

Abstract
The aryl hydrocarbon receptor (AhR) is a ligand-dependent transcription factor that mediates many of the biological and toxicological actions of structurally diverse chemicals, including the ubiquitous environmental contaminant 2,3,7,8-tetrachlorodibenzo-p-dioxin. Here, we have examined the ability of diuron, a widely used herbicide, and several structurally related substituted phenylureas to bind to and activate/inhibit the AhR and AhR signal transduction. Diuron induced CYP1A1 mRNA levels in mouse hepatoma (Hepa1c1c7) cells and AhR-dependent luciferase reporter gene expression in stably transfected mouse, rat, guinea pig, and human cell lines. In addition, ligand binding and gel retardation analysis demonstrated the ability of diuron to competitively bind to and stimulate AhR transformation and DNA binding in vitro and in intact cells. Several structurally related substituted phenylureas competitively bound to the guinea pig hepatic cytosolic AhR, inhibited 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced AhR-dependent luciferase reporter gene expression in a species-specific manner and stimulated AhR transformation and DNA binding, consistent with their role as partial AhR agonists. These results demonstrate not only that diuron and related substituted phenylureas are AhR ligands but also that exposure to these chemicals could induce/inhibit AhR-dependent biological effects.
Keywords: 2,3,7,8-Tetrachlorodibenzo-p-dioxin; TCDD; Ah Receptor; Diuron; Urea Herbicide
INTRODUCTION
The aryl hydrocarbon receptor (AhR) is a basic helix–loop–helix PAS-containing transcription factor, which activates gene expression in a ligand-dependent manner [1–4]. Exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD, dioxin), the prototypical and most potent AhR ligand, results in a wide variety of species- and tissue-specific toxic and biological responses, the majority of which are AhR dependent [5,6]. Following ligand binding, the cytosolic AhR protein complex containing two molecules of hsp90, the X-associated protein 2, and the co-chaperone p23 [7,8] translocates into the nucleus, the ligand-bound AhR is released from its associated protein subunits upon dimerization with the Arnt (Ah receptor nuclear translocator) protein and is converted into its high-affinity DNA binding form [1,2,9]. Binding of the heteromeric ligand:AhR:Arnt complex to its specific DNA recognition site, the dioxin response element (DRE), upstream of cytochrome P4501A1 (CYP1A1), and other AhR-responsive genes stimulates their transcription [3,4,10].
The best characterized high-affinity ligands for the AhR include a variety of halogenated aromatic hydrocarbons (HAHs), such as the polychlorinated dibenzo-p-dioxin, dibenzofurans, and biphenyls, as well as numerous polycyclic aromatic hydrocarbons (PAHs), such as benzo(a)pyrene, 3-methylcholanthrene, and others [11,12]. Recently, our laboratory and others have identified and characterized a relatively large number of natural and synthetic AhR ligands (agonist and antagonists) whose structure and physicochemical characteristics are dramatically different from that of the prototypical HAH and PAH AhR ligands [11–13]. While the relative potency of these diverse ligands in intact cells and animals are typically much lower than that of the HAHs and PAHs (predominantly due to differences in their affinity, intrinsic efficacy, and metabolic stability [11,13–16]). These results demonstrate that the AhR has an extremely promiscuous ligand binding pocket and raise questions as to the actual spectrum of chemicals that can bind to and activate the AhR and AhR-dependent signal transduction pathway.
Accordingly, we have carried out an extensive high-throughput bioassay screening analysis of a wide variety of chemicals and chemical mixtures in an attempt to identify and characterize novel AhR ligands and extend our understanding of the structural diversity. Chemical structure screening of currently used pesticides revealed some structural similarity between the phenylurea herbicide diuron and TCDD. Although two previous studies demonstrated that exposure of rats to diuron could induce ethoxyresorufin O-deethylase, a CYP1A1-associated enzyme, activity and other biological and toxicological effects [17,18], involvement of the AhR was not examined. These observations, combined with the widespread and relatively heavy use of this herbicide [19] and the potential for human and animal exposure, led us to examine the ability of diuron and selected structurally related phenylurea compounds to directly interact with and activate the AhR and AhR signal transduction pathway.
MATERIALS AND METHODS
Chemicals
TCDD, 2,3,7,8-tetrachlorodibenzofuran (TCDF), and [3H]-TCDD (37 Ci/mmol) were obtained from S. Safe (Texas A&M University, College Station, TX). [32P]-ATP (6000 Ci/mmol) was purchased from Amersham (Arlington Heights, IL) and DMSO from Aldrich (St. Louis, MO). Diuron (N-(3,4-dichlorophenyl)-N′,N′-dimethylurea) and related phenylurea compounds (Figure 1) were either purchased or synthesized with purity >99% [20,21]. Cell culture reagents and media were purchased from Gibco/BRL (Grand Island, NY) and G418 was from Gemini Bio-Products (Woodland, CA).
FIGURE 1.

Structures of chemicals examined in this study.
Cell Culture, Chemical Treatment, and AhR-Dependent Luciferase Reporter Gene Expression
Recombinant rat (H4L1.1c4), mouse (H1L1.1c2), human (HG2L6.1c3) hepatoma cells, and guinea pig intestinal adenocarcinoma (G16L1.1c8) were grown and maintained as previously described [22]. H4L1.1c4, H1L1.1c2, and G16L1.1c8 cells contain the stably integrated DRE-driven firefly luciferase reporter plasmid pGudLuc1.1 [22], HG2L6.1c3 cells contain pGudLuc6.1 [23] and transcriptional activation of those plasmids occurs in a ligand-, dose-, and AhR-dependent manner [22–24]. Cells were plated into white, clear-bottomed 96-well tissue culture dishes at 75,000 cells per well and allowed to attach for 24 h. Cells were incubated with carrier solvent DMSO (1% final solvent concentration), TCDD (1 nM) or the indicated compound (for measurement of agonist activity) or the indicated compound plus 1 nM TCDD (for measurement of antagonist activity) for either 4 h (H4L1.1c4, H1L1.1c2, and G16L1.1c8 cells) or 24 h (H4L1.1c4 and HG2L6.1c3 cells) at 37°C. For luciferase measurement, sample wells were washed twice with phosphate-buffered saline, followed by addition of cell lysis buffer (Promega) and shaking of the plates for 20 min at room temperature to allow cell lysis. Measurement of luciferase activity in each well was carried out using an Anthos Lucy2 (Durham, NC) microplate luminometer with automatic injection of Promega stabilized luciferase reagent. Luciferase activity in each well was expressed relative to that induced by 1 nM TCDD.
