

Abstract
Cytochrome (cyt) P450s comprise the enzyme superfamily responsible for human oxidative metabolism of a majority of drugs and xenobiotics. Electronic delivery of electrons to cyt P450s could be used to drive the natural catalytic cycle for fundamental investigations, stereo- and regioselective synthesis, and biosensors. We describe herein nm-thick films on electrodes featuring excess human cyt P450s and cyt P450 reductase (CPR) microsomes that efficiently mimic the natural catalytic pathway for the first time. Redox potentials, electron-transfer rates, CO-binding, and substrate conversion rates confirmed that electrons are delivered from the electrode to CPR, which transfers them to cyt P450. The film system enabled electrochemical probing of the interaction between cyt P450 and CPR for the first time. Agreement of film voltammetry data with theoretical simulations support a pathway featuring a key equilibrium redox reaction in the natural catalytic pathway between reduced CPR and cyt P450 occurring within a CPR-cyt P450 complex uniquely poised for substrate conversion.
Introduction
Cytochrome (cyt) P450 enzymes comprise a superfamily of iron heme monooxygenases that catalyze a majority of oxidative metabolism in human liver.1,2 Fifty-seven human membrane-bound cyt P450s have been identified with catalytic profiles encompassing carbon hydroxylation, epoxidation, dealkylation, heteroatom oxygenation, ester cleavage, cyclization, and isomerization.3 Cyt P450 reactions also play a critical role in drug development and toxicity, since reactive metabolites can trigger toxic effects.4 Thus, cyt P450 biosensors have been used to monitor drug metabolism5,6 and are key components of toxicity screening arrays that predict whether metabolites react with DNA.7 These versatile enzymes have also generated considerable interest for chemical synthesis since they catalyze reactions with exquisite stereo- and regioselectivity.3,8,9
The proposed catalytic cycle of cyt P450s involves a complex pathway that features donation of electrons from NADPH to CPR for subsequent delivery to cyt P450-FeIII. The reduced form of the enzyme that results then couples with dioxygen,1,10 producing an active ferryloxy radical cation +•(P450-FeIV=O) that transfers oxygen to bound substrate (RH) to form product (ROH). (Supplemental Fig. S1). Cytochrome b5 serves as the second electron donor for some cyt P450s.11,12
Using highly purified enzyme, Hill et al. achieved the first cyt P450cam electrochemistry in solution.13 Davis and Hill used scanning probe microscopy to characterize cytP450cam monolayers on Au suitable for direct voltammetry.14 However, an unmet challenge in cyt P450 research has been to deliver electrons from electrode to CPR to cyt P450s to drive the natural catalytic cycle.15–18 Such “bioelectronic” initiation would enable electrochemical probing of the catalytic pathway, and simplify synthetic and biosensor applications by replacing electron donation from expensive NADPH to the enzyme. While efficient electrochemical catalysis via the natural human cyt P450 cycle was not achieved previously, attempts have been made at electrode-driven catalysis.16,19–21 Vilker et al.17 designed an electrochemical bioreactor that supplied electrons to bacterial cyt P450cam via redox partner putidaredoxin. Systems like this with all proteins in solution are limited by slow protein diffusion, and are subject to electrode fouling.21 These drawbacks are overcome by protein film voltammetry, in which enzymes are immobilized directly on an electrode in a film that maintains its native state, so that biocatalytic activity does not depend on inefficient enzyme diffusion.22 In related work, stable, catalytic films fabricated layer-by-layer (LbL) of polyions and many enzymes have been demonstrated.23–26 Sligar et al. developed nanoscale lipid bilayers called “nanodiscs” to encapsulate cyt P450s for catalytic studies.27,28
In the late 1990s, we reported the first direct voltammetry of bacterial cyt P450cam in surfactant films29 and polyion LbL films,30 both of which facilitate direct electron exchange between electrodes and native cyt P450cam. We recently showed that voltammetric reduction and peroxide-driven oxidation of human cyt P450s in LbL polyion films is controlled by iron spin state and secondary structure.31 Other reports have also described direct electrochemistry and biocatalysis using cyt P450 films.18–21 However, electrochemical biocatalysis with cyt P450 films has thus far been accomplished utilizing enzyme-catalyzed reduction of oxygen to hydrogen peroxide which reacts with cyt P450FeIII to form the active ferryloxy species to oxidize substrates.21,32,33 A few studies using electrochemical peroxide-driven cyt P450 turnover showed comparable rates to standard NADPH solution assays.34,35 However, peroxide-driven cyt P450 electrocatalysis is often less efficient than the natural catalytic pathway19,20 and does not provide a system to voltammetrically probe key interactions between cyt P450s and CPR in the natural catalytic pathway.
There are two previous indications that cyt P450 biocatalysis might be achieved by delivering electrons from electrodes to CPR. In 2005, we reported in a short communication the direct voltammetry of thin films of genetically engineered cyt P450 1A2 and 3A4 “supersomes” on electrodes and suggested that electrons entered the films via CPR.36 The cyt P450s in these films oxidized styrene, possibly via electron transfer from CPR, but with low catalytic activity. Recently, Mie et al. reported better catalytic turnover for testosterone conversion by using natural microsomes containing cyt P450 3A4 and CPR immobilized on hydrophobic electrodes, and compared them to microsomes having only cyt P450 3A4 with no CPR.37 These authors suggested that increased catalytic efficiency could be due to electron transfer from electrode to CPR to cyt P450 3A4. However, they claimed direct electron transfer to cyt P450 3A4 in the same films, suggesting a mixed, unnatural catalytic pathway. Furthermore, conclusive evidence for electron transfer from electrode to CPR to cyt P450 3A4 was lacking.37
Strategies to improve the catalytic activity of cyt P450s include genetic engineering,38 and cyt P450-reductase fusion proteins.39 Notably, Gilardi et al.39 genetically fused reductase domains of CPR and flavodoxin with cyt P450 3A4 and observed improved catalytic activity in films on electrodes.39 They proposed that the catalytic enhancement in these synthetic systems was due to longer lived cyt P450 iron-peroxy species in the fusion proteins achieved by controlling the electron transfer rate to cyt P450 3A4 heme.39 However, there has been no definitive evidence for any reconstituted system that efficient cyt P450 biocatalysis can be achieved on electrodes by the natural catalytic pathway involving CPR.
