

Abstract
The current standard of care for hepatitis C virus (HCV) infection – combination therapy with pegylated interferon and ribavirin – elicits sustained responses in only ∼50% of the patients treated. No alternatives exist for patients who do not respond to combination therapy. Addition of ribavirin substantially improves response rates to interferon and lowers relapse rates following the cessation of therapy, suggesting that increasing ribavirin exposure may further improve treatment response. A key limitation, however, is the toxic side-effect of ribavirin, hemolytic anemia, which often necessitates a reduction of ribavirin dosage and compromises treatment response. Maximizing treatment response thus requires striking a balance between the antiviral and hemolytic activities of ribavirin. Current models of viral kinetics describe the enhancement of treatment response due to ribavirin. Ribavirin-induced anemia, however, remains poorly understood and precludes rational optimization of combination therapy. Here, we develop a new mathematical model of the population dynamics of erythrocytes that quantitatively describes ribavirin-induced anemia in HCV patients. Based on the assumption that ribavirin accumulation decreases erythrocyte lifespan in a dose-dependent manner, model predictions capture several independent experimental observations of the accumulation of ribavirin in erythrocytes and the resulting decline of hemoglobin in HCV patients undergoing combination therapy, estimate the reduced erythrocyte lifespan during therapy, and describe inter-patient variations in the severity of ribavirin-induced anemia. Further, model predictions estimate the threshold ribavirin exposure beyond which anemia becomes intolerable and suggest guidelines for the usage of growth hormones, such as erythropoietin, that stimulate erythrocyte production and avert the reduction of ribavirin dosage, thereby improving treatment response. Our model thus facilitates, in conjunction with models of viral kinetics, the rational identification of treatment protocols that maximize treatment response while curtailing side effects.
Author Summary
The treatment of HCV infection poses a major global health-care challenge today. The current standard of care, combination therapy with interferon and ribavirin, works in only about half of the patients treated. Because no alternatives are available yet for patients in whom combination therapy fails, identifying ways to improve response to combination therapy is critical. Increasing exposure to ribavirin does improve response but is associated with the severe side-effect, anemia. One way to maximize treatment response therefore is to increase ribavirin exposure to levels just below where anemia becomes intolerable. A second way is to supplement combination therapy with growth hormones, such as erythropoietin, that increase the production of red blood cells (erythrocytes) and compensate for ribavirin-induced anemia. Rational optimization of combination therapy thus relies on a quantitative description of ribavirin-induced anemia, which is currently lacking. Here, we develop a model of the population dynamics of erythrocytes in individuals exposed to ribavirin that quantitatively describes ribavirin-induced anemia. Model predictions capture several independent observations of ribavirin-induced anemia in HCV patients undergoing combination therapy, estimate the threshold ribavirin exposure beyond which anemia becomes intolerable, suggest guidelines for the usage of growth hormones, and facilitate rational optimization of therapy.
Introduction
130–170 million people worldwide are currently infected with hepatitis C virus (HCV) [1]. Over 70% of HCV infections become chronic and if untreated may lead to cirrhosis and hepatocellular carcinoma, necessitating liver transplantation [1]. The standard of care for HCV infection involves combination therapy with pegylated interferon and ribavirin [2]. Ribavirin alone does not elicit a lasting antiviral response [3]–[6], yet it substantially improves treatment response in combination with interferon [7]–[11]. For instance, whereas ∼29% of the patients treated with interferon exhibited a sustained virological response (SVR), the response rate increased to ∼56% upon addition of ribavirin [8]. Ribavirin, however, is associated with the side-effect, hemolytic anemia, which often renders therapy intolerable [4], [12]–[15]. With the standard ribavirin dosage of 1000–1200 mg/day, 54% of the patients treated experienced a decline in the hemoglobin (Hb) level of over 3 g/dL, and 10% of the men and 7% of the women treated experienced an Hb decline of over 5 g/dL (normal Hb range: 14–16 g/dL) [15]. This drop in Hb often necessitates a reduction of ribavirin dosage, which significantly compromises treatment response [7], [13], [14], [16]. The probability of achieving SVR is estimated to decrease from ∼65% to ∼45% when ribavirin dosage is reduced from ∼15 mg/kg to ∼7 mg/kg of body weight, in combination with pegylated interferon at 1.5 µg/kg of body weight [7]. Patients receiving fewer than 60% of the planned ribavirin doses had lower response rates [14], indicating that lower cumulative ribavirin exposure results in poorer treatment response [13], [16]. The rates of relapse of infection following the end of treatment also increased upon lowering ribavirin dosage [14], [16]. In a recent clinical trial where interferon was employed with telaprevir, a promising new inhibitor of HCV protease, response rates were lowest in patients who were not administered ribavirin [17], underscoring the importance of ribavirin in achieving SVR.
Alternatives for patients who do not respond to combination therapy do not exist yet [2], [18]. Significant efforts are underway therefore to identify treatment protocols that maximize response rates to combination therapy while curtailing side-effects [16], [19]–[25]. A particularly promising strategy is to supplement combination therapy with growth hormones, such as erythropoietin, that stimulate erythropoiesis and thus avert the reduction of ribavirin dosage, potentially improving treatment response [26]–[31]. The predominant mechanism(s) of the anti-HCV activity of ribavirin remain to be established [32]–[34]. Mathematical models of viral kinetics have been developed that describe the antiviral activity of interferon and the enhancement of treatment response rates due to ribavirin, and are being extended to predict the impact of new antiviral drugs [34]–[42]. Ribavirin-induced anemia, on the other hand, remains poorly understood [13], [24], [25], [43]–[47] and precludes rational optimization of combination therapy.
Here, we construct a mathematical model of the population dynamics of erythrocytes that quantitatively describes ribavirin-induced anemia and informs future strategies for improving outcomes of combination therapy. Model predictions capture experimental observations of the accumulation of ribavirin in erythrocytes and the ensuing Hb decline in HCV patients following the onset of combination therapy, estimate the enhanced turnover rate of erythrocytes during therapy and the threshold ribavirin exposure beyond which anemia is intolerable, present guidelines for the optimal usage of growth hormone supplements, and provide a framework, in conjunction with models of viral kinetics, for rational optimization of combination therapy.
Results
Model formulation
Prior to the onset of treatment with ribavirin, the population of erythrocytes (RBCs) in an HCV infected individual is constant; a balance exists between RBC production and death (Fig. 1). Following the onset of treatment, ribavirin administered orally gets rapidly transported from the plasma to RBCs, where it is phosphorylated to its mono-, di- and tri-phosphate analogs (RMP, RDP, and RTP) [48]. Phosphorylated analogs are neither easily metabolized nor transported out of RBCs [48]. Consequently, ribavirin accumulates inside RBCs in the form of its phosphorylated analogs; the total intracellular concentration of ribavirin can be >100-fold its extracellular concentration [47]. This dramatic accumulation of ribavirin may induce oxidative damage and result in enhanced extra vascular death of RBCs [12]. Indeed, RBC lifespan decreased from 107±22 d in HCV patients not exposed to ribavirin to 39±13 d in HCV patients undergoing treatment with ribavirin [49], [50]. The shortened RBC lifespan creates an imbalance between RBC production and death and results in a decline in the RBC population. Accordingly, Hb levels drop and patients become anemic. We construct a mathematical model to describe this dynamics of ribavirin-induced anemia (Methods).
Figure 1. Schematic of the population dynamics of RBCs in HCV infected individuals.

A, Before the onset of combination therapy, the production and death rates of RBCs are balanced and the population of RBCs in plasma is constant. B, Following the onset of therapy, ribavirin enters RBCs and increases their death rates. Cells born at different times are exposed to ribavirin for different durations and hence carry different concentrations of ribavirin. The enhanced death rate lowers the RBC population, which triggers an increase in the RBC production from the bone marrow by a negative feedback through the hormone erythropoietin.
