

Abstract
An organism requires a range of biomolecules for its growth. By definition, these are essential molecules which constitute the basic metabolic requirements of an organism. A small organic molecule with chemical similarity to that of an essential metabolite may bind to the enzyme that catalyzes its production and inhibit it, likely resulting in the stasis or death of the organism. Here, we report a high-throughput approach for identifying essential metabolites of an organism using genetic and biochemical approaches and then implement computational approaches to identify metabolite mimics. We generated and genotyped 5,126 Mycobacterium tuberculosis mutants and performed a statistical analysis to determine putative essential genes. The essential molecules of M. tuberculosis were classified as products of enzymes that are encoded by genes in this list. Although incomplete, as many enzymes of M. tuberculosis have yet to be identified and characterized, this is the first report of a large number of essential molecules of the organism. We identified essential metabolites of three distinct metabolic pathways in M. tuberculosis and selected molecules with chemical similarity using cheminformatics strategies that illustrate a variety of different pharmacophores. Our approach is aimed at systematic identification of essential molecules and their mimics as a blueprint for development of effective chemical probes of M. tuberculosis metabolism, with the ultimate goal of seeking drugs that can kill this pathogen. As an illustration of this approach, we report that compounds JFD01307SC and l-methionine-S-sulfoximine, which share chemical similarity with an essential molecule of M. tuberculosis, inhibited the growth of this organism at micromolar concentrations.
IMPORTANCE
The estimate that more lives may have been lost in 2009 due to tuberculosis (TB) than in any year in history is alarming. Approximately 9.2 million new cases and 1.8 million deaths due to TB were reported in 2008. The widespread prevalence of Mycobacterium tuberculosis strains that are resistant to drugs currently used to treat TB means that new drugs are urgently needed to treat these infections. Here, we have identified pathways for the biosynthesis of essential metabolites and associated enzymes in M. tuberculosis using a genetics-based approach. Small molecules that mimic these essential metabolites were identified, and some of them were shown to inhibit the growth of M. tuberculosis. Therefore, we illustrate an approach based on genetics to develop inhibitors that have the potential to be advanced as candidate drugs for treating TB.
INTRODUCTION
The availability of whole-genome sequences of bacterial pathogens was rapidly followed by high-throughput identification of genes essential for their proliferation. One of the main goals of these studies was to identify proteins that could be targeted for inhibition of growth, with the ultimate aim of developing drugs to treat infections with the pathogens. Mycobacterium tuberculosis is the etiological agent of tuberculosis (TB), an infectious disease of which ~9 million new cases and ~1.7 million mortalities were reported for the year 2009 (1). Seminal studies of the global burden of TB conducted by the WHO have declared drug-susceptible M. tuberculosis or readily treatable TB a global health emergency (2, 3). New and effective drugs are required, as existing drugs have become suboptimal in many settings due to the requirement for protracted periods of treatment and the emergence of multiple- and extensively drug resistant strains of M. tuberculosis (4, 5). Identification of genes that encode essential proteins is a first step toward determining targets whose inhibition may result in the arrest of growth or killing of M. tuberculosis.
In a seminal study, Sassetti and colleagues used a high-throughput method based on a whole-genome microarray to identify essential genes of M. tuberculosis by random mutagenesis (6). This essential gene set of M. tuberculosis represents the basic requirement for the growth and proliferation of the organism in a defined medium in vitro. The availability of these data facilitated further investigations to map the metabolic pathways and metabolites required by M. tuberculosis for proliferation. Given our focus on the development of drugs against TB based on targeting the essential metabolic machinery of M. tuberculosis, we herein provide a proof of concept for how one may transition from an essential gene to an enzyme to an essential metabolite to a potential inhibitor of the enzyme with whole-cell viability.
M. tuberculosis, like any organism, requires an array of biomolecules for its survival. These biomolecules either are obtained from the environment or are synthesized de novo. An enzyme that catalyzes the biosynthesis of an essential metabolite is itself essential, if we assume that there are no alternative enzymes or metabolic pathways capable of synthesizing the same metabolite. Therefore, inhibiting essential enzymes is an effective way to kill or arrest the growth of an organism.
Many drugs that are highly successful in human clinical use mimic a substrate or a product of essential enzymes. For example, folic acid is an essential biomolecule which needs to be synthesized de novo by many bacteria, and dihydropteroate synthase, an enzyme in the folic acid biosynthesis pathway, synthesizes dihydrofolate from p-aminobenzoate (7). Sulfonamide-based drugs are structural analogs of p-aminobenzoate and act by inhibiting dihydropteroate synthase (8). Many bacterial infections are effectively treated with sulfonamides, as they mimic an essential substrate and competitively inhibit an essential enzyme. Another well-known class of drugs that mimics an essential biomolecule is the β-lactam group of antibiotics (9). They possess a core four-membered ring that mimics the structure of the terminal d-alanyl-d-alanine subunit of the peptidoglycan layer in bacterial cell walls. This peptidoglycan layer is essential for the survival of bacteria, as it is required for maintenance of cell structure (10). Transpeptidases required for cross-linking of peptide chains to network the peptidoglycan layer use terminal d-alanyl-d-alanine as a substrate. By mimicking the natural substrate, β-lactams engage the transpeptidases and inhibit their actions, leading to destruction of the peptidoglycan layer and the eventual death of the organism (11). There are numerous other examples of inhibition of essential enzymes by mimicking their substrates.
In this study, we identified essential metabolites of M. tuberculosis using the strategy summarized in Fig. 1. We began by identifying genes and associated enzymes that are essential for the growth and proliferation of the bacterium. For this we used the Himar1 transposon to disrupt genes of M. tuberculosis. Nearly all genes are susceptible to disruption, as the transposon requires only the TA dinucleotide for insertion (12). The transposon is delivered into M. tuberculosis cells using suicide mycobacteriophage, and the transduced mutants are obtained from drug-containing plates (13, 14). A genome walking technique was used to locate the site of transposon insertion and to determine the identity of the disrupted gene (15). Genes whose disruptions do not compromise the growth of the bacilli are classified as nonessential.
FIG 1 .

Schematic for identification of essential molecules and their mimics. The genome of an organism is mutagenized to saturation, and nonessential genes whose loss does not compromise the ability of an organism to proliferate are identified. The genes whose loss could not be tolerated are essential and code for essential proteins that have structural or regulatory functions or are enzymes. The metabolite(s) that is produced by an essential enzyme is determined, and its molecular mimic(s) with pharmacological potential is identified using cheminformatics. These mimics may bind to the essential enzyme, inhibit its function, and eventually kill the organism.
Our method is based on sequencing of the site of mutagenesis to identify nonessential genes and, subsequently, the essential genes (15). We then identify metabolites that are essential for the growth and proliferation of M. tuberculosis as the products of reactions catalyzed by enzymes encoded by essential genes. Our current collection contains 5,126 independent, genotyped, and archived mutants with disruptions in both intra- and intergenic regions. A statistical analysis was performed to predict the essentiality of the genes that we were unable to disrupt. Here, we report that the pathways for peptidoglycan, tetrapyrrole, and chorismate biosynthesis are essential for the growth of M. tuberculosis. The products of the enzymes that comprise these pathways are identified as essential metabolites. We then discuss a cheminformatics strategy to identify mimics of the essential M. tuberculosis metabolites that may be rational competitive inhibitors of the essential enzymes and may thereby kill or inhibit the growth of M. tuberculosis. These mimics are starting points for further development of molecules with increased-potency activity against M. tuberculosis for use as chemical probes of metabolism or as antitubercular agents.
RESULTS
Identification of nonessential and essential genes.