Herein, we describe fabrication of LbL films made by combining pure cyt P450s with CPR microsomes on electrodes to achieve a large ratio of cyt P450 to CPR (Figure 1), as in the human liver.1,2,40 Electrons are injected into the film from the electrode to accurately mimic the natural cyt P450 catalytic cycle at high catalytic turnover. We provide unambiguous evidence for electron transfer from electrode to CPR to cyt P450 from measured redox potentials, electron transfer rates, enzyme turnover rates, and carbon monoxide (CO) binding. Results suggest dynamic participation of a CPR-cyt P450 complex in a key equilibrium redox process facilitating efficient catalytic turnover of the excess cyt P450s. In addition, the electrode-driven turnover rate for a model oxidation reaction was as good as or better than when NADPH was utilized.
Figure 1. Schematic representation of cyt P450/CPR bioelectronic films on pyrolytic graphite electrodes.
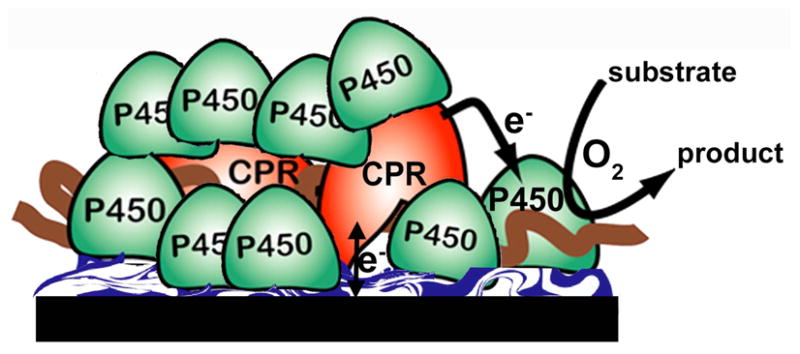
Films are assembled layer-by-layer (LbL) on pyrolytic graphite disk electrodes from CPR microsomes and pure cyt P450s on a polyion bed. Electrons are delivered from the electrode to CPR, which then transfers them to cyt P450 to convert substrates as in the natural catalytic pathway.
Experimental
Enzymes and Chemicals
Human cyt P450s 1A241 and 2E142 (clones of which were available to us) were expressed from DH5α Escherichia coli containing the relevant cDNA, and purified according to the literature. Microsomes from BD Biosciences (CA, USA) contained human cyt P450-reductase and cyt b5 (CPR+b5) but no cyt P450 [Protein content: 5 mg mL−1 in potassium phosphate (pH 7.4) buffer; CPR activity: 790 nmol min−1 mg−1 protein that corresponds to 0.26 nmol CPR mg−1 protein43; cyt b5: 2.5 nmol mg−1 protein], or containing cyt P450-reductase alone (denoted as msCPR) [Protein content: 10 mg mL−1 in potassium phosphate (pH 7.4) buffer; CPR activity: 2000 nmol min−1 mg−1 protein which is 0.67 nmol CPR mg−1 protein43]. Purified human cyt b5, horse heart myoglobin (Mb), poly (diallyldimethyl ammonium chloride) (PDDA), and poly(sodium 4-styrene sulfonate) (PSS) were from Sigma. 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and 4-hydroxy-1-(3-pyridyl)-1-butanone (HPB) were from Toronto research chemicals (North York, ON, Canada). H2O2 formed from oxygen reduction in the electrode driven biocatalysis was quantitated using peroxide assay kit (No. 23285) from Thermoscientific Inc., IL, USA. Standard NADPH assay was performed using the assay composition (1.3 mM NADP, 3.3 mM glucose 6-phosphate, 3.3 mM MgCl2 and 1 U mL−1 glucose 6-phosphate dehydrogenase) followed by the supplier (BD-Biosciences Inc.) with 2 mM NNK in aerobic 50 mM phosphate buffer, pH 7.0 (0.1 M NaCl) at 4 °C (Table 2).
Table 2.
Relative turnover rates for electrolysis and NADPH driven NNK oxidation in aerobic 50 mM phosphate buffer plus 0.1 M NaCl, pH 7.0.
| Product | Electrolysis (− 650 mV vs SCE) | NADPH assay (see Methods) | ||
|---|---|---|---|---|
| P450 2E1/CPR+b5 | Polyion/CPR+b5 | Polyion/P450 2E1 | P450 2E1/CPR+b5 | |
| HPB found (nmol) | 0.24 ± 0.04 | 0.003 ± 0.001 | 0.03 ± 0.01 | 0.14 ± 0.03 |
| Turnover rate h−1 (mol HPB)−1 (mol P450)−1 | 57 ± 9 | ~ 0.7 | 7 ± 2.5 | 34 ± 6 |
| H2O2 found (mM) | 0.75 ± 0.12 | 0.73 ± 0.10 | 0.30 ± 0.06 | - |
Construction of cyt P450/CPR films
Films consisting of six bilayers of purified human cyt P450 (1A2 or 2E1) and microsomal cyt P450-reductase containing cyt b5 (CPR+b5) or microsomal reductase alone (msCPR) were constructed on basal plane pyrolytic graphite disk electrodes (PG, Advanced Ceramics, 0.2 cm2 area) as layer-by-layer (LbL) films23 using techniques described previously.30,34,36 Similarly 6-bilayer films of Mb and CPR+b5, PDDA and CPR+b5, cyt P450 1A2 and cyt b5, PDDA and cyt b5 were assembled as controls. A precursor PDDA/PSS bilayer was adsorbed on all electrodes before assembling protein layers to avoid enzyme denaturation from direct contact with the electrode surface. The following solutions were used to grow the films: (a) purified human cyt P450s 1A2 and 2E1 (1 mg mL−1 in 50 mM phosphate buffer, pH 7.0); (b) 3 mg mL−1 PSS (polyanion) plus 0.5 M NaCl in deionized water; (c) 2 mg mL−1 PDDA (polycation) plus 0.05 M NaCl in deionized water; (d) 3 mg mL−1 myoglobin in 10 mM sodium acetate buffer, pH 4.5; (e) Human CPR+b5 or msCPR microsomes (BD Biosciences Inc.) as supplied in potassium phosphate buffer (pH 7.4). Under these conditions, the enzymes have net positive charges whereas microsomes have net negative charges due to the phospholipids and the slightly acidic pI of CPR and cyt b5.