RT-PCR Analysis of Endogenous Gene CYP1A1 Induction
Forward and reverse RT-PCR primers that amplify a 280 bp product between exon 5 and exon 7 of the mouse CYP1A1 gene were synthesized and contained the following sequences: mCYP1A1 FP, 5′-GCCTTCATTCTGGAGACCTTCC-3′; and mCYP1A1 RP, 5′-CAATGGTCTCTCCGATGC-3′ [24]. A highly conserved region of a constitutively expressed housekeeping gene, rig/S15, which encodes a small ribosomal subunit protein, was used as an internal control and amplification (S15 FP, 5′-TTCCGCAAGTTCACCTACC-3′; and S15 RP, 5′-CGGGCCGGCCATGCTTTACG-3′) produced a product of 361 bp [25]. Confluent mouse hepatoma cells (Hepa1c1c7) were treated with 1% carrier solvent (DMSO), 1 nM TCDD or 2 μM diuron for 3.5 h, respectively, prior to mRNA isolation using TRIzol (Gibco/BRL Life Technologies). Single stranded cDNA was synthesized using Superscript II Rnase H− Reverse Transcriptase (Gibco/BRL Life Technologies) and used for PCR amplification. PCR reactions were conducted in final volume of 50 μL consisting of 1.5 mM MgCl2, 2.5 U Platinum Taq (Gibco/BRL), 100 ng cDNA, and 2 μM each of forward and reverse primer. The reaction cycles for CYP1A1 amplification were 94°C for 2 min, 30 s at 94°C, 60 s at 55°C, and 60 s at 72°C and were repeated for 28 cycles, ending with a 10 min elongation step at 72°C. S15 primers were used as an internal control and amplification of S15 was carried out using the CYP1A1 cycle format as above. All PCR reactions were performed in a Stratagene Robocycler hot top PCR machine and visual analysis of the products (25 μL) was performed after agarose gel electrophoresis.
Preparation of Cytosol and Nuclear Extracts
Male Hartley guinea pigs (250–300 g), obtained from Charles River Breeding Laboratories (Wilmington, DE), were exposed to 12 h of light and 12 h of dark daily and were allowed free access to food and water. Hepatic cytosol was prepared in HEDG buffer (25 mM Hepes (pH 7.5), 1 mM EDTA, 1 mM DTT, and 10% (v/v) glycerol) as previously described [24]. Nuclear extracts were prepared from confluent plates of mouse hepatoma (Hepa1c1c7) cells incubated with DMSO or the indicated chemical for 1 h at 37°C as described [24]. The resulting cytosol and nuclear extracts were stored frozen at −80°C until use. Protein concentrations were determined by dye binding [26] using bovine serum albumin as the standard.
DNA and Ligand Binding Analysis
DNA binding analysis of in vitro transformed cytosolic or nuclear AhR complexes was carried out using gel retardation analysis as we have described in detail [24]. A complementary pair of synthetic oligonucleotides containing the sequences 5′-GATCTGGCTCTTCTCACGCAACTCCG-3′ and 5′-GATCCGGAGTTGCGTGAGAAGAGCCA-3′ (corresponding to the AhR binding site of murine CYP1A1 DRE3 and designated as the DRE oligonucleotide) was synthesized, purified, annealed, and radiolabeled with [32P]-ATP [24]. For in vitro transformation and DNA binding, guinea pig cytosol (8 mg of protein/mL) was incubated with TCDD (1 pM–20 nM), diuron (0.1–50 μM) or carrier solvent (DMSO) for 2 h at 20°C, followed by gel retardation analysis. DNA binding of in vitro transformed AhR complexes or nuclear AhR complexes isolated from cells that had been incubated with DMSO (1 μL/mL), TCDD (1 nM) or diuron (5 μM) was carried out using the procedure previously described [24]. The amount of [32P]-labeled DRE present in the TCDD-inducible protein–DNA complex was measured using a Molecular Dynamics Phosphorimager, and the amount of radioactivity in the inducible protein–DNA complex minus that present in the same position in the DMSO sample lane allowed calculation of the amount of TCDD:AhR:DRE complex. The amount of AhR–DRE complex formation induced by diuron or other compounds was expressed relative to that of TCDD. For ligand binding guinea pig, hepatic cytosol (2 mg protein/mL) was incubated with 2 nM [3H]-TCDD in the absence or presence of 200 nM TCDF or 100 μM of the indicated chemical and [3H]-TCDD specific binding or competitive ligand binding determined using the hydroxyapatite binding assay [24]. At least triplicate binding analyses were performed for each chemical and binding was expressed as the percent displacement of [3H]-TCDD specific binding per mg cytosolic protein.
RESULTS
Chemical structure screening of currently used pesticides revealed some structural similarity between the phenylurea herbicide diuron and TCDD (Figure 1). Although diuron was previously reported to induce EROD activity in rat liver [17,18], involvement of the AhR was not evaluated. We first examined the AhR agonist activity of diuron by testing its ability to stimulate AhR-dependent reporter gene expression in recombinant rat hepatoma (H4IIe) cells (H4L1.1c4) that contain the stably transfected DRE-luciferase reporter plasmid pGudLuc1.1 [22]. Dose-dependent induction of luciferase by diuron at 4 h was observed in this cell line to levels greater than 90% of that induced by 1 nM TCDD (Figure 2). The EC50 for induction by diuron was ~8 μM, approximately 66,000-fold higher than observed for TCDD (EC50: 0.12 nM). The parallel dose–response curves are also consistent with the role of the AhR in this response. These results combined with the lack of induction by diuron at concentrations ≤1 μM (Figure 2) indicate that it is a relatively weak AhR agonist when compared to TCDD and other potent HAH and PAH ligands [5,11,12]. The lower magnitude of induction of luciferase activity by diuron at 24 h (Figure 2) results from metabolism of diuron to a form that does not activate the AhR.
FIGURE 2.

Dose-dependent induction of luciferase activity by TCDD and diuron in rat H4L1.1c4 cells. Cells were incubated with the indicated concentration of diuron for 4 h or 24 h and luciferase activity determined as described in Materials and Methods section. Values are expressed in the figure as the percentage of maximal TCDD induction and represent the mean ± SD of triplicate determinations. All concentrations of TCDD ≥10−11 M and of diuron ≥10−5 M were significantly greater than DMSO-treated sample at p < 0.01 as determined by Student’s t-test. Actual DMSO and maximal TCDD induced luciferase activity in relative light units (RLUs) were 14.3 ± 1.2 and 362.54 ± 10.9 at 4 h and 3.9 ± 0.7, 870.6 ± 56.4 at 24 h, respectively.
