response of the guinea-pig aorta to a high concentration of 17-phenyl PGE2 under EP3 receptor blockade with (DG)-3ap. Given that 17-phenyl PGE2 is a potent EP1 receptor agonist (Lawrence et al., 1992), this protocol ought to have identified even a relatively insensitive EP1 receptor system. Consequently, we propose that EP1 agonism does not confound our analysis of the EP3 receptor system in the aorta. This supposition may not apply to the Jadhav et al. (2004) study on pig cerebral artery, in which DX-DM PGE2 was claimed to act as a selective EP3 receptor agonist on the basis that its established contraction was not suppressed by 30–300 µM AH-6809. First, DX-DM PGE2 at the high concentration used (3 µM) elicited near maximal contraction of the cerebral artery preparation. Second, this high concentration would be expected to cause considerable EP1 (and TP) receptor activation based on our guinea-pig trachea (and aorta) data. Third, AH-6809 in high micromolar concentration may not specifically block EP1 receptors. The low specificity of AH-6809 observed in our study (Figure 2C) may be due to functional antagonism driven by cAMP, as AH-6809 inhibited cAMP PDE in rat lung mast cells with IC50 of 26 µM (unpublished observations in Keery and Lumley, 1988). Correspondingly, 30 µM AH-6809 enhanced cAMP production by iloprost (IP receptor agonist) in washed platelet suspensions from man, rat and rabbit (Armstrong and Jones, 1988). So, in toto, strong (and potentially interactive) excitatory inputs from EP1, EP3 and TP receptors may have been opposed by both genuine EP1 receptor antagonism and functional antagonism; a model of this complexity is fraught with interpretative dangers.
Relevance of self-synergism to prostanoid receptor characterization
On a simple level, it may not be realized that the relative potencies of agonists for the receptor of interest are being distorted through self-synergism operating through a second receptor. Thus EP3/TP self-synergism is a plausible explanation for the very high contractile activity of DX-DM PGE2, relative to sulprostone or 17-phenyl PGE2 in pig cerebral artery (Jadhav et al., 2004). The confounding influence of self-synergism involving silent and overt contractile systems has been reported previously. For example, the selective α2-adrenoceptor agonist rilmenidine enhanced the response to noradrenaline less than that to phenylephrine in rat tail artery, indicating that α1/α2-adrenoceptor self-synergism was already operating with the former non-selective agonist (Xiao and Rand, 1989). A similar explanation was advanced for the ability of angiotensin II (strong agonist) to enhance responses to the selective α2 -adrenoceptor agonist UK-14304, but not to noradrenaline, in rabbit saphenous artery (Dunn et al., 1991).
Taken to an extreme degree, there is the possibility of receptor misidentification. Cao et al. (2002) inferred the presence of a FP system in the non-pregnant pig uterus on the basis of the moderately potent contractile activity of the PGF2α analogue cloprostenol (EC50∼15 nM). However, the pig uterus preparation contains both EP3 (Okada et al., 2000) and TP (Cao et al., 2004) contractile systems and, as we have shown on guinea-pig aorta (Figure 3B), cloprostenol [(+)-isomer] can achieve considerable contractile activity through EP3/TP self-synergism. The judicious use of selective prostanoid receptor antagonists ought to clarify these situations.
In relation to characterizing TP antagonists, comprehensive block of TP receptors by BMS-180291 in the phenylephrine-primed guinea-pig aorta revealed EP3 agonism for U-46619 at concentrations ≥500 nM, resulting in smaller right shifts than predicted for simple competition using the Schild equation (Figure 5). In a non-primed preparation, the potential EP3/TP self-synergism of U-46619 is less likely to alter the competition profile as BMS-180291 will annul the TP receptor arm of the synergism. However, Zhang et al. (1996) reported that BMS-180291 failed to achieve dose ratios greater than 20 for antagonism of U-46619 in the non-primed guinea-pig aorta; addition of a second TP receptor antagonist (SQ-29548, 100 nM, predicted dose ratio = 35) did not cause a further right shift of the U-46619 curve. They proposed that U-46619 (≥200 nM) activated a second ‘contractile’ receptor. In view of our earlier comments on EP3 receptor isoforms, this receptor may be an EP3 receptor mediating overt contraction as opposed to the silent system found in the current study. Unfortunately, the activity of a recognized EP3 receptor agonist was not reported by Zhang and colleagues.