Resulting PDDA/PSS(/1A2/CPR+b5)6 films on PG electrodes for cyt P450 1A2 are denoted as cyt P450 1A2/CPR+b5. Spectroscopic studies were done on films constructed on aminosilane functionalized fused silica slides (Sigma) with covalently attached poly(acrylic acid) as reported previously.31 Film characterization using quartz crystal microbalance (QCM) was similar to that reported previously for P450/polyion31 and microsomes/polyion films.36
Electrochemical and biocatalytic studies
A CHI 660A electrochemical analyzer was used for cyclic and rotating disk voltammetry (1000 rpm). The electrochemical cell employed a saturated calomel reference electrode (SCE), a Pt-wire counter electrode, and working electrode disk of ordinary basal plane pyrolytic graphite coated with appropriate films. Simulations of the film voltammetry were done using CH Instruments digital simulation software.
Enzyme-catalyzed constant potential electrolysis was done as reported previously.33,34,36 In brief, PDDA/PSS/P450 2E1/CPR+b5 or PDDA/PSS/P450 2E1/PSS (no CPR+b5) or (PDDA/PSS)2 LbL films were made on carbon-cloth cathodes (Zoltek Corp., 2 × 1 cm2), and reacted in a stirred electrolysis cell with 2 mM NNK in 1 mL air-saturated 50 mM potassium phosphate buffer, pH 7.0 at 4°C with an applied potential −650 mV vs. SCE to drive the biocatalysis. Similar PDDA/PSS/P450 2E1/CPR+b5 assemblies on carbon cloth were used for standard NADPH assays for NNK conversion using identical conditions without applied potential. The reaction mixture from electrochemical biocatalytic conversion of NNK to HPB was analyzed by capillary liquid chromatography-mass spectrometry (LC-MS), and quantified using standard HPB as reported previously.50 Structure was confirmed using the mass spectrometry detector in the same system.
Results
Film fabrication and characterization
Films of purified human cyt P450s and microsomes containing CPR were assembled by alternate layer-by-layer (LbL) electrostatic adsorption on pyrolytic graphite (PG) electrodes.23,31,36 A 1 nm precursor polyion bilayer was first deposited onto the 0.2 cm2 PG disk by successive adsorption of poly(diallyldimethyl ammonium chloride) (PDDA, polycation), and poly(styrene sulfonate) (PSS, polyanion). On this anionic underlayer,23 cationic enzyme layers [enzyme = cyt P450 1A2 or 2E1] were adsorbed alternately with anionic microsomes containing CPR and cyt b5 to make films of architecture PDDA/PSS/(enzyme/CPR+b5)6 [denoted as cyt P450/CPR+b5]. While these 6-bilayer films are fabricated one layer at a time, final structures of LbL films feature extensive layer intermixing,23 facilitating performance in the present application by bringing CPR and cyt P450s close together. Quartz crystal microbalance (QCM) weighing revealed fabrication of stable, reproducible films (Supplemental Figure S2). PDDA/PSS/(enzyme/CPR+b5)6 films had an average thickness ~30 nm (Supplemental Table S1), and ~100-fold more cyt P450 than CPR and 15-fold more cyt P450 than cyt b5. Similarly for PDDA/PSS/(enzyme/msCPR)6 films [msCPR= microsomes containing CPR but not cyt b5], the amount of cyt P450 was ~50-fold larger than msCPR (Table S1).
We previously reported spectroscopic signatures confirming the structural integrity of cyt P450s31 1A2 and 2E1 and CPR36,46 in polyion films. In the present work, Cyt P450-FeIII in PDDA/PSS/(P450 1A2/CPR+b5)6 films on fused silica slides were reduced by sodium dithionite followed by adding CO to form the P450-FeII-CO complex. This gave a characteristic difference absorbance band at 450 nm confirming the native conformation44 of cyt P450s (Figure 2a).
Figure 2. CO difference spectra of PDDA/PSS(/cyt P450 1A2/CPR+b5)6 films on aminosilane fused silica slides covalently cross-linked with poly(acrylic acid).