To examine the species specificity of diuron as an AhR agonist, we also tested its ability to induce DRE-luciferase gene expression in stably transfected, guinea pig (G16L1.1c8), mouse (H1L1.1c2), and human (HG2L6.1c3) cell lines. Interestingly, while induction was also observed at 4 h in guinea pig and mouse cells or at 24 h in human cells (Figure 3), the maximum induction levels obtained were only 20–30% of that of 1 nM TCDD, significantly lower than that observed in the rat cell line. The observed plateaus of induction activity in these cell lines at concentrations ≥10 μM could be due to cell toxicity (no visual changes in cell morphology or death were observed) or extremely rapid degradation of diuron in these cells. Alternatively, diuron may only be a partial agonist with significantly lower inducing efficacy in these cell lines. Irrespective of the reason(s), these results revealed a significant species- and/or cell-specific difference in induction of AhR-dependent gene expression by diuron.
FIGURE 3.

Dose-dependent induction of luciferase activity by TCDD and diuron in guinea pig, mouse, and human cell lines. Guinea pig (G16L1.1c8), mouse (H1L1.1c2), or human (HG2L1.1c3) cells were incubated with the indicated concentration of diuron for 4 h, and luciferase activity was determined as described in Materials and Methods section. Values are expressed in the figure as the percentage of maximal TCDD induction and represent the mean ±SD of triplicate determinations. The asterisk (*) indicates those values significantly greater than the DMSO-treated sample at p < 0.01 as determined by Student’s t-test. Actual DMSO and maximal TCDD induced luciferase activity in relative light units (RLUs) were 24.7 ± 3.9 and 108.1 ± 8.2 in G16L1.1c8 cells, 1.2 ± 0.3 and 49.8 ± 6.5 in H1L1.1c2 cells, and 4.2 ± 0.7 and 126.6 ± 10.7 in HG2L1.1c3 cells, respectively.
To confirm the ability of diuron to induce expression of an endogenous AhR-responsive gene, in addition to the stably transfected DRE-luciferase reporter, we examined its effect on CYP1A1 expression (i.e., mRNA levels) using RT-PCR. Incubation of mouse hepatoma (hepa1c1c7) cells with diuron for 3.5 h increased CYP1A1 mRNA levels, albeit to a lower level than that induced by TCDD (Figure 4), but these data are consistent with the reporter gene induction results and the AhR agonist activity of diuron.
FIGURE 4.

Diuron increases CYP1A1 mRNA levels in mouse hepatoma cells. Hepa1c1c7 cells were incubated with DMSO (1%, final concentration), TCDD (1 nM), or diuron (2 μM) for 3.5 h at 37°C, mRNA was extracted, subjected to RT-PCR and the resulting products were visualized by agarose gel electrophoresis. PCR amplification of S15 from the same sample was included as the loading control.
While the above results indicate that diuron can induce AhR-responsive gene expression in cells and are consistent with the involvement of the AhR in this response, they do not directly demonstrate this nor prove that this response is a direct action of diuron. Although significant induction was observed at 4 h, it is possible that diuron itself is not an AhR ligand but is metabolically converted into an AhR ligand and/or stimulates AhR activation by an indirect mechanism. To directly test this possibility, we examined the ability of diuron to stimulate AhR transformation and DNA binding in vitro using gel retardation analysis. Guinea pig hepatic cytosol that had been incubated for 2 h at 20°C with increased concentrations of diuron (0.1–50 μM) produced a dose-dependent AhR:DRE complex formation (Figure 5a). Phosphorimager analysis revealed that diuron induced significant AhR:DRE complex formation at all concentrations ≥1 μM with maximum complex formation (~40% of that induced by TCDD) observed with 50 μM diuron (Figure 5b). To confirm that diuron could also stimulate AhR transformation and DNA binding in intact cells, nuclear extracts were prepared from mouse hepatoma (Hepa1c1c7) cells that had been incubated with DMSO, 1 nM TCDD, or 5 μM diuron, and the presence of transformed nuclear ligand:AhR complex was determined using gel retardation analysis (Figure 6). Nuclear extracts from diuron-treated cells produced an inducible protein–DNA complex that migrated to the same position as that induced by TCDD and the lower amount of complex formation was consistent with the reduced potency of diuron in these cells. These results demonstrate the ability of diuron to stimulate AhR transformation and DNA binding in in vitro and in intact cells and support direct activation of the AhR by diuron. To confirm that diuron is a ligand for the AhR, we examined its ability to compete with [3H]-TCDD for binding to the AhR. Ligand binding experiments (Table 1) revealed that diuron could competitively bind to the AhR, displacing a maximum of ~70% of [3H]-TCDD specific binding in these experiments conditions.
FIGURE 5.

Dose-dependent stimulation of AhR transformation and DNA binding by TCDD and diuron in vitro. Guinea pig hepatic cytosol (8 mg protein/mL) was incubated with DMSO (20 μL/mL, final concentration) or increasing concentrations of TCDD (1, 10, 100 pM and 1, 10, 20 nM) or diuron (0.1, 0.5, 1, 5, 10, 20, 50 μM) for 2 h at 20°C. Protein–DNA complexes were resolved by gel retardation analysis (a) and the amount of induced protein–DNA complex formation determined by phosphorimager analysis (b). Values are expressed in the figure as the percentage of maximal TCDD induction and represent the mean ± SD of triplicate determinations. Induced complex formation at all concentrations of TCDD ≥10−11 M and of diuron ≥10−7 M were significantly greater than the DMSO-treated sample at p < 0.01 as determined by Student’s t-test. The arrow indicates the position of the AhR:DRE complex.
FIGURE 6.

Diuron stimulates nuclear accumulation of transformed AhR complexes. Nuclear extracts (7 μg protein) from mouse hepatoma (hepa1c1c7) cells that had been incubated for 1 h with DMSO (1μL/mL, final concentration), TCDD (1 nM), or diuron (5 μM) at 37°C were analyzed by gel retardation analysis. The arrow indicates the position of the AhR:DRE complex.
TABLE 1.
Competitive Binding of Diuron and Other Substituted Phenylureas to the Guinea Pig Hepatic Cytosolic AhR
| Competitor | Concentration (μM) | [3H]-TCDD Specific Binding (Percent of Displacement)a |
|---|---|---|
| TCDF | 0.2 | 100.0 ± 19.7 |
| Diuron | 100 | 70.8 ± 18.9b |
| Monuron | 100 | 15.9 ± 14.9 |
| CPU | 100 | 78.5 ± 21.1b |
| CDCPU | 100 | 124.5 ± 10.8b |
| DDCPU | 1 | 110.3 ± 36.4b |
Guinea pig cytosol was incubated with 2 nM [3H]-TCDD in the absence or presence of the indicated competitor chemical for 2 h at 20°C and specific binding of [3H]-TCDD was determined using the hydroxyapatite binding assay as described under the Materials and Methods section.
