

Abstract
Hypoxia, a fundamental stimulus in biology and medicine, uses reactive oxygen species (ROS) as second messengers. A surprising target of hypoxia-generated ROS is specific bases within hypoxic response elements (HREs) of the VEGF and other hypoxia-inducible genes. Oxidative modifications coincide with the onset of mRNA accumulation and are localized to transcriptionally active mono-nucleosomes. The oxidative base modifications are removed by the base excision DNA repair pathway for which one of its components, the bifunctional transcriptional co-activator and DNA endonuclease Ref-1/Ape1, is critical for transcription complex assembly. Mimicking the effect of hypoxia by introducing an abasic site in an oligonucleotide model of the VEGF HRE, altered transcription factor binding, enhanced sequence flexibility, and engendered more robust reporter gene expression. These observations suggest that controlled DNA “damage” and repair, mediated by ROS used as second messengers and critically involving the base excision pathway of DNA repair, respectively, are important for hypoxia-induced transcriptional activation.
Keywords: Hypoxia, oxygen radicals, oxidative DNA modifications, VEGF promoter, transcriptional regulation
1. Introduction
Hypoxia, a fundamental stimulus in biology and medicine, changes expression of about 400+ genes, depending on the cell type and nature of the microarray, which collectively serve to allow the cell and tissue in which it resides to adapt to the low oxygen environment. Not surprisingly, considerable effort has been directed at understanding the mechanisms governing transcription in hypoxia, and it is now widely appreciated that a family of transcription factors - the Hifs - are master regulators of adaptive gene expression in response to low environmental oxygen (Rey and Semenza, 2010). Hypoxia also causes genomic instability, which has been incriminated in aging related disorders, including acquisition of mutations underlying carcinogenesis or the emergence of metastatic cancer cell phenotypes (Bristow and Hill, 2008; Young et al., 1988; Yuan and Glazer, 1998). Exactly how hypoxia leads to genetic instability is not well appreciated. Herein we will review evidence that an oxidant stress associated with hypoxic signaling targets specific sequences of hypoxia-inducible genes for oxidative base modifications and their subsequent repair. We also will compare this paradigm for controlled DNA “damage” and repair in hypoxic signaling to hormone receptor-dependent stimuli using a similar pathway for transcriptional regulation that has recently been incriminated in the genomic instability linked to carcinogenesis. From this vantage point, we will propose a model in which controlled DNA damage and repair in hypoxia is important for normal transcriptional regulation, and that this physiologic process may be inextricably linked to genomic instability characterizing age-related diseases such as cancer and others oxygen radical-dependent disorders.
2. Reactive oxygen species (ROS) in hypoxic signaling
After a period of considerable controversy, the consensus has emerged that hypoxia, like other physiologic signals modulating cell proliferation and differentiation, employs ROS as second messengers. In pulmonary vascular and other cell types, for example, hypoxia promotes fluorescence of oxidant sensitive probes (Guzy et al., 2005; Killilea et al., 2000). Antioxidants also suppress activation of transcription in hypoxia, at least in part by modulating accumulation of Hif-1 (Bell et al., 2007b; Emerling et al., 2005). Sources of ROS involved in hypoxic signaling remain somewhat unclear; both membrane NADPH oxidase as well as mitochondria, and specifically the inner-membrane aspect of complex III, have been incriminated as ROS sources in hypoxic signaling (Bell et al., 2007a; Liu et al., 2006; Marshall et al., 1996). Interactions between ROS sources in hypoxia also seem likely, with mitochondria-to-membrane and membrane-to-mitochondria signaling pathways potentially operative (Ricci et al., 2008; Ward, 2008; Weissmann et al., 2006).
The idea that ROS function as second messengers in hypoxic signal transduction was somewhat slow to achieve consensus in part because the idea that low oxygen stimulates an increase in oxidant production is counter-intuitive and because there was occasional difficulty reproducing the initially-reported effects of mitochondrial depletion and antioxidant interventions in terms of various hypoxia-related outcome measures (Reeve et al., 2001; Vaux et al., 2001; Wolin et al., 2005). As an alternate strategy to determine if hypoxia increases oxidant stress we searched for hypoxia-induced oxidative DNA damage, initially concentrating on modifications in the mitochondrial (mt) DNA since mitochondria are a likely source of ROS generation in hypoxia, and since, in comparison to nuclear DNA, mtDNA is about 30-fold more sensitive to oxidative damage (Yakes and Van Houten, 1997). Primary cultures of rat pulmonary artery endothelial cells (PAECs) were selected as a model system because this cell type is directly responsive to hypoxia and contributes to remodeling of the pulmonary circulation in part by secreting growth factors that influence survival, differentiation, and proliferative events at downstream cellular targets.
3. Oxidative base modifications in mitochondrial and nuclear DNA in hypoxic pulmonary artery endothelial cells
These initial experiments used quantitative Southern blot analyses to detect oxidative base modifications in a 10.6 kb sequences of the mitochondrial genome harboring the “common deletion sequence” known to be prone to oxidative damage in various mitochondrial diseases. A 4.1 Kb sequence of the VEGF gene including the promoter and proximal coding region was examined as a negative control (Grishko et al., 2001). The results were surprising. Counter to expectations, while the mtDNA coding region was devoid of hypoxia-induced base modifications – and indeed, lesion density trended downward in hypoxia - there were transient oxidative base modifications in a functionally significant promoter sequence of the hypoxia-inducible VEGF gene. Prominent, time-dependent lesions were detected by alkali treatment of the DNA, indicating the presence of strand breaks and/or apyrimidinic/apurinic (AP) sites on the deoxyribose backbone, and by the DNA repair enzyme, Endo III, indicating the presence of pyrimidine base oxidation products.
Additional studies employed a simple PCR-based strategy to detect sequence-specific oxidative base modifications. We focused on hypoxic response elements (HREs), other promoter sequences, and coding regions not only of the VEGF gene, but also other hypoxia-regulated and non-regulated genes (Pastukh et al., 2007). Hypoxia caused oxidative base modifications in the HREs of hypoxia-inducible genes that coincided with the onset of mRNA accumulation. Importantly, the lesions were repaired over 6-24h despite the persistence of hypoxia and elevated mRNA abundances, as depicted for the VEGF HRE in Figure 1. As discussed below, removal of oxidative lesions in DNA is performed by the base excision repair (BER) pathway, for which at least one and perhaps more of its participating enzymes are components of the hypoxia-inducible transcriptional complex. Companion studies also demonstrated that promoters and coding regions of genes down-regulated in hypoxia or unaffected by hypoxia exposure failed to display changes in oxidative lesion density. Consistent with observations by Schumacker, Chandel and colleagues (Bell et al., 2007a; Bell et al., 2007b; Emerling et al., 2005; Schroedl et al., 2002; Waypa et al., 2001), both the hypoxia-induced oxidative base modifications and DCF fluorescence were suppressed by the specific complex III inhibitor, myxothiazol, thus supporting involvement of mitochondria as a source of the DNA damaging ROS generated in hypoxic PAECs.
