

Abstract
Degeneration of the hippocampus is associated with Alzheimer’s disease, and occurs very early in the progression of the disease. Current options for treating the cognitive symptoms associated with Alzheimer’s are inadequate, giving urgency to the search for novel therapeutic strategies. Pharmacologic agents that safely enhance hippocampal neurogenesis may provide new therapeutic approaches. We discovered the first synthetic molecule, named P7C3, which protects newborn neurons from apopotic cell death, and thus promotes neurogenesis in mice and rats in the subgranular zone of the hippocampal dentate gyrus, the site of normal neurogenesis in adult mammals. We describe the results of a medicinal chemistry campaign to optimize the potency, toxicity profile and stability of P7C3. Systematic variation of nearly every position of the lead compound revealed elements conducive towards increases in activity and regions subject to modification. We have discovered compounds that are orally available, non-toxic, stable in mice, rats and cell culture, and capable of penetrating the blood-brain barrier. The most potent compounds are active at nanomolar concentrations. Finally, we have identified derivatives that may facilitate mode-of-action studies through affinity chromatography or photocrosslinking.
Introduction
Alzheimer’s disease (AD) accounts for about 80% of all cases of dementia, and is the fifth leading cause of death in individuals aged 65 or older. AD is the most prevalent and devastating neurodegenerative disorder today. The greatest risk factor for developing this disease is advancing age. By 2025, the majority of baby boomers will have reached age 65 and the number of people in this age group with AD is predicted to expand by about 50% to nearly 8 million. By 2050, the number of afflicted individuals in the US is expected to have risen to between 11 and 16 million. People with AD are high users of health care, long-term care, and hospice services, and total payments for these types of care from all sources currently exceed $177 billion per year in the US.1
Pathologic changes in the brains of patients with AD include the accumulation of extracellular neuritic plaques between neurons, and neurofibrillary tangles within neurons.2 Numerous clinical trials aimed at reducing these plaques in Alzheimer’s patients have failed to elicit cognitive improvement, even when the brain tissue of patients has been cleared of plaques. Disappointingly, the most recent data from late-stage trials of a γ-secretase inhibitor, Eli Lilly’s semagacestat, were sufficiently discouraging to halt the trial.3 Currently, several biological and small molecule drug candidates that target the production or accumulation of Aβ42 plaques are in late-stage clinical trials. Other therapies for AD include acetylcholinesterase inhibitors (Aricept, Exelon, Razadyne and Cognex) and an NMDA antagonist (Namenda). These treatments offer some symptomatic relief, but no lasting benefit as they fail to address the underlying biology of AD.
An alternative approach to treating AD focuses on promoting hippocampal neurogenesis. AD is characterized by neurodegeneration in the cerebral cortex, hippocampus and other subcortical structures.4 Accordingly, we hypothesized that small molecules that retard neuronal death or increase neurogenesis may represent novel therapeutic agents. Currently no drugs operate through this mechanism. Nonetheless, groundbreaking studies starting in the 1960’s demonstrated that neurogenesis in adult mammals occurs in two regions of the brain, the subventricular zone (SVZ) and the subgranular zone (SGZ) of the dentate gyrus within the hippocampus.5,6,7 This process is important for maintenance of the normal structure and function of the hippocampus, and thus essential to hippocampal-dependent memory and learning.8 In particular, neurogenesis in the dentate gyrus serves to produce neurons that integrate locally in the granular layer of the dentate gyrus, which displays lifelong structural and functional plasticity. This process appears to be active within all mammalian species, including humans.9 In adult mice, neurogenesis is a month-long process involving proliferation of neuronal precursor cells and subsequent maturation. During this process the vast majority of the newly formed hippocampal neural precursor cells undergo apoptosis prior to maturation whereas the surviving cells mostly become incorporated as fully functional neurons.
The process of neurogenesis in adult mice can be affected by both environmental and chemical stimuli. Neurogenesis increases in adult mice when they are housed in an enriched environment containing toys, nesting materials and exercise equipment.10,11 Similarly, neurogenesis is enhanced when mice are free to exercise voluntarily.12,13 Provocatively, engagement in enriching educational activities and exercise also delays the onset and progression of AD.14 Antidepressants enhance neurogenesis in adult mice and humans,15 and allopregnanolone, a metabolite of progesterone that is synthesized in the CNS, promotes neurogenesis and learning in a mouse model of AD.16 In the latter instance, enhancement of neurogenesis was associated with amelioration of cognitive deficits.17 Finally, deficits in neurogenesis precede the formation of plaques and tangles associated with AD in mouse models of that disease.18 Taken together, the evidence suggests that drugs that promote hippocampal neurogenesis might provide effective treatment strategies for patients with Alzheimer’s disease.
To identify small molecules capable of promoting neurogenesis, we screened a collection of 2000 structurally unrelated small molecules assembled from an in-house compound library (Fig 1).19 Initially, molecules were screened by directly injecting mixtures of 10 compounds intracerebroventricularly (ICV) into the left lateral ventricle of living mice. Compounds were introduced over 7 days at a concentration of 10 μM each (12 μL/day at a constant rate). With this dosing, we estimated that compounds would attain intracerebroventricular concentrations in the low-micromolar to mid-nanomolar concentration range. During compound infusion, the thymidine analog bromodeoxyuridine (BrdU, 50 mg/kg) was administered daily to mark newly born hippocampal neural precursor cells. After 1 week of compound administration, the animals were sacrificed, 40 μm brain slices were prepared, and tissue was processed for immunohistochemical staining with an antibody to BrdU to identify newborn cells. Neurogenesis was quantified by counting BrdU+ cells in the hippocampal dentate gyrus in the brain hemisphere opposite to the side of compound infusion. The number of positive cells was normalized against the volume of the dentate gyrus.
Fig 1.

In vivo screen for pro-neurogenic compounds. Drugs were infused directly into the brain of ive mice over 7 days. Subsequent sectioning and immunohisochemical staining revealed newly formed neurons (black cells on right section).