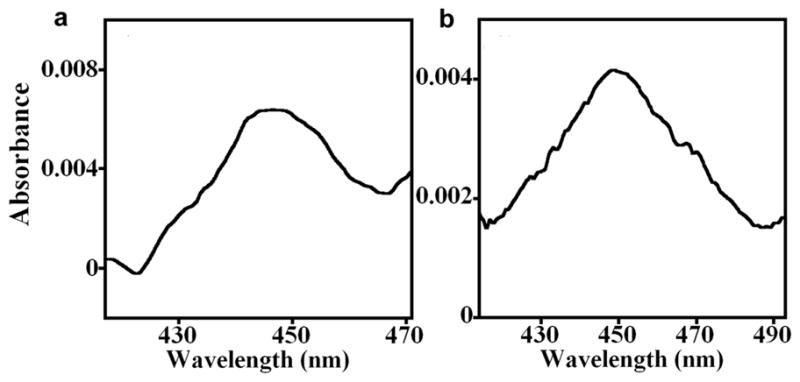
The cyt P450 heme in the PDDA/PSS(/cyt P450 1A2/CPR+b5)6 films were reduced directly with (a) 10 mM sodium dithionite, or (b) indirectly via CPR using 1 mM NADPH in anaerobic 50 mM phosphate buffer, pH 7.0, then purged 3 min with carbon monoxide (CO). Difference absorbance spectra with bands near 450 nm characteristic of native cyt P450s were acquired vs. similar PDDA/PSS(/cyt P450 1A2/CPR+b5)6 films that were reduced with sodium dithionite or NADPH, respectively, with no CO present. Denatured cyt P450s would give bands at 420 nm.
Bands at 450 nm were also obtained when electrons were delivered to P450-FeIII-heme in the films via CPR utilizing NADPH as electron donor in CO-saturated buffer (Figure 2b). Control PDDA/PSS/(PDDA/CPR+b5)6 films with no cyt P450 gave no difference absorbance bands when treated with CO and reduced by NAPDH or sodium dithionite (data not shown), showing that CPR or cyt b5 do not contribute to the 450 nm band.
Voltammetric studies
Fig. 3A shows background subtracted cyclic voltammograms (CV) of PDDA/PSS/(cyt P450 1A2/CPR+b5)6 films that feature nearly reversible reduction-oxidation peak pairs characteristic of electrode surface-confined redox proteins.21,22 Similar results were found for PDDA/PSS/(cyt P450 2E1/CPR+b5)6 films (Supplemental Fig. S4). Fig. 3B shows reversible CVs of the cyt P450 alone with much broader reduction-oxidation peaks centered at more positive potentials in films. Control PDDA/PSS films with no protein gave no peaks (Fig. 3Ba).
Figure 3. Experimental and simulated voltammetry.

Data for enzyme films showing influence of scan rate in anaerobic pH 7.0 buffer + 0.1 M NaCl. A. Experimental background- subtracted CVs: a, PDDA/PSS(/PDDA/CPR+b5)6 films at 0.3 Vs−1 with no cyt P450, and b–d, PDDA/PSS (/cyt P450 1A2/CPR+b5)6 films at scan rates (b) 0.1, (c) 0.2, and (d) 0.3 Vs−1. B. Experimental background- subtracted CVs: a, PDDA(/PSS/PDDA)4 film, and b–c, PDDA(/PSS/cyt P450 1A2)4 films at (b) 0.1 and (c) 0.2 V s−1. C. Digitally simulated CVs corresponding to a, reversible electron transfer for PDDA/PSS(/PDDA/CPR+b5)6 film at 0.3 Vs−1, and b–d, the ErCEo-model and parameters in Scheme 1 (eqs 1–3) for PDDA/PSS (/cyt P450 1A2/CPR+b5)6 films at scan rates (b) 0.1, (c) 0.2, and (d) 0.3 V s−1 showing excellent agreement with the experimental CVs in Fig. 3A. D. Influence of scan rate on oxidation (blue circles) and reduction (red diamonds) peak potentials for PDDA/PSS(/cyt P450 1A2/CPR+b5)6 films plotted with the peak potentials (lines) simulated using the ErCEo model.
From the CVs, we obtained formal potentials as the midpoint between oxidation-reduction peaks, the influence of CO complexation with cyt P450s on peak potential, and electrochemical heterogeneous electron transfer (hET) rate constants (ks, Table 1). Peak separation varied with CV scan rate for cyt P450/CPR+b5 or cyt P450/msCPR films in good agreement with Butler-Volmer theory (Fig. S3),45 and enabled initial estimates of ks (see Supplemental). Results show that films containing cyt P450s and CPR with or without cyt b5 have formal potentials near −250 mV vs NHE and ks near 40 s−1, as do films having only msCPR or CPR+b5 (Table 1).
Table 1.
Electrochemical parameters of enzyme films in anaerobic 50 mM phosphate buffer plus 0.1 M NaCl, pH 7.0.
| Film Assembly | Eo′ vs. NHEa with N2 (mV) | + Eo′ shift a with CO (mV) | ks (s−1)b |
|---|---|---|---|
| PEI(/PSS/P450 1A2)4c | −90 ± 10 | 36 ± 4 | 2.3 ± 0.4 |
| PEI(/PSS/P450 2E1)4c | −110 ± 9 | 50 ± 4 | 18 ± 3 |
| PDDA/PSS(/1A2/CPR+b5)6 | −230 ± 5 | No shift | 40 ± 6 |
| PDDA/PSS(/2E1/CPR+b5)6 | −250 ± 6 | No shift | 38 ± 6 |
| PDDA/PSS(/PDDA/CPR+b5)6 | −250 ± 6 | No shift | 42 ± 6 |
| PDDA/PSS(/2E1/msCPR)6 | −250 ± 8 | No shift | 40 ± 5 |
| PDDA/PSS(/1A2/b5)6 | −130 ± 5 | 35 ± 4 | 19 ± 3 |
avg±SD for n = 3 electrodes.
ks estimated from thin-film Butler-Volmer model (see Supplementary file).
From ref. 31. Where PEI = poly(ethyleneimine).