FIGURE 1.
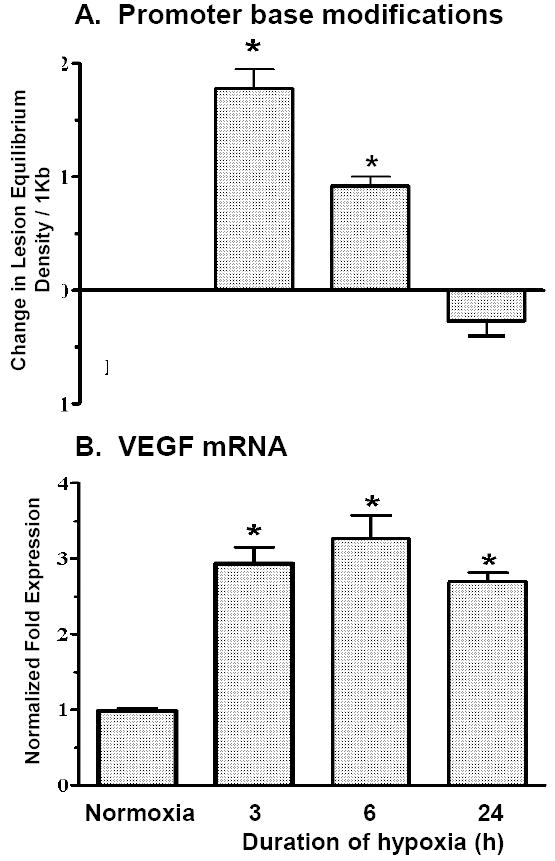
(A) Calculated increases in equilibrium density of Fpg-detectable purine base lesions evoked by hypoxia in the VEGF HRE. Oxidative base lesions were not detected in other sequences of the VEGF gene examined. (B) Increases in VEGF mRNA expressed as a function of the duration of hypoxic exposure. *Different from normoxia at p<0.05. Adapted from reference (Pastukh et al., 2007).
4. The conspicuous spatial relation between sequences harboring hypoxia-induced DNA “damage” and transcriptional regulation
The observation that hypoxia-induced oxidative base modifications were clustered in the HREs of hypoxia-inducible genes suggested that the damage could be functionally significant. To address this possibility, we first sought to determine whether hypoxia-induced base modifications were restricted to transcriptionally active nucleosomes. In this regard, promoter DNA is “packaged” into nucleosomes, wherein histones H2A, H2B, H3, and H4 and histone “variants” combine to form the nucleosome core particle around which is wrapped ≈ 147 bp of promoter DNA. Traditional concepts emphasize that promoter DNA of inactive genes is compacted within the nucleosome which thereby obstructs transcription factor binding (Li et al., 2007; Workman, 2006). However, upon stimulation, recruitment of chromatin remodeling enzymes catalyze eviction or repositioning of nucleosomes such that promoter DNA becomes more accessible to transcriptional regulators. To explore the idea that hypoxia-induced oxidative base modifications were linked to transcriptional activation, we determined if VEGF HRE sequences targeted for base modifications in hypoxia were associated with transcriptionally active nucleosomes (Ruchko et al., 2009). Nucleosome fractions derived from normoxic and hypoxic PAECs were prepared by micrococcal nuclease digestion, and the distribution of VEGF HRE sequences within the fractions was determined by Southern blot analysis. While 3h culture in hypoxia had no effect on the distribution of total DNA within micrococcal nuclease-derived nucleosomal fractions - a finding not unexpected since the transcriptional activity of only a small fraction of the genome is impacted by hypoxia - there was a subtle but reproducible enrichment in the abundance of HRE sequences detected in mono- and di-nucleosome fractions accompanied by a similar diminution in the abundance of HRE sequences residing in more compacted, micrococcal nuclease-resistant nucleosome structures. The PCR method noted above to detect purine base oxidation products in VEGF HRE sequences was applied to DNA excised from the nucleosome ladder and revealed that the hypoxia-induced oxidative base modifications were localized exclusively in mono-nucleosome fractions. These observations indicate that the onset of VEGF mRNA accumulation is accompanied by redistribution of a small but consistent portion of VEGF HRE sequences into transcriptionally active mono-nucleosomes and that about 25% of the HRE sequences located in the mono-nucleosome fraction display oxidative base modifications. We also found that the complex III inhibitor, myxothiazol, while blocking both hypoxia-induced lesion formation in VEGF HRE and VEGF mRNA expression, failed to prevent the hypoxia-mediated accumulation of VEGF HRE sequences in the mono-nucleosome fraction. Thus, introduction of base modification products per se does not seem to trigger chromatin remodeling events leading to acquisition of micrococcal nuclease cleavage sensitivity in hypoxic VEGF HRE sequences.
Hypoxia causes ROS-dependent accumulation of Hif-1 which binds along with other transcriptional co-activators and chromatin remodeling enzymes to the HRE sequence in hypoxic-inducible genes (Ebert and Bunn, 1998; Rey and Semenza, 2010). Based on the recognition that oxidative modifications at key nucleotides in this sequence could impact transcription complex assembly, we used ligation-mediated (LM) PCR to interrogate base modifications at single nucleotide resolution throughout the HRE of the VEGF promoter (Grishko et al., 2001; Ziel et al., 2005). As a negative control, we used LM PCR to search for oxidative damage to a 200 bp sequence of mtDNA harboring the common deletion sequence. Consistent with our earlier experience using quantitative Southern blot analysis, even the more sensitive LM PCR method was incapable of detecting oxidized bases in the mtDNA common deletion sequence in hypoxic PAECs. However, a guanine at the 3’ end of the Hif-1 DNA recognition sequence in the VEGF HRE was modified at high frequency while bases elsewhere in the HRE were modified at comparatively lower frequencies (Figure 2). This particular guanine also was targeted for high frequency oxidative modification by other stimuli using ROS as second messengers, including angiotensin II, PDGF, and thrombin.
FIGURE 2.