One of the molecules that we discovered with this approach was named P7C3, as it was identified as Compound 3 (C3) from Pool 7 (P7). Mice treated with P7C3 had approximately twice the number of BrdU+ cells as mice treated with vehicle alone (artificial cerebral spinal fluid, CSF), and a similar number as mice treated with a positive control, fibroblast growth factor 2 (FGF-2). Unlike some antidepressant drugs that have been reported to enhance neurogenesis after two to three weeks of treatment,20 P7C3 affects neurogenesis within days of administration. Additional studies have revealed that P7C3 maintains activity when a 1 μM solution was infused ICV (12 μL/day for 7 days); it displays maximal neurogenesis activity with an oral dose of 5 mg/kg and measurable activity at 1 mg/kg. The molecular target of P7C3 is not known. However, time-course experiments revealed that P7C3 does not stimulate proliferation of neural precursor cells. Rather, P7C3 increases the number of newborn neurons that survive the month-long maturation process. The mature neurons express neuron-specific proteins and appear fully functional.19 Accordingly, P7C3 enhances neurogenesis – i.e. is proneurogenic – by virtue of its neuroprotective activity. P7C3 was found to repair the structure and function of the dentate gyrus in mice deficient in the npas3 gene, a genotype characterized by an absence of hippocampal neurogenesis.21 Furthermore, it also enhanced neurogenesis in the dentate gyrus, impeded neuron cell death, and preserved cognitive capacity as a function of terminal aging in rats. In contrast, no effect was observed on neurogenesis in middle-aged rats, which do not display impaired neurogenesis.22 Finally, P7C3 is not toxic to embryonic, weaning or adult mice.
Here we describe efforts to understand the structural requirements for activity and to improve P7C3’s potency, stability and toxicity profile. To the best of our knowledge, this is the first class of small molecules designed to promote neurogenesis, and as such may represent lead compounds for the treatment of debilitating neurodegenerative diseases.
Results
P7C3 contains a dibromocarbazole connected to an aniline with a 2-hydroxypropyl linker. We set out to determine which positions of P7C3 would tolerate modification, systematically optimize each subsection of P7C3, and identify derivatives that could be used for affinity chromatography. Several commercially available analogs of P7C3 provided early indications of the structure/activity relationship. The bromines on the carbazole appear particularly important, as the derivatives with dichloro (1) and parent (2) carbazole did not appear active at the concentrations tested (Scheme 1, Fig 1). We tested derivatives possessing methyl or methoxy substitution on the aniline ring, and discovered that methyl (3–5) or ortho-methoxy substitutions appeared to decrease activity in our assay. In contrast, a methoxy group was well tolerated in the meta or para position (7, 8). A long-term objective is to identify the molecular target of P7C3, and to this end we planned to synthesize active derivatives that could be used as reagents for affinity chromatography. In that context, the observation that an O-alkyl group did not compromise activity was encouraging. Indeed, many of the synthetic analogs (see below) were prepared from m-anisidine with this consideration in mind. We tested two commercially available analogs possessing saturated heterocyclic rings rather than an aniline (9, 10). Previous work from a group at Serono Pharmaceuticals revealed that several carbazoles inhibited Bid-mediated release of cytochrome c, and, as a consequence, were anti-apoptotic.23 The most active compound from the Serono study (9) proved moderately active in our assay while a less active Serono compound (10) was also less active in our experiments.19 Finally, we evaluated the activity of dimebon (11), a γ-tetrahydrocarboline originally used as an antihistamine in Russia. This drug has recently been reported to block apoptosis of cortical neurons exposed to amyloid-beta peptide.24
Scheme 1.

P7C3 and commercially available analogs.
Anecdotal evidence of its efficacy against AD and age related cognitive decline25 prompted Medivation and Pfizer to conduct clinical trials in the U.S. to determine dimebon’s efficacy in treating several forms of neurodegenerative disease. While phase II data appeared exceptionally encouraging,26 unpublished results from a phase III study showed that dimebon offered no benefit for patients suffering from AD.27 Provocatively, dimebon is active in our neurogenesis assay, but substantially less so than P7C3.
Synthetically, P7C3 can emerge from a union of a carbazole, an aniline and an epoxide (Scheme 2). This convergent synthetic approach allows the independent variation of the two rings. Furthermore, additional processing of P7C3 and its derivatives provided access to a range of derivatives. Throughout the manuscript, compounds with numbers highlighted in green showed activity as good as or better than our positive control, FGF-2 (see below). As shown in Scheme 2, a wide range of analogs was prepared by first condensing 2- or 3-ring heterocycles with epibromohydrin.28 The resulting epoxides 13 were then opened with various anilines, thiols or heterocycles. We found that ring-opening with anilines generally required Lewis acid activation of the epoxides (BiCl329 or LiBr30) whereas alcohols, indole and carbazoles required deprotonation of the nucleophile with NaH. Stronger nucleophiles such as sodium azide, hydrazine, ammonia, thiophenol or aminopyridines reacted with the terminal epoxides upon simple heating. In a slight modification of this sequence, dibromocarbazole was allowed to react with arylglycidyl ethers to provide aryl ethers 42 and 43 directly. Similarly, m-anisidine was first condensed with epoxy-2-methyl-propyl chloride and then exposed to the carbazole to yield the tertiary alcohol 46.31 This synthetic approach provided access to a wide range of structural diversity, although the overall yields ranged from rather abysmal to nearly quantitative. However, none of the syntheses has been optimized. Complete experimental details are provided as supporting information.
Scheme 2.

Synthesis of deriviatives of P7C3.
Several of the analogs prepared according to Scheme 2 were subject to additional transformations. For instance, the thioethers 24 and 44 were oxidized with either NaIO4 or m-CPBA to provide either a diastereomeric mixture of sulfoxides (25) or the sulfones (26, 45), respectively. Phthalimide 39 was deprotected and acylated to provide derivative 41. Similarly, P7C3 analog 7 was subject to further processing (Scheme 3). For example, methylation was O-selective at low conversion, yielding ether 47. Urea formation and acylation were facile, providing 48 and 49 in quantitative yield. Oxidation with SO3·Py produced ketone 50 and, unexpectedly, thioether 51. A potential mechanism for this unusual and, to our knowledge, unprecedented, oxidation is provided in the supporting information. In brief, an α-ketoimine may be trapped by methanethiol released from DMSO.
Scheme 3.

Modification of analog 7.
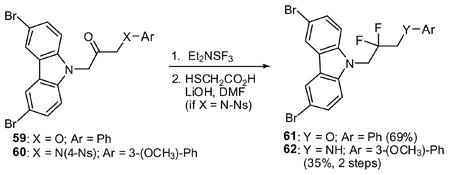
Fluorination appeared to improve activity (see below), so a series of fluorinated analogs were prepared, as shown in Table 1. We used either DAST (Et2NSF3) or its morpholino analog. Reactions were generally cleaner using the latter reagent. For aniline-containing starting materials, the NH proved problematic. In these cases, protection of the nitrogen with a 4-nitrobenzene sulfonyl (4-Ns) group32 greatly improved the fluorination. Similarly, the difluorides 61 and 62 were prepared from the corresponding ketones (eq 1).
Table 1.
Fluorination of alcohols.