Model predictions
Population dynamics of RBCs during treatment with ribavirin
We present model predictions in terms of the cumulative population, 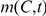 , which is the population of RBCs at time t following the onset of treatment in which the concentration of ribavirin phosphorylated analogs, RXP, is less than or equal to C (Fig. 2A). (RXP comprises RMP, RDP, and RTP.) At the start of treatment
, which is the population of RBCs at time t following the onset of treatment in which the concentration of ribavirin phosphorylated analogs, RXP, is less than or equal to C (Fig. 2A). (RXP comprises RMP, RDP, and RTP.) At the start of treatment 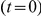 , no cells contain RXP, and
, no cells contain RXP, and 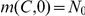 for all C, where N
0 is the steady population of RBCs prior to the onset of treatment. In other words, the population of cells carrying RXP at concentrations smaller than or equal to C is N
0 for all
for all C, where N
0 is the steady population of RBCs prior to the onset of treatment. In other words, the population of cells carrying RXP at concentrations smaller than or equal to C is N
0 for all 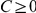 . At
. At 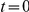 , the production and death rates of RBCs are in balance (Fig. 2B), the hemoglobin level, Hb = Hb
0, and the average intracellular concentration of ribavirin,
, the production and death rates of RBCs are in balance (Fig. 2B), the hemoglobin level, Hb = Hb
0, and the average intracellular concentration of ribavirin, 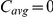 (Fig. 2C). With time, ribavirin accumulates inside cells. At any time
(Fig. 2C). With time, ribavirin accumulates inside cells. At any time 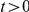 , a distribution of RXP concentrations across cells emerges with cells exposed to ribavirin longer possessing higher concentrations of RXP. Thus, cells present from the start of treatment possess the highest concentration of RXP. At
, a distribution of RXP concentrations across cells emerges with cells exposed to ribavirin longer possessing higher concentrations of RXP. Thus, cells present from the start of treatment possess the highest concentration of RXP. At 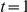 d, for instance, the latter cells possess RXP at the concentration
d, for instance, the latter cells possess RXP at the concentration 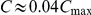 , where C
max =
, where C
max = 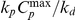 is the maximum intracellular concentration of RXP for a given extracellular concentration of ribavirin
is the maximum intracellular concentration of RXP for a given extracellular concentration of ribavirin 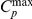 (Fig. 2A). Cells born after the onset of treatment possess RXP at concentrations smaller than
(Fig. 2A). Cells born after the onset of treatment possess RXP at concentrations smaller than 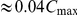 at
at 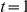 d. Because most of the cells present at
d. Because most of the cells present at 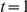 d are from the population that existed at the start of therapy,
d are from the population that existed at the start of therapy, 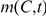 exhibits a sharp rise at
exhibits a sharp rise at 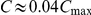 and reaches the value N(t = 1) (Fig. 2A). With time, the sharp rise in
and reaches the value N(t = 1) (Fig. 2A). With time, the sharp rise in 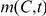 shifts to higher values of
shifts to higher values of 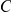 (Fig. 2A) indicating greater accumulation of RXP. Accordingly,
(Fig. 2A) indicating greater accumulation of RXP. Accordingly, 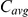 increases (Fig. 2C). Cells die at increasing rates as intracellular RXP accumulates (Fig. 2B). The population of RBCs, N(t), and, hence,
increases (Fig. 2C). Cells die at increasing rates as intracellular RXP accumulates (Fig. 2B). The population of RBCs, N(t), and, hence, 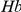 correspondingly decrease (Fig. 2C). New cells are continuously produced at a rate that increases as the RBC population declines (Fig. 2B). Eventually, a new balance between RBC production and death is attained,
correspondingly decrease (Fig. 2C). New cells are continuously produced at a rate that increases as the RBC population declines (Fig. 2B). Eventually, a new balance between RBC production and death is attained, 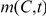 reaches a steady distribution,
reaches a steady distribution, 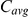 reaches an asymptotic maximum
reaches an asymptotic maximum 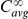 , and
, and 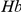 attains a new, lower steady value
attains a new, lower steady value 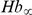 .
.
Figure 2. Model predictions of the dynamics of ribavirin-induced anemia.

A, The cumulative population, m(C,t), of RBCs as a function of the intracellular concentration of RXP, C, at different times, t, following the onset of combination therapy, predicted by Eqs. (1)–(3). Parameters employed are listed in Table 1. m(C,t) is normalized by N
0 and C by C
max = 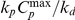 . B, The production rate, P(t), and the death rate
. B, The production rate, P(t), and the death rate 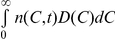 of RBCs. C, The corresponding change in Hb (red) and C
avg (green).
of RBCs. C, The corresponding change in Hb (red) and C
avg (green).
Factors that influence the severity of ribavirin-induced anemia
An increase in the steady state plasma concentration of ribavirin, 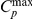 , which may be achieved with a higher dosage, results in an increase in
, which may be achieved with a higher dosage, results in an increase in 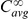 and a decrease in
and a decrease in 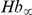 , illustrating the more severe anemia that results with increased ribavirin exposure. For instance, increasing
, illustrating the more severe anemia that results with increased ribavirin exposure. For instance, increasing 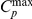 from 5 µM to 15 µM increases the total drop in Hb,
from 5 µM to 15 µM increases the total drop in Hb, 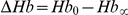 , from ∼2 g/dL to ∼5 g/dL (Fig. 3A). An increase in the intracellular phosphorylation rate,
, from ∼2 g/dL to ∼5 g/dL (Fig. 3A). An increase in the intracellular phosphorylation rate, 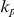 , (or a decrease in the loss rate of intracellular RXP,
, (or a decrease in the loss rate of intracellular RXP, 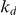 ) results in a higher
) results in a higher 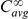 for the same
for the same 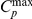 , which in turn elevates cell death rates and lowers
, which in turn elevates cell death rates and lowers 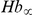 . Thus,
. Thus, 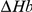 increases from ∼2.5 g/dL when
increases from ∼2.5 g/dL when 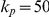 d−1 to ∼3.5 g/dL when
d−1 to ∼3.5 g/dL when 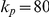 d−1 (Fig. 3B), illustrating that differences in the intracellular metabolism of ribavirin may contribute to inter-patient variations in the severity of ribavirin-induced anemia. Enhancing the RBC production rate, by increasing
d−1 (Fig. 3B), illustrating that differences in the intracellular metabolism of ribavirin may contribute to inter-patient variations in the severity of ribavirin-induced anemia. Enhancing the RBC production rate, by increasing 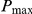 (see Eq. (3)), (or lowering the sensitivity of the RBC death rate to ribavirin accumulation, by increasing
(see Eq. (3)), (or lowering the sensitivity of the RBC death rate to ribavirin accumulation, by increasing 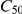 or decreasing
or decreasing  (see Eq. (2))), reduces
(see Eq. (2))), reduces 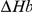 (Fig. 3C). Administration of growth hormones, such as erythropoietin, enhances the RBC production rate, which reduces
(Fig. 3C). Administration of growth hormones, such as erythropoietin, enhances the RBC production rate, which reduces 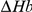 for the same ribavirin exposure and thus improves the tolerability of ribavirin. Similarly, decrease in the intracellular inosine triphosphatase level, observed recently in some patients [51], may interfere with RTP activity and lower the sensitivity of the RBC death rate to ribavirin, which also reduces
for the same ribavirin exposure and thus improves the tolerability of ribavirin. Similarly, decrease in the intracellular inosine triphosphatase level, observed recently in some patients [51], may interfere with RTP activity and lower the sensitivity of the RBC death rate to ribavirin, which also reduces 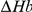 and improves the tolerability of ribavirin.
and improves the tolerability of ribavirin.
Figure 3. Factors influencing the dynamics of ribavirin-induced anemia.

Changes in Hb (red) and C
avg (green) predicted by Eqs. (1)–(3) for different parameter values: A, 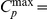 5 µM (solid lines), 10 µM (dashed lines) and 15 µM (dotted lines). B,
5 µM (solid lines), 10 µM (dashed lines) and 15 µM (dotted lines). B, 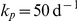 (solid lines), 65 d−1 (dashed lines), 80 d−1 (dotted lines). C,
(solid lines), 65 d−1 (dashed lines), 80 d−1 (dotted lines). C, 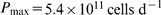 (solid lines),
(solid lines), 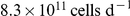 (dashed lines),
(dashed lines), 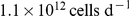 (dotted lines). The other parameters are mentioned in Table 1.
(dotted lines). The other parameters are mentioned in Table 1.