We created a library of 5,126 unique transposon insertion mutants of M. tuberculosis. This collection represents the disruption and therefore loss of function of 2,246 unique genes. As we were able to isolate viable mutants, we define the genes disrupted in these mutants to be nonessential for in vitro growth. In order to ensure that the function of any gene classified as nonessential was truly disrupted by mutagenesis, all genes with far-distal insertions, as defined by the 5′ 80%/3′ 100-bp test (which qualifies a transposon insertion in the proximal [5′] 80% of the gene or upstream of the distal [3′] 100 bp of the gene to be able to effectively disrupt the gene), were excluded from the nonessential gene list (15). Two hundred twelve genes were found with distal insertions and removed from the nonessential gene list (see Table S1 in the supplemental material). Similarly, if a disrupted gene had an isoenzyme that catalyzed the same reaction, the function of the disrupted gene would not be affected since the isoenzyme would carry out the reaction in the mutant. In these cases, the identification of a mutant with a single insertion in a gene with an isoenzyme was irrelevant to whether that gene’s function is essential for in vitro growth. Twenty-four disrupted genes were found to have isoenzymes and were removed from the nonessential list. Finally, genes that were not disrupted were reclassified as nonessential if they belonged to an operon with an insertion proximal to the gene of interest due to a likely polar effect on such genes. A total of 534 nondisrupted genes were classified as nonessential because their operons contained a proximal insertion. This filtering process thus left us with essential genes under this specific in vitro growth condition.
The metabolic requirement and availability of nutrients during an infection differ from the in vitro conditions that were used to identify essential genes in our study. Certain metabolites that have to be synthesized de novo during growth in vitro may be available in macrophages or in the extracellular environment during an infection, allowing the organism to bypass the requirement to synthesize the metabolites that may be identified as essential in our study and vice versa. Therefore, our method clearly does not identify all the metabolites required for growth and proliferation during an infection. However, as our understanding grows with respect to how the metabolism of M. tuberculosis changes given its environment, we anticipate application of our methodology to derive state-specific essential genes.
Definition of the essential molecules of M. tuberculosis and exemplary pathways.
Approximately 40% of M. tuberculosis genes are annotated as “hypothetical,” as they encode proteins whose functions have yet to be characterized (16). In this study, we focused on the genes that encode enzymes with homology to known enzymes, allowing facile identification of their products, which we hereby define as essential metabolites. At this time, we count 401 essential enzyme-encoding genes of M. tuberculosis via our genetic method, which should afford as many nonredundant essential metabolites of M. tuberculosis. Rather than exhaustively enumerate each of these, we have instead chosen to discuss three pathways where each of the steps processes an essential metabolite, thus highlighting the critical role they play in M. tuberculosis metabolism. Essential metabolites of these pathways, namely, peptidoglycan, chorismate, and tetrapyrrole biosynthesis, their potential molecular mimics, and their respective chemical similarities and known pharmacological information are presented (see Table S2 in the supplemental material).
The peptidoglycan biosynthetic pathway begins with an assembly of sugar and amino acid residues to generate a disaccharyl-pentapeptide (or tetrapeptide). The associated reactions occur internal to the plasma membrane and are catalyzed by a series of enzymes. This pathway and associated putative enzymes are shown in Fig. 2A. The core of the pathway begins with conversion of UDP-N-acetyl-d-glucosamine to UDP-N-acetyl-d-glucosamine-enol pyruvate and ends with generation of disaccharyl-pentapeptide, GlcNAc-MurNAc-l-Ala1-d-iGln2-mesoDapNH23-d-Ala4-d-Ala5 (17). Figure 2A also lists putative genes that encode enzymes associated with de novo biosynthesis of amino acid residues that comprise the pentapeptide chain. These 23 genes that constitute the peptidoglycan biosynthesis pathway contain a total of 416 eligible TA transposition sites that may be disrupted by the transposon. However, none of these sites could sustain a viable insertion disruption, as they were not represented among the 5,126 independent mutants in our collection. Figure 2B depicts a composite locus for all genes that comprise the peptidoglycan biosynthesis pathway and another random locus that is identical in size and TA density. According to our mutagenesis data, the random locus sustained 36 transposition disruptions, whereas none were observed in the loci for the peptidoglycan biosynthesis pathway. We used these data to assess the essentiality of the pathway given that there are 63,956 total TA sites in the open reading frames of the M. tuberculosis genome. The P value for the likelihood that the pathway for peptidoglycan biosynthesis is essential is 3.52 × 10−15. Therefore, we conclude that the pathway for peptidoglycan biosynthesis is essential for the growth and survival of M. tuberculosis. Disruption of the above-mentioned genes that constitute the peptidoglycan biosynthesis pathway was also not observed in a comprehensive mutagenesis study undertaken by Sassetti et al. (6). The constituent essential metabolites of this pathway are shown in Table 1.
FIG 2 .

(A) Genes and metabolites that comprise the pathway for the biosynthesis of the building blocks of peptidoglycan in M. tuberculosis; (B) composite of 23 genes of M. tuberculosis of the peptidoglycan biosynthesis pathway (right) and a depiction of random loci with identical sizes and TA densities. The arrows show the number of transposon insertions that each locus was able to sustain.
TABLE 1 .
Representative essential molecules of the peptidoglycan biosynthesis pathway in M. tuberculosis and their mimics
| Essential molecule | Molecule ID, % similarity, MIC90 for M. tuberculosis (μg/ml), and structure of an analoga |
|
|---|---|---|
UDP-GlcNAc-enol pyruvate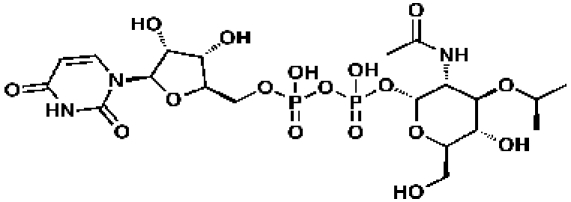
|
48239, 52, 0.237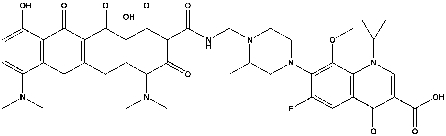
|
351369, 57, 0.13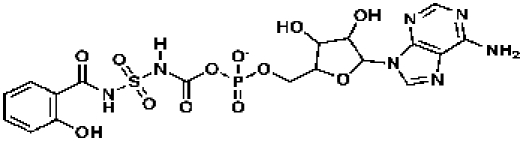
|
meso-Diaminopimelate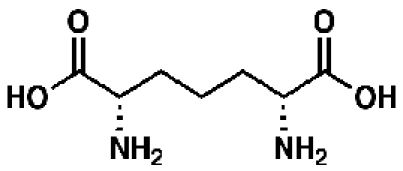
|
92585, 52, 0.21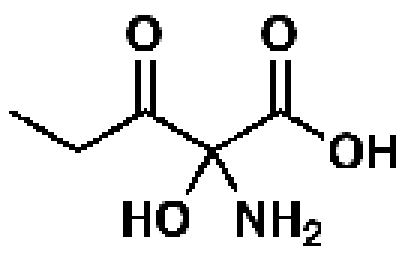
|
341652, 62, 43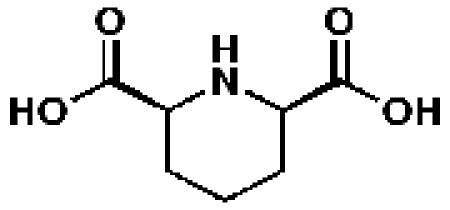
|
d-Alanyl-d-alanine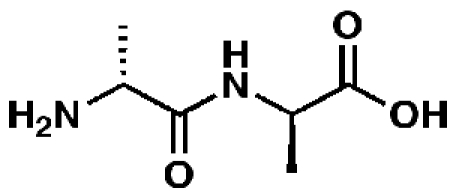
|
58257, 51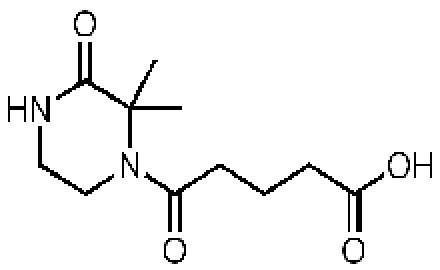
|
344159, 53, 29.5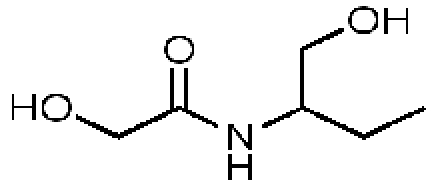
|
Representative essential molecules of the pathway for peptidoglycan biosynthesis in M. tuberculosis and examples of existing analogs. An identification provided by the CDD database (molecule ID), percent similarity to the corresponding essential molecule, and MIC90 for M. tuberculosis are shown. The MIC90 data are compiled in the CDD database from various laboratories. The percent inhibition of molecule 58257 at a 10 µM concentration was 8.98%.