Implications of prostanoid self-synergism in patho-physiological situations
Understanding the nature of the interaction between different prostanoid receptor systems is important, especially in the light of the recent intense interest in the clinical development of prostanoid receptor antagonists (see Jones et al., 2009). In human platelets, EP3 receptor agonists including PGE2 markedly enhance aggregation due to a variety of agents (ADP, U-46619, PAF), while showing no response on their own (Mattthews and Jones, 1993). EP3 receptor antagonists [relatives of (DG)-3ap] have been proposed for suppression of platelet activation associated with deterioration of atherosclerotic plaques (Heptinstall et al., 2008). Activation of EP3 and TP receptors mediating contraction of human blood vessels (see Jones et al., 2009) may impinge on these events, but the nature of their (synergistic) interaction has received little attention. It seems unlikely that PGE2 (or PGF2α) would be generated in sufficient concentration in patho-physiological situations to activate TP receptors and hence exert EP3/TP self-synergism. The reverse situation whereby TXA2 co-activates EP3 and TP receptors is also unlikely. Notwithstanding, the EP1 and EP3 agonist potencies of PGE2 are much closer. EP1 receptors efficiently activate the Gq/PLC/Ca2+ release pathway (Funk et al., 1993; Breyer et al., 1996) and mediate strong contraction of smooth muscle. EP1/EP3 receptor self-synergism in blood vessels may be worthy of future investigation.
Acknowledgments
The gift of BMS-180291 from Bristol-Myers Squibb, USA is much appreciated. A research grant from Allergan Inc, USA is also gratefully acknowledged.
Glossary
Abbreviations
- FA
free acid form of a C1-ester or C1-amide prostanoid
- nH
Hill slope for a sigmoidal log concentration–response curve
- pA2
negative logarithm of the molar concentration of antagonist producing a dose ratio of 2
- pEC50
negative logarithm of the agonist concentration inducing 50% maximum response
- PGE2
prostaglandin E2
- pIC50
negative logarithm of the molar concentration producing 50% inhibition of an established response
- TXA2
thromboxane A2
Conflicts of interest
None.
Supporting Information
Teaching Materials; Figs 1–5 as PowerPoint slide.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariens EJ, van Rossum J, Koopman PC. Receptor reserve and threshold phenomena. I. Theory and experiments with autonomic drugs tested on isolated organs. Arch Int Pharmacodyn Ther. 1960;127:459–478. [PubMed] [Google Scholar]
- Armstrong RA, Jones RL. Potentiation by AH6809 of iloprost-induced rises in platelet cyclic AMP levels. Br J Pharmacol. 1988;94(Suppl):404P. [Google Scholar]
- Banerjee AK, Tuffin DP, Walker JL. Pharmacological effects of (+/−)-11-deoxy, 16-phenoxy-prostaglandin E1 derivatives in the cardiovascular system. Br J Pharmacol. 1985;84:71–80. [PMC free article] [PubMed] [Google Scholar]
- Belley M, Gallant M, Roy B, Houde K, Lachance N, Labelle M, et al. Structure-activity relationship studies on ortho-substituted cinnamic acids, a new class of selective EP3 antagonists. Bioorg Med Chem Lett. 2005;15:527–530. doi: 10.1016/j.bmcl.2004.11.051. [DOI] [PubMed] [Google Scholar]
- Blaylock NA, Wilson VG. Pharmacological characterization of noradrenaline-induced contractions of the porcine isolated palmar lateral vein and palmar common digital artery. Br J Pharmacol. 1995;114:694–702. doi: 10.1111/j.1476-5381.1995.tb17194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breyer MD, Jacobson HR, Breyer RM. Functional and molecular aspects of renal prostaglandin receptors. J Am Soc Nephrol. 1996;7:8–17. doi: 10.1681/ASN.V718. [DOI] [PubMed] [Google Scholar]
- Cao J, Shayibuzhati M, Tajima T, Kitazawa T, Taneike T. In vitro pharmacological characterization of the prostanoid receptor population in the non-pregnant porcine myometrium. Eur J Pharmacol. 2002;442:115–123. doi: 10.1016/s0014-2999(02)01489-9. [DOI] [PubMed] [Google Scholar]