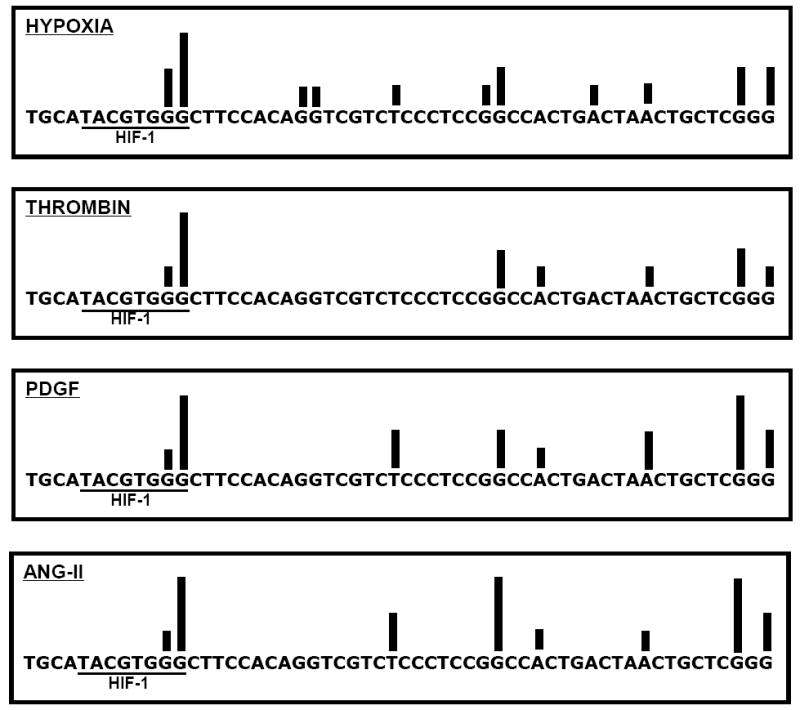
Ligation-mediated PCR-generated “map” of alkali-detectable modifications evoked by hypoxia, PDGF, angiotensin II, and thrombin in the coding strand of the hypoxic response element of the VEGF promoter in pulmonary artery endothelial cells. Position of bars reflects damage to specific nucleotides, designated by the letters A,T,G, and C, while height of bars indicates relative prevalence of damage as determined by hybridization intensities normalized to a scale of 1-6 where 1 corresponds to the least and 6 corresponds to the most intense hybridization. Note that the most commonly oxidized base was a guanine at the 3’ end of the HIF-1 DNA recognition sequence. Each bar reflects the mean of at least 4 experiments. Adapted from reference (Ziel et al., 2005).
An interesting question pertaining to the above findings is how the effects of ROS are targeted in a gene and sequence-specific manner. In this context, it has been known for many years that multiplets of guanine – the most commonly oxidized base - are prone to damage because of their low oxidation potential. In addition, R-G-T-R and R-G-G-G sequences have been shown to associate with iron more avidly than other sequences, which could confer increased sensitivity to oxidative damage via localized Fenton chemistry (Rai et al., 2001; Rai et al., 2005). It has also been demonstrated that oxidant attack initiated at distant sites can “tunnel” for surprisingly long distances (hundreds of Angstroms) through the DNA π-stack – a phenomenon termed “long range DNA charge transport” - and exit the duplex at the terminal guanine in guanine repeats, thereby “focusing” oxidative damage at the extreme 5’ guanine (Genereux et al., 2010; Merino et al., 2008). In a biological context, it may be significant that guanine repeats and R-T-G-R sequences are found in comparatively high density in promoter regulatory elements, particularly in transcription factor binding and regulatory G-quadruplex sequences (Goni et al., 2004; Wu et al., 2007). The VEGF promoter also harbors a G-quadruplex near the transcription start site (Qin and Hurley, 2008), but this sequence has not yet been examined for the presence of hypoxia-induced base oxidation products. Collectively, these considerations point to the intriguing idea that hypoxia-generated ROS cause gene and sequence-specific oxidative modifications in the VEGF HRE in part because of the relatively density of damage-prone guanine repeats and because of local sequence context.
5. Evidence that hypoxia-induced oxidative base modifications alter functional properties of the VEGF HRE
The above findings that hypoxia causes oxidative base modifications in the HRE of the inducible VEGF promoter raise the intriguing possibility that introduction of such modifications and/or their ensuing repair could impact hypoxic transcriptional activation by altering the functional properties of the sequence. To explore this idea, we constructed an oligonucleotide model of the wild type (WT) VEGF HRE and another in which the hypoxia-modified guanine - located at the extreme 3’ end of the Hif-1 DNA recognition sequence - was replaced by a tetrahydrofuran to create a stable AP site. We then compared the WT and AP-modified sequences in terms of their abilities to associate with transcriptional proteins present in normoxic and hypoxic nuclear extracts, to modulate local sequence topology and flexibility in response to nuclear protein binding, and finally, to drive hypoxia-induced reporter gene expression in intact PAECs.
To determine if the presence of base oxidation products impacted protein-DNA interactions, we used the oligonucleotide models of the WT and AP-modified VEGF HREs in concert with DNA affinity precipitation and Western blot analyses (Ziel et al., 2005). Relative to the WT HRE, the sequence harboring an abasic site at the hypoxia-modified guanine bound more HIF-1 and more Ref-1/Ape1; other probe-associated transcriptional regulators were unaffected by the presence of the base damage product (Figure 3; top panel).
FIGURE 3.
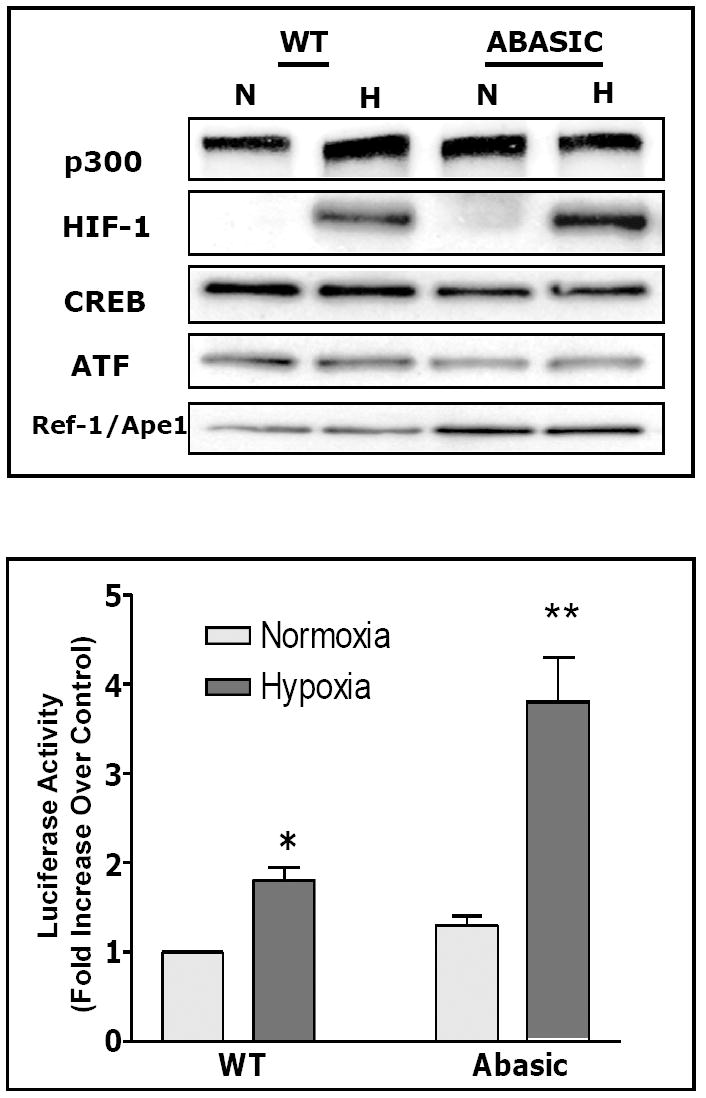
(Top panel) Western blot analysis of proteins associating with the hypoxic response element of the VEGF promoter as recovered by DNA affinity precipitation using WT and AP-modified (Abasic) oligonucleotide probes for the VEGF hypoxic response element. Note increased abundance of HIF-1 and Ref-1/Ape1 associated with the AP-modified oligonucleotide probe in comparison to the WT probe. Adapted from reference (Ziel et al., 2005). (Bottom panel) PAECs were transiently transfected with constructs composed of the 56-mer wild type (WT) or AP-modified (Abasic) hypoxic response elements of the rat VEGF promoter and a luciferase reporter. After 18h culture in normoxia or hypoxia, cells were harvested and luciferase activity determined. Note that the hypoxia-induced increase in reporter gene activity was augmented when the HRE included a model AP site at the hypoxia-modified guanine. Adapted from reference (Ziel et al., 2005).