The chorismate pathway is common to the biosynthesis of l-tryptophan, l-tyrosine, and folic acid and consists of seven genes, aroA to -C, -E, -G, -K, and -Q in M. tuberculosis. The aroC, -K, -B, and -Q genes are located in a putative operon, while aroA, -E, and -G are scattered around the genome. Each of these genes has multiple TA sites and is thus susceptible to insertion disruption by Himar1 transposon. However, we were unable to isolate viable mutants with disruption of any of these genes despite our attempts, resulting in 5,126 unique mutants. Our statistical analysis for the probability of this observation yielded a P value of 0.007. Therefore, we conclude that the pathway for chorismate biosynthesis is essential for the survival and growth of M. tuberculosis, and its essential molecules are shown in Table 2. It should be noted that a previous study showed that M. tuberculosis aroK is essential, as the investigators were unable to isolate a mutant with a deletion of this gene (18).
TABLE 2 .
Essential molecules of the chorismate biosynthesis pathway in M. tuberculosis and their mimics
| Essential molecule | Molecule ID, % similarity, MIC90 for M. tuberculosis (μg/ml), and structure of an analoga | ||
|---|---|---|---|
3-Dehydroquinate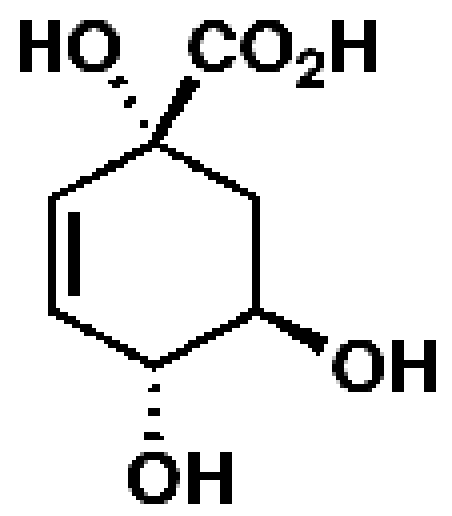
|
17699, 55, 14.7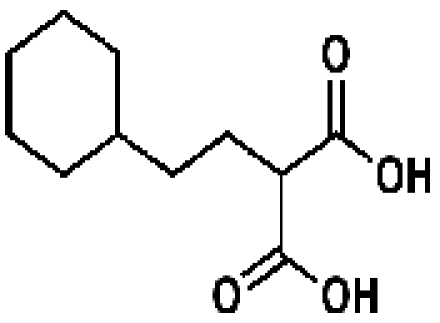
|
52283. 56, 2.07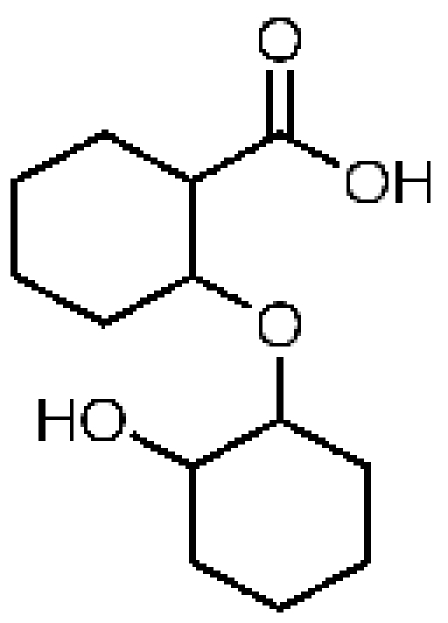
|
110597, 70, 7.1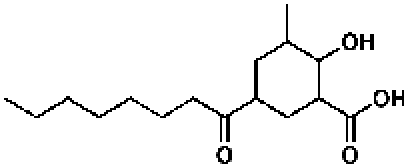
|
3-Dehydro-shikimate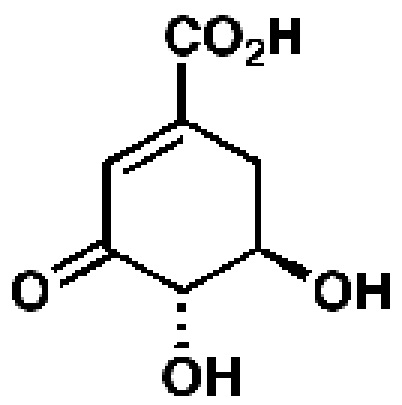
|
209508, 50, 12.0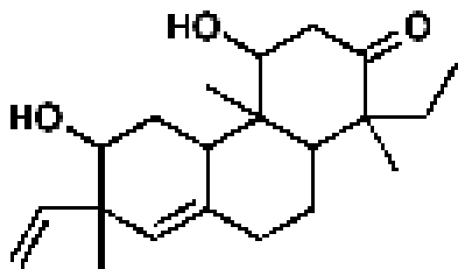
|
398410, 52, 2.3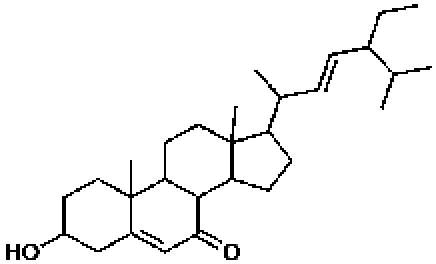
|
398407, 52, 4.6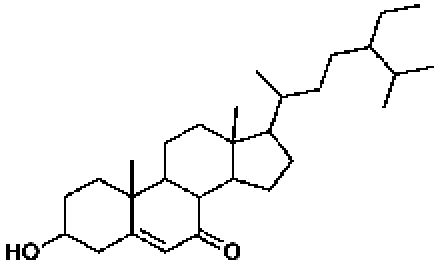
|
Shikimate-3-phosphate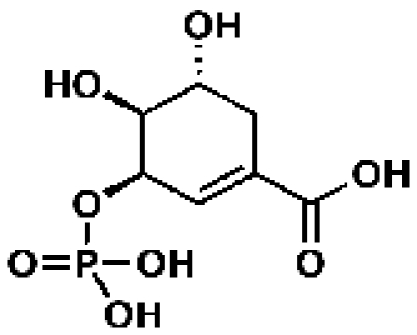
|
50320, 41, 73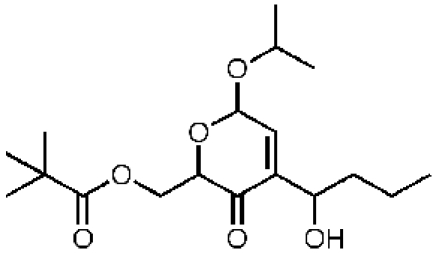
|
170440, 42, 14.4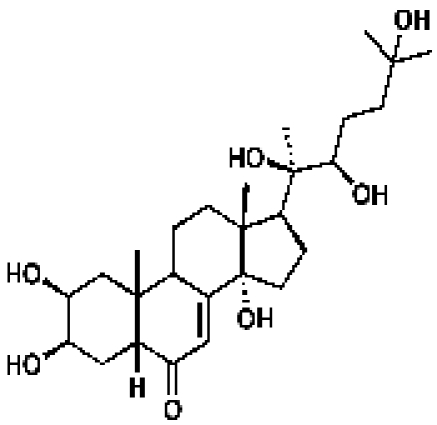
|
340891, 43, 10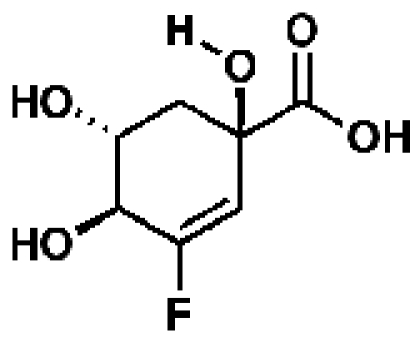
|
5-Enolpyruvyl-shikimate-3-phosphate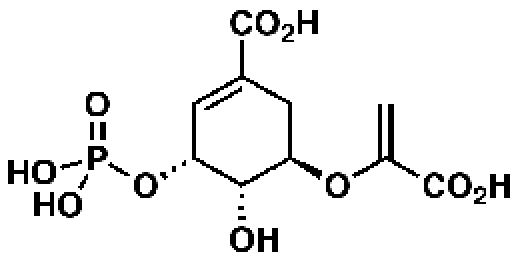
|
340430, 52, 3.1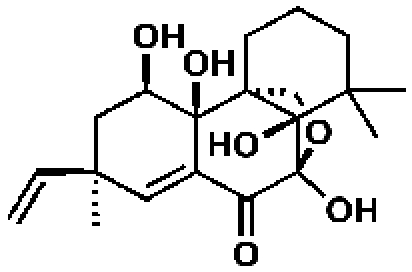
|
163046, 55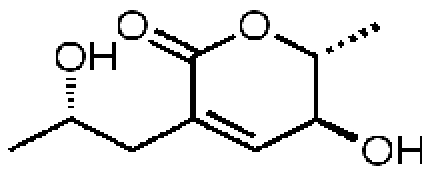
|
168409, 56
|
Chorismate
|
167369, 45. 0.98
|
340733, 50, 26
|
53251, 50, 10.16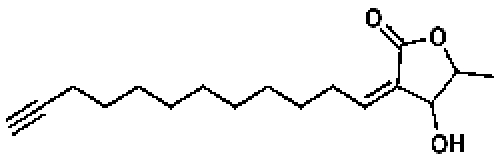
|
Representative essential molecules of the pathway for chorismate biosynthesis in M. tuberculosis and examples of existing analogs. An identification provided by the CDD database, percent similarity to the corresponding essential molecule, and MIC90 for M. tuberculosis are shown. The MIC90 data are compiled in the CDD database from various laboratories. The percent inhibition of molecule 163046 at a 10 µM concentration was 8.8%, and that for molecule 168409 was 48.6%.