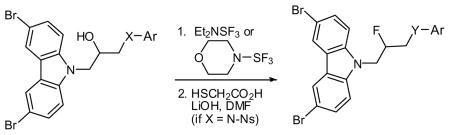 | |||||
|---|---|---|---|---|---|
| Entry | X | Ar | Y | Product | Yield (%)a |
| 1 | N(4-Ns)b | Ph | NH | 52 | 44 |
| 2 | N(4-Ns) | 3-(OCH3)-Ph | NH | 53 | 88 |
| 3 | N(4-Ns) | 4-(OCH3)-Ph | NH | 54 | 43 |
| 4 | N(4-Ns) | 4-(OCH2CO2Et)-Ph | NH | 55 | 61 |
| 5 | NCH3 | 3-(OCH3)-Ph | NCH3 | 56 | 71 |
| 6 | O | Ph | O | 57 | 97 |
| 7 | S | Ph | S(O)2c | 58 | 28 |
Isolated yield over one or two steps.
Ns = nitrobenzene sulfonyl.
After oxidation with m-CPBA
 |
(1) |
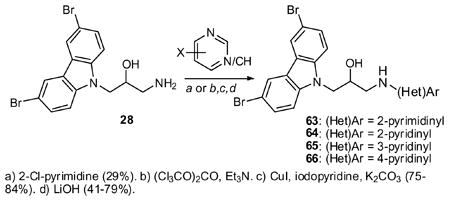
To explore the SAR on the right hand fragment, we replaced the N-phenyl ring with heterocyclic rings as shown in eq 2. The primary amine 28 condensed with 2-chloropyrimidine to provide analog 63. Introduction of pyridine rings required initial conversion to a urea. Subsequent Cu-catalyzed cross coupling with iodo-pyridines33 and hydrolysis of the cyclic urea yielded aminopyridines 64–66. Five-membered heterocyclic rings were incorporated as shown in Scheme 4. Thus, hydrazine 30 was condensed with a diketone to yield pyrazole 67 while azide 31 underwent Cu-catalyzed cycloaddition to yield the corresponding triazole (68).34
Scheme 4.

a) 2,4-pentanedione, EtOH, 70 °C (45%). b) 4-pentynol, Cu(SO4), NaAscorbate, H2O/tBuOH (54%)
 |
(2) |
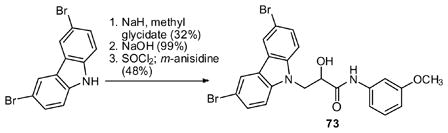
Several analogs were prepared with modified linking groups between the carbazole and aniline. As shown in Scheme 5, epoxide openings and alkylations provided derivatives without the aniline NH (69), without the central OH (70) or with additional methylenes in the linker (71, 72). Similarly, the secondary aniline was replaced with a secondary amide (73) through the following sequence: ring opening of methyl glycidate with dibromocarbazole, hydrolysis of the ester, and coupling to aniline by way of the acid chloride (eq 3).
Scheme 5.

Synthesis of analogs with modified linkers.
Ester 73 was originally conceived as a precursor to optically active derivatives of P7C3. Our plan was to perform the ring opening with enantioenriched methyl glycidate, and, following amidation, reduce scalemic 73 to yield optically active 7. In the event, however, we found that the ester generated in this manner was racemic, with the optical purity presumably being a casualty of the basic reaction conditions. Similarly, ring-opening of optically active epichlorohydrin yielded racemic epoxide 13 (heterocycle = Br2-carbazole). In this case, epichlorohydrin was likely racemized by adventitious chloride ion.35 Ultimately, using the known Mitsunobu reaction of dibromocarbazole with optically active glycidol,23 we were able to prepare both enantiomers of epoxide 13 in >99% ee, and from them access the single enantiomers of both 7 and the sulfone 26.36 Likewise, both enantiomers of the aryl ether 42 were prepared from optically active phenyl glycidyl ether. Interestingly, when these alcohols were subjected to the fluorination reaction conditions shown in Table 1, the resulting fluorides were found to have <20% ee. Partial reaction flux through an aziridinium ion likely provides a mechanism for retention of configuration that competes with the usual invertive pathway.
 |
(3) |
Finally, to provide a reagent for mechanism-of-action studies, the coumarin-tagged37 derivative 76 was prepared from anisidine derivative 7. Thus, cleavage of the methyl ether (→ 74), alkylation (→ 75) and conjugation to a coumarin derivative provided the desired reagent in acceptable yield (Scheme 6).
Scheme 6.

Synthesis of a coumarin-tagged derivative
Biological Data
All of the analogs were tested in live mice for their proneurogenic effect. Following the procedure described above, 10 μM solutions were administered continuously for 7 days (12 μL/day) by means of subcutaneous osmotic minipumps that delivered compound through a canula implanted into the left lateral ventricle. We estimate that the in vivo concentrations were in the low micromolar to mid-nanomolar range.19 The results are shown in Figure 3 grouped by the region of P7C3 that each analog modifies. Thus, derivatives of the carbazole, the propyl linker, the heteroatom or the aromatic ring were tested (Scheme 7). Some compounds represent multiple changes from P7C3.
Fig 3. Analysis of synthetic analogs.

In vivo analysis of synthetic analogs of P7C3. Data are expressed as mean ± SEM, 4 mice/compound except compound 56 (1 mouse). Compounds were infused ICV as a 10 μM solution introduced at a constant rate (12 μL/day) over 7 days. Brackets indicate portion of analog that differs from P7C3.
Scheme 7.
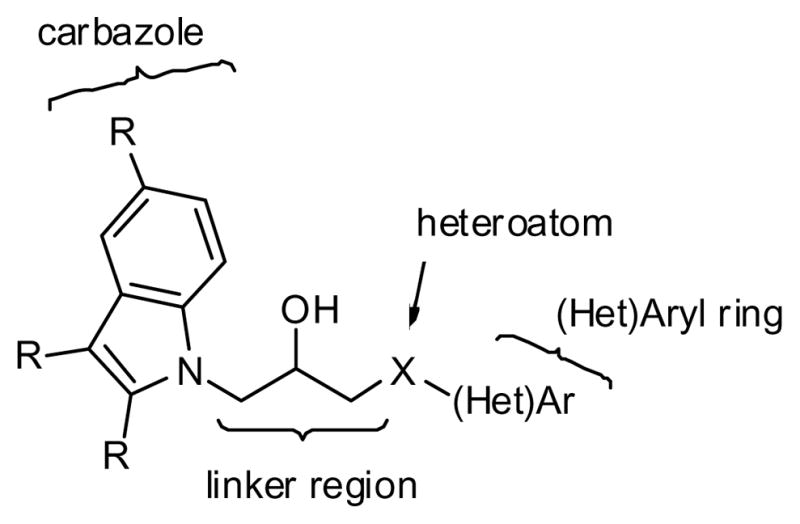
Regions of P7C3 subject to modification as labeled in Fig 3.