Values are expressed as the mean ± SD of the percent displacement of [3H]-TCDD specific binding from triplicate determinations.
Values represent a significant decrease in [3H]-TCDD specific binding at p < 0.01 as determined by Student’s t-test.
The above results demonstrate that diuron is an AhR agonist, albeit relatively weak compared to TCDD. In order to gain insights into structural characteristics of diuron involved in AhR activation, we examined the ability of several structurally related compounds, monuron, CPU, CDCPU, and DDCPU (Figure 1) to bind to and activate AhR-dependent reporter gene expression. Ligand binding experiments revealed that CPU, CDCPU, and DDCPU, but not monuron, could competitively bind to the guinea pig AhR, displacing over 80% of [3H]-TCDD specific binding (Table 1). DDCPU was used in these binding studies at 1 μM concentration because of its limited solubility at higher concentrations. The ability of each compound to induce luciferase reporter gene activity in the various cell lines was then examined. While diuron could induce reporter gene expression in the tested cell lines at concentrations above 1 μM (Figures 2 and 3), monuron was inactive at 1 μM (Table 2) and 10 μM (data not shown). While no induction was observed with CPU, CDCPU, or DDCPU when used at 1 μM (the maximal concentration that could be tested that did not produce significant cell death), the ability of these compounds to bind to the AhR but not induce significant AhR-dependent luciferase gene expression suggests that they may act as AhR antagonists or partial agonists. To examine this possibility, we determined their ability to reduce TCDD-dependent luciferase induction in the same cell lines. As expected, TCDD inducible luciferase gene expression was significantly reduced by CPU, CDCPU and diuron in rat and guinea pig cell lines and by DDCPU only in the rat cell line; no significant inhibition was observed in the mouse cell line (Table 2). While these results confirm the ability of CPU, CDCPU, and DDCPU to interact with the AhR and inhibit AhR-dependent gene expression in a species-specific manner, they do not confirm whether these compounds are antagonists or partial agonists. While we cannot easily resolve this issue in intact cells due to the toxicity elicited by these compounds at concentrations >1 μM, demonstration of their ability to stimulate AhR transformation and DNA binding in vitro would be consistent with their activity as partial AhR agonists. Accordingly, the ability of each of the compounds at elevated concentrations (200 μM) to stimulate AhR transformation and DNA binding in vitro was examined (Figure 7). Interestingly, while diuron, monuron, CDCPU, and DDCPU could stimulate AhR:DRE complex formation to 10–30% of that produced by TCDD in these experiments, CPU was inactive. Taken together, these results are consistent with CDCPU and DDCPU as partial AhR agonists and CPU as an antagonist.
TABLE 2.
Induction or Inhibition of AhR-Dependent Luciferase Reporter Gene Expression in H4L1.1c4, G16L1.1c8, and H1L1.1c2 Cells by Substituted Phenylureas
| Treatment |
Luciferase Activity in Different Cell Lines (Percent of TCDD)a |
|||
|---|---|---|---|---|
| Chemical | Concentration | H4L1.1c4 | G16L1.1c8 | H1L1.1c2 |
| Induction of luciferase | ||||
| TCDD | 1 nM | 100.0 ± 8.5b | 100.0 ± 5.3 | 100.0 ± 9.7 |
| Diuron | 1 μM | 1.0 ± 0.9 | 8.1 ± 2.4 | 2.3 ± 0.7 |
| Monuron | 1 μM | 2.9 ± 1.8 | 0.5 ± 0.3 | 0.7 ± 0.4 |
| CPU | 1 μM | 0.1 ± 1.7 | 0.8 ± 1.0 | 0.3 ± 0.4 |
| CDCPU | 1 μM | 0.8 ± 0.4 | 2.1 ± 1.5 | 1.5 ± 0.9 |
| DDCPU | 1 μM | 0.1 ± 0.2 | 1.4 ± 0.8 | 3.2 ± 1.3 |
| Inhibition of TCDD induction of luciferase | ||||
| DMSO | 1 nM | 100.0 ± 8.5 | 100.0 ± 5.3 | 100.0 ± 9.7 |
| Diuron | 1 μM | 72.4 ± 2.0c | 83.6 ± 4.4c | 109.1 ± 18.9 |
| Monuron | 1 μM | 81.9 ± 6.0 | 105.3 ± 15.0 | 127.2 ± 11.3 |
| CPU | 1 μM | 61.4 ± 0.7c | 48.4 ± 11.0c | 90.4 ± 6.6 |
| CDCPU | 1 μM | 64.6 ± 3.2c | 73.5 ± 13.0c | 85.7 ± 7.2 |
| DDCPU | 1 μM | 75.1 ± 2.5c | 100.8 ± 6.6 | 96.4 ± 12.5 |
Cells were incubated with the indicated concentration of chemical (induction) or simultaneously incubated with 1 nM TCDD and the indicated chemical (inhibition) for 4 h and luciferase activity determined as described in Materials and Methods section.
Actual DMSO and TCDD induced luciferase relative light units (RLU) for each cell line were: 24.1 ± 1.8 and 502.2 ± 23.6 in H4L1.1c4 cells; 23.4 ± 3.3 and 110.6 ± 8.9 in G16L1.1c8 cells; 2.4 ± 0.8 and 65.6 ± 9.5 in H1L1.1c2 cells, respectively.
Values represent the mean ± SD of triplicate determinations of luciferase activity expressed as a percentage of the maximal luciferase activity induced by TCDD.
Values are significantly different from the TCDD-treated sample at p < 0.01 as determined by Student’s t-test.
FIGURE 7.

Stimulation of AhR transformation and DNA binding by TCDD and phenylurea compounds in vitro. Guinea pig hepatic cytosol (8 mg protein/mL) was incubated with DMSO (20 μL/mL, final concentration), 20 nM TCDD, or 200 μM of the indicated chemical for 2 h at 20°C. Protein–DNA complexes were resolved by gel retardation analysis. The arrow indicates the position of the AhR:DRE complex.
DISCUSSION
The majority of the high-affinity AhR ligands that have been identified and characterized to date include HAHs, PAHs, and related compounds [5,11,12]. Structure–activity relationship analysis using a large number of these compounds has suggested that the AhR ligand binding pocket binds planar ligands with maximal dimensions of 14 Å × 12 Å × 5 Å and that high affinity ligand binding appears dependent upon key electronic and thermodynamics properties of the ligand, such as lipophilicity, electron affinity, entropy, and molecular electrostatic potential and charge distribution patterns [27–30]. However, our laboratory and others have identified and characterized a relatively large number of chemicals, whose structural and physiochemical characteristics differ dramatically from HAH and PAH ligands, yet they can bind to and/or activate AhR-dependent gene expression [11–13]. These results strongly support our hypothesis that the AhR has a very promiscuous ligand binding site, with the range of structural diversity of AhR ligands remaining to be established.