M. tuberculosis possesses six conserved genes that are dedicated to tetrapyrrole biosynthesis. The genes hemA to -D, hemL, and gltS are located in three regions in the chromosome. The hemA to -D genes comprise a putative operon. The loci containing hemABCD, adjacent genes, and the sites at which the Himar1 transposon has inserted are shown (see Fig. S1 in the supplemental material). None of the genes that comprise the tetrapyrrole biosynthesis pathway have sustained a transposon insertion. The P value for not observing a transposon insertion in any of the genes in our collection of 5,126 genotyped mutations is 7 × 10−5. Therefore, we conclude with high confidence that the pathway for tetrapyrrole biosynthesis is essential in M. tuberculosis. The essential molecules of this pathway are listed elsewhere (see Table S3 in the supplemental material).
Search for mimics of essential metabolites using cheminformatics strategies.
The essential metabolites of M. tuberculosis represent a significant resource in the search for chemical probes/tools of metabolism that may inspire novel antitubercular therapeutics. While the specific biological activity of an essential molecule is critical to these applications, in most cases, one expects a lack of drug-like properties to limit utility. A number of strategies can be considered for identifying a set of more druggable small molecules that mimic an essential molecule. Principally, all strategies should involve utilization of the neighborhood principle (19, 20), which states that structurally similar molecules should exhibit similar properties and, in particular, biological activities. This approach has been used frequently in drug discovery (21–24) and in other areas, such as therapeutic drug monitoring (25). Different computational metrics may be used to assess the similarity of two molecules, including one-dimensional (1-D), 2-D, and 3-D methods (24, 26). Quite simply, for example, one can use Tanimoto similarity (24) to find 2-D similar molecules meeting a given level of similarity (the higher the better) to the identified essential metabolite. Another approach is to generate a 3-D pharmacophore around a molecule (27), perhaps with some molecular shape restriction to form a 3-D query, which could be used to search large multiconformer libraries of compounds. Examples of such compound libraries to be searched could include molecules with antitubercular activity, at the whole-cell and/or enzyme or target level, such as those available in the Collaborative Drug Discovery (CDD) TB database, consisting of many individual data sets of compounds tested against TB, malaria, and other diseases (27, 28). Searching metabolite databases, such as the Kyoto Encyclopedia of Genes and Genomes (KEGG) (29, 30) and LipidMaps (31), or databases composed of commercially available molecules like those found in ChemSpider (32), eMolecules, and ZINC (33) could also be fruitful for identifying similar molecules for potential testing.
We have chosen for our first attempts in essential molecule mimicry to rely on 2-D Tanimoto similarity. The choice of Tanimoto similarity rests not only on its relative simplicity but also on our previously reported finding that azaserine potently inhibits M. tuberculosis glutamate synthase (GltB) (15). GltB is an essential enzyme which catalyzes the formation of l-glutamate from l-glutamine and 2-ketoglutarate. An analysis of the 2-D similarity of l-glutamate and azaserine using the ChemAxon hashed-fingerprint methodology in the CDD TB database yields a Tanimoto value of 33%. We hypothesized that azaserine may bind and inhibit glutamate synthase by mimicking the natural substrate of this enzyme. In order to test this hypothesis, we created an M. tuberculosis strain overexpressing gltB and determined the MIC of azaserine using the broth dilution method (34). Quantitative reverse transcription-PCR (RT-PCR) revealed a 3.2-fold increase in the expression of gltB in the recombinant strain overexpressing this gene. The MICs for wild-type M. tuberculosis CDC1551 and the strain overexpressing gltB were 0.25 to 0.5 µg/ml and 1.0 to 2.0 µg/ml, respectively. As a control, we also determined the MIC of isoniazid, as GltB is not known to be the target for this drug; wild-type M. tuberculosis CDC1551 and gltB-overexpressing strains exhibited identical susceptibilities (MICs of 0.03 to 0.06 µg/ml). While overexpression mutants may be subject to higher-order metabolic perturbations due to feedback or compensations in connected pathways, we assert that the 4-fold increase in MIC due to increased expression of gltB is supportive of the idea that azaserine, a mimic of the natural substrate of GltB, inhibits this enzyme.
Examining a more sophisticated and tunable informatics strategy, we turned to 3-D pharmacophore searching. Specifically, we generated a common-feature pharmacophore model for l-glutamate in Discovery Studio 2.5.5. The pharmacophore consisted of a positive ionizable feature for the α-amino group, a negative ionizable feature for the α-carboxyl moiety, and a hydrogen bond acceptor feature for the side chain carboxylate (see Fig. S2 in the supplemental material). The constraint around the side chain carboxylate was “relaxed” from a negative ionizable feature to a hydrogen bond acceptor feature in order to avoid returning diacids as hits, which in our experience fail to exhibit whole-cell activity due to the lack of M. tuberculosis permeability. Azaserine, in addition to exhibiting Tanimoto similarity to l-glutamate, favorably fits this pharmacophore model, possessing chemical moieties mapping to all three pharmacophoric elements and a FitValue (reflecting goodness of fit to the model) of 2.1. This 3-D model was utilized to search conformer databases of commercial compounds chosen from Asinex, Maybridge, and Microsource. The pharmacophore model was selective, because, for example, only 130 out of 2,642 FDA-approved drugs and 199 out of 57,181 Maybridge compounds fit and were scored with a FitValue. Visual inspection of hits afforded 12 molecules for purchase that were assayed (Table 3 and see Table S4 in the supplemental material). As a collection, all of these putative mimics maintain the α-carboxyl group in close spatial proximity to a basic amine, while displaying a variety of polar functionalities with hydrogen bond-accepting ability. Among these compounds, JFD01307SC and l-methionine-S-sulfoximine showed activity against M. tuberculosis, with MICs in the range of 8 to 16 µg/ml and 8 to 12 µg/ml, respectively. To our surprise, we observed the susceptibility of the M. tuberculosis strain overexpressing gltB to the two compounds to be similar to that of the wild-type strain. We hypothesize that the observed activity of JFD01307SC and l-methionine-S-sulfoximine may have resulted from a target that is similar to GltB, or the compounds are metabolized into more than one active molecule. l-Methionine-S-sulfoximine is known to inhibit glutamine synthase in humans and M. tuberculosis (35). Given the structural similarity of glutamine and glutamate and their shared biosynthetic pathway in M. tuberculosis, JFD01307SC and l-methionine-S-sulfoximine may target enzymes involved in glutamine biosynthesis.
TABLE 3 .