Inter-patient variations
Inter-patient variability in the severity of anemia may arise from variations in the intracellular uptake, accumulation, or metabolism of ribavirin, as well as in the dependence of the RBC lifespan on ribavirin accumulation and in the sensitivity of the RBC production rate to changes in Hb. To obtain a measure of this inter-patient variability, we calculated  using 500 different combinations of the values of the parameters,
using 500 different combinations of the values of the parameters, 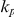 ,
, 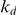 ,
, 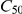 and
and  , for a range of values of
, for a range of values of 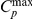 . The parameter values for each combination were chosen randomly from Gaussian distributions based on the mean values and confidence levels of the respective parameters obtained from comparisons of our model predictions with patient data (see below). Indeed, we find that inter-patient variations in
. The parameter values for each combination were chosen randomly from Gaussian distributions based on the mean values and confidence levels of the respective parameters obtained from comparisons of our model predictions with patient data (see below). Indeed, we find that inter-patient variations in 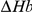 may be substantial for any ribavirin exposure (Fig. 4A) or intracellular accumulation (Fig. 4B). Further, the variation increases with increase in ribavirin exposure and intracellular accumulation.
may be substantial for any ribavirin exposure (Fig. 4A) or intracellular accumulation (Fig. 4B). Further, the variation increases with increase in ribavirin exposure and intracellular accumulation.
Figure 4. Model predictions of the variations in the severity of ribavirin-induced anemia.

The reduction in Hb (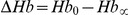 ) predicted by Eqs. (1)–(3) as a function of A,
) predicted by Eqs. (1)–(3) as a function of A, 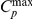 , and B,
, and B, 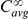 , for 500 different combinations of the parameter values (mean±s.d.)
, for 500 different combinations of the parameter values (mean±s.d.) 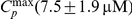 ,
, 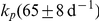 ,
, 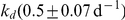 ,
, 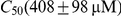 , and
, and 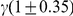 . The standard deviations on the parameter values correspond to the 95% confidence limits obtained from the best-fits to patient data (see Fig. 5). The other parameters are mentioned in Table 1.
. The standard deviations on the parameter values correspond to the 95% confidence limits obtained from the best-fits to patient data (see Fig. 5). The other parameters are mentioned in Table 1.
Below, we compare our model predictions with experiments.
Comparisons of model predictions with patient data
We consider a recent study of the time-evolution of 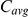 and
and 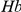 in 19 Japanese patients following the onset of combination therapy [47]. In this latter study, no reduction of ribavirin dosage is reported. The patients were divided into two groups based on whether
in 19 Japanese patients following the onset of combination therapy [47]. In this latter study, no reduction of ribavirin dosage is reported. The patients were divided into two groups based on whether 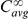 <1000 µM (7 patients) or
<1000 µM (7 patients) or 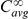 >1000 µM (12 patients); the data are reported as the average within each group. We fit model predictions of
>1000 µM (12 patients); the data are reported as the average within each group. We fit model predictions of 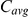 and
and 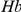 to the data of the former 7 patients using
to the data of the former 7 patients using 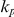 ,
, 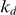 ,
, 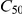 and
and  as adjustable parameters. (Interferon may also induce anemia, but does so to a much smaller extent than ribavirin [13]. We therefore assume that the Hb decline in patients undergoing combination therapy is primarily due to ribavirin.) We fix the remaining parameters based on previous studies or from analysis of independent experiments (Methods). Model predictions provide good fits to the data and yield estimates of
as adjustable parameters. (Interferon may also induce anemia, but does so to a much smaller extent than ribavirin [13]. We therefore assume that the Hb decline in patients undergoing combination therapy is primarily due to ribavirin.) We fix the remaining parameters based on previous studies or from analysis of independent experiments (Methods). Model predictions provide good fits to the data and yield estimates of 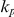 ,
, 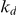 ,
, 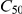 and
and  (Fig. 5A). The fits suggest that our model is able to describe the underlying dynamics of ribavirin-induced anemia in HCV patients.
(Fig. 5A). The fits suggest that our model is able to describe the underlying dynamics of ribavirin-induced anemia in HCV patients.
Figure 5. Comparisons of model predictions with experiments.

A, Best-fits of model predictions (lines) of Hb (red) and C
avg (green) with experimental data (symbols) from 7 Japanese patients with 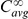 <1000 µM [47]. We let Hb
0 = 14.4 g/dL and
<1000 µM [47]. We let Hb
0 = 14.4 g/dL and 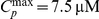 following the mean reported values for these patients [47]. We use
following the mean reported values for these patients [47]. We use  ,
, 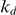 ,
, 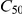 , and
, and  as adjustable parameters. The remaining parameters are mentioned in Table 1. The resulting best-fit parameter estimates (95% CI) are
as adjustable parameters. The remaining parameters are mentioned in Table 1. The resulting best-fit parameter estimates (95% CI) are 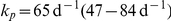 ,
, 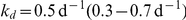 ,
, 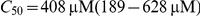 , and
, and 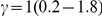 . Dashed lines show 95% confidence intervals on the predictions. B, Comparisons of our predictions (lines) of Hb (red) and C
avg (green) using the parameters above with data (symbols) from 12 Japanese patients with
. Dashed lines show 95% confidence intervals on the predictions. B, Comparisons of our predictions (lines) of Hb (red) and C
avg (green) using the parameters above with data (symbols) from 12 Japanese patients with 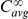 >1000 µM (solid circles) [47]. In the latter patients, the mean Hb
0≈15 g/dL and
>1000 µM (solid circles) [47]. In the latter patients, the mean Hb
0≈15 g/dL and 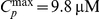 . We also show comparisons with independent data from 20 patients [29] (open circles) with mean Hb
0 = 15.1 g/dL. Solid lines are predictions with Hb
0 = 15.1 g/dL and
. We also show comparisons with independent data from 20 patients [29] (open circles) with mean Hb
0 = 15.1 g/dL. Solid lines are predictions with Hb
0 = 15.1 g/dL and 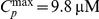 , and the mean parameters mentioned above, and dashed lines represent standard deviations. C, D, Model predictions (lines) and experimental observations (symbols) of the reduction in Hb (
, and the mean parameters mentioned above, and dashed lines represent standard deviations. C, D, Model predictions (lines) and experimental observations (symbols) of the reduction in Hb (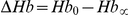 ) under combination therapy as a function of C,
) under combination therapy as a function of C, 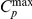 , and D,
, and D, 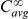 , in 19 Japanese patients [47]. Solid lines represent predictions with the mean parameter values above and dashed lines represent standard deviations obtained from several hundred realizations of our model predictions for different combinations of the values of
, in 19 Japanese patients [47]. Solid lines represent predictions with the mean parameter values above and dashed lines represent standard deviations obtained from several hundred realizations of our model predictions for different combinations of the values of 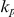 ,
, 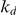 ,
, 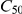 , and
, and  generated randomly from distributions based on the best-fit parameter estimates and 95% confidence limits mentioned above.
generated randomly from distributions based on the best-fit parameter estimates and 95% confidence limits mentioned above.
Interestingly, with the same parameter values, our model captures changes in Hb and 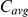 from the other 12 Japanese patients, as well as an independent data set of Hb decline in another group of HCV patients undergoing combination therapy [29] (Fig. 5B), validating our best-fit parameter estimates. Further, with the same parameter values, we estimate that the RBC lifespan is 38 days (95% CI: 19–55 days) in Japanese patients with
from the other 12 Japanese patients, as well as an independent data set of Hb decline in another group of HCV patients undergoing combination therapy [29] (Fig. 5B), validating our best-fit parameter estimates. Further, with the same parameter values, we estimate that the RBC lifespan is 38 days (95% CI: 19–55 days) in Japanese patients with 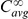 <1000 µM and 33 days (95% CI: 14–53 days) in Japanese patients with
<1000 µM and 33 days (95% CI: 14–53 days) in Japanese patients with 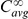 >1000 µM. These estimates of the RBC lifespan are in close agreement with independent estimates, 39±13 days, from measurements of alveolar carbon monoxide [49], [50], presenting another successful test of our model. Finally, we find that our predictions of the dependence of
>1000 µM. These estimates of the RBC lifespan are in close agreement with independent estimates, 39±13 days, from measurements of alveolar carbon monoxide [49], [50], presenting another successful test of our model. Finally, we find that our predictions of the dependence of 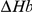 on
on 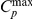 and
and 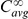 using the same parameters above are also in agreement with observations in the Japanese patients [47] (Fig. 5C,D). Our model thus presents a robust description of ribavirin-induced anemia in HCV patients undergoing combination therapy.
using the same parameters above are also in agreement with observations in the Japanese patients [47] (Fig. 5C,D). Our model thus presents a robust description of ribavirin-induced anemia in HCV patients undergoing combination therapy.