Here, we have demonstrated the ability of diuron and several structurally related substituted phenylureas to bind to and activate/inhibit the AhR and AhR signal transduction pathway. Interestingly, our results not only indicate that diuron is a transient inducer (with metabolism likely contributing to its decreased inducing potency over time) but also reveal that it is a significantly more efficacious inducer in rat cells than in the mouse, guinea pig, or human cell lines tested. Although our analysis confirms the ability of diuron to bind to and activate the AhR and AhR signal transduction pathway, the reduced efficacy of gene induction may result from differences in diuron metabolism between these cells lines. Alternatively, the reported ability of diuron and other substituted ureas to affect cell signaling pathway and enzymes, including peroxisome proliferators-activated receptor α (PPAR α) [31], p38 MAP kinase [32], epoxide hydrolase [20], and other cellular protein and factors [33,34], and the documented ability of some cell signaling pathways to regulate AhR-dependent gene expression (i.e., PKC and NF-kB [35–37]) suggest that species- and/or cell-specific differences in these or other targets could contribute to the observed differences in AhR ligand efficacy (i.e., the magnitude of the induction response). Given previously documented species differences in the ability of ligands to bind to and/or activate the AhR and AhR signal transduction pathway [38–40], it is also possible that the observed species-specific differences in diuron efficacy may be related to differences in its ability to bind to and stimulate AhR transformation and DNA binding and/or to activate AhR-dependent gene expression. We also found differences in the relatively efficacy of diuron to activate AhR-dependent gene expression in guinea pig cells as compared to its ability to bind to and stimulate transformation and DNA binding of the guinea pig AhR in vitro. This is not surprising, given that we have previously found most AhR agonists to be significantly more potent in in vitro, cell-free AhR assays when compared to cell-based induction assays [15,16]. In cell-free AhR assays (ligand and DNA binding), the agonist has direct access to the AhR in the incubation condition and if it can bind, it will, resulting in a positive response. For a compound to be positive in cell-based bioassays, it must enter the cell, avoiding sequestration (by membranes, lipids, proteins, and organelles) and metabolism (by reduction enzymes such as cytochrome P450s), and bind to the AhR, stimulating AhR nuclear localization, transformation and DNA binding, and induction of gene expression, all within the time frame of the bioassays. Decreased concentrations of ligand at the AhR within the cell can lead to a decrease in the relative potency of an AhR ligand, especially for those ligands that are metabolically labile or at relatively low concentrations due to decreased cellular uptake.
Diuron can bind to the AhR and stimulate its transformation and DNA binding in vitro and in intact cells and it is a relatively efficacious inducer of AhR-dependent gene expression. However, its ability to antagonize induction of gene expression by a full AhR agonist (TCDD) would suggest that diuron is not a full agonist but a partial AhR agonist. In addition, the intrinsic efficacy of diuron as a partial agonist could vary significantly within different species and would be consistent with our results. On the other hand, while monuron did not compete with TCDD for binding to the AhR or stimulate AhR-dependent gene expression, it did stimulate AhR transformation and DNA binding in vitro at a relatively high concentration (200 μM) (Figure 7). These results could suggest that monuron is a very weak partial agonist with a much lower affinity than that of the other compounds (hence our inability to detect competitive binding by monuron). The inability to demonstrate competitive binding between [3H]-TCDD, an extremely high affinity full agonist of the AhR and weak AhR agonists (confirmed by functional analysis) has been previously documented [13,41–43]. While CPU, CDCPU, and DDCPU can compete with [3H]-TCDD for binding to the AhR, the inability to use concentrations >1 μM in cell culture (a concentration necessary for induction of gene expression by diuron) precluded us from directly confirming their agonist activity. However, demonstration that CDCPU and DDCPU could also stimulate AhR transformation and DNA binding in vitro (Figure 7) supports their identity as partial agonists. In contrast, although CPU can competitively bind to the AhR and inhibit induction of AhR-dependent gene expression by TCDD, its inability to stimulate transformation and DNA binding of the AhR in vitro is more consistent with CPU being an AhR antagonist.
From a structural perspective, the inability of monuron to bind to and activate the AhR supports the importance of the two lateral chlorines present on monophenylureas for more efficient binding to and activation of the AhR. In contrast, for the diphenylureas, the presence of two lateral chlorines on at least one ring (with at least one chlorine on the other ring) appears to be necessary for agonist activity, while results with CPU suggests the presence of a single lateral chlorine on each ring confers antagonist activity. Interestingly, comparison of three-dimensional structures [44] of TCDD and DDCPU revealed that the four chlorines on DDCPU molecule are present very similar relative positions as those of TCDD, even though the DDCPU molecule itself is not planar (Figure 8). This spatial configuration may be sufficient to allow DDCPU (as well as CPU and CDCPU) to bind to and activate the AhR. However, since these compounds are significantly less active than TCDD, more than simply the relative positions of the chlorines in a molecule are important for AhR agonist activity. Presumably, DDCPU and the other antagonistic substituted phenylureas bind within the AhR ligand binding pocket in such a way that AhR does not allow appropriate conformational changes to occur to allow conversion of the AhR into its fully functional form. This hypothesis is consistent with the large amount of information available on ligand-dependent steroid hormone receptors, wherein different ligands (i.e., ligands that are now typically referred to as selective receptor modulators) have been shown to alter the conformation of a particular receptor protein leading to differences in its functionality and interactions with coregulator proteins [45–47]. In light of a recent study reporting the ability of different ligands to bind to distinctly different regions within the AhR binding pocket [48], combined with proteolysis experiments demonstrating significant structural conformation differences in the AhR when occupied by different ligands (i.e., agonists and antagonists) [49], the ligand-dependent differences in AhR responses we observe between and within a species are not unexpected.
FIGURE 8.