Examples of biomolecules that are essential for the metabolism and survival of M. tuberculosis and their structural analogsa
| Essential molecule and structure | Structural analog | % similarity | Activity against M. tuberculosis | Reference or source |
|---|---|---|---|---|
l-Glutamate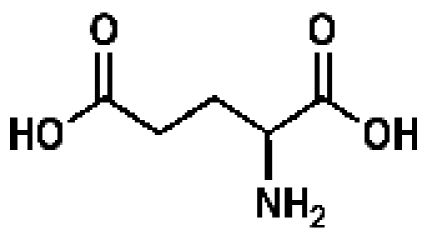
|
Azaserine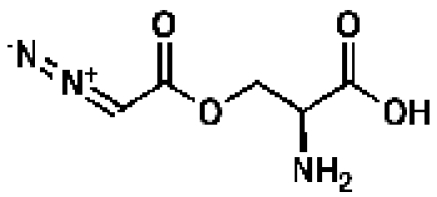
|
32 | MIC < 1 µg/ml | 15 |
l-Methionine-S-sulfoximineb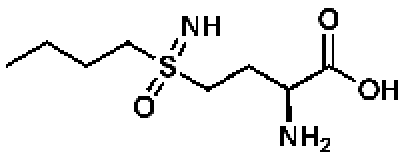
|
30 | MIC = 8–12 µg/ml | ||
JFD01307SCb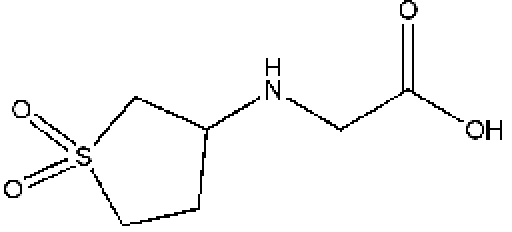
|
22 | MIC = 8–16 µg/ml | ||
UDP-N-acetylmuramoyl-l-alanyl-d-glutamyl-meso-2,6-diaminopimelyl-d-alanyl-d-alanine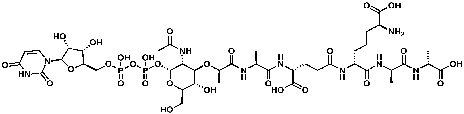
|
CDD-343074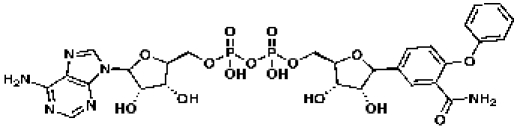
|
58 | 50 | |
Bleomycin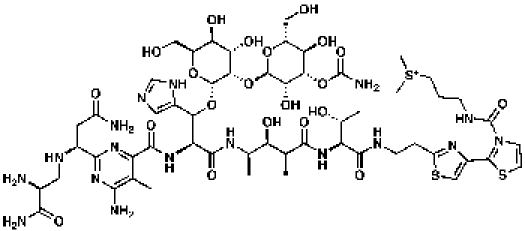
|
57 | MIC = 0.1 µg/ml | CDD database | |
Hydroxymethylbilane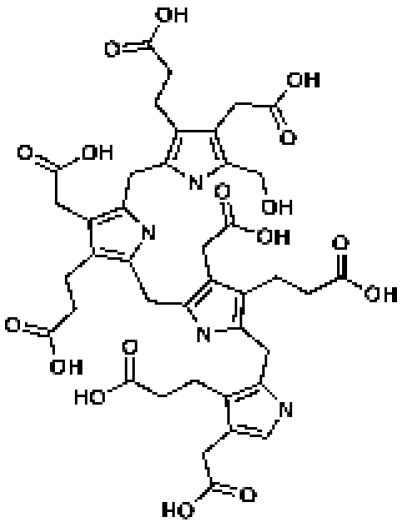
|
ChemDiv 4286-0015d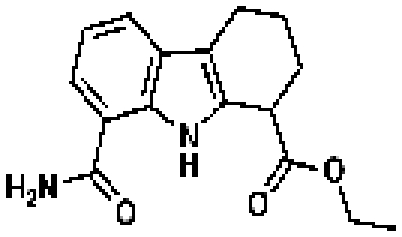
|
45 | IC50 = 3.38 µMc | CDD database |
Porfimer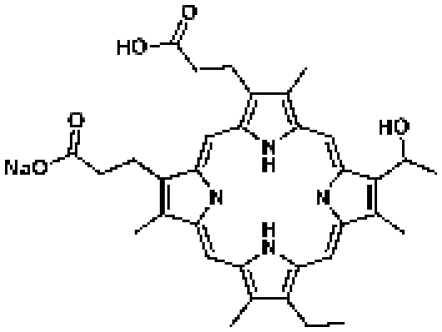
|
45 | CDD database |
Examples of biomolecules essential for the metabolism and survival of M. tuberculosis and their structural analogs are shown as a proof of the principle that molecular mimics with medicinal properties can be rapidly identified. Some of these molecules have known activities against M. tuberculosis. CDD stands for Collaborative Drug Discovery, a drug database and mining software. % similarity was calculated in the CDD database using ChemAxon fingerprints and Tanimoto similarity.
Molecules identified by 3-D pharmacophore searching.
IC50, 50% inhibitory concentration.
MLSMR data set for dose response.
Essential metabolites of the peptidoglycan, chorismate, and tetrapyrrole pathways and their mimics.
The Tanimoto similarity searching methodology was utilized to search the Collaborative Drug Discovery TB database for mimics of the essential molecules of the peptidoglycan, chorismate, and tetrapyrrole biosynthetic pathways. The results for these searches are shown in Tables 1, 2 and 3. Clearly, the mimics exhibit a range of chemical structures that in many cases do not appear to be obvious structural analogs of the essential molecules. For example, the glycopeptide antibiotic bleomycin has 57% molecular similarity to UDP-N-acetylmuramoyl-l-alanyl-d-glutamyl-meso-2,6-diaminopimelyl-d-alanyl-d-alanine, commonly known as disaccharyl pentapeptide, and a MIC of 0.1 µg/ml (36). This study reported that guinea pigs treated with bleomycin had smaller lesions and fewer acid-fast bacilli than untreated guinea pigs and a prolonged survival time. Future efforts will examine the inhibition of Rv2153c (MT2212) by bleomycin in addition to its whole-cell activity against an M. tuberculosis strain overexpressing this gene.
DISCUSSION
A powerful and widely accepted method for developing chemical probes for and inhibitors of a target protein is to determine the structure of the protein and identify or design molecules that fit the active site of the protein. Structure-based optimization of these starting points by leveraging X-ray crystal structures of the target essential gene product bound to the essential metabolite or its close analogs would provide a starting point for developing more potent molecules (37, 38). However, these enzyme inhibitors may still lack whole-cell activity and in vivo efficacy. This approach also requires the structure of the protein to be known and available. We assume that the fundamental knowledge of the shape of the substrate for an essential enzyme can be used to infer details of the binding site requirements (39). We herein have proposed a strategy for leveraging the essential molecules of M. tuberculosis to arrive at potent antitubercular small molecules with significant promise as chemical probes that may ultimately inspire preclinical drug candidates. Optimally, this involves choosing mimics from a database of small-molecule inhibitors of M. tuberculosis growth, such as the CDD TB database.
This is a systematic approach for drug discovery which begins with identification of targets in the pathogen. As selection of candidate inhibitory compounds is based on the identities of verified targets, the pathways affected and the likely mechanism of action are known a priori. While we cannot discount the potential for small-molecule analogs of essential metabolites exhibiting affinity for other mycobacterial targets, attempts to maintain significant similarity to the essential metabolite should effectively minimize this promiscuity. This might also be predicted by understanding the 2-D or 3-D pharmacophore similarity to other metabolites of the essential target mimic. This is a significant advantage, since a drug with a novel mechanism of action is a necessity for killing those strains of M. tuberculosis that are resistant to one or more current TB drugs.