Films containing cyt P450s but no CPR had redox potentials near −100 mV vs NHE and >50% smaller ks values (Table 1). These differences from the cyt P450/msCPR films strongly support the view that electrons are transferred to CPR sites in the cyt P450/msCPR films and not directly to cyt P450s. Films with only cyt b5 gave small reversible redox peak pairs with formal potential −30±5 mV vs NHE, and did not significantly influence the voltammetry of CPR in cyt P450/CPR+b5 films. Formal potentials of cyt P450/CPR+b5 or cyt P450/msCPR films were in good agreement with those reported for films of CPR in pure human CPR,46 cyt P450 1A2 supersomes,36 and rat liver microsomes in polyion films.47 It should be realized that these CV potentials are somewhat dependent on film properties and electrode materials,21 and do not represent exact redox potentials in vivo, although interrelations between different proteins will be similar.
Simulations of voltammetry
Digital simulations helped elucidate the multistep reaction pathway corresponding to the observed voltammetric shapes. Assuming that electrons enter the film via CPR, and realizing that product formation (see below) indicated that CPR delivers electrons to cyt P450s, we formulated a stepwise electrochemical-chemical-electrochemical (ErCEo) simulation model (eqs 1–3). Here, Er denotes electrochemical reduction of CPR and Eo electrochemical re-oxidation. Scheme 1 duplicates key features within the natural catalytic mechanism. CPR is the initial electron acceptor (eq 1), characterized by electrochemical reduction rate constant ks,red. Eq 2 is reversible electron exchange between CPR and cyt P450 species, where kf is the forward and kb is the reverse second order chemical rate constant. CPRox and CPRred denote oxidized and reduced forms. Eq 3 represents oxidation of CPRred characterized by electrochemical rate constant ks,ox.
Scheme 1.

ErCEo Simulation model for CVs of cyt P450 1A2/CPR films with best fit parameters.
By exploring a wide range of parameter space involving variation of chemical and electrochemical rate constants in Scheme 1, we established that kb ≥ 5 kf (eq 2) with oxidation rate constant 40 s−1 (eq 3) and reduction rate constant 0.4 s−1 (eq 1) gave excellent agreement with CV shapes (Fig. 3C, cf. Fig. 3A). In addition, simulations gave good fits to reduction-oxidation peak potential vs. scan rate (Fig. 3D), often called “trumpet plots”.22 No other simulation parameter sets fit the experimental data nearly as well (see Supplementary). While Butler-Volmer theory assumes that ks (Table 1) describes oxidation and reduction steps at the formal potential, simulations using an electrochemical oxidation rate constant ~100 times larger than the reduction rate constant31 were necessary to obtain good fits to the somewhat unsymmetrical trumpet plots of the cyt P450 1A2/CPR (Fig. 3D) and cyt P450 2E1/CPR films (Fig. S4).
Electrode-driven biocatalysis
We used the oxidation of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) to evaluate catalytic efficiency. NNK metabolism by human cyt P450s 1A2 and 2E1 yields 4-hydroxy-1-(3-pyridyl)-1-butanone (HPB).48 Using rotating disk voltammetry (RDV), we found that cyt P450 1A2/CPR+b5 films on PG electrodes gave an increase in plateau current with increasing NNK concentration (Fig. 4a) characteristic of a catalytic process.21,22 Similarly, cyt P450 2E1/CPR+b5 films using cyt P450 2E1 enzyme gave increased plateau current with increasing NNK concentration. This electrode-driven natural cyt P450 catalytic NNK oxidation by cyt P450/CPR+b5 films is illustrated in Scheme 2.
Figure 4. Cyt P450/CPR films in electrochemical biocatalysis.

Influence of increasing NNK concentration on rotating disk voltammograms (1000 rpm) in aerobic 50 mM phosphate buffer, pH 7.0 containing 0.1 M NaCl of a, PDDA/PSS(/cyt P450 1A2/CPR+b5)6 and b, Control PDDA/PSS(/Mb/CPR+b5)6 films on PG electrodes.
Scheme 2.

Electrode-driven cyt P450 catalytic cycle involving electron supply by electrode to CPR and then to cyt P450.
A control film containing heme protein myoglobin (Mb) that does not oxidize NNK49 did not give increased plateau current with increasing NNK concentration (Fig. 4b) supporting cyt P450-specificity of NNK conversion in cyt P450/CPR films. In Fig. 4b, the basic steady state current is due to electrochemical catalytic reduction of oxygen by Mb,21,30,32,33 and the small decrease in plateau current reflects damage to Mb by production of H2O2.32,33
Metabolite identification and enzyme turnover
As electrolysis requires considerable enzyme to obtain sufficient product for analysis, we used only cyt P450 2E1 for this study. Electrolysis was done at an applied potential −650 mV vs. SCE, in the mass transport controlled plateau region (cf. Fig. 4a). A single bilayer PDDA/PSS/cyt P450 2E1/CPR+b5 films on a 2 cm2 carbon cloth cathode was sufficient to produce enough HPB from NNK for identification and quantitation of the HPB product by LC-MS50 (Figure 5).
Figure 5. Identification of 4-hydroxy-1-(3-pyridyl)-1-butanone (HPB).

A. Capillary LC-UV chromatogram (210–300 nm) of reaction mixture after 1 h. electrolysis (−650 mV vs. SCE at 4°C) using P450 2E1/CPR+b5 LbL films on carbon cloth (2 cm2) in 2 mM NNK in aerobic pH 7.0 + 0.1 M NaCl. The HPB peak was identified using commercial HPB standard (Table 2); B. Selected reaction monitoring (SRM)-MS/MS chromatogram monitoring HPB mass transition from m/z 166.1 ([M+H]+) to m/z 79.2 (pyridine fragment); C. Product ion spectrum of HPB (m/z 166.1) with inserted fragmentation pattern.
Notable results include a 1.5-fold larger turnover rate for electrode-driven vs. NADPH-driven biocatalysis, a much poorer turnover rate using the cyt P450 film alone, and negligible turnover for CPR film alone (Table 2). The amount of peroxide found suggests that the electrode-driven reaction in P450 2E1/CPR+b5 films produces very little hydrogen peroxide from the uncoupled peroxide shunt (Fig. S1), since the value is similar to that for CPR+b5 alone, and for polyion films with no protein which gave 0.73 ± 0.10 mM. These results suggest that all the peroxide is produced by electrolysis of oxygen, and not utilized during the cyt P450 catalytic cycle.