Clinical implications
Our model has several clinical implications. First, it enables estimation of the threshold ribavirin exposure beyond which anemia is intolerable. Current treatment guidelines recommend a reduction of ribavirin dosage when Hb decreases below 10 g/dL. We apply our model to predict 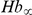 as a function of
as a function of 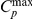 . We find that on average (when
. We find that on average (when 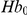 = 14.4 g/dL)
= 14.4 g/dL) 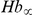 <10 g/dL when
<10 g/dL when 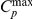 >13 µM (Fig. 6A). Thus, steady state plasma concentrations above 13 µM would render ribavirin therapy intolerable. While the dependence of the peak plasma concentration on dosage following a single ribavirin dose has been determined [48], the dependence of
>13 µM (Fig. 6A). Thus, steady state plasma concentrations above 13 µM would render ribavirin therapy intolerable. While the dependence of the peak plasma concentration on dosage following a single ribavirin dose has been determined [48], the dependence of  on dosage remains to be established. A description of the multiple dose pharmacokinetics of ribavirin, which also remains elusive [6], [34], [48], [52], [53], would establish the dosage corresponding to
on dosage remains to be established. A description of the multiple dose pharmacokinetics of ribavirin, which also remains elusive [6], [34], [48], [52], [53], would establish the dosage corresponding to 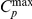 of 13 µM that would render ribavirin intolerable.
of 13 µM that would render ribavirin intolerable.
Figure 6. Model predictions of threshold ribavirin exposure and requisite RBC production.

A, Prediction of 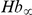 as a function of
as a function of 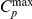 . The arrow indicates the threshold
. The arrow indicates the threshold 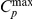 above which
above which 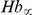 <10 g/dL. B, Predictions of the RBC production rate during therapy (black) and the production rate required to maintain
<10 g/dL. B, Predictions of the RBC production rate during therapy (black) and the production rate required to maintain 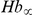 of 10 g/dL (pink) and 12 g/dL (blue) as functions of
of 10 g/dL (pink) and 12 g/dL (blue) as functions of 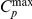 . The arrows indicate the desired production rates when
. The arrows indicate the desired production rates when 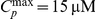 . Parameter values employed are mentioned in Table 1.
. Parameter values employed are mentioned in Table 1.
Second, when 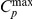 is above the threshold, our model allows estimation of the increase in RBC production, which may be achieved by administration of exogenous growth hormones such as recombinant erythropoietin, necessary to avert the currently recommended reduction of dosage. Because growth hormones also have side-effects [29], [54], one strategy is to use them at levels just enough to increase
is above the threshold, our model allows estimation of the increase in RBC production, which may be achieved by administration of exogenous growth hormones such as recombinant erythropoietin, necessary to avert the currently recommended reduction of dosage. Because growth hormones also have side-effects [29], [54], one strategy is to use them at levels just enough to increase 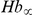 to 10–12 g/dL (rather than the pretreatment level), which renders ribavirin tolerable [16]. We apply our model to predict the level of RBC production necessary for achieving
to 10–12 g/dL (rather than the pretreatment level), which renders ribavirin tolerable [16]. We apply our model to predict the level of RBC production necessary for achieving 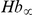 of 10–12 g/dL for different values of
of 10–12 g/dL for different values of 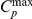 (Fig. 6B). Thus, when
(Fig. 6B). Thus, when 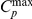 = 15 µM, RBC production rates of 8.44 and 10.2 million cells s−1 are necessary for ensuring
= 15 µM, RBC production rates of 8.44 and 10.2 million cells s−1 are necessary for ensuring 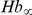 of 10 and 12 g/dL, respectively. Increase in endogenous erythropoietin levels during therapy, also observed experimentally [45], [55], results in an enhanced production rate of 8.1 million cells s−1, which is 3.5-fold higher than the basal production rate (here 2.3 million cells s−1 in the absence of ribavirin) but inadequate to achieve the desired
of 10 and 12 g/dL, respectively. Increase in endogenous erythropoietin levels during therapy, also observed experimentally [45], [55], results in an enhanced production rate of 8.1 million cells s−1, which is 3.5-fold higher than the basal production rate (here 2.3 million cells s−1 in the absence of ribavirin) but inadequate to achieve the desired 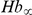 . Hormone supplements may be employed to provide the balance of 0.34 or 2.1 million cells s−1 increase in the RBC production rate to ensure
. Hormone supplements may be employed to provide the balance of 0.34 or 2.1 million cells s−1 increase in the RBC production rate to ensure 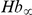 of 10 or 12 g/dL, respectively. This deficiency in RBC production that hormone supplements must compensate increases with ribavirin exposure (Fig. 6B).
of 10 or 12 g/dL, respectively. This deficiency in RBC production that hormone supplements must compensate increases with ribavirin exposure (Fig. 6B).
Discussion
The ability to enhance treatment response rates renders ribavirin central to the treatment of HCV infection. Maximizing the benefit of ribavirin to patients requires striking the right balance between its antiviral activity and its treatment-limiting side-effect, hemolytic anemia. Rational approaches to therapy optimization thus rely on quantitative descriptions of both the antiviral and the hemolytic activities of ribavirin. Extant mathematical models predict the enhancement in treatment response due to ribavirin [34]–[42]. Ribavirin-induced anemia, however, remains poorly described and limits our ability to maximize treatment response. Here, we fill this gap by constructing a model of the population dynamics of RBCs that quantitatively describes ribavirin-induced anemia. By assuming that intracellular accumulation of ribavirin enhances RBC death rate in a dose-dependent manner, our model captures several independent observations of ribavirin-induced anemia in HCV patients undergoing combination therapy. In particular, our model predicts the dynamics of the accumulation of ribavirin in RBCs and the resulting decline of Hb in patients following the onset of therapy, estimates the reduced lifespan of RBCs during therapy, and describes inter-patient variations in the severity of anemia, thus presenting a robust description of ribavirin-induced anemia, which, in conjunction with models of viral kinetics, may facilitate identification of treatment protocols that maximize the impact of ribavirin in the treatment of HCV infection.
Our model has clinical implications. First, it allows estimation of the threshold ribavirin exposure beyond which ribavirin-induced anemia becomes intolerable. For instance, with model parameters that describe ribavirin-induced anemia in the patients we considered (Fig. 5), we estimate that steady state plasma ribavirin concentrations above 13 µM would render ribavirin therapy intolerable. Determining dosage levels corresponding to this steady state plasma concentration requires knowledge of the pharmacokinetics of ribavirin, which is currently lacking [6], [34], [48], [52], [53]. Ribavirin pharmacokinetics is peculiar because of an unusually long elimination phase that follows rapid absorption and distribution phases upon oral dosing [48]. Standard absorption-elimination models of drug pharmacokinetics are unable to describe this long elimination phase. Models that include additional compartments have been proposed to capture the three-phase pharmacokinetics of ribavirin [52], but the biological origin of these compartments remains unclear. An additional complication is that the half-life of the elimination phase increases from 79 h following a single dose to 274–298 h following multiple dosing [48], suggesting that parameters that describe single dose pharmacokinetics may not apply to multiple dose pharmacokinetics. In the absence of rigorous models of ribavirin pharmacokinetics, one may have to rely on empirical relationships between the dosage and the resulting steady state plasma concentration following multiple dosing (e.g., [56]) to establish the dosage that would ensure tolerability of ribavirin while maximizing treatment response.
Second, our model suggests guidelines for the usage of hormone supplements, such as erythropoietin, which enhance RBC production and improve the tolerability of ribavirin. For instance, we predict that when ribavirin accumulates to a plasma concentration of 15 µM, the associated enhanced RBC death rate elicits a natural response that increases RBC production 3.5-fold, from 2.3 to 8.1 million cells s−1. This response, however, is inadequate to suppress ribavirin-induced anemia adequately and renders ribavirin intolerable. We estimate then that growth hormone supplements must increase RBC production rate by an additional 0.34–2.1 million cells s−1 to render ribavirin tolerable. This compensation that hormone supplements must provide increases with ribavirin accumulation. Identifying the dosage of the growth hormones that induces the necessary RBC production requires knowledge of the dose-response relationships and of the pharmacokinetics of the growth hormones, which are yet to be fully elucidated [26]–[31].