The transcriptional co-activator and base excision DNA repair enzyme, Ref-1/Ape1, facilitates DNA binding and transcriptional activity of a number of transactivating factors, including Hif-1 (Evans et al., 2000; Gray et al., 2005; Tell et al., 2009). Ref-1/Ape1 seems to be required for assembly of the hypoxia-inducible transcriptional complex insofar as immuno-depletion of the protein from nuclear extract prevented probe association with Hif-1, p300, ATF and CREB (Ziel et al., 2004). As shown by co-immunoprecipitation experiments, Ref-1/Ape1 interacted with Hif-1 and p300, but not ATF/CREB. However, when Ref-1/Ape was immunoprecipitated from the oligonucleotide, both Hif-1 and p300 remained probe-associated while ATF/CREB co-immunoprecipitated. These findings suggest that Ref-1/Ape1 is a component of the hypoxia-inducible transcriptional complex forming on the HRE of the VEGF promoter and that the presence of Ref-1/Ape1 in the transcriptional complex is required for the apparent high affinity association between Hif-1 and its DNA recognition sequence. These observations raise the interesting prospect that formation of an AP site, through the DNA glycosylase-mediated excision of an oxidatively damaged base, increases Hif-1-DNA binding by optimizing the Ref-1/Ape1-dependent assembly of the transcriptional complex. As discussed below, emerging evidence supports this idea.
It has been known for many years that the presence of base oxidation products in model DNA sequences distorts local sequence topology and mechanical properties. Accordingly, the hypoxia-induced formation of base oxidation products in the VEGF HRE might be predicted to alter mechanical attributes of this sequence as well, an occurrence which could profoundly impact its functional properties. An extension to these older concepts, based on the finding that the strand-cleaving Ref-1/Ape1 binds to the HRE when an AP site is present, is that the AP endonuclease activity of the bifunctional protein creates a strand break at the AP site which also impacts sequence topology and bendability. To examine these ideas, WT and AP site-containing VEGF HRE duplex oligonucleotides were labeled at two specific positions, both downstream of the AP site replacing the hypoxia-modified guanine, with fluorescein and Texas red, respectively, to enable lifetime and steady-state fluorescence resonance energy transfer (FRET)-based quantification of inter-site distances (Breit et al., 2008). In initial experiments, examination of Coomassie-stained gels and Western analyses showed that the total number of nuclear proteins as well as the presence of specific transcriptional activator and co-activator proteins, respectively, bound to either WT or abasically modified HRE oligonucleotides were not altered by introduction of the two fluorophores. Subsequent FRET analyses demonstrated first, that Hif-1 present in nuclear extract was required for changes in FRET in both the WT and AP-modified HRE oligonucleotides, and second, that whereas the WT modified HRE sequences were comparatively stiff in the absence of nuclear proteins, the AP-modified HRE sequence, but not the WT sequence, acquired a significant degree of flexibility when incubated with nuclear proteins from either normoxic or hypoxic PAECs. Mean distances between the two fluorophores decreased slightly when the AP-modified oligonucleotide was incubated with hypoxic nuclear extract in comparison to normoxic extract, while no changes in inter-site distance were noted for the WT HRE oligonucleotide. Thus, hypoxia-inducible DNA binding proteins, probably Hif-1, are capable of altering the conformation of a model VEGF HRE, but only when an abasic site was present. Perhaps most interestingly, the increased flexibility of the abasically modified HRE oligonucleotide required formation of a Ref-1/Ape1-mediated single-strand DNA break. Collectively, these observations point to the prospect that hypoxia-induced oxidative base modifications may cause dynamic changes in the functional properties of the VEGF HRE in part through Ref-1/Ape1-mediated strand break formation and attendant changes in local sequence flexibility.
In light of the above observations, it was reasonable to predict that increased Hif-1 DNA binding associated with the presence of an AP site – an intermediate step in BER-mediated restoration of DNA integrity - would enhance reporter gene expression. Consistent with this suspicion, the AP-modified VEGF HRE ligated to a luciferase reporter gene engendered 2-3 times more expression in hypoxic PAECs than the WT HRE (Figure 3; bottom panel) (Ziel et al., 2005). These observations support the idea that hypoxia-induced base modifications alter transcriptional complex assembly and promoter function in a model system, but they have not yet been confirmed in vivo.
6. Interesting similarities between hypoxia-induced DNA modifications and other transcriptional regulators
The observation that ROS generated during signaling may target sequences in inducible genes for oxidative base modifications and repair has been made not only for hypoxia, but for receptor-mediated stimuli as well. As previously noted, thrombin, PDGF, and angiotensin II also cause oxidative base modifications in the HRE of the VEGF promoter in PAECs (Ziel et al., 2005). In addition, while appearing in the literature somewhat more recently, estrogen and androgen receptor-mediated oxidative DNA damage and repair also are known to occur in various tumor cell lines; in these instances, associations between base damage and repair, transcription, and gene rearrangements underlying carcinogenesis and metastatic behavior have been explored in a comparatively thorough manner, with intriguing results.