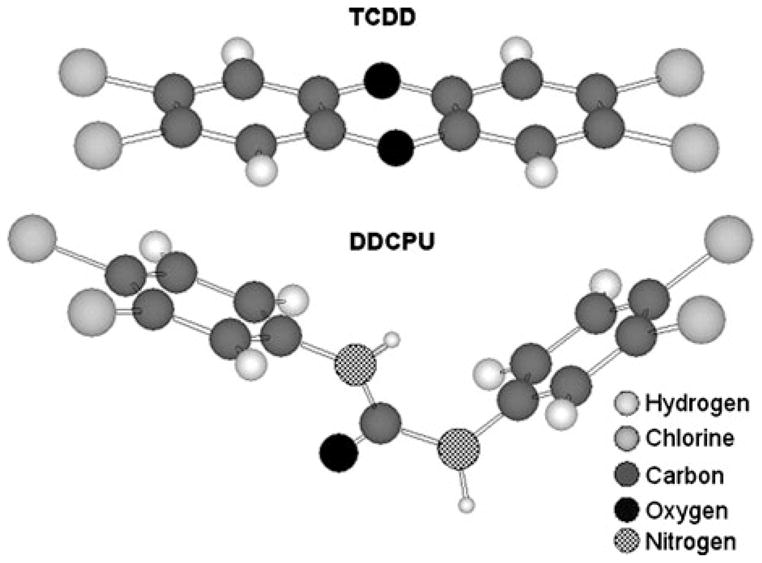
Comparison of three-dimensional structures of TCDD versus DDCPU molecules. Models were generated using ChemFinder online Chem3D modeling program [44].
Diuron has been used extensively in agriculture for more than 50 years to inhibit the growth of weeds in terrestrial environment [19] and more widely used as an antifouling agent in aquatic environment [50]. Exposure of animals to diuron is reported to cause a variety of biological and toxicological effects, such as increased mortality, growth retardation, changes in spleen and bone marrow, alternations in blood chemistry, abnormal blood pigment, and anemia [19]. While diuron and other substituted phenylureas can affect cellular signaling pathways (PPAR α [31], p38 MAP kinase [32], and apoptosis [33,34]) in addition to activating the AhR, the direct connection of these processes with the adverse effects of diuron and related compounds remains to be determined. However, the results present here combined with previous studies demonstrating the ability of diuron to induce CYP1A1-dependent ethoxyresorufin O-deethylase activity in rat [17,18] are consistent with diuron being an AhR agonist in vitro and in vivo. It is unclear whether diuron or other substituted phenylureas would be able to activate the AhR and AhR signal transduction pathway in humans in vivo. Given the lower affinity of the human AhR for ligands [51,52], the lack of significant induction of AhR-dependent reporter gene activity in our recombinant human cell line and the fact that diuron does not accumulate within the body because of its relatively rapid metabolism [53], it is unlikely that under normal exposure scenarios that serum or tissue levels of diuron would reach concentrations necessary to activate the AhR pathway. However, it is possible that transient activation of the AhR may occur in individuals that may become highly exposed to diuron in agricultural application situations, where it is still relatively heavily used [19,53]. Similarly, chronic exposure of wildlife species to diuron in terrestrial and aquatic environments, where concentrations of diuron have been reported as high as 7 μg/L of diuron in water and 1.5 μg/g of diuron in sediment [50] may lead to activation of the AhR pathway. Overall, the AhR-dependent effect of diuron on humans and wildlife remains to be determined.
Acknowledgments
Contract Grant Sponsor: National Institutes of Environmental Health Sciences.
Contract Grant Numbers: ES07685, ES04699, and ES05707.
The authors acknowledge generous contributions from the American Taxpayers and the California Agricultural Experiment Station.
References
- 1.Hankinson O. The aryl hydrocarbon receptor complex. Annu Rev Pharmacol Toxicol. 1995;35:307–340. doi: 10.1146/annurev.pa.35.040195.001515. [DOI] [PubMed] [Google Scholar]
- 2.Schmidt JV, Bradfield CA. Ah receptor signaling pathways. Annu Rev Cell Dev Biol. 1996;12:55–89. doi: 10.1146/annurev.cellbio.12.1.55. [DOI] [PubMed] [Google Scholar]
- 3.Denison MS, Elferink CF, Phelan D. The Ah receptor signal transduction pathway. In: Denison MS, Helferich WG, editors. Toxicant–receptor interactions in the modulation of signal transduction and gene expression. Philadelphia: Taylor & Francis; 1998. pp. 3–33. [Google Scholar]
- 4.Whitlock JP., Jr Induction of cytochrome P4501A1. Annu Rev Pharmacol Toxicol. 1999;39:103–125. doi: 10.1146/annurev.pharmtox.39.1.103. [DOI] [PubMed] [Google Scholar]
- 5.Safe S. Polychlorinated biphenyls (PCBs), dibenzo-p-dioxins (PCDDs), dibenzofurans (PCDFs), and related compounds: Environmental and mechanistic considerations which support the development of toxic equivalency factors (TEFs) Crit Rev Toxicol. 1990;21:51–88. doi: 10.3109/10408449009089873. [DOI] [PubMed] [Google Scholar]
- 6.Safe S. Modulation of gene expression and endocrine response pathways by 2,3,7,8-tetrachlorodibenzo-p-dioxin and related compounds. Pharmacol Therap. 1995;67:247–281. doi: 10.1016/0163-7258(95)00017-b. [DOI] [PubMed] [Google Scholar]
- 7.Meyer BK, Pray-Grant MG, Vanden Heuvel JP, Perdew GH. Hepatitis B virus X-associated protein 2 is a subunit of the unliganded aryl hydrocarbon receptor core complex and exhibits transcriptional enhancer activity. Molec Cell Biol. 1998;18:978–988. doi: 10.1128/mcb.18.2.978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kazlauskas A, Poellinger L, Pongratz I. Evidence that the co-chaperone p23 regulates ligand responsiveness of the dioxin (aryl hydrocarbon) receptor. J Biol Chem. 1999;274:13519–13524. doi: 10.1074/jbc.274.19.13519. [DOI] [PubMed] [Google Scholar]
- 9.Heid SE, Pollenz RS, Swanson HI. Role of heat shock protein 90 dissociation in mediating agonist-induced activation of the aryl hydrocarbon receptor. Mol Pharmacol. 2000;57(1):82–92. [PubMed] [Google Scholar]
- 10.Denison MS, Fisher JM, Whitlock JP., Jr The DNA recognition site for the dioxin-Ah receptor complex: Nucleotide sequence and functional analysis. J Biol Chem. 1988;263:17721–17724. [PubMed] [Google Scholar]
- 11.Denison MS, Seidel SD, Rogers WJ, Ziccardi M, Winter GM, Heath-Pagliuso S. Natural and synthetic ligands for the Ah receptor. In: Puga A, Wallace KB, editors. Molecular biology approaches to toxicology. Philadelphia: Taylor & Francis; 1998. pp. 393–410. [Google Scholar]