The examples of azaserine, JFD01307SC, and l-methionine-S-sulfoximine provide a proof of principle that mimics of essential metabolites, as identified through cheminformatic methods, may represent effective molecules with anti-TB activity in vitro. It should be noted that gabapentin and nine additional molecules with significant Tanimoto and/or 3-D-pharmacophore-assessed similarity to l-glutamate (see Table S4 in the supplemental material) did not show activity against wild-type M. tuberculosis and the gltB-overexpressing strain in our study, illustrating our conclusion that molecular mimicry, as judged by Tanimoto similarity or fit (technically defined as the FitValue in Discovery Studio) to a given pharmacophore model for a metabolite, does not singularly meet the criteria for effective inhibition of the putative enzyme. Further studies are necessary to probe the relationship between activity against the intended biological target and the Tanimoto similarity between the associated metabolite and its mimics as well as the calculated fit of mimics to the metabolite pharmacophore model. Clearly, the degree of Tanimoto similarity is dependent on the molecular fingerprints chosen. We observed a range of pairwise Tanimoto similarities (25.6% to 48.8%) for l-glutamate and azaserine using FCFP_6 or Molecular Design Limited (MDL) fingerprints, respectively. A best practice may involve sampling a subset of available fingerprint algorithms or using a methodology, like that from ChemAxon, as done in this study, where similarity scores consistently neared the midrange. Additionally, it is evident that pharmacophore models are tunable, given the chemical features one chooses to include or ignore. Thus, it is our anticipation that optimization of pharmacophore search models will be necessary to further leverage our methodology for choosing mimics of essential metabolites. This is neither unexpected nor completely dissimilar from what we have learned in high-throughput screening campaigns, where the choice of compound library and assay are critical for success.
These data illustrate that a single cheminformatics method is not universally applicable in predicting the specificity of a synthetic molecule for the target essential enzyme. Approximately 40% of genes of M. tuberculosis have yet to be functionally annotated (16). Our lack of information on the pathways constituted by enzymes encoded by these hypothetical genes means that a large number of metabolites cannot be accounted for at this stage of study. Identification of all metabolites of M. tuberculosis is a major hurdle that diminishes the ability of our approach to identify a molecular mimic with high specificity to the target essential enzyme. The potential does exist, however, for the implementation of medicinal chemistry techniques to optimize these initially chosen molecular mimics for selectivity as well as potency. These medicinal chemistry operations would be part of a more global optimization of the chemical structure of the mimic, with regard to pharmacokinetic, pharmacodynamic, and safety profiles, in order to transition from chemical probe to lead molecule and ultimately to drug candidate. Additionally, cheminformatic techniques may have to be developed and leveraged to choose mimics for their similarity to a given metabolite and dissimilarity to all other metabolites.
The time-tested method of screening for growth inhibitors has also been a useful approach in finding TB drugs (for example, the recent discoveries of R207910 and OPC-67683), but often the molecular target of the inhibitor remains unknown at the end of a successful screen (40, 41). Indeed, Andries et al. resorted to whole-genome sequencing of strains susceptible and resistant to R207910 in order to ascertain that its target is likely to be a subunit of M. tuberculosis ATP synthase encoded by atpE (40). Our approach obviates the initial requirement for identification of a molecular target(s) and thereby accelerates the drug discovery process, reducing the associated costs. It should be noted that it is unlikely that the molecular mimicry method shown here would have identified R207910, as this compound targets the Fo subunit of ATP synthase. This subunit in itself is not an enzyme and does not generate an essential metabolite. Another limitation of our approach is that a molecule that mimics an essential metabolite and inhibits the corresponding essential enzyme may not be active against the whole-cell organism for numerous reasons, including lack of penetration, active efflux or metabolism, and clearance by the organism or the host.
Our approach also allows for selective targeting of the pathogen. Selection of pathways and targets that are present only in the pathogen reduces the probability of toxicity to the host. According to our analysis using the BioCyc pathway/genome database (PGDB), the three pathways studied in this work, namely, biosynthesis of peptidoglycan, tetrapyrroles, and chorismate, are absent in the human host. Yet it should be noted that there is a possibility for cross toxicity to the host itself or for cross toxicity from unintended toxicity to the resident microbiome in the host. Mycobacterium bovis, another organism that belongs to the same genus as M. tuberculosis, also possesses the pathways for biosynthesis of peptidoglycan, tetrapyrroles, and chorismate. This organism causes bovine tuberculosis and, thus, is of major concern to the cattle industry. Using the BioCyc database, we compared the genomes of M. bovis and Bos taurus, domestic cattle, and found that the three pathways are present only in the pathogen. Therefore, developing drugs to target these pathways is likely to have selective toxicity to the pathogen when administered to the host to treat mycobacterial infections.
We have elaborated an intermediate strategy between high-throughput screening and rational structure-based drug design, leveraging knowledge of the essential M. tuberculosis genes and consequently essential pathways and their associated enzymes to arrive at essential molecules and their analogs. Our definition of the essential molecules of M. tuberculosis and their partial enumeration are clearly reliant on the assumptions made to establish the essential genes of M. tuberculosis (6, 15). We assert that a collection of these essential molecules and of others that have yet to be identified is what ultimately is required for determining the essential metabolic functions of the organism. A perturbation in the concentration or availability of these molecules is likely to adversely affect the organism’s viability. A rational way to alter the critical concentrations of these essential molecules in vivo is to utilize a compound that mimics its structural and chemical properties as a competitive inhibitor of the essential enzyme. An analog that is able to strongly bind to the active site of the enzyme and thereby prevent interaction with the native essential molecules is likely to competitively inhibit the function and eventually lead to stasis or the death of the organism. A continued evolution of this set of genes and pathways may be requisite to more accurately and comprehensively reveal the essential targets under a given set of growth conditions deemed to be relevant for a specific disease state. Of course, a scenario in which an approved drug is found to be a metabolite mimic could present itself, and through repurposing, the mimic could represent a novel antitubercular agent with little if any need for optimization prior to clinical trials.
MATERIALS AND METHODS
Bacterial strains, mutagenesis, and gene essentiality.
A clinical isolate of M. tuberculosis, CDC1551, was used as the host organism. It was grown in Middlebrook 7H9 medium (Difco) supplemented with 0.2% glycerol, 10% (vol/vol) oleic acid-albumin-dextrose-catalase (OADC; Becton Dickinson), and 0.05% Tween 80 at 37°C. Mutagenesis using the Himar1 transposon, isolation, and genotyping of mutants were undertaken as described in our previous work (15). Any gene in which we identified a transposon insertion disruption was deemed nonessential for in vitro survival. Once these initial nonessential and essential gene lists were established, three filter operations were performed to refine both groups. The first filter, designated here the 5′ 80%/3′ 100-bp test, returned nonessential genes with distal insertions within 20% (for small genes) or 100 bp of the 3′ end to the essential list because such insertions may not significantly affect the product’s function. The second filter, called the operon test, reclassified essential genes as nonessential if they belonged to an operon that was disrupted in a more proximal gene. Therefore, such genes were not essential for the in vitro growth of those mutants. The 4,590-bp gltB open reading frame was cloned from M. tuberculosis CDC1551 genomic DNA by PCR using primers MT3974-P1 (5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTTAAGGAGGTGTGGGTATGACGCCTAAG-3′) and MT3974-P2 (5′-GGGGACCACTTTGTACAAGAAAGCTGGGTATCAGCCATGCGCAGCCGCCATG-3′). A fully sequenced and mutation-free clone was recombined into the Escherichia coli-M. tuberculosis Gateway-compatible knock-in shuttle vector pGS400K (a variant of pGS400H which features kanamycin selection [42]). M. tuberculosis CDC1551 was transformed with the resulting plasmid, pGS400K-gltB(tb), by using standard techniques (43), and transformants were obtained after 3 to 4 weeks of growth on 7H10 plates (Becton Dickinson) containing 20 µg/ml kanamycin (Sigma). A gltB knock-in strain was grown in Middlebrook 7H9 broth, total RNA was isolated using standard techniques (44), and the expression of gltB was quantified by real-time RT-PCR.
Determination of essential metabolic pathways, essential molecules, and analogs.
The BioCyc PGDB also included a database of predicted metabolic pathways in M. tuberculosis, along with the genes involved in each pathway (17). This database was used as a starting point for determining the essentiality of each pathway. The BioCyc PGDB was used to map the putative reactions catalyzed by enzymes that were assessed to be essential in our analysis. The database collects the molecular structures and formulas of the reactants and products for each enzyme. This information was used to generate a database of putative essential molecules of M. tuberculosis which comprise the reactants and products of enzymes that are essential for the growth and survival of the organism.
Determination of MICs.