In MCF-7 cells, for example, the estrogen-stimulated binding of liganded estrogen receptor (ER)-α to estrogen responsive elements (EREs) of the pS2 and Bcl2 genes causes the recruitment of histone demethylase, LSD-1, and attendant local generation of hydrogen peroxide. The hydrogen peroxide so produced seems to cause formation of base oxidation products in EREs with the subsequent recruitment of BER enzymes, strand cleavage, and repair, all of which appear to be required for transcriptional activation (Perillo et al., 2008). The specific mechanism of strand cleavage in ERE sequences has not been established; although the first two steps of the BER pathway involve enzymes with DNA strand cleaving activities – Ogg1 and Ref-1/Ape1, respectively (Almeida and Sobol, 2007) another strand cleaving enzyme, topoisomerase IIβ, also is known to be recruited to the sequence (Ju et al., 2006). Regardless of the specific mechanism, ROS-triggered strand breaks seem to be important for long-range changes in DNA topology, perhaps needed for the nuclear molecular motor-dependent repositioning of genes into distinct interchromatin granules serving as “transcription factories” (Hu et al., 2008).
In an important extension of these observations, it recently has been reported that androgen receptor signaling in prostate cancer cells also engenders DNA strand cleavage, possibly mediated by topoisomerase IIβ (Haffner et al., 2010). Androgen receptor-dependent strand cleavage occurring in isolation or in concert with other genotoxic stresses (e.g., radiation) may cause double strand DNA break formation (Lin et al., 2009). Most provocatively, such breaks, if occurring in genes located on different chromosomes but brought into close proximity for transcriptional activation – perhaps in so-called inter-chromatin granules – markedly increase the potential for translocation and rearrangement of sequences bracketed by the strand breaks (Lin et al., 2009; Nunez et al., 2008).
7. New concepts for the role of ROS in hypoxic transcriptional activation
Our observations show that hypoxia, like certain other physiologic signals, leads to ROS generation that targets specific DNA sequences for oxidative base modifications. The full physiologic and pathologic significance of these findings have yet to be defined. However, accumulating evidence suggests that such ROS-mediated base modifications could be important for regulating hypoxia-induced gene expression (Gillespie and Wilson, 2007). The general model, depicted in Figure 4, is founded on observations that hypoxia increases generation of ROS, probably from mitochondrial complex III as suggested by Chandel and coworkers (Bell et al., 2007a). The ROS so generated are vectored away from the mitochondrial matrix and its genome into the cytosol, as recently shown by Waypa and Schumacker (Waypa et al., 2010), thus explaining why hypoxia fails to cause widespread oxidative damage to the mtDNA. We believe that mitochondrial derived ROS directly or indirectly cause targeted base modifications in at least some hypoxia-inducible nuclear genes. Mechanisms by which HRE sequences are targeted are unknown, but could relate to sequence context, sequence binding of iron, or DNA charge transport phenomenon, as discussed previously. We further suspect that base modifications targeted to HREs serve to activate the BER pathway (Almeida and Sobol, 2007), which is responsible for formation of a strand break as part of the process needed to fully repair the oxidatively modified base. Though it is not yet established that a sequence-specific strand break occurs in living hypoxic cells, in our model oligonucleotide system, Ref-1/Ape1, a component of the multi-protein, hypoxia-inducible transcriptional complex and the enzyme mediating the second step in the BER pathway, is responsible for creating a single strand break at an AP replacing the hypoxia-modified guanine (Breit et al., 2008).
FIGURE 4.
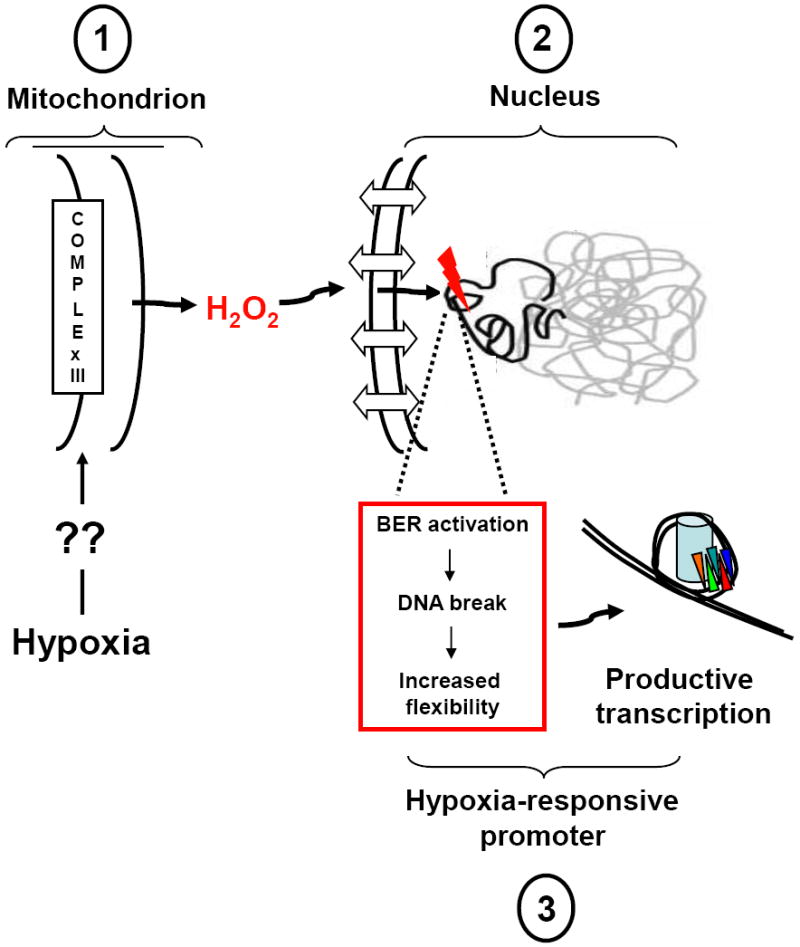
Proposed model for the role of hypoxia-generated ROS in transcriptional regulation. Key steps are: (1) By an unknown sensing and transduction mechanism, environmental hypoxia triggers mitochondrial superoxide anion production from the Uo site of complex III, thus vectoring the ROS away from the matrix-located mitochondrial genome from an oxidant stress. Superoxide anion released into the inner-membrane space is converted by resident superoxide dismutase to the comparatively long-lived signaling molecule, hydrogen peroxide, which diffuses into and accumulates in the cytosol. (2) Hydrogen peroxide generated from peri-nuclear mitochondria diffuses into the nucleus where it participates in Fenton reactions with DNA-bound iron, thus forming the highly reactive hydroxyl radical in the immediate vicinity of target DNA sequences. The reason why specific HRE sequences are targeted for oxidative base modifications is unclear, and may relate to those sequences being prepositioned in an un-protected, open conformation, to the location of DNA-bound iron in specific sequences, or to DNA charge transport which effectively tunnels ROS-mediated damage initiated elsewhere to guanine repeat sequences where the 5’ guanine is most sensitive to oxidant damage. (3) The oxidatively damaged base leads to processive recruitment of enzymes participating in the base excision pathway of DNA repair (BER). Importantly, the DNA glycosylases recognizing the oxidatively damaged base (e.g., Ogg1 in the case of oxidatively damaged purines) as well as the second enzyme in the BER pathway (Ref-1/Ape1), display strand cleaving activity that would result formation of a transient strand break in the HRE at the site of the initial oxidative damage. The strand break so produced would result in profound changes in local sequence topology and flexibility that are postulated to impact DNA-nucleosome apposition or long-range DNA conformation, thereby permitting the onset of productive transcription. Please see text for additional details and references.