Third, genetic variations that resulted in a deficiency in the enzyme inosine triphosphatase (ITPA) were recently found to protect HCV patients against ribavirin-induced anemia [51]. Deficiency in ITPA causes an increase in inosine triphosphate levels in RBCs, which is thought to interfere with RTP activity and thereby suppress the hemolytic potential of ribavirin. Because deficiency in ITPA is a clinically benign condition, therapeutic intervention to suppress ITPA presents a promising new strategy to curtail ribavirin-induced anemia without compromising the antiviral activity of ribavirin [51]. Our model may be adapted to inform the development of such an intervention strategy. In our model, the dependence of the death rate of RBCs on ribavirin accumulation, determined by Eq. (2) (Methods), would now be a function of the ITPA level. Thus, experiments that determine how variations in the ITPA level both in the absence and in the presence of ribavirin influence RBC lifespan would provide the necessary inputs for our model to account explicitly for the role of ITPA in ribavirin-induced anemia. The resulting model would enable determination of the minimal inhibition of ITPA necessary to maintain ribavirin-induced anemia within tolerable limits. Conversely, using information of the ITPA level intrinsic to a patient, the model can be applied to predict the maximum ribavirin dosage that the patient can tolerate, thus presenting an avenue for personalizing the treatment of HCV infection.
Methods
Model development
We consider the RBC population in an individual at time t following the onset of treatment with ribavirin (t = 0) (Fig. 1). RBCs produced at different times in the interval from 0 to t will have been exposed to ribavirin for different durations and accordingly have different intracellular levels of ribavirin. We define 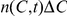 as the population of RBCs that contain ribavirin phosphorylated analogs, RXP, which comprises RMP, RDP, and RTP, at concentrations between
as the population of RBCs that contain ribavirin phosphorylated analogs, RXP, which comprises RMP, RDP, and RTP, at concentrations between 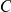 and
and 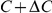 at time
at time  .
. 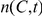 is thus the number density of RBCs containing RXP at concentration C at time t. The time evolution of
is thus the number density of RBCs containing RXP at concentration C at time t. The time evolution of 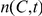 is governed by the following equation (Text S1)
is governed by the following equation (Text S1)
| (1) |
The first term on the right-hand-side in Eq. (1) represents the change in 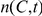 due to intracellular phosphorylation of ribavirin.
due to intracellular phosphorylation of ribavirin. 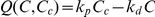 is the net rate of increase of C due to phosphorylation,
is the net rate of increase of C due to phosphorylation, 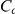 is the intracellular concentration of (unphosphorylated) ribavirin,
is the intracellular concentration of (unphosphorylated) ribavirin, 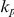 is the phosphorylation rate and
is the phosphorylation rate and 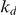 is the rate of loss, including by possible slow dephosphorylation, of RXP. In vitro studies of ribavirin uptake by RBCs observe rapid (<10 min) equilibration of intracellular and extracellular ribavirin [53], [57]. We assume therefore that
is the rate of loss, including by possible slow dephosphorylation, of RXP. In vitro studies of ribavirin uptake by RBCs observe rapid (<10 min) equilibration of intracellular and extracellular ribavirin [53], [57]. We assume therefore that 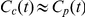 , the concentration of ribavirin in plasma. With twice daily oral administration of ribavirin,
, the concentration of ribavirin in plasma. With twice daily oral administration of ribavirin, 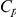 rises from zero at
rises from zero at 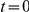 and reaches an asymptotic maximum,
and reaches an asymptotic maximum, 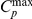 , so that
, so that 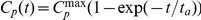 , where
, where 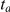 is the characteristic timescale of the accumulation of ribavirin in plasma [6], [38].
is the characteristic timescale of the accumulation of ribavirin in plasma [6], [38].
The second term on the right-hand side of Eq. (1) accounts for the loss of RBCs due to their death. We assume that the death rate, D, of RBCs increases with 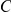 as follows
as follows
| (2) |
where 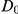 is the death rate of RBCs in the absence of ribavirin,
is the death rate of RBCs in the absence of ribavirin, 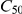 is that value of
is that value of 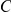 at which the death rate doubles (or the lifespan halves) compared to that in the absence of ribavirin, and
at which the death rate doubles (or the lifespan halves) compared to that in the absence of ribavirin, and  , analogous to the Hill coefficient, determines the sensitivity of
, analogous to the Hill coefficient, determines the sensitivity of 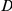 to changes in
to changes in 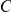 . (A saturable form for D(C) appears inconsistent with available data; see Text S2, Fig. S1.)
. (A saturable form for D(C) appears inconsistent with available data; see Text S2, Fig. S1.)
Equation (1) is constrained by the initial condition that 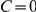 in all cells at the start of therapy, so that
in all cells at the start of therapy, so that 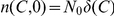 , where N
0 is the population of RBCs at t = 0, and
, where N
0 is the population of RBCs at t = 0, and 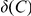 is the Dirac delta function, which satisfies
is the Dirac delta function, which satisfies 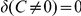 and
and 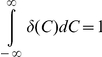 . In other words, the Dirac delta function ensures that no cells have RXP at non-zero concentrations at t = 0. A second constraint on Eq. (1) is imposed by the boundary condition that when
. In other words, the Dirac delta function ensures that no cells have RXP at non-zero concentrations at t = 0. A second constraint on Eq. (1) is imposed by the boundary condition that when  >0, newborn cells contain no RXP so that
>0, newborn cells contain no RXP so that 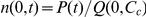 (Text S1) where
(Text S1) where
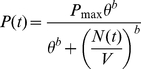 |
(3) |
is the rate of production of RBCs at time t.
The production of RBCs by the bone marrow is regulated by a negative feedback mechanism involving the hormone erythropoietin [58]. Recent studies on modeling erythropoiesis elucidate the complexities involved in a quantitative description of this feedback mechanism [59]–[65]. Here, we employ Eq. (3) to capture the essential features of this negative feedback: As the population of RBCs, 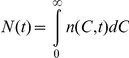 , decreases, P increases.
, decreases, P increases. 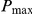 is the maximum production rate of RBCs, which occurs when N is vanishingly small,
is the maximum production rate of RBCs, which occurs when N is vanishingly small, 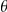 is that value of the RBC population per unit volume of blood (
is that value of the RBC population per unit volume of blood (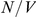 ) at which
) at which 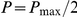 ,
, 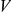 is the volume of blood, and
is the volume of blood, and 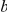 , analogous to the Hill coefficient, determines the sensitivity of
, analogous to the Hill coefficient, determines the sensitivity of 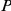 to changes in
to changes in 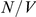 . Eq. (3) provides good fits to independent measurements of the dynamics of the recovery of RBCs following phlebotomy (Text S3, Fig. S2).
. Eq. (3) provides good fits to independent measurements of the dynamics of the recovery of RBCs following phlebotomy (Text S3, Fig. S2).
Equations (1)–(3) present a model of the population dynamics of RBCs in individuals undergoing treatment with ribavirin. We solve the equations (see below) and obtain the population density, 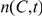 , and the corresponding cumulative population,
, and the corresponding cumulative population, 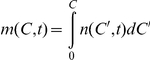 , using which we predict the time-evolution of the hemoglobin level in blood,
, using which we predict the time-evolution of the hemoglobin level in blood, 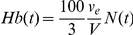 (where
(where 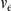 is the volume of a single erythrocyte); the average concentration of ribavirin in RBCs,
is the volume of a single erythrocyte); the average concentration of ribavirin in RBCs, 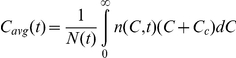 ; and the average RBC lifespan,
; and the average RBC lifespan, 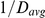 , where
, where 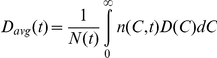 is the average death rate of RBCs.
is the average death rate of RBCs.