Discussion
Results above demonstrate for the first time a thin film on an electrode featuring excess purified human cyt P450s and CPR microsomes (Fig. 1) that bioelectronically mimics the natural cyt P450 oxidation cycle. Amounts of cyt P450s used in these reconstituted molecular assemblies were ~100 fold larger than the CPR, in accord with the excess of cyt P450s over CPR in microsomal membranes.51,52
Taken together, electrochemical redox potentials, ks values, and CO complexation results (Table 1) unequivocally demonstrate that electrons are injected from the electrode to CPR sites in the cyt P450/CPR+b5 or cyt P450/msCPR films. Formal potentials (−230 to −250 mV vs SHE) and ks values represent those of CPR (average ks ~42 s−1) when CPR and cyt P450s are present together (average ks ~ 40 s−1, Table 1). While the characteristic 450 nm absorbance band of native cyt-P450-CO is found in the presence of CO when cyt P450s are reduced (Figure 2), there is no midpoint potential shift in the CVs of cyt P450s/CPR+b5 or cyt P450/msCPR films upon CO binding (Table 1). Cyt P450s in polyion films without CPR have ~50 mV potential shifts upon CO binding (Table 1, Fig. S1). While the spectra confirm native cyt P450s in all films, the voltammetry shows that the electrode does not address cyt P450 in cyt P450/CPR films. All these results are consistent with electron transfer from electrode to CPR rather than directly to cyt P450.
Efficient electrode-driven biocatalytic oxidation of NNK (Table 2) by cyt P450 2E1/CPR+b5 films shows that electrons delivered electrochemically to CPR are transferred to cyt P450s as in the natural catalytic pathway. Electrode-driven NNK oxidation was about 1.5 times more efficient than the standard NADPH-driven reaction. While we cannot be certain that electron flow is equivalent in the two experiments, electrochemical biocatalysis in cyt P450/CPR+b5 films is at least as efficient as the NADPH-driven reaction in these films.
As mentioned, cyt P450 films without CPR utilize electrochemical biocatalysis of reduction of oxygen to peroxide to activate the enzyme to oxidize substrates.21,32–34 Table 2 shows that this peroxide-initiated pathway in cyt P450 2E1 films without CPR is much less efficient for NNK conversion (Table 2) than the cyt P450/CPR+b5 films. For cyt P450 2E1/CPR+b5 films, electrode driven biocatalysis does not utilize H2O2. This is supported by the finding that control films of polyion/CPR+b5 or polyion without protein produced equivalent amounts of H2O2 to that of cyt P450 2E1/CPR+b5 films (Table 2). This is consistent with electrochemically-driven cyt P450/CPR+b5 biocatalysis following the natural CPR mediated catalytic pathway (Fig. S1).
Simulations of CVs using the ErCEo model (eqs 1–3) were in excellent agreement with experimental voltammetry for the cyt P450/CPR films containing cyt P450 1A2 (Fig. 3A,C,D) or 2E1 (Supplemental Fig. S4A,B). The model is consistent with the natural catalytic pathway, and provides a means to probe the key equilibrium electron exchange between cyt P450 and CPR (eq 2). The best fit condition kb ≥5kf for this chemical step indicates that the equilibrium (in absence of O2) with constant K = kf/kb≤0.2 lies to the left, in favor of CPRred. Moreover, results of the simulation explain why CPRox is reduced electrochemically in cyt P450/CPR films instead of cyt P450, even though without CPR, cyt P450s have the more positive reduction potential (Table 1). The equilibrium in eq 2 is controlled by the thermodynamics of the protein electron transfers22 in Scheme 1, and when kb ≥5kf, the redox potential difference (ΔEo) between CPR and cyt P450 is given by the expression ΔEo = [RT/nF] ln(K). Thus, a value of K ≤ 0.2 in this expression indicates that the cyt P450 redox potential is shifted more negative than that of CPRox by ~40 mV or more, and CPRox is more easily reduced than the cyt P450s.
Thus, the shift in redox potential of cyt P450s in the cyt P450/CPR films compared to cyt P450 films without CPR is around 140 mV (see Table 1). The most likely cause of this large potential shift is complexation of cyt P450s with CPR, which has been proposed as a key feature of cyt P450 catalytic cycles for decades.1,51,53,54 The equilibrium in eq 2 most probably involves transient cyt P450/CPR complexes, and also influences the need for individual electrochemical oxidation and reduction rate constants in the simulations to fit the CV data. This latter requirement reflects the fact that the CPR redox couple does not only involve a simple cofactor oxidation state change, but probably features structural reorganization as well. For example, if CPRox is reduced within a CPRox/cyt P450 complex, the reduction product may undergo a rapid structural reorganization to yield a new complex within which CPR is oxidized. Interestingly, individual electrochemical oxidation and reduction rate constants are also required to fit CV data for cyt P450 1A2 and 2E1 without CPR in polyion films, implying rapid structural re-organizations of oxidized and reduced forms of the enzymes accompanying electron transfer.31 Also, the possibility of a heterogeneous population of complexed CPR and free CPR cannot be ruled out.
A K-value ≤ 0.2 for eq 2 suggests that most CPR in the electrochemical experiment is stored as CPRred. However, when oxygen and substrate are present, equilibrium will be driven to the right by rapid removal of cyt P450-FeII via reaction with dioxygen and subsequent catalytic steps.1 This will cause more CPRred to be formed by electrochemical reduction to drive the catalysis (Fig. 4A). It is tempting to envision a similar process in the natural NADPH-driven catalytic cycle, where storage of reducing power in CPRred could partly explain how CPR can address such a large excess of cyt P450s.
