

Abstract
Heme is vital to our aerobic universe. Heme cellular content is finely tuned through an exquisite control of synthesis and degradation. Heme deficiency is deleterious to cells, whereas excess heme is toxic. Most of the cellular heme serves as the prosthetic moiety of functionally diverse hemoproteins, including cytochromes P450 (P450s). In the liver, P450s are its major consumers with >50% of hepatic heme committed to their synthesis. Prosthetic heme is the sine qua non of P450 catalytic biotransformation of both endo- and xenobiotics. This well-recognized functional role notwithstanding, heme also regulates P450 protein synthesis, assembly, repair and disposal. These less well-appreciated aspects are reviewed herein.
Introduction
Heme plays a pivotal role in our lives as the prosthetic moiety of several vital hemoproteins [hemoglobin, mitochondrial cytochromes, myoglobin, catalase, peroxidases, tryptophan 2,3-dioxygenase (TDO), cystathionine β-synthase, nitric oxide synthases (NOS) and cytochromes P450 (P450s; CYPs); (Padmanaban et al., 1989; Ortiz de Montellano, 2009; Furuyama et al., 2007; Fig. 1). It also serves as an activator of various O2/gas-sensors such as heme oxygenase 2 (HO-2), heme-regulated inhibitor (HRI), Slo I BK channels and other biologically relevant proteins such as neudesin, and Toll-like receptor 4 (Kimura et al., 2008; Figueiredo et al., 2007; Furuyama et al., 2007). In addition to this critical prosthetic role, it also plays important regulatory roles in cell signalling (Zhu et al., 2002a; Mense & Zhang, 2006), and in transcriptional, translational and posttranslational events (Padmanaban et al., 1989; Ortiz de Montellano, 2009; Furuyama et al., 2007).
Fig. 1. Prosthetic and regulatory roles of cellular heme.

Heme concentration in the liver is exquisitely controlled by a very finely tuned balance between its synthesis and degradation that maintains the cellular “free” or regulatory heme pool at ≈ 0.1 μM (Granick et al., 1975). Unlike prosthetically incorporated heme, this “free” heme pool is not really free in solution but is loosely bound to cellular proteins and thus readily available for the regulation of heme-dependent cellular processes. As discussed, the major fraction of heme synthesized in the liver is prosthetically incorporated into P450s and other cellular hemoproteins. Heme in excess of cellular needs is disposed via HO-1, whose induction accelerates the heme breakdown process. Adapted from Fig. 9 (Furuyama et al., 2007).
Over 50% of the heme synthesized in the liver is committed to the synthesis of P450 enzymes, making these hepatic hemoproteins the major consumers of hepatic heme (Meyer, 2007). P450s are endoplasmic reticulum (ER)-anchored hemoproteins responsible for the metabolism of numerous endogenous and foreign compounds. As the prosthetic moiety of all P450s, heme is responsible for the remarkable and often exquisite, catalytic prowess of these enzymes. This mechanistic feature of the P450 heme moiety is well recognized and reviewed elsewhere in several excellent articles (Ortiz de Montellano & De Vos, 2004; Guengerich, 2007; Zhu & Silverman, 2008). Herein, we will focus instead on a few of the other less well appreciated, albeit essential aspects of the interplay between hepatic heme metabolism and P450 protein regulation and cell biology, and the generally reciprocal nature of this relationship. Specifically we will address 3 topics: (1) Influence of heme synthesis and degradation on hepatic P450 heme content; (2) heme-protein interactions in P450 assembly and repair; and (3) heme regulation of P450 protein synthesis and disposal.
1. Influence of heme synthesis and degradation on hepatic P450 heme content
Heme is continuously made and destroyed, the rate of its synthesis being balanced by the rate of its disposal so as to ensure that its supply is adequate to meet various physiological needs without a significant accumulation in excess of this requirement. This is critical because free heme, i.e. heme not appropriately bound and shielded by hemoproteins or special heme-binding proteins is a powerful pro-oxidant agent and therefore potentially toxic. For these reasons, both its synthesis and degradation are exquisitely regulated through the “free” or “regulatory” heme pool, a pool that because of its small size, dynamic properties and ability to readily exchange with heme-containing proteins reflects the overall status of cellular heme content. In this context, “free heme” signifies heme loosely bound to intracellular proteins and thus not committed to any particular hemoprotein.
1.1.1 Enzymes of the heme synthetic pathway
Heme synthesis occurs in all mammalian cells. Eight enzymes are involved in the biosynthesis of heme from glycine and succinyl CoA (Fig. 2). Four of these enzymes are found in the mitochondrion and the others in the cytosol. The genes of all these enzymes have been cloned and the proteins expressed and crystallized. Their structures have been used to examine the catalytic mechanisms (Detailed discussion of these mechanisms is beyond the scope of this review, but information can be found in recent reviews (Ajioka et al., 2006; Heinemann et al., 2008; Layer et al., 2010).