Solution of model equations using the method of characteristics
Equation (1) along with the initial and boundary conditions is equivalent to the following set of differential equations obtained using the method of characteristics (Text S4)
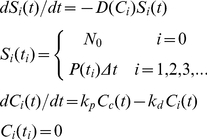 |
(4) |
where Si(t) is the subpopulation of cells born within an interval 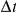 of
of 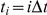 that survive at time t. Ci(t) is the concentration of RXP in the latter cells at time t. We solve Eq. (4) along with Eqs. (2) and (3) with
that survive at time t. Ci(t) is the concentration of RXP in the latter cells at time t. We solve Eq. (4) along with Eqs. (2) and (3) with 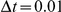 d using a program written in MATLAB (Text S5). We validate our solution methodology against an analytical solution that can be obtained in the limiting case when the RBC death rate is independent of RXP accumulation (Text S6, Fig. S3). We also ensure that
d using a program written in MATLAB (Text S5). We validate our solution methodology against an analytical solution that can be obtained in the limiting case when the RBC death rate is independent of RXP accumulation (Text S6, Fig. S3). We also ensure that 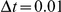 d allows accurate integration of Eq. (4) without compromising computational efficiency (Fig. S4). From the solution, we calculate the quantities of interest, viz.,
d allows accurate integration of Eq. (4) without compromising computational efficiency (Fig. S4). From the solution, we calculate the quantities of interest, viz., 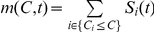 ,
, 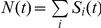 ,
, 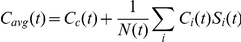 , and
, and 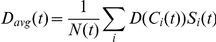 .
.
Model parameters
We employ the following values of the model parameters unless stated otherwise. The average RBC lifespan in normal man is ∼120 days [49], [66], which corresponds to 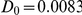 d−1. We let b = 7 following earlier studies [59] and obtain
d−1. We let b = 7 following earlier studies [59] and obtain 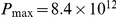 cells d−1 from an independent analysis of blood loss experiments (Text S3). We fix
cells d−1 from an independent analysis of blood loss experiments (Text S3). We fix 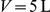 and
and 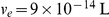 [67]. Using
[67]. Using 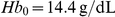 [47], we get
[47], we get 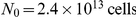 . We obtain
. We obtain 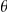 from the initial steady state
from the initial steady state 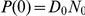 . Further, we let
. Further, we let 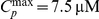 [47] and because ribavirin accumulates in plasma to its maximum concentration in ∼4 weeks, we set
[47] and because ribavirin accumulates in plasma to its maximum concentration in ∼4 weeks, we set 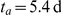 [6], [38]. The remaining parameter values
[6], [38]. The remaining parameter values 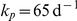 ,
, 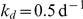 ,
, 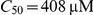 , and
, and 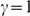 are obtained from best-fits of our model predictions to experimental data (Fig. 5A). We summarize model parameters and their values in Table 1.
are obtained from best-fits of our model predictions to experimental data (Fig. 5A). We summarize model parameters and their values in Table 1.
Table 1. Summary of model parameters and their values employed.
| Parameter | Description | Value (95% CI)a | Source |
| V | Volume of blood | 5 L | [67] |
| ve | Volume of one RBC | 9×10−14 L | [67] |
| ta | Characteristic timescale of accumulation of RBV in plasma | 5.4 d | [6], [38] |
| b | Coefficient determining sensitivity of RBC production rate to changes in number per volume of RBCs | 7 | [59] |
| D 0 | Death rate of RBCs in the absence of ribavirin | 8.3×10−3 d−1 | [49], [66] |
| Hb 0 | Initial Hb level | 14.4 g/dL | [47] |
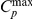
|
Steady state plasma RBV concentration | 7.5 µM | [47] |
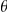
|
RBC population per unit volume at which RBC production rate is half maximal | Determined from pretreatment steady state, 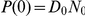
|
|
| Pmax | Maximum production rate of RBCs | 8.4×1012 cells d−1 | Best-fit (Text S3) |
| kp | Phosphorylation rate of ribavirin in RBCs | 65 (47–84) d−1 | Best-fit (Fig. 5A) |
| kd | Loss rate of ribavirin phosphorylated analogs in RBCs | 0.5 (0.3–0.7) d−1 | Best-fit (Fig. 5A) |
| C 50 | Concentration of ribavirin phosphorylated analogs at which RBC death rate is twice the pretreatment value | 408 (189–628) µM | Best-fit (Fig. 5A) |
| γ | Coefficient determining sensitivity of RBC death rate to changes in concentration of ribavirin phosphorylated analogs | 1 (0.2–1.8) | Best-fit (Fig. 5A) |
Typical values employed. Variations are indicated in the text and in figure legends.
Fits of model predictions to patient data
We fit model predictions to experimental data (Fig. 5A) using the nonlinear regression tool NLINFIT in MATLAB.
Supporting Information
Hemoglobin reduction as a function of the intracellular ribavirin concentration.
(0.09 MB PDF)
Analysis of RBC recovery following phlebotomy.
(0.41 MB PDF)
Validation of the solution methodology.
(0.39 MB PDF)
Sensitivity of the numerical solution to the integration time step.
(0.37 MB PDF)
Derivation of equation (1) and its boundary condition.
(0.29 MB PDF)
Dependence of RBC death rate on intracellular ribavirin concentration.
(0.19 MB PDF)
Analysis of phlebotomy experiments.
(0.28 MB PDF)
Solution of model equations using the method of characteristics.
(0.30 MB PDF)
MATLAB program for solving model equations.
(0.19 MB PDF)
Validation of the solution methodology.
(0.20 MB PDF)
Acknowledgments
We thank Alan S. Perelson and Ruy M. Ribeiro for helpful comments.
Footnotes
The authors have declared that no competing interests exist.
This work was supported by the Department of Biotechnology Centre of Excellence for Research on Hepatitis C Virus, India. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Lavanchy D. The global burden of hepatitis C. Liver Int. 2009;29:74–81. doi: 10.1111/j.1478-3231.2008.01934.x. [DOI] [PubMed] [Google Scholar]
- 2.Ghany MG, Strader DB, Thomas DL, Seeff LB. Diagnosis, management, and treatment of hepatitis C: An update. Hepatology. 2009;49:1335–1374. doi: 10.1002/hep.22759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dusheiko G, Main J, Thomas H, Reichard O, Lee C, et al. Ribavirin treatment for patients with chronic hepatitis C: results of a placebo-controlled study. J Hepatol. 1996;25:591–598. doi: 10.1016/s0168-8278(96)80225-x. [DOI] [PubMed] [Google Scholar]
- 4.Bodenheimer HC, Lindsay KL, Davis GL, Lewis JH, Thung SN, et al. Tolerance and efficacy of oral ribavirin treatment of chronic hepatitis C: A multicenter trial. Hepatology. 1997;26:473–477. doi: 10.1002/hep.510260231. [DOI] [PubMed] [Google Scholar]
- 5.Zoulim F, Haem J, Ahmed SS, Chossegros P, Habersetzer F, et al. Ribavirin monotherapy in patients with chronic hepatitis C: a retrospective study of 95 patients. J Viral Hepat. 1998;5:193–198. doi: 10.1046/j.1365-2893.1998.00099.x. [DOI] [PubMed] [Google Scholar]
- 6.Pawlotsky JM, Dahari H, Neumann AU, Hezode C, Germanidis G, et al. Antiviral action of ribavirin in chronic hepatitis C. Gastroenterology. 2004;126:703–714. doi: 10.1053/j.gastro.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 7.Manns MP, McHutchison JG, Gordon SC, Rustgi VK, Shiffman M, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet. 2001;358:958–965. doi: 10.1016/s0140-6736(01)06102-5. [DOI] [PubMed] [Google Scholar]