Conclusions
Data presented above demonstrate novel human cyt P450/CPR microsomes films that for the first time efficiently mimic the natural catalytic pathway of this superfamily of metabolic monooxygenases on electrodes. Intriguing dynamic features of the earlier proposed CPR-cyt P450 complexes involved in cyt P450 biocatalysis have been unraveled by voltammetrically monitoring electrons flowing from electrode to CPR to cyt P450 in the films. Results provide clear evidence of the long-postulated complex between cyt P450 and CPR in the catalytic cycle. This novel bioelectronic system also has potential for applications in drug discovery and development by monitoring substrate metabolism and enzyme inhibition. Other applications include efficient new biosensors or arrays for toxicity screening and bioreactors for chemical synthesis.
Supplementary Material
Acknowledgments
This work was supported financially by U.S. PHS grant ES03154 from the National Institute of Environmental Health Sciences (NIEHS), NIH.
Footnotes
Supporting Information Available. Four additional figures and two tables giving the P450 catalytic cycle, full QCM data, agreement of experimental rate constant with Butler-Volmer model, electrochemical parameters of control myoglobin/CPR films, and experimental CVs and trumpet plots with simulations for cyt P450 2E1/CPR films are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References and notes
- 1.Ortiz de Montellano PR. Cytochrome P450: Structure, Mechanism, and Biochemistry. 3. Kluwer/Plenum; New York: 2005. [Google Scholar]
- 2.Schenkman JB, Greim H, editors. Cytochrome P450. Springer-Verlag; Berlin: 1993. [Google Scholar]
- 3.Coon MJ. Annu Rev Pharmacol Toxicol. 2005;45:1–25. doi: 10.1146/annurev.pharmtox.45.120403.100030. [DOI] [PubMed] [Google Scholar]
- 4.Kramer JA, Sagartz JE, Morris DL. Nature Rev Drug Discov. 2007;6:636–649. doi: 10.1038/nrd2378. [DOI] [PubMed] [Google Scholar]
- 5.Joseph S, Rusling JF, Lvov YM, Friedberg T, Fuhr U. Biochem Pharmacol. 2003;65:1817–1826. doi: 10.1016/s0006-2952(03)00186-2. [DOI] [PubMed] [Google Scholar]
- 6.Bistolas N, Wollenberger U, Jung C, Scheller FW. Biosens Bioelec. 2005;20:2408–2423. doi: 10.1016/j.bios.2004.11.023. [DOI] [PubMed] [Google Scholar]
- 7.Rusling JF, Hvastkovs EG, Schenkman JB, Nassar A, Hollenburg PF, Scatina J, Wiley NJ, editors. Drug Metabolism Handbook. 2009. pp. 307–340. [Google Scholar]
- 8.Koeller KM, Wong CH. Nature. 2001;409:232–240. doi: 10.1038/35051706. [DOI] [PubMed] [Google Scholar]
- 9.Urlacher VB, Eiben S. Trends Biotechnol. 2006;24:324–330. doi: 10.1016/j.tibtech.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 10.Guengerich FP. Chem Res Toxicol. 2008;21:70–83. doi: 10.1021/tx700079z. [DOI] [PubMed] [Google Scholar]
- 11.Schenkman JB, Jansson I. Pharmacology & Therapeutics. 2003;97:139–152. doi: 10.1016/s0163-7258(02)00327-3. [DOI] [PubMed] [Google Scholar]
- 12.Shimada T, Mernaugh RL, Guengerich FP. Arch Biochem Biophys. 2005;435:207–216. doi: 10.1016/j.abb.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 13.Kazlauskaite J, Westlake ACG, Wong L-L, Hill HAO. Chem Commun. 1996:2189–2190. [Google Scholar]
- 14.Davis JJ, Hill HAO. Chem Comm. 2002:393–401. [Google Scholar]
- 15.Vilker VL, Khan F, Shen D, Baizer MM, Nobe K, Dryhurst G, Niki K, editors. Redox chemistry and interfacial behavior of biological molecules. Plenum Press; New York: 1988. pp. 105–112. [Google Scholar]
- 16.Nobe K, Baizer MM, Shen DH, Vilker VL, Cho AK, Little RD, Weinberg NL, editors. Electro organic synthesis: festschrift for Manuel M Baizer. Marcel Dekker Inc; New York: 1991. pp. 265–271. [Google Scholar]
- 17.Reipa V, Mayhew MP, Vilker VL. Proc Natl Acad Sci USA. 1997;94:13554–13558. doi: 10.1073/pnas.94.25.13554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mak LH, Sadeghi SJ, Fantuzzi A, Gilardi G. Anal Chem. 2010;82:5357–5362. doi: 10.1021/ac101072h. [DOI] [PubMed] [Google Scholar]
- 19.Fleming BD, Johnson DL, Bond AM, Martin LL. Expert Opin Drug Metab Toxicol. 2006;2:581–589. doi: 10.1517/17425255.2.4.581. [DOI] [PubMed] [Google Scholar]
- 20.Dodhia VR, Gilardi G, Davis J, editors. Engineering the Bioelectronic Interface: Applications to analyte biosensing and protein detection. RSC publications; 2009. pp. 153–189. [Google Scholar]
- 21.Rusling JF, Wang B, Yun S-E, Bartlett PN. Bioelectrochemistry. John Wiley; N. Y: 2008. Electrochemistry of Redox Enzymes; pp. 39–86. [Google Scholar]