Fig. 2. Heme biosynthetic and degradative pathways and regulation by heme and cytochrome P450.

Heme synthesis is regulated mainly by negative feedback control exercised by the free heme pool on the first enzyme of the pathway 5-aminolevulinate synthase (ALAS1) through at least three different mechanisms (see text). The size of the free heme pool is dependent on the rate of synthesis, degradation and incorporation of heme into hemoproteins including P450s. Note that URO-gen, Copro-gen and Proto-gen intermediates are not porphyrins, but rather the reduced porphyrinogens. Abbreviations: Gly, glycine; Succ CoA, succinyl CoA; PBG, porphobilinogen; HMB, hydroxymethylbilane; URO-gen, uroporphyrinogen; Copro-gen, coproporphyrinogen; Proto-gen, protoporphyrinogen; PROTOpn, protoporphyrin; HO, heme oxygenase; ApoP450, apocytochrome P450
5-Aminolevulinate synthase (ALAS1)
This mitochondrial enzyme catalyzes the first step in heme biosynthesis in non-erythroid cells, namely the condensation of glycine and succinyl CoA to form 5-aminolevulinic acid (ALA). The enzyme requires pyridoxal phosphate as a cofactor.
ALA dehydrase (ALAD)
This enzyme catalyzes the condensation of two molecules of ALA released from mitochondria to form the monopyrrole, porphobilinogen (PBG). The activity of this enzyme normally greatly exceeds that of ALAS1. But when ALAS1 is highly induced, excretion of ALA and PBG is readily detected indicating that the ability of ALAD to metabolize excess ALA is limited. This enzyme can be inhibited by lead or succinylacetone (SA), an ALA analog.
PBG deaminase (PBGD) and uroporphyrinogen III synthase (UROS)
PBGD catalyzes the formation of a linear tetrapyrrole, hydroxymethylbilane (HMB), from four molecules of PBG. UROS catalyzes inversion of the HMB D ring, followed by closure to form the macrocycle, uroporphyrinogen III. When UROS is absent or limiting, HMB can spontaneously be converted to the macrocycle uroporphyrinogen I in which there is no inversion of the D pyrrole ring. The name “uroporphyrinogen” stands for the reduced form of the porphyrin, which is colorless. Various amounts of uroporphyrins I and III are found in the urine due to some oxidation of the two porphyrinogens (Elder, 1998).
Uroporphyrinogen decarboxylase (UROD)
The next enzyme in the pathway converts the acetate side-chains of uroporphyrinogen III to methyl groups via decarboxylation to form coproporphyrinogen III.
Coproporphyrinogen oxidase (CPO)
This mitochondrial enzyme catalyzes the oxidative decarboxylation of the propionate side-chains of two of the coproporphyrinogen III pyrroles to vinyl groups to form protoporphyrinogen IX.
Protoporphyrinogen oxidase (PPO) and ferrochelatase (FC)
PPO catalyzes the six electron oxidation of protoporphyrinogen to the porphyrin, protoporphyrin IX (PROTO), which is the substrate of the other mitochondrial enzyme FC that inserts ferrous iron to form heme. Other metals can be inserted (cobalt, zinc) and the resultant metalloporphyrins are found in animals administered large amounts of salts of these metals. Since holoP450s with the heme replaced by these metalloporphyrins are not functional, this approach represents a way to manipulate the amount of functional P450s.
1.1.2. Regulation of heme biosynthesis via negative feedback on ALAS1
Hepatic ALAS1 differs from the isoform in erythropoietic cells (ALAS2), in that the latter is regulated principally by the availability of inorganic iron (Ponka, 1999). By contrast, the regulation of ALAS1 is via negative feedback by heme, the end product of the heme synthetic pathway. The pool of heme that controls this regulation, the so-called “regulatory or free heme pool” is a dynamic pool whose size is determined not only by the rate of heme synthesis (that could be significantly impaired when one of the intermediary enzymes in the pathway is decreased by inherited mutations or inhibited by endogenous or exogenous chemicals; see below), but also by the rate of heme degradation (Fig. 2).