We determined the MICs of all compounds in this study using the broth dilution method (34). For this, we inoculated 105 wild-type M. tuberculosis bacilli and bacilli of the gltB knock-in strain separately in 2.5 ml of 7H9 broth without detergent and added the compounds by 2-fold serial dilutions. We incubated these cultures at 37°C and evaluated them for granulation, a measure of growth, by visual inspection at 7 and 14 days. For those samples with diminished growth compared to that of a no-drug control, we determined numbers of CFU.
CDD database.
Structural analogs or mimics of the putative essential molecules were then compiled by conducting similarity searches using the CDD TB database (Collaborative Drug Discovery Inc., Burlingame, CA), which has been described previously (27), with applications for collaborative research (28). This consists of several TB screening data sets collected and uploaded from SDF files and mapped to custom protocols, including the data sets of the NIAID (n = 3,748), GVK BIO (n = 2,880) (45), TAACF (n = 812), and MLSMR (n = 220,463) (45), known TB drugs (n = 13) (38), TB candidates of Ballell et al. (n = 48) (46), and SAR data collated from the TB literature (n = 6,770) (27). In addition, the CDD database also includes FDA-approved drugs (n = 2,815) and vendor molecule libraries (27).
3-D pharmacophore.
3-D computational molecular modeling studies were carried out using Catalyst in Discovery Studio 2.5.5 (Accelrys, San Diego, CA). A common-feature (HipHop) pharmacophore model was generated around l-glutamate. Such pharmacophores attempt to describe the arrangement of key features that are important for biological activity and have been widely described (47, 48). Ten pharmacophores were created in Discovery Studio (note that all had slightly different features, etc.). Pharmacophore 4 consisted of a negative ionizable, a positive ionizable, and a hydrogen bond acceptor feature (Fig. S2).
The database of FDA-approved drugs (n = 2,815) from the CDD database was downloaded as an SDF file and converted to a 3-D Catalyst database after generating up to 100 molecule conformations with the FAST conformer generation method, with a maximum energy threshold of 20 kcal/mol. In addition, the Maybridge vendor database (n = 57,181) was generated in three dimensions in the same way. The common-feature pharmacophore model was then applied to screen these databases using the FAST search method in the same manner as previously described (49). The quality of the molecule mapping to the pharmacophore was determined by the FitValue, which is dependent on the proximity of a compound to the pharmacophore feature centroids and the weights assigned to each centroid, where a higher FitValue represents a better fit. Compounds were then selected for purchase and testing.
ACKNOWLEDGMENTS
G.L. and W.R.B. acknowledge support of NIAID grants AI 30036, AI 37856, AI 79590, and AI 36973. The CDD TB database, along with introductory training, was provided freely to M. tuberculosis researchers through October 2010 thanks to funding from the Bill and Melinda Gates Foundation (grant 49852 [Collaborative Drug Discovery for TB through a novel database of SAR data optimized to promote data archiving and sharing]).
S.E. gratefully acknowledges Accelrys Inc. for providing Discovery Studio.
Footnotes
G.L., N.W., and W.R.B. designed the genetics and microbiology experiments. G.L., N.W., and S.T.N. conducted the experiments and analyzed data. J.S.F. and S.E. performed medicinal chemistry and cheminformatics evaluations, respectively. G.L. wrote the manuscript with help from the coauthors.
Citation Lamichhane, G., J. S. Freundlich, S. Ekins, N. Wickramaratne, S. T. Nolan, et al. 2011. Essential metabolites of Mycobacterium tuberculosis and their mimics. mBio 2(1):e00301-10. doi:10.1128/mBio.00301-10.
SUPPLEMENTAL MATERIAL
REFERENCES
- 1. WHO 2009. Global tuberculosis control, p. 4–5 WHO, Geneva, Switzerland [Google Scholar]
- 2. Corbett E. L., Watt C. J., Walker N., Maher D., Williams B. G., Raviglione M. C., Dye C. 2003. The growing burden of tuberculosis: global trends and interactions with the HIV epidemic. Arch. Intern. Med. 163:1009–1021 [DOI] [PubMed] [Google Scholar]
- 3. Dye C., Scheele S., Dolin P., Pathania V., Raviglione M. C. 1999. Consensus statement. Global burden of tuberculosis: estimated incidence, prevalence, and mortality by country. WHO Global Surveillance and Monitoring Project. JAMA 282:677–686 [DOI] [PubMed] [Google Scholar]
- 4. Fauci A. S. 2008. Multidrug-resistant and extensively drug-resistant tuberculosis: the National Institute of Allergy and Infectious Diseases Research agenda and recommendations for priority research. J. Infect. Dis. 197:1493–1498 [DOI] [PubMed] [Google Scholar]
- 5. Jassal M., Bishai W. R. 2009. Extensively drug-resistant tuberculosis. Lancet Infect. Dis. 9:19–30 [DOI] [PubMed] [Google Scholar]
- 6. Sassetti C. M., Boyd D. H., Rubin E. J. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48:77–84 [DOI] [PubMed] [Google Scholar]
- 7. Lascelles J., Woods D. D. 1952. The synthesis of folic acid by Bacterium coll and Staphylococcus aureus and its inhibition by sulphonamides. Br. J. Exp. Pathol. 33:288–303 [PMC free article] [PubMed] [Google Scholar]
- 8. Achari A., Somers D. O., Champness J. N., Bryant P. K., Rosemond J., Stammers D. K. 1997. Crystal structure of the anti-bacterial sulfonamide drug target dihydropteroate synthase. Nat. Struct. Biol. 4:490–497 [DOI] [PubMed] [Google Scholar]
- 9. Tipper D. J., Strominger J. L. 1965. Mechanism of action of penicillins: a proposal based on their structural similarity to acyl-d-alanyl-d-alanine. Proc. Natl. Acad. Sci. U. S. A. 54:1133–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Denome S. A., Elf P. K., Henderson T. A., Nelson D. E., Young K. D. 1999. Escherichia coli mutants lacking all possible combinations of eight penicillin binding proteins: viability, characteristics, and implications for peptidoglycan synthesis. J. Bacteriol. 181:3981–3993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Waxman D. J., Strominger J. L. 1983. Penicillin-binding proteins and the mechanism of action of beta-lactam antibiotics. Annu. Rev. Biochem. 52:825–869 [DOI] [PubMed] [Google Scholar]
- 12. Akerley B. J., Rubin E. J., Camilli A., Lampe D. J., Robertson H. M., Mekalanos J. J. 1998. Systematic identification of essential genes by in vitro mariner mutagenesis. Proc. Natl. Acad. Sci. U. S. A. 95:8927–8932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Murry J. P., Sassetti C. M., Lane J. M., Xie Z., Rubin E. J. 2008. Transposon site hybridization in Mycobacterium tuberculosis. Methods Mol. Biol. 416:45–59 [DOI] [PubMed] [Google Scholar]
- 14. Rubin E. J., Akerley B. J., Novik V. N., Lampe D. J., Husson R. N., Mekalanos J. J. 1999. In vivo transposition of mariner-based elements in enteric bacteria and mycobacteria. Proc. Natl. Acad. Sci. U. S. A. 96:1645–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lamichhane G., Zignol M., Blades N. J., Geiman D. E., Dougherty A., Grosset J., Broman K. W., Bishai W. R. 2003. A postgenomic method for predicting essential genes at subsaturation levels of mutagenesis: application to Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 100:7213–7218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cole S. T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S. V., Eiglmeier K., Gas S., Barry C. E., III, Tekaia F., Badcock K., Basham D., Brown D., Chillingworth T., Connor R., Davies R., Devlin K., Feltwell T., Gentles S., Hamlin N., Holroyd S., Hornsby T., Jagels K., Krogh A., McLean J., Moule S., Murphy L., Oliver K., Osborne J., Quail M. A., Rajandream M. A., Rogers J., Rutter S., Seeger K., Skelton J., Squares R., Squares S., Sulston J. E., Taylor K., Whitehead S., Barrell B. G. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537–544 [DOI] [PubMed] [Google Scholar]