We propose that formation and repair of strand breaks in HREs is a key event by which hypoxia-induced ROS stress impacts transcriptional activity. Our data suggest that breaks could introduce a “hinge” or flexible region in the sequence, which would have consequences for short and long-range sequence topologies. By enhancing local sequence flexibility, the break may facilitate dissociation between nucleosome core particles and promoter DNA, thereby minimizing structural impediments to transcription factor and/or chromatin remodeling enzyme binding (Haince et al., 2006). A dynamic hinge region also could influence long-range DNA conformational changes enabling distant sequences to be brought into close proximity with minimal energy expenditure. This process could involve nuclear molecular motors, as proposed for the repositioning of specific sequences of androgen-responsive genes into transcription factories (Hu et al., 2008). Considerably more work will be required to explore these ideas.
In addition to sequence flexibility, hypoxia-induced oxidative base modifications and the attendant formation of DNA repair-related strand breaks might influence other dynamic properties of DNA. For example, strand breaks are known to block transmission of supercoiling forces caused by RNA polymerase binding to transcription start sites, essentially isolating sequences up or down-stream from tortional strains linked to transcription initiation (Kouzine et al., 2008). It is thus reasonable to consider that hypoxia-induced formation of base oxidation products and strand break-repair intermediates in HREs could serve to buffer these functionally important sequences from mechanical stresses induced by events closer to transcription start sites, while their subsequent repair and re-ligation by the closing steps in BER would then permit transmission of mechanical forces from the transcription start site to upstream HRE sequences, or visa versa.
The biology of hypoxia-induced oxidant stress targeted to HRE sequences also may be impacted by long range DNA charge transport. As noted previously, in intact DNA sequences, oxidant stress triggered at a specific location is capable of “tunneling” or “hopping” through the π–stack of duplex DNA to exit the sequence at guanine multiplets; charge transport can occur for distances up to 200 Angstroms away from the initial site of oxidant stress (Genereux et al., 2010; Merino et al., 2008). Interestingly, guanine repeats are found either within, or in the vicinity of, Hif DNA recognition sequences in many promoters. According to the above-described model for long range DNA charge transport, the 5’ guanine in these guanine repeats would be expected to serve as a “sink” for oxidant stress initiated at remote sites. In this context, our data show that while low frequency, hypoxia-induced oxidative damage can be detected at many bases in the VEGF HRE, the most commonly damaged base is the 5’ guanine of a guanine triplet at the 5’ end of the Hif-1 DNA recognition sequence. It is thus tempting to speculate that ROS generated in hypoxic PAECs may target multiple bases in the VEGF HRE for oxidative modification, perhaps because of the sequence is located in or repositioned into a specific nuclear microdomain(s) enriched in ROS, but that the damage is “focused” on the 5’ end of the Hif-1 DNA recognition sequence because of charge transport to the uniquely sensitive guanine. This contention, too, will require much additional study.
The DNA charge transport concept also holds that some of the charge can exit the duplex at the sensitive 5’ guanine sites to recruit and redox activate bound proteins (Genereux et al., 2010; Merino et al., 2008). Indeed, this has already been shown to occur in bacterial systems for DNA-bound SoxR (Gorodetsky et al., 2008), a well-characterized iron-sulfur containing protein that functions as a transcriptional regulator of oxidant stress-response genes, for the bacterial BER glycosylases MutY and endonuclease III (Boal et al., 2009), which are also iron-sulfur containing proteins, and for the redox active mammalian transcription factor, p53 (Augustyn et al., 2007). All of these proteins, when bound to duplex DNA at positions remote from the initial site of oxidation, are recruited to the vicinity of sensitive 5’ guanines in guanine repeats and redox activated. These events, directly attributable to long range DNA charge transport, have been touted as means of localizing key DNA binding proteins to functionally important sequences where their activities can be discretely regulated by redox reactions emanating from the DNA to which they are bound. Applying this model to the involvement of ROS in hypoxic transcriptional regulation raises the intriguing possibility that the oxidative DNA damage localized to the Hif-1 DNA recognition sequence in the VEGF HRE (and perhaps other HRE sequences as well) functions to recruit the transcriptional co-activator and BER repair enzyme, Ref-1/Ape1, and govern its redox state via local reactions with the DNA sequence to which it is bound. In this manner, Ref-1/Ape1 would be functionally and spatially positioned such that it could dictate proper assembly of the hypoxia-inducible transcriptional complex, an activity supported by previous reports (Gray et al., 2005; Ziel, et al., 2004).
There is, admittedly, much speculation in the above discussion of the role of hypoxia-induced ROS stress in transcriptional regulation. A considerable degree of inferential information is drawn from ligand-dependent transcriptional regulatory systems – estrogen and androgen-dependent-mediated gene regulation, for example – for which the ability to extrapolate to hypoxia may be limited. The contribution of long range DNA charge transport to transcriptional regulation also is poorly understood at present. But, the general observations described herein - that hypoxia-induced ROS generation, now well entrenched in the literature, targets nuclear DNA for oxidative modifications - clearly has important implications. Even if the notion that such modifications are important for transcriptional regulation is misguided, the fact that nuclear genes are oxidatively threatened by mechanisms responding to a fundamental stimulus like hypoxia links physiological signaling to initiation of genomic instability, a prospect which has considerable implications for aging and for age-related diseases such as cancer and other disorders in which hypoxia plays a pathogenic role.