ALAS1 is regulated by heme, through at the least three different mechanisms that influence the content of this enzyme. Accumulation of the mature protein in the mitochondrion can be blocked by heme binding to the nascent precursor protein in the cytosol (Lathrop & Timko, 1993; Dailey et al., 2005). Heme also regulates the stability of the relatively short-lived ALAS1 mRNA (Drew & Ades, 1986; Hamilton et al., 1988; Cable et al., 1994). In addition, recent evidence indicates that heme may regulate ALAS1 transcription (Yamamoto et al., 1982; Srivastava et al., 1988; Kolluri et al, 2005) as well as affect the stability of the mature protein (Zheng et al 2008). However, the relative sensitivity of ALAS1 to heme in each of these different regulatory mechanisms remains to be established. Tissue culture studies are required to determine the heme concentration response of each of these mechanisms. Available data suggest that relatively higher concentrations of heme are required to inhibit ALAS1 transcription and thus, this mode of ALAS1 regulation by heme may be relatively less effective than the other known heme-mediated mechanisms of ALAS1 regulation (Cable et al., 2000; Kolluri et al., 2005; Sinclair et al., 1988). Recent results in mice have suggested that heme destabilization of ALAS1 mRNA may be tissue-specific with the largest effects observed in liver, Harderian gland and testes (Okano et al., 2010).
Lipophilic chemicals and drugs such as phenobarbital (PB) that also induce the synthesis of several P450s, upregulate the enzyme especially in the liver. ALAS1 upregulation by lipophilic drugs was first thought to occur solely through modulation of heme repression (May et al., 1986). Accordingly, drugs such as PB upregulated ALAS1 by increasing the synthesis of apoP450s resulting in the depletion of the free heme pool. This hypothesis predicted that if heme synthesis is inhibited or its rate of degradation stimulated, then there ought to be an increase in ALAS1, even in the absence of lipophilic drugs. Some studies have shown this to be the case in chick hepatocyte cultures and in rat brain after treatment with inhibitors such as succinylacetone (SA) and N-methyl protoporphyrin (De Matteis & Marks, 1983; De Matteis & Ray, 1982). Similarly, an increase in hepatic ALAS1 activity - again in the absence of lipophilic drugs - has been reported in rats after stimulation of liver HO-1 (see Section 1.2.4). Other studies with the chick hepatocyte culture using both enzyme activities and ALAS1 mRNA as endpoints, have failed to detect such induction of ALAS1 by treatment with inhibitors of heme synthesis alone (Hamilton et al., 1988; Ryan & Ades, 1989). Furthermore in this culture system, genetic overexpression of apoP450s failed to increase ALAS1 mRNA in the absence of lipophilic drugs (Jover et al., 1996).
Although relatively small increases of ALAS1 activity are observed when either PB or an inhibitor of heme biosynthesis are given on their own, a marked potentiation of the ALAS1 induction is observed when these two treatments are combined, reflecting the individual contribution of two separate mechanisms (De Matteis & Gibbs, 1972; Maxwell & Meyer, 1976). In addition, drugs such as PB directly activate ALAS1 transcription, independent of their modulatory effects on the free heme pool (Hamilton et al., 1988; Ryan & Ades, 1989). However, when this enhancement of ALAS1 transcription is combined with chemically induced depletion of the free heme pool (by another agent), collectively these separate effects can potentiate ALAS1 induction (Granick et al., 1975; Hamilton et al., 1988; Ryan & Ades, 1989, Jover et al., 2000). The ability of lipophilic drugs to stimulate ALAS1 transcription independently of their effects on the free heme pool was suggested in early experiments (De Matteis & Gibbs, 1975; Anderson, 1978; Whiting & Granick, 1976), and later supported by direct ALAS1 and CYP2H mRNA measurements (Hamilton et al., 1988; Ryan & Ades, 1989). Such effects of the lipophilic drugs on ALAS1 transcription were subsequently shown to be mediated by xenobiotic sensing nuclear receptors (Fraser et al., 2002; Fraser et al., 2003). Accordingly, ALAS1 transcription is regulated via binding of the constitutive androstane receptor (CAR) and pregnane X receptor (PXR) at two enhancer elements 16 and 20 kb upstream of its transcriptional start site in human, mouse and rat liver (Podvinec et al., 2004), whereas in the chicken liver it is regulated via CAR/chicken xenobiotic sensing receptor (CXR) (Fraser et al., 2002). Some of these aspects are more extensively reviewed elsewhere (Dierks, 1990; Ades, 1990).