- 17. Caspi R., Foerster H., Fulcher C. A., Kaipa P., Krummenacker M., Latendresse M., Paley S., Rhee S. Y., Shearer A. G., Tissier C., Walk T. C., Zhang P., Karp P. D. 2008. The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 36:D623–D631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Parish T., Stoker N. G. 2002. The common aromatic amino acid biosynthesis pathway is essential in Mycobacterium tuberculosis. Microbiology 148:3069–3077 [DOI] [PubMed] [Google Scholar]
- 19. Johnson M., Lajiness M., Maggiora G. 1989. Molecular similarity: a basis for designing drug screening programs. Prog. Clin. Biol. Res. 291:167–171 [PubMed] [Google Scholar]
- 20. Johnson M., Maggiora G. 1990. Concepts and applications of molecular similarity. Wiley Interscience, Hoboken, NY [Google Scholar]
- 21. Horvath D., Jeandenans C. 2003. Neighborhood behavior of in silico structural spaces with respect to in vitro activity spaces—a benchmark for neighborhood behavior assessment of different in silico similarity metrics. J. Chem. Inf. Comput. Sci. 43:691–698 [DOI] [PubMed] [Google Scholar]
- 22. Kaiser D., Zdrazil G., Ecker G. F. 2005. Similarity based descriptors (SIBAR)—a tool for safe exchange of chemical information. J. Comput. Aided Mol. Des. 19:687–692 [DOI] [PubMed] [Google Scholar]
- 23. Wang N., DeLisle R. K., Diller D. J. 2005. Fast small molecule similarity searching with multiple alignment profiles of molecules represented in one-dimension. J. Med. Chem. 48:6980–6990 [DOI] [PubMed] [Google Scholar]
- 24. Willett P., Barnard J. M., Downs G. M. 1998. Chemical similarity searching. J. Chem. Inf. Comput. Sci. 38:983–996 [Google Scholar]
- 25. Krasowski M. D., Siam M. G., Iyer M., Pizon A. F., Giannoutsos S., Ekins S. 2009. Chemoinformatic methods for predicting interference in drug of abuse/toxicology immunoassays. Clin. Chem. 55:1203–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Klein C., Kaiser D., Kopp S., Chiba P., Ecker G. F. 2002. Similarity based SAR (SIBAR) as tool for early ADME profiling. J. Comput. Aided Mol. Des. 16:785–793 [DOI] [PubMed] [Google Scholar]
- 27. Ekins S., Bradford J., Dole K., Spektor A., Gregory K., Blondeau D., Hohman M., Bunin B. A. 2010. A collaborative database and computational models for tuberculosis drug discovery. Mol. Biosyst. 10:840–851 [DOI] [PubMed] [Google Scholar]
- 28. Hohman M., Gregory K., Chibale K., Smith P. J., Ekins S., Bunin B. 2009. Novel web-based tools combining chemistry informatics, biology and social networks for drug discovery. Drug Discov. Today 14:261–270 [DOI] [PubMed] [Google Scholar]
- 29. Kanehisa M., Goto S. 2000. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Karp P. D., Ouzounis C. A., Moore-Kochlacs C., Goldovsky L., Kaipa P., Ahren D., Tsoka S., Darzentas N., Kunin V., Lopez-Bigas N. 2005. Expansion of the BioCyc collection of pathway/genome databases to 160 genomes. Nucleic Acids Res. 33:6083–6089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sud M., Fahy E., Cotter D., Brown A., Dennis E. A., Glass C. K., Merrill A. H., Jr., Murphy R. C., Raetz C. R., Russell D. W., Subramaniam S. 2007. LMSD: LIPID MAPS structure database. Nucleic Acids Res. 35:D527–D532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Williams A. J. 2008. A perspective of publicly accessible/open-access chemistry databases. Drug Discov. Today 13:495–501 [DOI] [PubMed] [Google Scholar]
- 33. Irwin J. J., Shoichet B. K. 2005. ZINC—a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 45:177–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wiegand I., Hilpert K., Hancock R. E. 2008. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 3:163–175 [DOI] [PubMed] [Google Scholar]
- 35. Harth G., Horwitz M. A. 1999. An inhibitor of exported Mycobacterium tuberculosis glutamine synthetase selectively blocks the growth of pathogenic mycobacteria in axenic culture and in human monocytes: extracellular proteins as potential novel drug targets. J. Exp. Med. 189:1425–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ertem E., Yuce K., Karakartal G., Onal O., Yuce G. 1990. The antituberculous effect of bleomycin. J. Antimicrob. Chemother. 26:862–863 [DOI] [PubMed] [Google Scholar]
- 37. Cho Y., Ioerger T. R., Sacchettini J. C. 2008. Discovery of novel nitrobenzothiazole inhibitors for Mycobacterium tuberculosis ATP phosphoribosyl transferase (HisG) through virtual screening. J. Med. Chem. 51:5984–5992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sacchettini J. C., Rubin E. J., Freundlich J. S. 2008. Drugs versus bugs: in pursuit of the persistent predator Mycobacterium tuberculosis. Nat. Rev. Microbiol. 6:41–52 [DOI] [PubMed] [Google Scholar]
- 39. Kortagere S., Krasowski M. D., Ekins S. 2009. The importance of discerning shape in molecular pharmacology. Trends Pharmacol. Sci. 30:138–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Andries K., Verhasselt P., Guillemont J., Gohlmann H. W., Neefs J. M., Winkler H., Van Gestel J., Timmerman P., Zhu M., Lee E., Williams P., de Chaffoy D., Huitric E., Hoffner S., Cambau E., Truffot-Pernot C., Lounis N., Jarlier V. 2005. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307:223–227 [DOI] [PubMed] [Google Scholar]
- 41. Matsumoto M., Hashizume H., Tomishige T., Kawasaki M., Tsubouchi H., Sasaki H., Shimokawa Y., Komatsu M. 2006. OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med. 3:e466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Davis S. L., Be N. A., Lamichhane G., Nimmagadda S., Pomper M. G., Bishai W. R., Jain S. K. 2009. Bacterial thymidine kinase as a non-invasive imaging reporter for Mycobacterium tuberculosis in live animals. PLoS One 4:e6297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Larsen M. H. 2000. Appendix 1. Some common methods in mycobacterial genetics. Electroporation—M. tuberculosis and BCG, p. 320. In Molecular genetics of mycobacteria. ASM Press, Washington, DC [Google Scholar]
- 44. Larsen M. H. 2000. Appendix 1. Some common methods in mycobacterial genetics. RNA preparation—Trizol, p. 317. In Hatfull G. F., Jacobs W. R., Jr., Molecular genetics of mycobacteria. ASM Press, Washington, DC [Google Scholar]
- 45. Prathipati P., Ma N. L., Keller T. H. 2008. Global Bayesian models for the prioritization of antitubercular agents. J. Chem. Inf. Model. 48:2362–2370 [DOI] [PubMed] [Google Scholar]
- 46. Ballell L., Field R. A., Duncan K., Young R. J. 2005. New small-molecule synthetic antimycobacterials. Antimicrob. Agents Chemother. 49:2153–2163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Clement O. O., Mehl A. T. 2000. HipHop: pharmacophore based on multiple common-feature alignments, p. 69–84 In Guner O. F., Pharmacophore perception, development, and use in drug design. IUL, San Diego, CA. [Google Scholar]
- 48. Ekins S., Chang C., Mani S., Krasowski M. D., Reschly E. J., Iyer M., Kholodovych V., Ai N., Welsh W. J., Sinz M., Swaan P. W., Patel R., Bachmann K. 2007. Human pregnane X receptor antagonists and agonists define molecular requirements for different binding sites. Mol. Pharmacol. 72:592–603 [DOI] [PubMed] [Google Scholar]
- 49. Ekins S., Johnston J. S., Bahadduri P., D’Souza V. M., Ray A., Chang C., Swaan P. W. 2005. In vitro and pharmacophore-based discovery of novel hPEPT1 inhibitors. Pharmacol. Res. 22:512–517 [DOI] [PubMed] [Google Scholar]
- 50. Bonnac L., Gao G. Y., Chen L., Felczak K., Bennett E. M., Xu H., Kim T., Liu N., Oh H., Tonge P. J., Pankiewicz K. W. 2007. Synthesis of 4-phenoxybenzamide adenine dinucleotide as NAD analogue with inhibitory activity against enoyl-ACP reductase (InhA) of Mycobacterium tuberculosis. Bioorg. Med. Chem. Lett. 17:4588–4591 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.