- 12.Denison MS, Heath-Pagliuso S. The Ah receptor: A regulator of the biochemical and toxicological actions of structurally diverse chemicals. Bull Environ Contam Toxicol. 1998;61:557–568. doi: 10.1007/pl00002973. [DOI] [PubMed] [Google Scholar]
- 13.Denison MS, Nagy SR. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol. 2003;43:309–334. doi: 10.1146/annurev.pharmtox.43.100901.135828. [DOI] [PubMed] [Google Scholar]
- 14.Hestermann EV, Stegeman JJ, Hahn ME. Relative contributions of affinity and intrinsic efficacy to aryl hydrocarbon receptor ligand potency. Toxicol Appl Pharmacol. 2000;168(2):160–172. doi: 10.1006/taap.2000.9026. [DOI] [PubMed] [Google Scholar]
- 15.Seidel SD, Li V, Winter GM, Rogers WJ, Martinez EI, Denison MS. Ah receptor-based chemical screening bioassays: Application and limitations for the detection of Ah receptor agonists. Toxicol Sci. 2000;55(1):107–115. doi: 10.1093/toxsci/55.1.107. [DOI] [PubMed] [Google Scholar]
- 16.Nagy SR, Sanborn JR, Hammock BD, Denison MS. Development of a green fluorescent protein-based cell bioassay for the rapid and inexpensive detection and characterization of ah receptor agonists. Toxicol Sci. 2002;65(2):200–210. doi: 10.1093/toxsci/65.2.200. [DOI] [PubMed] [Google Scholar]
- 17.Schoket B, Vince I. Induction of rat hepatic drug metabolizing enzymes by substituted urea herbicides. Acta Pharmacol Toxicol. 1985;56:283–288. doi: 10.1111/j.1600-0773.1985.tb01291.x. [DOI] [PubMed] [Google Scholar]
- 18.Schoket B, Vince I. Dose-related induction of rat hepatic drug-metabolizing enzymes by diuron and chlorotoluron, two substituted phenylurea herbicides. Toxicol Lett. 1990;50:1–7. doi: 10.1016/0378-4274(90)90246-i. [DOI] [PubMed] [Google Scholar]
- 19.Liu J. Handbook of pesticide toxicology: Agents. Hardcover: Academic Press; 2001. Phenylurea herbicides; pp. 1521–1527. [Google Scholar]
- 20.Morisseau C, Goodrow MH, Dowdy D, Zheng J, Greene JF, Sanborn JR, Hammock BD. Potent urea and carbamate inhibitors of soluble epoxide hydrolase. Proc Natl Acad Sci USA. 1999;96:8849–8854. doi: 10.1073/pnas.96.16.8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Newman JW, Denton DL, Morisseau C, Koger CS, Wheelock CE, Hinton DE, Hammock BD. Evaluation of fish models of soluble epoxide hydrolase inhibition. Environ Health Perspect. 2001;109(1):61–66. doi: 10.1289/ehp.0110961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garrison PM, Tullis K, Aarts JMMJG, Brouwer A, Giesy JP, Denison MS. Species-specific recombinant cell lines as bioassay systems for the detection of 2,3,7,8-tetrachlorodibenzo-p-dioxin-like chemicals. Fundam Appl Toxicol. 1996;30(2):194–203. doi: 10.1006/faat.1996.0056. [DOI] [PubMed] [Google Scholar]
- 23.Han D, Nagy SR, Denison MS. Comparison of recombinant cell bioassays for the detection of Ah receptor agonists. Biofactors. 2004;20(1):11–22. doi: 10.1002/biof.5520200102. [DOI] [PubMed] [Google Scholar]
- 24.Denison MS, Rogers JM, Rushing SR, Jones CL, Tetangco SC, Heath-Pagliuso S. Analysis of the Ah receptor signal transduction pathway. In: Maines M, Costa LG, Reed DJ, Sassa S, Sipes IG, editors. Current protocols in toxicology. New York: John Wiley and Sons; 2002. pp. 4.8.1–4.8.45. [DOI] [PubMed] [Google Scholar]
- 25.Kitagawa M, Takasawa S, Kikuchi N, Itoh T, Teraoka H, Yamamoto H, Okamoto H. Rig encodes ribosomal protein S15. The primary structure of mammalian ribosomal protein S15. FEBS Lett. 1991;283(2):210–214. doi: 10.1016/0014-5793(91)80590-y. [DOI] [PubMed] [Google Scholar]
- 26.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 27.Waller CL, McKinney JD. Three-dimensional quantitative structure–activity relationships of dioxins and dioxin-like compounds: Model validation and Ah receptor characterization. Chem Res Toxicol. 1995;8:847–858. doi: 10.1021/tx00048a005. [DOI] [PubMed] [Google Scholar]
- 28.Bonati L, Fraschini E, Lasagni M, Modoni EP, Pitea D. A hypothesis on the mechanism of PCDD biological activity based on molecular electrostatic potential modeling, part 2. J Mol Struct (Theochem) 1995;340:83–95. [Google Scholar]
- 29.Fraschini E, Bonati L, Pitea D. Molecular polarizability as a tool for understanding the binding properties of polychlorinated dibenzo-p-dioxins: Definition of a reliable computational procedure. J Phys Chem. 1996;100:10564–10569. [Google Scholar]
- 30.Tuppurainen K, Ruuskanen J. Electronic eigenvalue (EEVA): A new QSAR/QSPR descriptor for electronic substituent effects based on molecular orbital energies. A QSAR approach to the Ah receptor binding affinity of polychlorinated biphenyls (PCBs), dibenzo-p-dioxins (PCDDs) and dibenzofurans (PCDFs) Chemosphere. 2000;41:843–848. doi: 10.1016/s0045-6535(99)00525-1. [DOI] [PubMed] [Google Scholar]
- 31.Fang X, Hu S, Watanabe T, Weintraub NL, Snyder GD, Yao J, Liu Y, Shyy JY, Hammock BD, Spector AA. Activation of peroxisome proliferator-activated receptor alpha by substituted urea-derived soluble epoxide hydrolase inhibitors. J Pharmacol Exp Ther. 2005;314(1):260–270. doi: 10.1124/jpet.105.085605. [DOI] [PubMed] [Google Scholar]
- 32.Regan J, Breitfelder S, Cirillo P, Gilmore T, Graham AG, Hickey E, Klaus B, Madwed J, Moriak M, Moss N, Pargellis C, Pav S, Proto A, Swinamer A, Tong L, Torcellini C. Pyrazole urea-based inhibitors of p38 MAP kinase: From lead compound to clinical candidate. J Med Chem. 2002;45(14):2994–3008. doi: 10.1021/jm020057r. [DOI] [PubMed] [Google Scholar]