1.1.3. Circadian regulation of heme biosynthesis
An additional consideration is the diurnal variation of heme synthesis through the circadian regulation of the ALAS1 and CAR genes (Kanno et al 2004) and consequently of P450 proteins. Indeed, ALAS1 mRNA expression is found to peak at the interphase of the light and dark cycles and this increase is followed by increased expression of several hepatic P450 (Cyp2B10, Cyp2E1 and Cyp3A11) mRNAs along with that of their redox partner NADPH-cytochrome P450 oxidoreductase (OR) in the dark cycle (Panda, et al., 2002; Zhang et al., 2009). Interestingly, this expression of ALAS1 gene coincided with that of genes for CAR and PXR, the nuclear receptors involved in the transcriptional regulation of not only ALAS1 as discussed above, but also of certain hepatic P450s such as PB-inducible CYPs 2B and CYPs 3A (Williams et al., 2005; Handschin & Meyer, 2003). Indeed, the circadian regulation of CAR by PAR-domain basic leucine zipper (PARbZip) transcription factors is reported to modulate basal and inducible xenobiotic detoxification (Gachon et al, 2006). Of note, is that heme biosynthesis and the circadian clock are apparently regulated reciprocally (Kaasik & Lee, 2004). Central to this reciprocal regulation is a heme-sensor REV-ERBα, an orphan nuclear receptor that binds heme reversibly and serves as a critical negative component of the circadian core clock (Raghuram et al., 2007; Yin et al., 2007). Heme acts as a physiological ligand of REV-ERBα, binding in a 1:1 stoichiometry, enhancing the thermal stability of this crucial component of the clock mechanism, and modulating its recruitment of a nuclear receptor corepressor NCoR complex (Raghuram et al., 2007; Yin et al., 2007). Heme thus regulates the circadian clock by leading to the repression of REV-ERBα target genes including BMAL1 that encodes a critical component of the circadian oscillator (Duez and Staels, 2008). Accordingly, heme depletion of cells results in a 3-fold increased expression of BMAL1 (Raghuram et al., 2007). Thus, in addition to serving as the essential prosthetic moiety of P450 enzymes, heme can also influence their function through circadian control of ALAS1, P450 and OR genes. The circadian control of genes of pharmacological interest (including ALAS1, CAR, P450s, OR, cholesterol-metabolizing enzymes including CYP7A1, and Phase II drug metabolizing enzymes) has been recently reviewed (Levi & Schibler, 2007; Paschos et al., 2009).
1.1.4. Inherited or chemically induced porphyries
Inherited mutations in some of the enzymes of the heme synthetic pathway, ALAD, PBGD, UROD, CPO, PPO and FC account for clinical diseases in which the symptoms are attributed to cellular heme deficiency and/or accumulation of toxic intermediates of the pathway, produced mainly in the liver (Anderson et al., 2005). In the acute porphyrias, clinically associated with inherited deficiencies of ALAD, PBGD, CPO or PPO, the major etiology is usually the consumption of drugs that can exacerbate the metabolic syndrome and precipitate the appearance of clinical symptoms. These drugs both increase the heme requirement for new P450 synthesis and/or directly stimulate the synthesis of ALAS1. Although well tolerated by normal individuals, when given to porphyric individuals whose liver heme synthesis and the feedback regulation of ALAS1 are impaired, these drugs will markedly increase ALAS1 activity and lead to the accumulation of toxic heme pathway intermediates. As discussed above, this synergistic interaction between drugs and heme biosynthetic defects has been reproduced in animal models wherein PB and other drugs powerfully induce ALAS1 in the presence of a chemically induced block in heme biosynthesis.
Three different classes of xenobiotics have been reported to induce hepatic porphyria in experimental animals (Meyer & Marver, 1971; Onisawa & Labbe, 1963; Sinclair & Granick, 1974; Granick & Urata, 1963; and references therein). With all these three chemical classes, hepatic P450 is involved in the mechanism of porphyria induction: 1) 2-Allyl-2-isopropylacetamide (AIA) and other olefinic suicide subtrates alkylate the heme prosthetic group of certain liver P450 enzymes at one of its pyrrole nitrogens (Ortiz de Montellano et al., 1978). As discussed in detail later, this modified heme product leaves the P450 active site and is replaced by fresh heme that undergoes fresh N-alkylation. As a result, the cellular heme concentration declines, leading to decreased ALAS1 feedback regulation and consequently increased synthesis of the enzyme. 2) The second group of drugs [3,5-dicarbethoxy-2,4,6-trimethyl-1,4-dihydropyridine (DDC) and its 4-ethyl analog, 3,5-dicarbethoxy-2,6-trimethyl-4-ethyl-1,4-dihydropyridine (4-ethylDDC)] also similarly N-alkylate the heme of certain P450 enzymes. In this case, however, the N-alkylated heme products [N-methyl- and N-ethylprotoporphyrin (NMPP, NEPP)] are also very powerful FC inhibitors (Tephly et al., 1979; De Matteis et al., 1980; Ortiz de Montellano et al., 1980; 1981), resulting in ALAS1 stimulation that is more pronounced and accompanied by accumulation of PROTO, the substrate of the inhibited FC enzyme. 3) The third group of chemicals that include tetrachlorodibenzo-p-dioxin (TCDD) and various polyhalogenated biphenyls, cause uroporphyrin accumulation by an as yet incompletely understood mechanism. An iron-dependent inhibition of UROD is involved (Smith & Francis, 1993; Smith et al., 2001), along with a rapid oxidation of uroporphyrinogen by liver CYP1A2 (which is inducible by these polyhalogenated chemicals) to uroporphyrin which cannot be metabolized and accumulates (Urquhart et al., 1988; Jacobs et al., 1989, Sinclair et al., 1998).