- Cao J, Wakatsuki A, Yoshida M, Kitazawa T, Taneike T. Thromboxane A2 (TP) receptor in the non-pregnant porcine myometrium and its role in regulation of spontaneous contractile activity. Eur J Pharmacol. 2004;485:317–327. doi: 10.1016/j.ejphar.2003.11.073. [DOI] [PubMed] [Google Scholar]
- Chen J, Senior J, Marshall K, Abbas F, Dinh H, Dinh T, et al. Studies using isolated uterine and other preparations show bimatoprost and prostanoid FP agonists have different activity profiles. Br J Pharmacol. 2005;144:493–501. doi: 10.1038/sj.bjp.0706044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ GJ, Jean-Jacques M. Mutual-effect amplification of contractile responses elicited by simultaneous activation of alpha-1 adrenergic and 5-hydroxytryptamine2 receptors in isolated rat aorta. J Pharmacol Exp Ther. 1991;256:553–561. [PubMed] [Google Scholar]
- Coleman RA, Kennedy I. Characterisation of the prostanoid receptors mediating contraction of guinea-pig isolated trachea. Prostaglandins. 1985;29:363–375. doi: 10.1016/0090-6980(85)90096-6. [DOI] [PubMed] [Google Scholar]
- Coleman RA, Kennedy I, Sheldrick RLG. AH6809, a prostanoid EP1 receptor blocking drug. Br J Pharmacol. 1985;85(Suppl):273P. [Google Scholar]
- Coleman RA, Kennedy I, Sheldrick RLG. Further evidence for the existence of three subtypes of PGE2-senstive (EP-) receptors. Br J Pharmacol. 1987;91(Suppl):407P. [Google Scholar]
- Coleman RA, Smith WL, Narumiya S. VIII International union of pharmacology classification of prostanoid receptors: properties distribution, and structure of the receptors and their subtypes. Pharmacol Rev. 1994;46:205–229. [PubMed] [Google Scholar]
- De La Lande IS, Cannell VA, Waterson JG. The interaction of serotonin and noradrenaline on the perfused artery. Br J Pharmacol. 1966;28:255–272. doi: 10.1111/j.1476-5381.1966.tb01893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn WR, McGrath JC, Wilson VG. Postjunctional α-adrenoceptors in the rabbit isolated distal saphenous artery: indirect sensitivity to prazosin of responses to noradrenaline mediated via post-junctional α2-adrenoceptors. Br J Pharmacol. 1991;103:1484–1492. doi: 10.1111/j.1476-5381.1991.tb09815.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvinsson L, Ekblad E, Håkanson R, Wahlestedt C. Neuropeptide Y potentiates the effect of various vasoconstrictor agents on rabbit blood vessels. Br J Pharmacol. 1984;83:519–523. doi: 10.1111/j.1476-5381.1984.tb16516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk CD, Furci L, FitzGerald GA, Grygorczyk R, Rochette C, Bayne MA, et al. Cloning and expression of a cDNA for the human prostaglandin E receptor EP1 subtype. J Biol Chem. 1993;268:26767–26772. [PubMed] [Google Scholar]
- Gallant M, Carrière MC, Chateauneuf A, Denis D, Gareau Y, Godbout C, et al. Structure-activity relationship of biaryl acylsulfonamide analogues on the human EP3 prostanoid receptor. Bioorg Med Chem Lett. 2002;12:2583–2586. doi: 10.1016/s0960-894x(02)00518-8. [DOI] [PubMed] [Google Scholar]
- Griffin BW, Williams GW, Crider JY, Sharif NA. FP prostaglandin receptors mediating inositol phosphates generation and calcium mobilization in Swiss 3T3 cells: a pharmacological study. J Pharmacol Exp Ther. 1997;281:845–854. [PubMed] [Google Scholar]
- Hallinan EA, Stapelfeld A, Savage MA, Reichman M. 8-Chlorodibenz[b,f][1,4]oxazepine-10(11H)-carboxylic acid, 2-[3-[2-(furanylmethyl)thio]-1-oxopropyl]hydrazide (SC-51322): a potent PGE2 antagonist and analgesic. Bioorg Med Chem Lett. 1994;4:509–514. [Google Scholar]
- Heptinstall S, Espinosa DI, Manolopoulos P, Glenn JR, White AE, Johnson A, et al. DG-041 inhibits the EP3 prostanoid receptor – a new target for inhibition of platelet function in atherosclerotic disease. Platelets. 2008;19:605–613. doi: 10.1080/09537100802351073. [DOI] [PubMed] [Google Scholar]