- 33.Wang Z, Cuddy M, Samuel T, Welsh K, Schimmer A, Hanaii F, Houghten R, Pinilla C, Reed JC. Cellular, biochemical, and genetic analysis of mechanism of small molecule IAP inhibitors. J Biol Chem. 2004;279(46):48168–48176. doi: 10.1074/jbc.M405022200. [DOI] [PubMed] [Google Scholar]
- 34.Schimmer AD, Welsh K, Pinilla C, Wang Z, Krajewska M, Bonneau MJ, Pedersen IM, Kitada S, Scott FL, Bailly-Maitre B, Glinsky G, Scudiero D, Sausville E, Salvesen G, Nefzi A, Ostresh JM, Houghten RA, Reed JC. Small-molecule antagonists of apoptosis suppressor XIAP exhibit broad antitumor activity. Cancer Cell. 2004;5(1):25–35. doi: 10.1016/s1535-6108(03)00332-5. [DOI] [PubMed] [Google Scholar]
- 35.Chen YH, Tukey RH. Protein kinase C modulates regulation of the CYP1A1 gene by the aryl hydrocarbon receptor. J Biol Chem. 1996;271(42):26261–26266. doi: 10.1074/jbc.271.42.26261. [DOI] [PubMed] [Google Scholar]
- 36.Long WP, Pray-Grant M, Tsai JC, Perdew GH. Protein kinase C activity is required for aryl hydrocarbon receptor pathway-mediated signal transduction. Mol Pharmacol. 1998;53(4):691–700. doi: 10.1124/mol.53.4.691. [DOI] [PubMed] [Google Scholar]
- 37.Tian Y, Ke S, Denison MS, Rabson AB, Gallo MA. Ah receptor and NF-kappaB interactions, a potential mechanism for dioxin toxicity. J Biol Chem. 1999;274(1):510–515. doi: 10.1074/jbc.274.1.510. [DOI] [PubMed] [Google Scholar]
- 38.Denison MS, Wilkinson CF. Identification of the Ah receptor in selected mammalian species and induction of aryl hydrocarbon hydroxylase. Eur J Biochem. 1985;147(2):429–435. doi: 10.1111/j.1432-1033.1985.tb08767.x. [DOI] [PubMed] [Google Scholar]
- 39.Denison MS, Vella LM, Okey AB. Structure and function of the Ah receptor for 2,3,7,8-tetrachlorodibenzo-p-dioxin: Species difference in molecular properties of the receptors from mouse and rat hepatic cytosols. J Biol Chem. 1986;261(9):3987–3995. [PubMed] [Google Scholar]
- 40.Denison MS, Pandini A, Nagy SR, Baldwin EP, Bonati L. Ligand binding and activation of the Ah receptor. Chem Biol Interact. 2002;141(1–2):3–24. doi: 10.1016/s0009-2797(02)00063-7. [DOI] [PubMed] [Google Scholar]
- 41.Gradelet S, Astorg P, Pineau T, Canivenc MC, Siess MH, Leclerc J, Lesca P. Ah receptor-dependent CYP1A induction by two carotenoids, canthaxanthin and beta-apo-8′-carotenal, with no affinity for the TCDD binding site. Biochem Pharmacol. 1997;54(2):307–315. doi: 10.1016/s0006-2952(97)00176-7. [DOI] [PubMed] [Google Scholar]
- 42.Fontaine F, Delescluse C, de Sousa G, Lesca P, Rahmani R. Cytochrome 1A1 induction by primaquine in human hepatocytes and HepG2 cells: Absence of binding to the aryl hydrocarbon receptor. Biochem Pharmacol. 1999;57(3):255–262. doi: 10.1016/s0006-2952(98)00304-9. [DOI] [PubMed] [Google Scholar]
- 43.Backlund M, Ingelman-Sundberg M. Regulation of aryl hydrocarbon receptor signal transduction by protein tyrosine kinases. Cell Signal. 2005;17(1):39–48. doi: 10.1016/j.cellsig.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 44.ChemFinder-Chem3D chemical structure database. ( http://chemfinder.cambridgesoft.com)
- 45.Katzenellenbogen BS, Sun J, Harrington WR, Kraichely DM, Ganessunker D, Katzenellenbogen JA. Structure–function relationships in estrogen receptors and the characterization of novel selective estrogen receptor modulators with unique pharmacological profiles. Ann N Y Acad Sci. 2001;949:6–15. doi: 10.1111/j.1749-6632.2001.tb03998.x. [DOI] [PubMed] [Google Scholar]
- 46.Lewis JS, Jordan VC. Selective estrogen receptor modulators (SERMS): Mechanisms of anticarcinogenesis and drug resistance. Mutat Res. 2005;591(1–2):247–263. doi: 10.1016/j.mrfmmm.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 47.Buijsman RC, Hermkens PH, van Rijn RD, Stock HT, Teerhuis NM. Non-steroidal steroid receptor modulators. Curr Med Chem. 2005;12(9):1017–1075. doi: 10.2174/0929867053764671. [DOI] [PubMed] [Google Scholar]
- 48.Backlund M, Ingelman-Sundberg M. Different structural requirements of the ligand binding domain of the aryl hydrocarbon receptor for high- and low-affinity ligand binding and receptor activation. Mol Pharmacol. 2004;65(2):416–425. doi: 10.1124/mol.65.2.416. [DOI] [PubMed] [Google Scholar]
- 49.Henry EC, Gasiewicz TA. Agonist but not antagonist ligands induce conformational change in the mouse aryl hydrocarbon receptor as detected by partial proteolysis. Mol Pharmacol. 2003;63(2):392–400. doi: 10.1124/mol.63.2.392. [DOI] [PubMed] [Google Scholar]
- 50.Konstantinou IK, Albanis TA. Worldwide occurrence and effects of antifouling paint booster biocides in the aquatic environment: A review. Environ Int. 2004;30(2):235–248. doi: 10.1016/S0160-4120(03)00176-4. [DOI] [PubMed] [Google Scholar]
- 51.Harper PA, Prokipcak RD, Bush LE, Golas CL, Okey AB. Detection and characterization of the Ah receptor for 2,3,7,8-tetrachlorodibenzo-p-dioxin in the human colon adenocarcinoma cell line LS180. Arch Biochem Biophys. 1991;290(1):27–36. doi: 10.1016/0003-9861(91)90587-9. [DOI] [PubMed] [Google Scholar]
- 52.Ema M, Ohe N, Suzuki M, Mimura J, Sogawa K, Ikawa S, Fujii-Kuriyama Y. Dioxin binding activities of polymorphic forms of mouse and human arylhydrocarbon receptors. J Biol Chem. 1994;269(44):27337–27343. [PubMed] [Google Scholar]
- 53.Giacomazzi S, Cochet N. Environmental impact of diuron transformation: A review. Chemosphere. 2004;56:1021–1032. doi: 10.1016/j.chemosphere.2004.04.061. [DOI] [PubMed] [Google Scholar]