1.2. Heme degradation and hepatic P450 heme content
1.2.1: Why is heme degradation necessary?
The physiological characteristics of the heme molecule, its ability to bind and activate molecular O2, its participation in redox and electron transfer reactions (where reactive radical species may be produced) and its interaction with hydrogen peroxide and organic peroxides, all make heme a highly versatile molecule in various biological processes. However, these intrinsic properties also make heme a center of chemical reactivity that may ultimately be responsible for its own demise, or for toxic alteration of other biologically important molecules. Thus in short, heme is a potentially toxic molecule endowed with a suicidal tendency. Nature has therefore deployed strategies to protect cells from any excessive heme toxicity that include: (i) Containment of heme in appropriate proteins to reduce or abolish its reactivity. Complexation of excess heme to specific proteins provides a rapid and efficient protective mechanism. These proteins include either apo-hemoproteins (i.e. apo-P450s) or heme-binding proteins that provide amino acid ligands for both the available axial positions of its Fe atom, thereby safely shielding it while diminishing its reactivity (Tolosano and Altruda, 2002; Langlois and Delanghe, 1996; Guéye et al., 2006; Morgan et al., 1993; Satoh et al., 1994); and (ii) the evolution of the heme oxygenase system, which not only degrades excess heme, but also is up-regulated when heme levels exceed beyond its normal requirement.
1.2.2: The heme oxygenase system
Nature has exploited the suicidal tendency of heme to devise an enzyme (heme oxygenase) that employs heme both as the catalyst and as the target of its oxygenation reaction directed specifically to one of its methene bridges. The cyclic tetrapyrrolic structure of heme is thus cleaved open and a linear tetrapyrrole, biliverdin IXa, is generated which is subsequently reduced to bilirubin IXa. The latter is devoid of the pro-oxidant reactivity of heme, acquiring instead marked antioxidant and free radical scavenging properties that afford protection from reactive molecules, including those produced by heme itself.
Since heme oxygenase was first described (Tenhunen et al. 1968, 1969), two genes each encoding a corresponding isoform of the enzyme have been identified: Heme oxygenase 1 (HO-1) is inducible, while heme oxygenase 2 (HO-2) is constitutive (Maines et al., 1986; Shibahara et al, 1985; Rotenberg and Maines, 1990). HO-1 is found to play a protective role in a plethora of human diseases, recorded in an ever-explosive recent literature (Abraham et al., 2009; Gozzelino et al., 2010; Mancuso & Barone, 2009; Morse et al., 2009; Peterson et al., 2009). A truncated fully active and water-soluble recombinant form of HO-1 has been crystallized and this structure has greatly facilitated elucidation of its catalytic mechanism (Wilks and Ortiz de Montellano, 1993; Wilks et al., 1995; Schuller et al., 1999). In common with P450s, HOs utilize NADPH-cytochrome P450 reductase as the source of reducing electrons, but they also exhibit quite significant functional differences. First, heme is both their prosthetic moiety and their substrate; second, 3 moles of oxygen and 7 electrons per cycle are utilized resulting in biliverdin IXa, CO and Fe. Biliverdin IXa is then reduced to bilirubin by a soluble enzyme, biliverdin reductase, which may also accelerate HO-1 catalytic turnover by direct binding and facilitating its catalysis through accelerated product release (Wang and Ortiz de Montellano, 2003). Bilirubin is glucuronidated and then excreted in the bile, CO is released and may act as a NO-like messenger, while Fe undergoes reutilization.
Bilirubin has been shown to possess marked antioxidant and free-radical scavenging properties (Stocker et al., 1987; Barañano et al., 2002), so the HO-1 pathway affords protection against oxidative stress injury not only because it destroys heme, a powerful pro-oxidant, but also because it produces an effective antioxidant (bilirubin). Apart from a few special cases (Regan et al., 2004; Duvigneau et al., 2008), HO-1 represents a first line of defence against various cell-damaging stresses, as suggested by its upregulation under many types of toxic conditions, as discussed below.