The bromines on P7C3 are particularly important: the dimethyl (14), dichloro (17) and diiodo (20) versions appear to have little activity, and the mono-bromo derivative (16) appears substantially less active in our assays. Nonetheless, dibromination is not required as the bromo-methyl-substituted compound (15) is highly active. Likewise, in a promising result for the synthesis of photocrosslinking agents, the azido-bromo-carbazole (18) retains significant activity. The substantial activity of the bis-nitrile (19) indicates that the bromines can be replaced entirely, and provides a lead compound with substantially reduced lipophilicity.
The carbazole ring system does not appear to be strictly required. For example, the β-carboline (38) is as active as P7C3. The tetrahydro-γ-carboline 33, a structural hybrid between dimebon and P7C3, shows intermediated activity, as does the dimethyl indole 27.
In the linker region, several compounds were improved by replacing the hydroxyl with a fluorine. Indeed, the most active compound prepared (53), is the fluorinated anisidine derivative. Similar trends were observed with the N-methyl derivative (35 vs. 56) and the sulfoxide-containing analogs (26 vs. 58). However, the effect of fluorines appears dependent on context. For example, while the activities of the m- and p-anisidine-containing analogs are improved by fluorination (7 vs. 53; 8 vs. 54), the parent compound with an aniline ring gains little from fluorination (P7C3 vs 52). Similarly, the derivative featuring a phenol is highly active with a 2-hydroxypropyl linker, but less active with a 2-fluoropropyl linker (42 vs. 57).
Two additional changes to the linker region were well tolerated. First, conversion to a tertiary alcohol (46) improved activity. Second, extending to a hydroxy butyl linker maintained high activity, so long as the extra methylene was inserted between the hydroxy group and the aniline (72). In contrast, inclusion of an additional methylene between the hydroxyl and the carbazole (71) essentially abrogated activity. Intermediate activity was observed for analogs possessing O- or N-methyl groups (35, 47), a ketone (50), an α-thioketone (51), an N-acyl group (49), a cyclic urea (48), an amide (73), or an unsubstituted propyl chain (70).
P7C3 features a nitrogen linking the propyl chain to the right-hand aromatic ring. We synthesized several congeners to explore the effectiveness of alternative heteroatoms. Encouragingly, the aniline could be replaced with an aryl ether (42, 43), an aryl thioether (24) or an aryl sulfone (26) and retain superior activity. Interestingly, while both the thioether and sulfone were highly active, the mixture of diastereomeric sulfoxides was equivalent to vehicle. Because C-S bonds are longer than C-N bonds, we speculated that smaller groups than Br might be tolerated on the carbazole. While not confirming our reasoning, it was heartening to observe substantial neuroprotective activity from a dichlorocarbazole possessing a sulfone linker (45). While several heteroatoms appear acceptable at this position, an analog with only a hydrocarbon linker (69) did not appear neuroprotective at the concentration tested.
The right-hand aromatic fragment tolerates substantial variation, although some surprising observations were made. Modest activity is retained even with simple NH2- or O-alkyl-terminated compounds (28, 29), i.e. compounds lacking a right-hand aromatic ring. As described above, a piperazine group effectively replaces the aniline. Heteroaromatic rings have proved less effective with two notable exceptions. The 2-aminopyridine (64) displays activity nearly as high as P7C3 itself, and the azapyridone (32) maintains robust activity. In contrast, the other pyridines, pyrazines, indoles, carbazoles, pyrazoles and triazoles were substantially less active in our assays. Substitution around the aniline ring of P7C3 was somewhat enigmatic. For instance, a 3-OMe (7) group had little effect on activity, but a minor extension to an OEt (21) group23 essentially abrogated activity. Likewise, a 3-N3 group (22) substantially decreased activity. Nonetheless, substantial activity was observed for a derivative designed to facilitate affinity chromatography and localization studies, the coumrain-linked analog (76),38 Meanwhile, the 4-OMe (8), 4-OEt39 and 4-N3 (23) derivatives were all highly active.
Finally, several enantiomeric pairs were tested. Specifically, we tested the anisidine analog of P7C3 (7), the aryl ether 42, and the sulfoxide 26. In all three cases evaluated, a significant difference in activity was noted between the two enantiomers. The same enantiomer displayed higher activity in all three cases, although the R/S designation varies due to differences in priority among the substituents.
Tertiary alcohol 46, fluoro-aniline 53 and fluoro-sulfone 58 were evaluated at a range of doses. In each instance, 10 – 0.01 μM solutions were infused over a 7 day period at a constant rate (12 μL/day) concurrent with IV administration of BrdU. The tertiary alcohol (46) roughly paralleled P7C3, showing substantial activity when a 1 μM solution was infused, but showing little effect from a 0.1 μM solution. In contrast, both fluorides maintained activity at 0.1 μM. Indeed, our most potent analog (53) remained active when 0.01 μM solution was infused, which corresponds to a total of 0.43 mg added over one week’s time.
Several of our most potent compounds were analyzed for pharmacokinetic properties and hallmarks of toxicity (Table 2). A competition assay was performed to determine if P7C3 or its derivatives bound to the hERG channel, as interference with hERG function can cause cardiac toxicity.40 Specifically, we measured what percentage of [3H]astemizole (Kd = 6.8 nM) was displaced with a 10 μM concentration of drug.41 P7C3 and (R)-7 showed significant displacement of the radioligand, and (S)-42 showed modest binding. Encouragingly, the other analogs tested appear to bind hERG only weakly.
Table 2.
Off-target, stability and toxicity data for highly active compounds.
| Compound | hERG binding (% displacement @ 10 μM)a | Histamine H2 binding (% displacement @ 10 μM)b | Toxicity towards HeLa cells | Half life in cell culture (Min) | ||
|---|---|---|---|---|---|---|
| GI50 (μM)c | LC50 (μM)d | Mouse hepatocytes | Human hepatocytes | |||
| P7C3 | 92 | 63 | 5.1 | 21.4 | >240 | >240 |
| (R)-7 | 75 | 88 | 23.5 | >30 | >240 | >240 |
| 26 | 24 | 5 | 16.1 | >30 | 224 | 43 |
| (S)-42 | 52 | 52 | 10.3 | >30 | >240 | >240 |
| 46 | 36 | 52 | 10.9 | >30 | >240 | >240 |
| 53 | 19 | 21 | >30 | >30 | >240 | >240 |
| 58 | 38 | −3 | 9.1 | 20.3 | 141 | 178 |
% displacement of [3H]astemizole (1.5 nM; Kd = 6.8 nM) from hERG with 10 μM drug.
% displacement of nM [125I]Aminopotentidine (0.1 nM; Kd = 0.45 nM) from histamine H2 receptor with 10 μM drug.