- 8.Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347:975–982. doi: 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- 9.McHutchison JG, Gordon SC, Schiff ER, Shiffman ML, Lee WM, et al. Interferon alfa-2b alone or in combination with ribavirin as initial treatment for chronic hepatitis C. N Engl J Med. 1998;339:1485–1492. doi: 10.1056/NEJM199811193392101. [DOI] [PubMed] [Google Scholar]
- 10.Reichard O, Norkrans G, Fryden A, Braconier JH, Sonnerborg A, et al. Randomized, double-blind, placebo-controlled trial of interferon alpha-2b with and without ribavirin for chronic hepatitis C. Lancet. 1998;351:83–87. doi: 10.1016/s0140-6736(97)06088-1. [DOI] [PubMed] [Google Scholar]
- 11.Poynard T, Marcellin P, Lee SS, Niederau C, Minuk GS, et al. Randomized trial of interferon alpha2b plus ribavirin for 48 weeks or for 24 weeks versus interferon alpha2b plus placebo for 48 weeks for treatment of chronic infection with hepatitis C virus. Lancet. 1998;352:1426–1432. doi: 10.1016/s0140-6736(98)07124-4. [DOI] [PubMed] [Google Scholar]
- 12.Franceschi LD, Fattovich G, Turrini F, Brugnara C, Manzato F, et al. Hemolytic anemia induced by ribavirin therapy in patients with chronic hepatitis C virus infection: Role of membrane oxidative damage. Hepatology. 2000;31:997–1004. doi: 10.1053/he.2000.5789. [DOI] [PubMed] [Google Scholar]
- 13.Sulkowski MS. Anemia in the treatment of hepatitis C virus Infection. Clin Infect Dis. 2003;37:s315–322. doi: 10.1086/376911. [DOI] [PubMed] [Google Scholar]
- 14.Reddy KR, Shiffman ML, Morgan TR, Zeuzem S, Hadziyannis S, et al. Impact of ribavirin dose reductions in hepatitis C virus genotype 1 patients completing peginterferon alfa-2a/ribavirin treatment. Clin Gastroenterol Hepatol. 2007;5:124–129. doi: 10.1016/j.cgh.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 15.Sulkowski MS, Wasserman R, Brooks L, Ball L, Gish R. Changes in haemoglobin during interferon alpha-2b plus ribavirin combination therapy for chronic hepatitis C virus infection. J Viral Hepat. 2004;11:243–250. doi: 10.1111/j.1365-2893.2004.00490.x. [DOI] [PubMed] [Google Scholar]
- 16.Reddy KR, Nelson DR, Zeuzem S. Ribavirin: Current role in the optimal clinical management of chronic hepatitis C. J Hepatol. 2009;50:402–411. doi: 10.1016/j.jhep.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 17.Hazode C, Forestier N, Dusheiko G, Ferenci P, Pol S, et al. Telaprevir and peginterferon with or without ribavirin for chronic HCV infection. N Engl J Med. 2009;360:1839–1850. doi: 10.1056/NEJMoa0807650. [DOI] [PubMed] [Google Scholar]
- 18.Lemon SM, McKeating JA, Pietschmann T, Frick DN, Glenn JS, et al. Development of novel therapies for hepatitis C. Antivir Res. 2010;86:79–92. doi: 10.1016/j.antiviral.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 19.Zeuzem S. Interferon-based therapy for chronic hepatitis C: current and future perspectives. Nat Clin Pract Gastroenterol Hepatol. 2008;5:610–622. doi: 10.1038/ncpgasthep1274. [DOI] [PubMed] [Google Scholar]
- 20.Jain MK, Zoellner C. Role of ribavirin in HCV treatment response: now and in the future. Expert Opin Pharmaco. 2010;11:673–683. doi: 10.1517/14656560903580001. [DOI] [PubMed] [Google Scholar]
- 21.Landaverde C, Saab S. Optimizing peginterferon and ribavirin administration in difficult-to-treat patient populations. Curr Hepatitis Rep. 2009;8:43–51. [Google Scholar]
- 22.Morello J, Rodriguez-Novoa S, Jimenez-Nacher I, Soriano V. Usefulness of monitoring ribavirin plasma concentrations to improve treatment response in patients with chronic hepatitis C. J Antimicrob Chemother. 2008;62:1174–1180. doi: 10.1093/jac/dkn421. [DOI] [PubMed] [Google Scholar]
- 23.Schmid M, Kreil A, Jessner W, Homoncik M, Datz C, et al. Suppression of haematopoiesis during therapy of chronic hepatitis C with different interferon alpha mono and combination therapy regimens. Gut. 2005;54:1014–1020. doi: 10.1136/gut.2004.057893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tod M, Farcy-Afif M, Stocco J, Boyer N, Bouton VR, et al. Pharmacokinetic/pharmacodynamic and time-to-event models of ribavirin-induced anaemia in chronic hepatitis C. Clin Pharmacokinet. 2005;44:417–428. doi: 10.2165/00003088-200544040-00006. [DOI] [PubMed] [Google Scholar]
- 25.DebRoy S, Kribs-Zaleta C, Mubayi A, Cardona-Meléndez GM, Medina-Rios L, et al. Evaluating treatment of hepatitis C for hemolytic anemia management. Math Biosci. 2010;225:141–155. doi: 10.1016/j.mbs.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 26.Dieterich DT, Wasserman R, Brau N, Hassanein TI, Bini EJ, et al. Once-weekly epoetin alfa improves anemia and facilitates maintenance of ribavirin dosing in hepatitis C virus-infected patients receiving ribavirin plus interferon alfa. Am J Gastroenterol. 2003;98:2491–2499. doi: 10.1111/j.1572-0241.2003.08700.x. [DOI] [PubMed] [Google Scholar]
- 27.Afdhal NH, Dieterich DT, Pockros PJ, Schiff ER, Shiffman ML, et al. Epoetin alfa maintains ribavirin dose in HCV-infected patients: a prospective, double-blind, randomized controlled study. Gastroenterology. 2004;126:1302–1311. doi: 10.1053/j.gastro.2004.01.027. [DOI] [PubMed] [Google Scholar]
- 28.Rio RAD, Post AB, Singer ME. Cost-effectiveness of hematologic growth factors for anemia occurring during hepatitis C combination therapy. Hepatology. 2006;44:1598–1606. doi: 10.1002/hep.21409. [DOI] [PubMed] [Google Scholar]
- 29.Homoncik M, Sieghart W, Formann E, Schmid M, Ferenci P, et al. Erythropoietin treatment is associated with more severe thrombocytopenia in patients with chronic hepatitis C undergoing antiviral therapy. Amer J Gastroenterol. 2006;101:2275–2282. doi: 10.1111/j.1572-0241.2006.00774.x. [DOI] [PubMed] [Google Scholar]
- 30.Shiffman ML, Salvatore J, Hubbard S, Price A, Sterling RK, et al. Treatment of chronic hepatitis C virus genotype 1 with peginterferon, ribavirin, and epoetin alpha. Hepatology. 2007;46:371–379. doi: 10.1002/hep.21712. [DOI] [PubMed] [Google Scholar]
- 31.Falasca F, Ucciferri C, Mancino P, Gorgoretti V, Pizzigallo E, et al. Use of epoetin beta during combination therapy of infection with hepatitis c virus with ribavirin improves a sustained viral response. J Med Virol. 2010;82:49–56. doi: 10.1002/jmv.21657. [DOI] [PubMed] [Google Scholar]
- 32.Feld JJ, Hoofnagle JH. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature. 2005;436:967–972. doi: 10.1038/nature04082. [DOI] [PubMed] [Google Scholar]
- 33.Parker WB. Metabolism and antiviral activity of ribavirin. Virus Res. 2005;107:165–171. doi: 10.1016/j.virusres.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 34.Dixit NM, Perelson AS. The metabolism, pharmacokinetics and mechanisms of antiviral activity of ribavirin against hepatitis C virus. Cell Mol Life Sci. 2006;63:832–842. doi: 10.1007/s00018-005-5455-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neumann AU, Lam NP, Dahari H, Gretch DR, Wiley TE, et al. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science. 1998;282:103–107. doi: 10.1126/science.282.5386.103. [DOI] [PubMed] [Google Scholar]
- 36.Herrmann E, Lee JH, Marinos G, Modi M, Zeuzem S. Effect of ribavirin on hepatitis C viral kinetics in patients treated with pegylated interferon. Hepatology. 2003;37:1351–1358. doi: 10.1053/jhep.2003.50218. [DOI] [PubMed] [Google Scholar]
- 37.Colombatto P, Civitano L, Oliveri F, Coco B, Ciccorossi P, et al. Sustained response to interferon-ribavirin combination therapy predicted by a model of hepatitis C virus dynamics using both HCV RNA and alanine aminotransferase. Antivir Ther. 2003;8:519–530. [PubMed] [Google Scholar]