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Almeida KH, Sobol RW. A unified view of base excision repair: lesion-dependent protein complexes regulated by post-translational modification. DNA Repair (Amst) 2007;6:695–711. doi: 10.1016/j.dnarep.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustyn KE, Merino EJ, Barton JK. A role for DNA-mediated charge transport in regulating p53: Oxidation of the DNA-bound protein from a distance. Proc Natl Acad Sci U S A. 2007;104:18907–12. doi: 10.1073/pnas.0709326104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell EL, Klimova TA, Eisenbart J, Moraes CT, Murphy MP, Budinger GR, Chandel NS. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J Cell Biol. 2007a;177:1029–36. doi: 10.1083/jcb.200609074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell EL, Klimova TA, Eisenbart J, Schumacker PT, Chandel NS. Mitochondrial reactive oxygen species trigger hypoxia-inducible factor-dependent extension of the replicative life span during hypoxia. Mol Cell Biol. 2007b;27:5737–45. doi: 10.1128/MCB.02265-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boal AK, Genereux JC, Sontz PA, Gralnick JA, Newman DK, Barton JK. Redox signaling between DNA repair proteins for efficient lesion detection. Proc Natl Acad Sci U S A. 2009;106:15237–42. doi: 10.1073/pnas.0908059106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breit JF, Ault-Ziel K, Al-Mehdi AB, Gillespie MN. Nuclear protein-induced bending and flexing of the hypoxic response element of the rat vascular endothelial growth factor promoter. Faseb J. 2008;22:19–29. doi: 10.1096/fj.07-8102com. [DOI] [PubMed] [Google Scholar]
- Bristow RG, Hill RP. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer. 2008;8:180–92. doi: 10.1038/nrc2344. [DOI] [PubMed] [Google Scholar]
- Ebert BL, Bunn HF. Regulation of transcription by hypoxia requires a multiprotein complex that includes hypoxia-inducible factor 1, an adjacent transcription factor, and p300/CREB binding protein. Mol Cell Biol. 1998;18:4089–96. doi: 10.1128/mcb.18.7.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerling BM, Platanias LC, Black E, Nebreda AR, Davis RJ, Chandel NS. Mitochondrial reactive oxygen species activation of p38 mitogen-activated protein kinase is required for hypoxia signaling. Mol Cell Biol. 2005;25:4853–62. doi: 10.1128/MCB.25.12.4853-4862.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans AR, Limp-Foster M, Kelley MR. Going APE over ref-1. Mutat Res. 2000;461:83–108. doi: 10.1016/s0921-8777(00)00046-x. [DOI] [PubMed] [Google Scholar]
- Genereux JC, Boal AK, Barton JK. DNA-mediated charge transport in redox sensing and signaling. J Am Chem Soc. 2010;132:891–905. doi: 10.1021/ja907669c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie MN, Wilson GL. Bending and breaking the code: dynamic changes in promoter integrity may underlie a new mechanism regulating gene expression. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1–3. doi: 10.1152/ajplung.00275.2006. [DOI] [PubMed] [Google Scholar]
- Goni JR, de la Cruz X, Orozco M. Triplex-forming oligonucleotide target sequences in the human genome. Nucleic Acids Res. 2004;32:354–60. doi: 10.1093/nar/gkh188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorodetsky AA, Dietrich LE, Lee PE, Demple B, Newman DK, Barton JK. DNA binding shifts the redox potential of the transcription factor SoxR. Proc Natl Acad Sci U S A. 2008;105:3684–9. doi: 10.1073/pnas.0800093105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray MJ, Zhang J, Ellis LM, Semenza GL, Evans DB, Watowich SS, Gallick GE. HIF-1alpha, STAT3, CBP/p300 and Ref-1/APE are components of a transcriptional complex that regulates Src-dependent hypoxia-induced expression of VEGF in pancreatic and prostate carcinomas. Oncogene. 2005;24:3110–20. doi: 10.1038/sj.onc.1208513. [DOI] [PubMed] [Google Scholar]
- Grishko V, Solomon M, Breit JF, Killilea DW, Ledoux SP, Wilson GL, Gillespie MN. Hypoxia promotes oxidative base modifications in the pulmonary artery endothelial cell VEGF gene. Faseb J. 2001;15:1267–9. doi: 10.1096/fj.00-0755fje. [DOI] [PubMed] [Google Scholar]
- Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, Schumacker PT. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1:401–8. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Haffner MC, Aryee MJ, Toubaji A, Esopi DM, Albadine R, Gurel B, Isaacs WB, Bova GS, Liu W, Xu J, Meeker AK, Netto G, De Marzo AM, Nelson WG, Yegnasubramanian S. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat Genet. 2010;42:668–75. doi: 10.1038/ng.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haince JF, Rouleau M, Poirier GG. Transcription. Gene expression needs a break to unwind before carrying on. Science. 2006;312:1752–3. doi: 10.1126/science.1129808. [DOI] [PubMed] [Google Scholar]
- Hu Q, Kwon YS, Nunez E, Cardamone MD, Hutt KR, Ohgi KA, Garcia-Bassets I, Rose DW, Glass CK, Rosenfeld MG, Fu XD. Enhancing nuclear receptor-induced transcription requires nuclear motor and LSD1-dependent gene networking in interchromatin granules. Proc Natl Acad Sci U S A. 2008;105:19199–204. doi: 10.1073/pnas.0810634105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju BG, Lunyak VV, Perissi V, Garcia-Bassets I, Rose DW, Glass CK, Rosenfeld MG. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science. 2006;312:1798–802. doi: 10.1126/science.1127196. [DOI] [PubMed] [Google Scholar]
- Killilea DW, Hester R, Balczon R, Babal P, Gillespie MN. Free radical production in hypoxic pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2000;279:L408–12. doi: 10.1152/ajplung.2000.279.2.L408. [DOI] [PubMed] [Google Scholar]
- Kouzine F, Sanford S, Elisha-Feil Z, Levens D. The functional response of upstream DNA to dynamic supercoiling in vivo. Nat Struct Mol Biol. 2008;15:146–54. doi: 10.1038/nsmb.1372. [DOI] [PubMed] [Google Scholar]
- Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–19. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- Lin C, Yang L, Tanasa B, Hutt K, Ju BG, Ohgi K, Zhang J, Rose DW, Fu XD, Glass CK, Rosenfeld MG. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell. 2009;139:1069–83. doi: 10.1016/j.cell.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JQ, Zelko IN, Erbynn EM, Sham JS, Folz RJ. Hypoxic pulmonary hypertension: role of superoxide and NADPH oxidase (gp91phox) Am J Physiol Lung Cell Mol Physiol. 2006;290:L2–10. doi: 10.1152/ajplung.00135.2005. [DOI] [PubMed] [Google Scholar]
- Marshall C, Mamary AJ, Verhoeven AJ, Marshall BE. Pulmonary artery NADPH-oxidase is activated in hypoxic pulmonary vasoconstriction. Am J Respir Cell Mol Biol. 1996;15:633–44. doi: 10.1165/ajrcmb.15.5.8918370. [DOI] [PubMed] [Google Scholar]