Concentration at which growth in cell number is 50% of control at 48 h.
Concentration at which cell numbers at 48 h equal 50% of cell number at t = 0.
The tetrahydrocarboline dimebon (11) had been used in Russia as an antihistamine prior to engendering enthusiasm regarding its potential as a treatment for Alzheimer’s disease. Accordingly, we performed a competition assay using [125I]aminopotentidine, a potent (Kd = 0.1 nM) ligand for the histamine H2 receptor.41 Using this assay, dimebon displaced 99% of the aminopotentidine at 10 μM. P7C3 and (R)-7 also display modest binding to this receptor. However, histamine binding and neuroprotection activity are clearly unrelated because several of our most potent analogs display weak or no binding. No analog showed significant binding to the histamine H1, H3 or H4 receptor.
To determine metabolic stability, compounds were incubated with either mouse or human hepatocytes at 2 μM. HPLC/MS/MS analysis of the culture media at several time points provided measurements of half-life. Gratifyingly, of the analogs tested, most were extremely stable. Compound 58, a fluorosulfone, had a half-life of 2–3 h. Compound 26, a β-hydroxy sulfoxide, was quite stable towards mouse hepatocytes, but was processed by human hepatocytes with a t1/2 < 1 h. All other analogs displaying high activity showed only marginal levels of metabolism after 4 h. While we did not determine the metabolites of the two sulfoxides, it is interesting to note that during their syntheses these compounds displayed a tendency to undergo β-elimination to yield vinyl sulfones. We conjecture that this reactivity profile in the flask is mirrored by their reactivity in cell culture.
As previously reported, P7C3 is orally available and displays favorable distribution into the brain (AUC brain: AUC plasma ratio of 3.7 for oral delivery). For comparison, brain exposure to 53 was determined after intraperitoneal (IP) and oral gavage (PO) administration. It was found to have good penetration, with a AUC brain:AUC plasma of 0.61 and 0.82 by IP and oral gavage administration, respectively. Treating at 20 mg/kg PO with fluoride 53 resulted in approximately 0.9 μM concentration in the brain (450 ng/g brain tissue) 4 hr after dosing.
Discussion
Neurodegenerative diseases remain an area of unmet medical need. The poignancy of medicine’s deficiency in this arena will only become more evident in the near future. Aging populations in most industrialized countries, and many developing ones, will confront an increasing incidence of neurodegenerative disorders.1 In that context, small molecules capable of fostering the growth of new neurons could facilitate the development of novel molecular therapeutics. P7C3 possesses this ability, and has been found to repair damaged neural circuitry in mice and improve cognition in aged rats. Accordingly, we became interested in improving the activity of P7C3 in terms of potency and toxicity as well as identifying reagents to aid studies of its mode of action.
We modified nearly every atom of P7C3 during this study. Our investigation has revealed several important characteristics of P7C3 and has led to the discovery of improved analogs. First, we found that P7C3 displays an encouraging combination of sensitivity and flexibility. That is, while effects are evident from modification to all sections of the molecule - the carbazole, the linker region, the aryl ring - none of these subunits is sacrosanct. For instance, highly active analogs were identified in which the carbazole had been replaced with a carboline, the hydroxy-propyl linker had been replaced either a fluoropropyl linker or a hydroxybutyl linker, or the aniline had been replaced with an aminopyridine, a phenol, a thiophenol or an aryl sulfone. This SAR should allow the identification of safe, effective and bioavailable analogs for further development.
Second, we have shown that the neuoroprotective activity of P7C3 and its analogs is unrelated to histamine receptor binding. No mechanism of action has been suggested for dimebon’s putative anti-AD activity, but a) it is active in our assay and b) it is a potent antagonist of the histamine receptor. We cannot claim with certainty that dimebon and the P7C3 class of molecules work by the same mechanism, but we can exclude the possibility that binding to the histamine receptor is required for proneuorogenic activity.
Third, we have prepared analogs of P7C3 that we anticipate will facilitate mechanism-of-action studies. Specifically, we have prepared two aryl azides (18, 23) that could be used for photoaffinity studies. Likewise, we have prepared an active derivative containing a coumarin moiety for use in pull-down and localization experiments. More broadly, we have revealed which sections of P7C3 accommodate alteration; this knowledge is expected to aid the preparation of other derivatives that will be useful in mode-of-action studies.
Finally, we have identified analogs displaying no observed toxicity, no binding of the hERG channel, good blood/brain distribution, oral availability and excellent metabolic stability. We are optimistic that this information will enable the discovery of the molecular target of P7C3, and will continue to facilitate the identification of compounds suitable for further development.
P7C3 was discovered using an unusual approach. Modern drug discovery relies on high-throughput screening against defined receptors or enzymes using in vitro or cell-based assays. In connection with neurodegenerative diseases, however, little consensus exists regarding which receptors or enzymes represent ideal targets. Furthermore, these diseases likely involve pathologies of multiple cell types in the complex setting of the central nervous system. Accordingly, devising simple, predictive assays has proven difficult, a challenge which manifests itself in the paucity of available therapeutics. We opted for an alternative strategy wherein we identified and optimized molecules capable of affecting a desired function, neurogenesis, within a living mammal. The risks of this approach were clear, among which were low throughput, an inability to separate binding to a target from pharmacokinetic effects, and ignorance of the molecular target. Nonetheless, the benefits of this experimental design appear to outweigh these limitations as we were able to simultaneously select for molecules that possessed activity within live mice and rats, desirable toxicity profiles and acceptable physical characteristics. It may be that this atavistic approach to drug discovery deserves consideration for other complex diseases.
Experimental Details
Neuroprotection Assay
Test compounds were evaluated as described previously.19 Compounds were infused intracerebroventricularly (i.c.v.) into the left lateral ventricle of four adult (12 week old) wild-type C57BL/J6 mice by means of surgically implanted Alzet osmotic minipumps that delivered solution into animals at a constant rate of 0.5 μl/hour for 7 days. Bromodeoxyuridine (BrdU) was injected intraperitoneally at 50 mg/kg/day for six days during pump infusion. Twenty-four hours after the final BrdU administration, mice were sacrificed by transcardial perfusion with 4% paraformaldehyde at pH 7.4, and their brains were processed for immunohistochemical detection of incorporated BrdU in the SGZ. Dissected brains were immersed in 4% paraformaldehyde overnight at 4 °C, then cryoprotected in sucrose before being sectioned into 40 μm thick free-floating sections. Unmasking of BrdU antigen was achieved through incubating tissue sections for two hours in 50% formamide/2X SSC at 65 °C, followed by a five minute wash in 2X SSC and subsequent incubation for thirty minutes in 2M HCl at 37 °C. Sections were processed for immunohistochemical staining with mouse monoclonal anti-BrdU (1:100). The number of BrdU+ cells in the entire dentate gyrus SGZ in the contralateral hemisphere (opposite side of surgically implanted pump) was quantified by counting BrdU+ cells within the SGZ and dentate gyrus in every fifth section throughout the entire hippocampus and then normalizing for dentate gyrus volume.