- Jadhav V, Jabre A, Lin SZ, Lee TJ. EP1- and EP3-receptors mediate prostaglandin E2-induced constriction of porcine large cerebral arteries. J Cereb Blood Flow Metab. 2004;24:1305–1316. doi: 10.1097/01.WCB.0000139446.61789.14. [DOI] [PubMed] [Google Scholar]
- Jones RL, Peesapati V, Wilson NH. Antagonism of the thromboxane-sensitive contractile systems of the rabbit aorta, dog saphenous vein and guinea-pig trachea. Br J Pharmacol. 1982;76:423–438. doi: 10.1111/j.1476-5381.1982.tb09236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RL, Qian Y, Chan KM, Yim AP. Characterization of a prostanoid EP3-receptor in guinea-pig aorta: partial agonist action of the non-prostanoid ONO-AP-324. Br J Pharmacol. 1998;125:1288–1296. doi: 10.1038/sj.bjp.0702189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RL, Shum WW, Gurney AM. Synergism between prostanoids and other vasoactive agents. J Card Surg. 2002;17:436–438. doi: 10.1111/j.1540-8191.2001.tb01174.x. [DOI] [PubMed] [Google Scholar]
- Jones RL, Giembycz MA, Woodward DF. Prostanoid receptor antagonists: development strategies and therapeutic applications. Br J Pharmacol. 2009;158:104–145. doi: 10.1111/j.1476-5381.2009.00317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RL, Woodward DF, Wang JW, Clark RL. Roles of affinity and lipophilicity in the slow kinetics of prostanoid receptor antagonists on isolated smooth muscle preparations. Br J Pharmacol. 2010 doi: 10.1111/j.1476-5381.2010.01087.x. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keery RJ, Lumley P. AH6809, a prostaglandin DP-receptor blocking drug on human platelets. Br J Pharmacol. 1988;94:745–754. doi: 10.1111/j.1476-5381.1988.tb11584.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiriyama M, Ushikubi F, Kobayashi T, Hirata M, Sugimoto Y, Narumiya S. Ligand binding specificities of the eight types and subtypes of the mouse prostanoid receptors expressed in Chinese hamster ovary cells. Br J Pharmacol. 1997;122:217–224. doi: 10.1038/sj.bjp.0701367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence RA, Jones RL, Wilson NH. Characterization of receptors involved in the direct and indirect actions of prostaglandins E and I on the guinea-pig ileum. Br J Pharmacol. 1992;105:271–278. doi: 10.1111/j.1476-5381.1992.tb14245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leff P. An analysis of amplifying and potentiating interactions between agonists. J Pharmacol Exp Ther. 1987;243:1035–1042. [PubMed] [Google Scholar]
- McGrath JC, Dunn WR, Templeton AG. Physiological modulation of alpha-adrenoceptor and 5HT receptor expression in blood vessels. Blood Vessels. 1990;27:146–152. doi: 10.1159/000158805. [DOI] [PubMed] [Google Scholar]
- Matsuda K, Ruff A, Morinelli TA, Mathur RS, Halushka PV. Testosterone increases thromboxane A2 receptor density and responsiveness in rat aortas and platelets. Am J Physiol. 1994;267:H887–H893. doi: 10.1152/ajpheart.1994.267.3.H887. [DOI] [PubMed] [Google Scholar]
- Mattthews JS, Jones RL. Potentiation of aggregation and inhibition of adenylate cyclase in human platelets by prostaglandin E analogues. Br J Pharmacol. 1993;108:363–369. doi: 10.1111/j.1476-5381.1993.tb12810.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molderings G, Malinowska B, Schlicker E. Inhibition of noradrenaline release in the rat vena cava via prostanoid receptors of the EP3-subtype. Br J Pharmacol. 1992;107:352–355. doi: 10.1111/j.1476-5381.1992.tb12750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molderings GJ, Colling E, Likungu J, Jakschik J, Göthert M. Modulation of noradrenaline release from the sympathetic nerves of the human saphenous vein and pulmonary artery by presynaptic EP3- and DP-receptors. Br J Pharmacol. 1994;111:733–738. doi: 10.1111/j.1476-5381.1994.tb14799.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison KJ, Vanhoutte PM. Characterization of muscarinic receptors that mediate contraction of guinea-pig isolated trachea to choline esters: effect of removing epithelium. Br J Pharmacol. 1992;106:672–676. doi: 10.1111/j.1476-5381.1992.tb14393.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naganawa A, Matsui T, Saito T, Ima M, Tatsumi T, Yamamoto S, et al. Discovery of heteroaryl sulfonamides as new EP1 receptor selective antagonists. Bioorg Med Chem. 2006;14:6628–6639. doi: 10.1016/j.bmc.2006.05.076. [DOI] [PubMed] [Google Scholar]