- Merino EJ, Boal AK, Barton JK. Biological contexts for DNA charge transport chemistry. Curr Opin Chem Biol. 2008;12:229–37. doi: 10.1016/j.cbpa.2008.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez E, Kwon YS, Hutt KR, Hu Q, Cardamone MD, Ohgi KA, Garcia-Bassets I, Rose DW, Glass CK, Rosenfeld MG, Fu XD. Nuclear receptor-enhanced transcription requires motor- and LSD1-dependent gene networking in interchromatin granules. Cell. 2008;134:189. doi: 10.1016/j.cell.2008.06.045. [DOI] [PubMed] [Google Scholar]
- Pastukh V, Ruchko M, Gorodnya O, Wilson GL, Gillespie MN. Sequence-specific oxidative base modifications in hypoxia-inducible genes. Free Radic Biol Med. 2007;43:1616–26. doi: 10.1016/j.freeradbiomed.2007.08.027. [DOI] [PubMed] [Google Scholar]
- Perillo B, Ombra MN, Bertoni A, Cuozzo C, Sacchetti S, Sasso A, Chiariotti L, Malorni A, Abbondanza C, Avvedimento EV. DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science. 2008;319:202–6. doi: 10.1126/science.1147674. [DOI] [PubMed] [Google Scholar]
- Qin Y, Hurley LH. Structures, folding patterns, and functions of intramolecular DNA G-quadruplexes found in eukaryotic promoter regions. Biochimie. 2008;90:1149–71. doi: 10.1016/j.biochi.2008.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai P, Cole TD, Wemmer DE, Linn S. Localization of Fe(2+) at an RTGR sequence within a DNA duplex explains preferential cleavage by Fe(2+) and H2O2. J Mol Biol. 2001;312:1089–101. doi: 10.1006/jmbi.2001.5010. [DOI] [PubMed] [Google Scholar]
- Rai P, Wemmer DE, Linn S. Preferential binding and structural distortion by Fe2+ at RGGG-containing DNA sequences correlates with enhanced oxidative cleavage at such sequences. Nucleic Acids Res. 2005;33:497–510. doi: 10.1093/nar/gki192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve HL, Michelakis E, Nelson DP, Weir EK, Archer SL. Alterations in a redox oxygen sensing mechanism in chronic hypoxia. J Appl Physiol. 2001;90:2249–56. doi: 10.1152/jappl.2001.90.6.2249. [DOI] [PubMed] [Google Scholar]
- Rey S, Semenza GL. Hypoxia-inducible factor-1-dependent mechanisms of vascularization and vascular remodelling. Cardiovasc Res. 2010;86:236–42. doi: 10.1093/cvr/cvq045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci C, Pastukh V, Leonard J, Turrens J, Wilson G, Schaffer D, Schaffer SW. Mitochondrial DNA damage triggers mitochondrial-superoxide generation and apoptosis. Am J Physiol Cell Physiol. 2008;294:C413–22. doi: 10.1152/ajpcell.00362.2007. [DOI] [PubMed] [Google Scholar]
- Ruchko MV, Gorodnya OM, Pastukh VM, Swiger BM, Middleton NS, Wilson GL, Gillespie MN. Hypoxia-induced oxidative base modifications in the VEGF hypoxia-response element are associated with transcriptionally active nucleosomes. Free Radic Biol Med. 2009;46:352–9. doi: 10.1016/j.freeradbiomed.2008.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroedl C, McClintock DS, Budinger GR, Chandel NS. Hypoxic but not anoxic stabilization of HIF-1alpha requires mitochondrial reactive oxygen species. Am J Physiol Lung Cell Mol Physiol. 2002;283:L922–31. doi: 10.1152/ajplung.00014.2002. [DOI] [PubMed] [Google Scholar]
- Tell G, Quadrifoglio F, Tribelli C, Kelley M. The many functions of APE1/Ref-1: Not only a DNA repair enzyme. Antiox Redox Signal. 2009;11:601–619. doi: 10.1089/ars.2008.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaux EC, Metzen E, Yeates KM, Ratcliffe PJ. Regulation of hypoxia-inducible factor is preserved in the absence of a functioning mitochondrial respiratory chain. Blood. 2001;98:296–302. doi: 10.1182/blood.v98.2.296. [DOI] [PubMed] [Google Scholar]
- Ward JP. A twist in the tail: synergism between mitochondria and NADPH oxidase in the hypoxia-induced elevation of reactive oxygen species in pulmonary artery. Free Radic Biol Med. 2008 doi: 10.1016/j.freeradbiomed.2008.08.015. [DOI] [PubMed] [Google Scholar]
- Waypa GB, Chandel NS, Schumacker PT. Model for hypoxic pulmonary vasoconstriction involving mitochondrial oxygen sensing. Circ Res. 2001;88:1259–66. doi: 10.1161/hh1201.091960. [DOI] [PubMed] [Google Scholar]
- Waypa GB, Marks JD, Guzy R, Mungai PT, Schriewer J, Dokic D, Schumacker PT. Hypoxia triggers subcellular compartmental redox signaling in vascular smooth muscle cells. Circ Res. 2010;106:526–35. doi: 10.1161/CIRCRESAHA.109.206334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissmann N, Zeller S, Schafer RU, Turowski C, Ay M, Quanz K, Ghofrani HA, Schermuly RT, Fink L, Seeger W, Grimminger F. Impact of mitochondria and NADPH oxidases on acute and sustained hypoxic pulmonary vasoconstriction. Am J Respir Cell Mol Biol. 2006;34:505–13. doi: 10.1165/rcmb.2005-0337OC. [DOI] [PubMed] [Google Scholar]
- Wolin MS, Ahmad M, Gupte SA. Oxidant and redox signaling in vascular oxygen sensing mechanisms: basic concepts, current controversies, and potential importance of cytosolic NADPH. Am J Physiol Lung Cell Mol Physiol. 2005;289:L159–73. doi: 10.1152/ajplung.00060.2005. [DOI] [PubMed] [Google Scholar]
- Workman JL. Nucleosome displacement in transcription. Genes Dev. 2006;20:2009–17. doi: 10.1101/gad.1435706. [DOI] [PubMed] [Google Scholar]
- Wu Q, Gaddis SS, MacLeod MC, Walborg EF, Thames HD, DiGiovanni J, Vasquez KM. High-affinity triplex-forming oligonucleotide target sequences in mammalian genomes. Mol Carcinog. 2007;46:15–23. doi: 10.1002/mc.20261. [DOI] [PubMed] [Google Scholar]
- Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci U S A. 1997;94:514–9. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young SD, Marshall RS, Hill RP. Hypoxia induces DNA overreplication and enhances metastatic potential of murine tumor cells. Proc Natl Acad Sci U S A. 1988;85:9533–7. doi: 10.1073/pnas.85.24.9533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Glazer PM. Mutagenesis induced by the tumor microenvironment. Mutat Res. 1998;400:439–46. doi: 10.1016/s0027-5107(98)00042-6. [DOI] [PubMed] [Google Scholar]
- Ziel KA, Campbell CC, Wilson GL, Gillespie MN. Ref-1/Ape is critical for formation of the hypoxia-inducible transcriptional complex on the hypoxic response element of the rat pulmonary artery endothelial cell VEGF gene. Faseb J. 2004;18:986–988. doi: 10.1096/fj.03-1160fje. [DOI] [PubMed] [Google Scholar]
- Ziel KA, Grishko V, Campbell CC, Breit JF, Wilson GL, Gillespie MN. Oxidants in signal transduction: Impact on DNA integrity and gene expression. FASEB J. 2005;19:387–394. doi: 10.1096/fj.04-2805com. [DOI] [PubMed] [Google Scholar]