Binding assays
Binding to hERG and histamine receptors was performed by MDS Pharma Services, now Ricera. Briefly, a competition binding assay was performed in which the level of specific binding of a radioligand, used at a low concentration (≤Kd), was determined in the presence of 10 μM test compound. Specific radioligands, the concentration utilized, and historical Kd values as measured by Ricera for each receptor and radioligand are as follows: HERG: 1.5 nM [3H]Astemizole (Kd 6.8 nM); Histamine H1: 1.2 nM [3H]Pyrilamine (Kd 1.1 nm); Histamine H2: 0.1 nM [125I]Aminopotentidine (Kd 0.45 nM); Histamine H3: 3 nM [3H] (R)-(−)-α-Methylhistamine (Kd 2.4 nM); Histamine H4: 8.2 nM [3H]histamine (Kd 5.7 nM).
Cell Viability Assays
HeLa cells were plated at optimally determined densities for either 96 or 384 well plates and compounds were added in serial five-fold dilutions the following day. Forty-eight hours later, cell viability was assessed by addition of CellTiter-Glo Reagent (Promega, Madison, WI). Growth Inhibitory 50 (GI50) values were calculated according to the formula 100 × (T−T0)/(C−T0) = 50, where T = optical density of test well after 48 hr exposure to compound; T0 = optical density at time zero; and C = control optical density. The LC50 (concentration of drug resulting in a 50% reduction in the measured protein/ATP at the end of the drug treatment as compared to that at the beginning) indicating a net loss of cells following treatment is calculated from [(T−T0)/T0] × 100 = −50.
Hepatocyte Metabolism Assays42
Male ICR/CD-1 mouse hepatocytes, InVitroGRO HI and HT Medium, and Celsis Torpedo Antibiotic Mix were purchased from Celsis/In Vitro Technologies (Baltimore, MD). Cryopreserved hepatocytes were thawed in HT Media containing antibiotics, resuspended in HI media at 2 ×106/ml and plated in 96 well plates at 0.05 ml (105 cells)/well. Compounds to be tested were dissolved in DMSO at 2 mM, further diluted to 4 μM in HI media, and added to the cells in 50 μl so that the final compound concentration was 2 μM. Two additional wells containing compound and no cells were plated to serve as time 0 (C0) and endpoint solvent control (Cep). The cells were then placed in a 37 °C, 5% CO2 incubator. At the timepoints indicated, the well contents were harvested and a 2-fold volume of methanol added to lyse the cells and precipitate proteins. The samples were incubated 10 min at RT and then spun at 15,000 × g for 5 min in a microcentrifuge. The supernatant was analyzed by LC/MS/MS. Metabolism of 7-ethoxycoumarin was used to monitor hepatocyte performance.
Pharmacokinetic analysis of P7C3 and 53
P7C3 was prepared for dosing by dissolving the compound in DMSO at 50 mg/ml. The compound was diluted to a final formulation of 3% DMSO/10% cremophor EL (Sigma, St. Louis, MO)/87.5% D5W (5% dextrose in water, pH 7.2). Adult mice were dosed IV, IP or via oral gavage (PO) in a total volume of 0.2 ml. Compound 53 was prepared in a similar fashion in 5% DMSO/10% cremophor EL/85% D5W and dosed either IP or PO. Animals were sacrificed at varying times after dosing by inhalation overdose of CO2. Whole blood was collected with an ACD solution (sodium citrate) coated syringe and needle. The blood was subsequently centrifuged at 9300 × g for 10 min to isolate plasma. Plasma was stored at −80 °C until analysis. Brains were isolated from mice immediately after sacrifice, rinsed extensively with PBS, blotted dry, weighed, and snap frozen in liquid nitrogen. Lysates were prepared by homogenizing the brain tissue in a 3-fold volume of PBS (weight of brain in g × 3 = volume of PBS in ml added). Total lysate volume was estimated as volume of PBS added + volume of brain in ml. One hundred μl of either plasma or brain was processed by addition of 200 μl of acetonitrile to precipitate plasma or tissue protein and release bound drug. In some experiments, this mixture was centrifuged at 16,100 × g for 5 min and the supernatant analyzed directly by HPLC/MS/MS while in other experiments the sample was additionally processed by passage over a solid phase extraction column. An additional 700 μl of PBS was added to the plasma or lysate acetonitrile mixture and the sample centrifuged at 16,100 × g for 5 min. Nine hundred μl of supernatant was mixed with 1 ml of PBS and passed over a Waters (Milford, MA) OASIS HLB solid phase extraction column primed by addition of 2 ml acetonitrile followed by 2 ml of water. The column was washed twice with 2 ml of 5% acetonitrile in H20 and compound was eluted by addition of 2 ml of acetonitrile. 500 μl of the eluant was added to 500 μl of H20 containing 0.2% formic acid (final formic acid 0.1%) and analyzed by HPLC/MS/MS as described above. Standard curves were prepared by addition of P7C3 to blank plasma or blank brain lysate. A value of 3-fold above the signal obtained from blank plasma or brain lysate was designated the limit of detection (LOD). The limit of quantitation (LOQ) was defined as the lowest concentration at which back calculation yielded a concentration within 20% of theoretical. The LOQ for both plasma and brain was 5 ng/ml. In general back calculation of points on both curves yielded values within 25% of theoretical over 4 orders of magnitude (10000 to 5 ng/ml). Pharmacokinetic parameters were calculated using the noncompartmental analysis tool of WinNonLin (Pharsight). Bioavailability was calculated as AUCoral/AUCiv × Doseiv/Doseoral × 100 (AUC is area under the concentration time curve). The Brain:Blood ratio was calculated using AUC values.
Supplementary Material
Fig 2. Analysis of commercial analogs.

In vivo analysis of commercially available analogs of P7C3. Data are expressed as mean ± SEM, 4 mice/compound. Compounds were infused ICV as a 10 μM solution introduced at a constant rate (12 μL/day) over 7 days.
Fig 4. Dose-Response of selected analogs.