1.2.3: Heme regulation of HO-1
Heme accumulating in excess of cellular needs triggers the induction of HO-1 for gradual disposal of this excess. The modulation of HO-1 gene transcription is finely regulated through antagonistic interactions and competition between Bach-1 and Nrf2 transcription factors for small Maf family proteins (sMafs) at the Nrf2/NF-E2-binding Maf-recognition element (MARE) of HO-1 gene promoter. These interactions are exquisitely controlled by heme (Ogawa et al., 2001; Sun et al., 2002). Accordingly, heme binds Bach-1 through C-terminal Cys-Pro heme-binding/heme regulatory motifs (HRMs), thus preventing its interactions with sMafs. This not only releases HO-1 MARE from its Bach-1 repression, but also simultaneously promotes its positive interactions with Nrf2-sMafs complexes that activate HO-1 gene transcription/expression. The mechanism of this response is quite complex, as it apparently involves not only recognition of heme itself, but also cellular sulfhydryl surveillance and/or consumption by various potentially toxic agents [xenobiotics, heavy metals and various oxidants, including reactive O2 species (Maines, 1992; Bauer and Bauer, 2004)].
1.2.4: HO-1 regulation of P450 content and function
HO-1 up-regulation not only lowers excessive heme, thus acting as a regulator of heme homeostasis, but also can often lead to heme depletion with associated decrease in hemoprotein content. The ensuing loss of functional hemoproteins is mostly due to the loss of their prosthetic group, but also in some cases to decreased levels of the protein moiety. Thus, HO-1 stimulation in the mitochondrial inner membrane (Converso et al., 2006) decreased mitochondrial heme content resulting in a significant decline in immunodetectable cytochrome oxidase subunits that was rectified by co-administration of a HO inhibitor. Similarly, CoCl2 and many other HO-1 inducers decrease liver P450 content and activity (Maines, 1992; Maines and Kappas, 1975), thereby suggesting that a shortage of cellular heme will either directly or indirectly lower the content and activity of cellular hemoproteins.
Indeed, a reequilibration between exchangeable pools of heme (from which “free” heme is drawn for degradation) and P450 heme is likely to occur under conditions of accelerated heme turnover, whether this is a consequence of species differences, nutritional factors, or HO-1 induction. Accordingly, a greater loss of radioactivity from 14C-ALA-prelabeled liver heme within the subsequent 24 h was found in fed mice compared to fed rats and in starved rats compared to fed rats (De Matteis, 1982). Such losses of radioactivity from 14C-ALA-prelabeled liver homogenates could be further enhanced by treatment with Fe-dextran or CoCl2, two powerful HO-1 inducers, along with a significant loss of P450 content (De Matteis, 1982). In these experiments, the losses of 14C-ALA-prelabeled liver radioactivity could all be correlated with the increased activity of liver ALAS1 at the end of the 24 h (Fig. 3). In separate studies carried out under similar conditions over a 21 h-period, the loss of radioactivity from 14C-ALA-prelabeled liver homogenates as well as crystalline heme isolated therefrom was monitored (De Matteis & Unseld, 1976). Comparable losses of radioactivity were found in homogenates (74%) and heme (77%) of fasted rats given an HO-1 inducer and in homogenates (44%) and heme (47%) of corresponding untreated controls. Together, this suggested that, under these conditions of accelerated liver heme turnover, the size of the “regulatory” heme pool (a pool with which P450 heme readily exchanges and equilibrates) was also significantly decreased. However, this mechanism may not necessarily operate in all cell types, as recently it was shown in cultured macrophages, that HO-1 increase did not result in increased degradation of intracellular heme (Sheftel et al., 2007).
Fig. 3. Correlation between loss of radioactivity from prelabeled liver heme and compensatory liver 5-aminolevulinate synthase (ALAS1) activity. Effect of species differences, nutritional state and heme oxygenase induction.