- 22.Armstrong FA, Heering HA, Hirst J. Chem Soc Rev. 1997;26:169–179. [Google Scholar]
- 23.Lvov Y, Nalwa RW, editors. Handbook of Surfaces and Interfaces of Materials. Vol. 3. Academic Press; San Diego: 2001. pp. 170–189. [Google Scholar]
- 24.Onda M, Lvov Y, Ariga K, Kunitake T. Biotechnol Bioeng. 1996;51:163–167. doi: 10.1002/(SICI)1097-0290(19960720)51:2<163::AID-BIT5>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 25.Onda M, Lvov Y, Ariga K, Kunitake T. J Ferment Bioeng. 1996;82:502–506. doi: 10.1002/(SICI)1097-0290(19960720)51:2<163::AID-BIT5>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 26.Onda M, Ariga K, Kunitake T. J Biosci Bioeng. 1999;87:69–75. doi: 10.1016/s1389-1723(99)80010-3. [DOI] [PubMed] [Google Scholar]
- 27.Denisov IG, Sligar SG. Biochim Biophys Acta. 2011;1814:223–229. doi: 10.1016/j.bbapap.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Das A, Sligar SG. Biochemistry. 2009;48:12104–12112. doi: 10.1021/bi9011435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Z, Nassar AEF, Lu Z, Schenkman JB, Rusling JF. J Chem Soc Faraday Trans. 1997;93:1769–1774. [Google Scholar]
- 30.Lvov YM, Lu Z, Schenkman JB, Zu X, Rusling JF. J Am Chem Soc. 1998;120:4073–4080. [Google Scholar]
- 31.Krishnan S, Abeykoon A, Schenkman JB, Rusling JF. J Am Chem Soc. 2009;131:16215–16224. doi: 10.1021/ja9065317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zu X, Lu Z, Zhang Z, Schenkman JB, Rusling JF. Langmuir. 1999;15:7372–7377. [Google Scholar]
- 33.Munge B, Estavillo C, Schenkman JB, Rusling JF. Chem Biochem. 2003;4:82–89. doi: 10.1002/cbic.200390018. [DOI] [PubMed] [Google Scholar]
- 34.Estavillo C, Lu Z, Jansson I, Schenkman JB, Rusling JF. Biophys Chem. 2003;104:291–296. doi: 10.1016/s0301-4622(02)00383-6. [DOI] [PubMed] [Google Scholar]
- 35.Rudakov YO, Shumyantseva VV, Bulko TV, Suprun EV, Kuznetsova GP, Samenkova NF, Archakov AI. J Inorg Biochem. 2008;102:2020–2025. doi: 10.1016/j.jinorgbio.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 36.Sultana N, Schenkman JB, Rusling JF. J Am Chem Soc. 2005;127:13460–13461. doi: 10.1021/ja0538334. [DOI] [PubMed] [Google Scholar]
- 37.Mie Y, Suzuki M, Komatsu Y. J Am Chem Soc. 2009;131:6646–6647. doi: 10.1021/ja809364r. [DOI] [PubMed] [Google Scholar]
- 38.Gillam EMJ. Chem Res Toxicol. 2008;21:220–231. doi: 10.1021/tx7002849. [DOI] [PubMed] [Google Scholar]
- 39.Dodhia VR, Sassone C, Fantuzzi A, Nardo GD, Sadeghi SJ, Gilardi G. Electrochem Commun. 2008;10:1744–1747. [Google Scholar]
- 40.Miwa GT, Lu AYH. Arch Biochem Biophys. 1984;234:161–166. doi: 10.1016/0003-9861(84)90337-0. [DOI] [PubMed] [Google Scholar]
- 41.Fisher CW, Caudle DL, Martin-Wixtrom C, Quattrochi LC, Tukey RH, Waterman MR, Estabrook RW. FASEB J. 1992;6:759–764. doi: 10.1096/fasebj.6.2.1537466. [DOI] [PubMed] [Google Scholar]
- 42.Gillam EMJ, Guo Z, Guengerich FP. Arch Biochem Biophys. 1994;312:59–66. doi: 10.1006/abbi.1994.1280. [DOI] [PubMed] [Google Scholar]
- 43.Guengerich FP, Martin MV, Sohl CD, Cheng Q. Nat Protoc. 2009;4:1245–1251. doi: 10.1038/nprot.2009.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Omura T, Sato R. J Biol Chem. 1964;239:2379–2385. [PubMed] [Google Scholar]
- 45.Laviron E. J Electroanal Chem. 1979;101:19–28. [Google Scholar]
- 46.Sultana N, Schenkman JB, Rusling JF. Electroanalysis. 2007;19:2499–2506. [Google Scholar]
- 47.Krishnan S, Rusling JF. Electrochem Commun. 2007;9:2359–2363. doi: 10.1016/j.elecom.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hecht SS. Chem Res Toxicol. 1998;11:559–603. doi: 10.1021/tx980005y. [DOI] [PubMed] [Google Scholar]
- 49.Krishnan S, Hvastkovs EG, Bajrami B, Schenkman JB, Rusling JF. Mol Biosyst. 2009;5:163–169. doi: 10.1039/b815910f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bajrami B, Krishnan S, Rusling JF. Drug Metab Lett. 2008;2:158–162. doi: 10.2174/187231208785425854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Backes WL, Kelley RW. Pharmacology & Therapeutics. 2003;98:221–233. doi: 10.1016/s0163-7258(03)00031-7. [DOI] [PubMed] [Google Scholar]
- 52.Estabrook RW, Franklin MR, Cohen B, Shigamatzu A, Hildebrandt AG. Metabolism. 1971;20:187–199. doi: 10.1016/0026-0495(71)90091-6. [DOI] [PubMed] [Google Scholar]
- 53.Peterson JA, Ebel RE, O’Keeffe DH, Matsubara T, Estabrook RW. J Biol Chem. 1976;251:4010–4016. [PubMed] [Google Scholar]
- 54.Taniguchi H, Imai Y, Sato R. Biochemistry. 1987;26:7084–7090. doi: 10.1021/bi00396a033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.