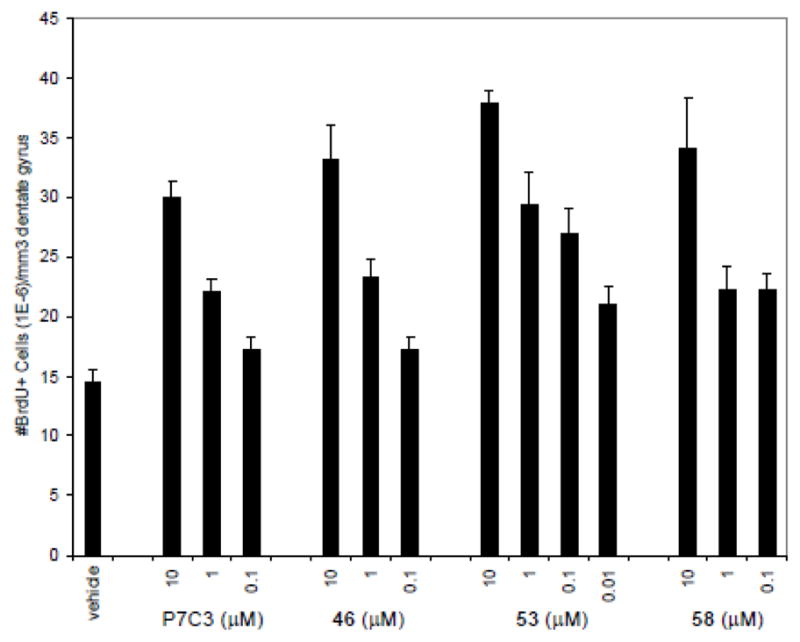
In vivo analysis of synthetic analogs of P7C3. Data are expressed as mean ± SEM, 4 mice/compound. Compounds were infused ICV at a constant rate (12 μL/day) over 7 days as a solution of the indicated concentration.
Acknowledgments
Funding provided by The Hartwell Foundation, Staglin Family fund, UT Southwestern High Risk/High impact research fund, National Alliance for Research on Schizophrenia and Depression, Morton H. Meyerson Family Tzedakah fund (A.A.P), NIH (1R01 MH087986, 5DP1OD000276, RO1MN59388, 5PO1CA95471), a Simons Foundation Autism Research Initiative grant, the Welch foundation (I-1612, I-1422). JMR is a fellow of the Alfred P. Sloan Foundation.
Footnotes
SUPPORTING INFORMATION Complete experimental details and characterization data for new compounds; full citations for abbreviated references.
References
- 1.Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA. Neurology. 2004;62:1645. doi: 10.1212/01.wnl.0000123018.01306.10. [DOI] [PubMed] [Google Scholar]
- 2.Jarvis LM. Chem & Eng News. 2010 April 5;:12. [Google Scholar]
- 3.Ledford H. Nature. 2010;466:1031. doi: 10.1038/4661031a. [DOI] [PubMed] [Google Scholar]
- 4.Chuang TT. Biochim Biophys Acta Mol Bas Disease. doi: 10.1016/j.bbadis.2009.12.008. [DOI] [Google Scholar]
- 5.(a) Altman J. Science. 1962;135:1127. doi: 10.1126/science.135.3509.1127. [DOI] [PubMed] [Google Scholar]; (b) Altman J, Das GD. J Comp Neurol. 1965;124:319. doi: 10.1002/cne.901240303. [DOI] [PubMed] [Google Scholar]
- 6.Paton J, Nottebohm F. Science. 1984;225:1046. doi: 10.1126/science.6474166. [DOI] [PubMed] [Google Scholar]
- 7.Gross CG. Nat Rev Neurosci. 2000;1:67. doi: 10.1038/35036235. [DOI] [PubMed] [Google Scholar]
- 8.(a) Gage FH, Kempermann G, Palmer TD, Peterson DA, Ray J. J Neurobiol. 1998;36:249. doi: 10.1002/(sici)1097-4695(199808)36:2<249::aid-neu11>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]; (b) Zhao C, Deng W, Gage FH. Cell. 2008;132:645. doi: 10.1016/j.cell.2008.01.033. [DOI] [PubMed] [Google Scholar]; (c) Aimone JB, Wiles J, Gage FH. Nat Neurosci. 2006;9:723. doi: 10.1038/nn1707. [DOI] [PubMed] [Google Scholar]
- 9.Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C, Peterson DA, Gage FH. Nat Med. 1998;4:1313. doi: 10.1038/3305. [DOI] [PubMed] [Google Scholar]
- 10.Kempermann G, Kuhn HG, Gage FH. Nature. 1997;386:493. doi: 10.1038/386493a0. [DOI] [PubMed] [Google Scholar]
- 11.Hu YS, Xu P, Pigino G, Brady ST, Larson J, Lazarov O. FASEB J. 2010;24:1667. doi: 10.1096/fj.09-136945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van PH, Kempermann G, Gage FH. Nat Neurosci. 1999;2:266. doi: 10.1038/6368. [DOI] [PubMed] [Google Scholar]
- 13.Brown J, Cooper-Kuhn CM, Kempermann G, van Pragg H, Winkler J, Gage FH, Kuhn HG. Eur J Neurosci. 2003;17:2042. doi: 10.1046/j.1460-9568.2003.02647.x. [DOI] [PubMed] [Google Scholar]
- 14.(a) Stern Y. Alzheimer Dis Assoc Disord. 2006;20:112. doi: 10.1097/01.wad.0000213815.20177.19. [DOI] [PubMed] [Google Scholar]; (b) Paradise M, Cooper C, Livingston G. Int Psychoger. 2009;21:25. doi: 10.1017/S1041610208008053. [DOI] [PubMed] [Google Scholar]; (c) Radak Z, Hart N, Sarga L, Koltai E, Atalay M, Ohno H, Boldogh I. J Alzheimer’s Dis. 2010;20:777. doi: 10.3233/JAD-2010-091531. [DOI] [PubMed] [Google Scholar]
- 15.(a) Schmidt HD, Duman RS. Behav Pharmacol. 2007;18:391. doi: 10.1097/FBP.0b013e3282ee2aa8. [DOI] [PubMed] [Google Scholar]; (b) Boldrini M, Underwood MD, Hen R, Rosoklija GB, Dwork AJ, John MJ, Arango V. Neuropsychopharmacology. 2009;34:2376. doi: 10.1038/npp.2009.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang JM, Singh C, Liu L, Irwin RW, Chen S, Chung EJ, Thompson RF, Brinton RD. Proc Natl Acad Sci U S A. 2010;107:6498. doi: 10.1073/pnas.1001422107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Brinton RD, Wang JM. Curr Alzheimer Res. 2006;3:185. doi: 10.2174/156720506777632817. [DOI] [PubMed] [Google Scholar]; (b) Wang JM, Irwin RW, Liu L, Chen S, Brinton RD. Curr Alzheimer Res. 2007;4:510. doi: 10.2174/156720507783018262. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Brinton RD, Wang JM. Curr Alzheimer Res. 2006;3:11. doi: 10.2174/156720506775697160. [DOI] [PubMed] [Google Scholar]; (d) Imbimbo BP, Giardino L, Sivilia S, Giuliani A, Gusciglio M, Pietrini V, Del Giudice E, D’Arrigo A, Leon A, Villetti G, Calza L. J Alzheimer’s Dis. 2010;20:159. doi: 10.3233/JAD-2010-1366. [DOI] [PubMed] [Google Scholar]
- 18.Hamilton LK, Aumont A, Julien C, Vadnais A, Calon F, Fernandes KJ. Eur J Neurosci. 2010 Aug 19; doi: 10.1111/j.1460-9568.2010.07379.x. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 19.(a) Pieper AA, Xie S, Capota E, Estill SJ, Zhong J, Long JM, Becker GL, Huntington P, Goldman SE, Shen, et al. Cell. 2010;142:39. doi: 10.1016/j.cell.2010.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McKnight SL, Pieper AA, Ready JM, De Brabander JK. 2010/020681 US Patent.