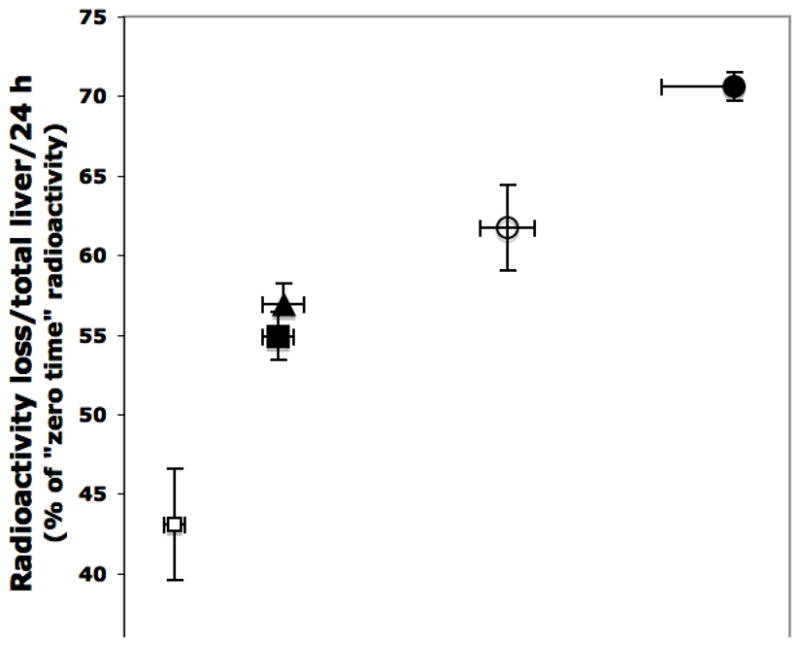
Fed mice or rats were given 1 μCi 5-amino [4-14C]levulinate per 100 g body weight and were either killed 3 h later (“zero time”) or kept fed or starved for further 24 h and then killed. Some rats were treated with dextran or with Fe-dextran (an inducer of heme oxygenase). The groups studied are indicated thus: fed rats (open squares); starved rats (solid squares); fed mice (open circles); starved rats treated with dextran (solid triangles); starved rats treated with Fe-dextran (closed circles). The liver ALAS1 values obtained at the end of the 24 h period are given as activity per total liver of 100 g body weight. The radioactivity lost from the total liver homogenate during the 24 h before killing was also calculated on a whole liver basis and is expressed as a percentage of the “zero time” radioactivity (De Matteis, 1982). The following statistical differences between groups applied to both radioactivity loss and ALAS activity: P < 0.02, when comparing values from fed and starved rats; P < 0.01, when comparing fed rats and fed mice; P < 0.001, when comparing treatment of rats with Fe-dextran and with dextran alone.
Additionally, the following findings also suggested that HO-1 induction contributes significantly to the loss of P450 heme observed after HO-1 inducers: a) The accelerated loss of radioactivity did not preceed, but instead clearly followed the induction of HO-1 (De Matteis & Unseld, 1976), at a time when the amount of bilirubin in the bile and its specific radioactivity were also increased (De Matteis & Gibbs, 1976). b) SKF525A, metyrapone and other P450 ligands largely prevented the loss of hepatic P450 caused by CoCl2 and other HO-1-inducers in vivo, without preventing the induction of HO-1 (Drummond et al., 1982; Hockin & Paine, 1983). Metyrapone could also protect from the time-dependent loss of P450 heme associated with increased HO-1 in cultured hepatocytes (Hockin and Paine, 1983), wherein, c) in the absence of such a protective P450 ligand, an increase in the heme-free apo-P450 could be documented by exogenous heme reconstitution of the microsomal P450 in vitro (Kim et al., 1995). Such a reconstitution of functional P450 through heme uptake by a heme-free apoP450 will be discussed in detail later.
These studies thus reveal that increased HO-1 activity has the potential to deplete P450 heme, most probably by reducing the size (and increasing the turnover) of exchangeable heme pools with which P450 heme equilibrates. The protection from degradation of P450 heme by stable ligands, in spite of a potentially increased recruitment by HO-1, may be explained by an increased affinity of the apoprotein for such ligated heme complexes (versus the affinity for heme itself) and/or more stable conformation of the ligated hemoprotein. It is as yet unclear whether HO-1 affects P450 content only after it is markedly induced/stimulated by exogenous chemicals, or whether it can play a similar role under more “physiological” conditions. It is intriguing in this context that Bakken et al. (1972) have documented that starvation can also stimulate HO-1 to some extent. Consistent with this, a correlation between loss of 14C-ALA-prelabeled liver radioactivity and hepatic ALAS1 activity also exists when fed and starved rats were compared (Fig. 3). Thus, it is plausible that under “physiological” conditions, that is, in the absence of exogenous HO-1 inducers, a similar mechanism operates to enhance liver heme turnover.
An apparent exception to this P450 response to HO-1 induction is murine CYP2A5, which rather than being decreased is markedly induced by oxidative stress, inflammation and administration of metals, situations that are often associated with HO-1 induction (Camus-Randon et al., 1996). It is plausible that the transcription of murine CYP2A5 gene is stimulated by oxidative stress in a Nrf2-dependent manner, as in the case of HO-1 gene (Nichols and Kirby, 2008). Possibly a marked increase in the corresponding apoprotein, with an unusually high affinity for its prosthetic heme, may explain the increase in the enzymatically active CYP2A5 holoenzyme in spite of cellular heme depletion. Alternatively, it is possible that CYP2A5 and HO-1 are localized differently in the liver cell/lobule. These aspects will require further investigation.