- O'Connell M, Zeller W, Burgeson J, Mishra RK, Ramirez J, Kiselyov AS, et al. Peri-substituted hexahydro-indolones as novel, potent and selective human EP3 receptor antagonists. Bioorg Med Chem Lett. 2009;19:778–782. doi: 10.1016/j.bmcl.2008.12.027. [DOI] [PubMed] [Google Scholar]
- Okada Y, Hara A, Ma H, Xiao CY, Takahata O, Kohgo Y, et al. Characterization of prostanoid receptors mediating contraction of the gastric fundus and ileum: studies using mice deficient in prostanoid receptors. Br J Pharmacol. 2000;131:745–755. doi: 10.1038/sj.bjp.0703627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlemper V, Medeiros R, Ferreira J, Campos MM, Calixto JB. Mechanisms underlying the relaxation response induced by bradykinin in the epithelium-intact guinea-pig trachea in vitro. Br J Pharmacol. 2005;145:740–750. doi: 10.1038/sj.bjp.0706222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessa WC, Halushka PV, Okwu A, Nasjletti A. Characterization of the vascular thromboxane A2/prostaglandin endoperoxide receptor in rabbit aorta. Regulation by dexamethasone. Circ Res. 1990;67:1562–1569. doi: 10.1161/01.res.67.6.1562. [DOI] [PubMed] [Google Scholar]
- Sharif NA, Xu SX, Williams GW, Crider JY, Griffin BW, Davis TL. Pharmacology of [3H]prostaglandin E1/[3H]prostaglandin E2 and [3H]prostaglandin F2α binding to EP3 and FP prostaglandin receptor binding sites in bovine corpus luteum: characterization and correlation with functional data. J Pharmacol Exp Ther. 1998;286:1094–1102. [PubMed] [Google Scholar]
- Sharif NA, Kelly CR, Crider JY. Agonist activity of bimatoprost, travoprost, latanoprost, unoprostone isopropyl ester and other prostaglandin analogs at the cloned human ciliary body FP prostaglandin receptor. J Ocul Pharmacol Ther. 2002;18:313–324. doi: 10.1089/10807680260218489. [DOI] [PubMed] [Google Scholar]
- Stupecky GL, Murray DL, Purdy RE. Vasoconstrictor threshold synergism and potentiation in the rabbit isolated thoracic aorta. J Pharmacol Exp Ther. 1986;238:802–828. [PubMed] [Google Scholar]
- Sugimoto Y, Narumiya S. Prostaglandin E receptors. J Biol Chem. 2007;282:11613–11617. doi: 10.1074/jbc.R600038200. [DOI] [PubMed] [Google Scholar]
- Wahlestedt C, Edvinsson L, Ekblad E, Håkanson R. Neuropeptide Y potentiates noradrenaline-evoked vasoconstriction: mode of action. J Pharmacol Exp Ther. 1985;234:735–741. [PubMed] [Google Scholar]
- Wallis SJ, Martin W. Conditions permitting suppression of stretch-induced and vasoconstrictor tone by basal nitric oxide in porcine cerebral artery. Br J Pharmacol. 2000;130:567–574. doi: 10.1038/sj.bjp.0703351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X-H, Rand MJ. α2-Adrenoceptor agonists enhance responses to certain other vasoconstrictor agonists in rat tail artery. Br J Pharmacol. 1989;96:539–546. doi: 10.1111/j.1476-5381.1989.tb11851.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildiz O, Smith JR, Purdy RE. Serotonin and vasoconstrictor synergism. Life Sci. 1998;62:1723–1732. doi: 10.1016/s0024-3205(97)01166-1. [DOI] [PubMed] [Google Scholar]
- Zhang R, Ogletree ML, Moreland S. Characterization of thromboxane A2/prostaglandin endoperoxide receptors in aorta. Eur J Pharmacol. 1996;317:91–96. doi: 10.1016/s0014-2999(96)00697-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.