- 20.Malberg JE, Eisch AJ, Nestler EJ, Duman RS. J Neurosci. 2000;20:9104. doi: 10.1523/JNEUROSCI.20-24-09104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pieper AA, Wu X, Han TW, Estill SJ, Dang Q, Wu LC, Reece-Fincanon S, Dudley CA, Richardson JA, Brat DJ, McKnight SL. Proc Natl Acad Sci USA. 2005;102:14052. doi: 10.1073/pnas.0506713102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.See Figure S1 in the supporting information.
- 23.Bombrun A, Gerber P, Casi G, Terradillos O, Antonsson B, Halazy S. J Med Chem. 2003;46:4365. doi: 10.1021/jm034107j. [DOI] [PubMed] [Google Scholar]
- 24.(a) Bachurin S, Bukatina E, Lermontova N, Tkachenko S, Afanasiev A, Grigoriev V, Grigorieva I, Ivanov Y, Sablin S, Zefirov N. Ann N Y Acad Sci. 2001;939:425. doi: 10.1111/j.1749-6632.2001.tb03654.x. [DOI] [PubMed] [Google Scholar]; (b) Bachurin SO, Shevtsova EP, Kireeva EG, Oxenkrug GF, Sablin SO. Ann N Y Acad Sci. 2003;993:334. doi: 10.1111/j.1749-6632.2003.tb07541.x. [DOI] [PubMed] [Google Scholar]
- 25.(a) O’Brien JT. Lancet Neurol. 2008;7:768. doi: 10.1016/S1474-4422(08)70177-7. [DOI] [PubMed] [Google Scholar]; (b) Burns A, Jacoby R. Lancet. 2008;372:179. doi: 10.1016/S0140-6736(08)61046-6. [DOI] [PubMed] [Google Scholar]
- 26.Doody RS, Gavrilova SI, Sano M, Thomas RG, Aisen PS, Bachurin SO, Seely L, Hung D. Lancet. 2008;372:207. doi: 10.1016/S0140-6736(08)61074-0. [DOI] [PubMed] [Google Scholar]
- 27.Miller G. Science. 2010;327:1309. doi: 10.1126/science.327.5971.1309. [DOI] [PubMed] [Google Scholar]
- 28.Asso V, Ghilardi E, Bertini S, Digiacomo M, Granchi C, Minutolo F, Rapposelli S, Bortolato A, Moro S, Macchia M. Chem Med Chem. 2008;3:1530. doi: 10.1002/cmdc.200800162. [DOI] [PubMed] [Google Scholar]
- 29.McCluskey A, Leitch SK, Garner J, Caden CE, Hill TA, Odell LR, Stewart SG. Tetrahedron Lett. 2005;46:8229. [Google Scholar]
- 30.Chakraborti AK, Rudrawar S, Kondaskar A. Eur J Org Chem. 2004:3597. doi: 10.1039/b400588k. [DOI] [PubMed] [Google Scholar]
- 31.Getautis V, Dashkyavichene M, Paulauskaite I, Stanisauskaite A. Chemistry of Heterocyclic Compounds. 2005;41:426. [Google Scholar]
- 32.Kan T, Fukuyama T. Chem Commun. 2004:353. doi: 10.1039/b311203a. [DOI] [PubMed] [Google Scholar]
- 33.Tatsumi R, Fujio M, Satoh H, Katayama J, Takanashi S-i, Hashimoto K, Tanaka H. J Med Chem. 2005;48:2678. doi: 10.1021/jm049188d. [DOI] [PubMed] [Google Scholar]
- 34.Himo F, Lovell T, Hilgraf R, Rostovtsev VV, Noodleman L, Sharpless KB, Fokin VV. J Am Chem Soc. 2004;127:210. doi: 10.1021/ja0471525. [DOI] [PubMed] [Google Scholar]
- 35.Furrow ME, Schaus SE, Jacobsen EN. J Org Chem. 1998;63:6776. doi: 10.1021/jo981332d. [DOI] [PubMed] [Google Scholar]
- 36.Alternatively, optically active analogs could be prepared by way of their diastereomeric Mosher’s esters or amides.
- 37.Alexander MD, Burkart MD, Leonard MS, Portonovo P, Liang B, Ding X, Joullié MM, Gulledge BM, Aggen JB, Chamberlin AR, Sandler J, Fenical W, Cui J, Gharpure SJ, Polosukhin A, Zhang H-R, Evans PA, Richardson AD, Harper MK, Ireland CM, Vong BG, Brady TP, Theodorakis EA, La Clair JJ. Chem Bio Chem. 2006;7:409. doi: 10.1002/cbic.200500466. [DOI] [PubMed] [Google Scholar]
- 38.BrdU+ cells/mm3 dentate gyrus = 22.9 × 106 ± 0.5 × 106
- 39.This analog is commercially available: BrdU+ cells/mm3 dentate gyrus = 24.3 × 106 ± 2.8 × 106
- 40.Sanguinetti MC, Tristani-Firouzi M. Nature. 2006;440:463. doi: 10.1038/nature04710. [DOI] [PubMed] [Google Scholar]
- 41.Binding to hERG and histamine receptors was performed by MDS Pharma Services, which is now owned by Ricera.
- 42.(a) McNaney CA, Drexler DM, Hnatyshyn S, Zvyaga TA, Knipe JO, Belcastro JV, Sanders M. Assay Drug Dev Technol. 2008;6:121. doi: 10.1089/adt.2007.103. [DOI] [PubMed] [Google Scholar]; (b) Drexler DM, Belcastro JV, Kickinson KE, Edinger KJ, Hnatyshy SY, Josephs JL, et al. Assay Drug Devel Technol. 2007;5:247. doi: 10.1089/adt.2006.038. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.