- 38.Dixit NM, Layden-Almer JE, Layden TJ, Perelson AS. Modelling how ribavirin improves interferon response rates in hepatitis C virus infection. Nature. 2004;432:922–924. doi: 10.1038/nature03153. [DOI] [PubMed] [Google Scholar]
- 39.Dahari H, Ribeiro RM, Perelson AS. Triphasic decline of hepatitis C virus RNA during antiviral therapy. Hepatology. 2007;46:16–21. doi: 10.1002/hep.21657. [DOI] [PubMed] [Google Scholar]
- 40.Dixit NM. Advances in the mathematical modelling of hepatitis C virus dynamics. J Indian I Sci. 2008;88:37–43. [Google Scholar]
- 41.Rong L, Dahari H, Ribeiro RM, Perelson AS. Rapid emergence of protease inhibitor resistance in hepatitis C virus. Sci Transl Med. 2010;2:30ra32. doi: 10.1126/scitranslmed.3000544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rong L, Perelson AS. Treatment of hepatitis C virus infection with interferon and small molecule direct antivirals: viral kinetics and modeling. Crit Rev Immunol. 2010;30:131–148. doi: 10.1615/critrevimmunol.v30.i2.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vlierberghe HV, Delanghe JR, Vos MD, Leroux-Roel G. Factors influencing ribavirin-induced hemolysis. J Hepatol. 2001;34:911–916. doi: 10.1016/s0168-8278(01)00029-0. [DOI] [PubMed] [Google Scholar]
- 44.Takaki S, Tsubota A, Hosaka T, Akuta N, Someya T, et al. Factors contributing to ribavirin dose reduction due to anemia during interferon alfa2b and ribavirin combination therapy for chronic hepatitis C. J Gastroenterol. 2004;39:668–673. doi: 10.1007/s00535-003-1363-9. [DOI] [PubMed] [Google Scholar]
- 45.Balan V, Schwartz D, Wu GY, Muir AJ, Ghalib R, et al. Erythropoietic response to anemia in chronic hepatitis C patients receiving combination pegylated interferon/ribavirin. Am J Gastroenterol. 2005;100:299–307. doi: 10.1111/j.1572-0241.2005.40757.x. [DOI] [PubMed] [Google Scholar]
- 46.Donnerer J, Grahovic M, Stelzl E, Kessler HH, Bankuti C, et al. Ribavirin levels and haemoglobin decline in early virological responders and non-responders to hepatitis C virus combination therapy. Pharmacology. 2006;76:136–140. doi: 10.1159/000090942. [DOI] [PubMed] [Google Scholar]
- 47.Inoue Y, Hommaa M, Matsuzaki Y, Shibata M, Matsumura T, et al. Erythrocyte ribavirin concentration for assessing hemoglobin reduction in interferon and ribavirin combination therapy. Hepatol Res. 2006;34:23–27. doi: 10.1016/j.hepres.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 48.Glue P. The clinical pharmacology of ribavirin. Semin Liv Dis. 1999;19:17–24. [PubMed] [Google Scholar]
- 49.Krishnan SM, Dixit NM. Estimation of red blood cell lifespan from alveolar carbon monoxide measurements. Transl Res. 2009;154:15–17. doi: 10.1016/j.trsl.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 50.Virtue MA, Furne JK, Ho SB, Levitt MD. Use of alveolar carbon monoxide to measure the effect of ribavirin on red blood cell survival. Am J Hematol. 2004;76:107–113. doi: 10.1002/ajh.20069. [DOI] [PubMed] [Google Scholar]
- 51.Fellay J, Thompson AJ, Ge D, Gumbs CE, Urban TJ, et al. ITPA gene variants protect against anaemia in patients treated for chronic hepatitis C. Nature. 2010;464:405–408. doi: 10.1038/nature08825. [DOI] [PubMed] [Google Scholar]
- 52.Preston SL, Drusano GL, Glue P, Nash J, Gupta SK, et al. Pharmacokinetics and absolute bioavailability of ribavirin in healthy volunteers as determined by stable-isotope methodology. Antimicrob Agents Chemother. 1999;43:2451–2456. doi: 10.1128/aac.43.10.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Endres CJ, Moss AM, Ke B, Govindarajan R, Choi DS, et al. The role of the equilibrative nucleoside transporter 1 (ENT1) in transport and metabolism of ribavirin by human and wild-type or Ent1-/- mouse erythrocytes. J Pharmacol Exp Ther. 2009;329:387–398. doi: 10.1124/jpet.108.145854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McHutchison JG, Manns MP, Brown RS, Reddy KR, Shiffman ML, et al. Strategies for managing anemia in hepatitis C patients undergoing antiviral therapy. Am J Gastroenterol. 2007;102:880–889. doi: 10.1111/j.1572-0241.2007.01139.x. [DOI] [PubMed] [Google Scholar]
- 55.Mangoni ED, Marrone A, Saviano D, Del Vecchio C, Utili R, et al. Normal erythropoietin response in chronic hepatitis C patients with ribavirin-induced anemia. Antivir Ther. 2003;8:57–63. [PubMed] [Google Scholar]
- 56.Glue P, Rouzier-Panis R, Raffanel C, Sabo R, Gupta SK, et al. A dose-ranging study of pegylated interferon alfa-2b and ribavirin in chronic hepatitis C. Hepatology. 2000;32:647–653. doi: 10.1053/jhep.2000.16661. [DOI] [PubMed] [Google Scholar]
- 57.Jarvis SM, Thorn JA, Glue P. Ribavirin uptake by human erythrocytes and the involvement of nitrobenzylthioinosine-sensitive (es)-nucleoside transporters. Br J Pharmacol. 1998;123:1587–1592. doi: 10.1038/sj.bjp.0701775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Spivak JL. The blood in systemic disorders. Lancet. 2000;355:1707–1712. doi: 10.1016/S0140-6736(00)02249-2. [DOI] [PubMed] [Google Scholar]
- 59.Mahaffy JM, Belair J, Mackey MC. Hematopoietic model with moving boundary condition and state dependent delay: applications in erythropoiesis. J Theor Biol. 1998;190:135–146. doi: 10.1006/jtbi.1997.0537. [DOI] [PubMed] [Google Scholar]
- 60.Veng-Pederson P, Chapel S, Schmidt PRL, Al-Huniti NH, Cook RT, et al. An integrated pharmacodynamic analysis of erythropoietin, reticulocyte, and hemoglobin responses in acute anemia. Pharm Res. 2002;19:1630–1635. doi: 10.1023/a:1020797110836. [DOI] [PubMed] [Google Scholar]
- 61.Colijn C, Mackey MC. A mathematical model of hematopoiesis–I. Periodic chronic myelogenous leukemia. J Theor Biol. 2005;237:117–132. doi: 10.1016/j.jtbi.2005.03.033. [DOI] [PubMed] [Google Scholar]
- 62.Roeder I. Quantitative stem cell biology: computational studies in the hematopoietic system. Curr Opin Hematol. 2006;13:222–228. doi: 10.1097/01.moh.0000231418.08031.48. [DOI] [PubMed] [Google Scholar]
- 63.Crauste F, Pujo-Menjouet L, Génieys S, Molina C, Gandrillon O. Adding self-renewal in committed erythroid progenitors improves the biological relevance of a mathematical model of erythropoiesis. J Theor Biol. 2008;250:322–338. doi: 10.1016/j.jtbi.2007.09.041. [DOI] [PubMed] [Google Scholar]
- 64.Krzyzanski W, Perez-Ruixo J, Vermeulen A. Basic pharmacodynamic models for agents that alter the lifespan distribution of natural cells. J Pharmacokinet Phar. 2008;35:349–377. doi: 10.1007/s10928-008-9092-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Savill NJ, Chadwick W, Reece SE. Quantitative analysis of mechanisms that govern red blood cell age structure and dynamics during anaemia. PLoS Comput Biol. 2009;5:e1000416. doi: 10.1371/journal.pcbi.1000416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Berk PD, Berlin NI, Blaschke TF, Bloomer JR, Howe RB. Bilirubin production as a measure of red-cell life-span. J Lab Clin Med. 1972;79:364–378. [PubMed] [Google Scholar]
- 67.Beck WS. Hematology. 1991. MIT Press, Cambridge, Massachusetts.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Hemoglobin reduction as a function of the intracellular ribavirin concentration.
(0.09 MB PDF)
Analysis of RBC recovery following phlebotomy.
(0.41 MB PDF)
Validation of the solution methodology.
(0.39 MB PDF)
Sensitivity of the numerical solution to the integration time step.
(0.37 MB PDF)
Derivation of equation (1) and its boundary condition.
(0.29 MB PDF)
Dependence of RBC death rate on intracellular ribavirin concentration.
(0.19 MB PDF)
Analysis of phlebotomy experiments.
(0.28 MB PDF)
Solution of model equations using the method of characteristics.
(0.30 MB PDF)
MATLAB program for solving model equations.
(0.19 MB PDF)
Validation of the solution methodology.
(0.20 MB PDF)