2. Heme-protein interactions in P450 assembly and reconstitution
2.1.1. Co-translational insertion of P450 into the ER-membranes
Before considering the heme uptake by apo-P450 and the reversible exchange of P450 heme with the other cellular heme pools, the mechanism of insertion of P450 into the ER-membranes and the potental role of heme in this process will be discussed briefly.
Several studies with rat or rabbit liver preparations have documented a co-translational incorporation of P450 into ER-membranes wherein the synthesis of the polypeptide and its insertion into the membranes occur simultaneously. Thus, Negishi et al. (1976) have found P450 protein to be synthesized on membrane-bound polysomes. Bar-Nun et al. (1980) have confirmed this by showing membrane-bound ribosomes to be ≈ 10-fold more effective than free ribosomes at synthesizing P450 in vitro: The polypeptide appears to be directly inserted into the ER-membranes, because it was not released on microsomal treatment with low detergent concentrations that released albumin and other adventitious proteins. In agreement with this, the hydrophobic amino-terminal domain of the P450 protein has been shown to function as an insertion signal for both the PB- (Alterman et al., 1980; Monier et al., 1998) and the 3-methylcholanthrene (3-MC)-inducible (Sakaguchi et al., 1987) liver P450s. These studies of liver P450 therefore suggest that synthesis of the apoprotein and its incorporation into the membranes are tightly coupled processes. On the other hand, the insertion of heme appears to be less tightly coupled to the synthesis of the apoprotein, in that the extent of heme saturation of P450 apoprotein varies depending on how much heme is available. When the heme supply is sufficiently high, heme binding can occur immediately after the synthesis and proper folding of the polypeptide [as described for catalase (Lazarow and deDuve, 1973)] in an amount sufficient to saturate the apoprotein with no significant heme-free apoprotein detected. Complete heme saturation of rat liver P450 has also been reported at different developmental stages, ranging from 2 days before birth and 4 days thereafter (Negishi and Kreibich, 1978), during which ER-membrane proliferation occurs in rat liver cells. This is also the case for total liver P450 of the adult rat under basal conditions, that is in the absence of inducers. However, when the heme supply is deficient or inhibited relative to the rate of apoprotein synthesis, heme saturation of the apoprotein will only be partial, so that a significant fraction of the microsomal apoprotein remains heme-free and capable of accepting exogenous heme (Correia and Meyer, 1975; Sadano and Omura, 1983; 1985) to reconstitute the holoenzyme.
An issue that has recently attracted interest is whether heme is necessary for the incorporation of the apo-P450 into the ER-membranes. The detection of heme-free apo-P450 in rat liver microsomes after PB- and 3-MC-induction (Sadano and Omura, 1985) and of even greater levels of heme-free apoprotein when an inhibitor of heme biosynthesis is coadministered with PB (Correia and Meyer, 1975), suggests that, at least in the liver, the P450 polypeptide can also be incorporated into the ER-membrane in a heme-deficient form. In an apparent contrast to the above findings, Meyer et al. (2002) have recently reported that CYP1A1 protein accumulates in the cytosol of brain and other extrahepatic tissues in a heme-free form. After addition of heme to cultured COS-1 (monkey kidney derived) cells, there is an apparent migration of the enzyme from the cytosol to the ER. Meyer et al. (2002) suggest that in extrahepatic tissues decreased heme availability reduces CYP1A1 function not only by allowing the apoprotein to accumulate in an inactive heme-deficient state, but also by limiting the incorporation of the enzyme into the ER where it can function. The intriguing hypothesis (Meyer et al., 2002) that in extrahepatic tissues heme has a functional role in the incorporation of CYP1A1 in the ER-membranes will need to be examined further.
The exact significance of cytosolic CYP1A1, present in relatively large amounts in extrahepatic tissues, is not yet clear. It is unknown whether it represents P450 synthesized on free polysomes, waiting [like cytochrome b5 (b5) and b5-reductase (Borgese and Meldolesi, 1980; Okada et al., 1982; Tanaka et al., 2003)] to be incorporated posttranslationally into the ER-membranes. Alternatively, in extrahepatic tissues, cytosolic CYP1A1 may have some unknown function or monitored in transit on its way to another organelle and/or disposal. These aspects will also require further investigation.
2.2.1. The heme-free P450 apoprotein can accept fresh heme to reconstitute the holoenzyme
Reversible heme-apoprotein association modulates P450 hemoprotein content and activity in a process governed by the concentration of the two reactants and by the affinity of a specific apoP450 for the heme prosthetic group. The original observations that heme could be taken up by a heme-free apo-P450 to reconstitute a functional holo-P450 enzyme were obtained by Correia and Meyer (1975). They showed that when rats were treated in vivo with PB, an inducer of CYP2B1/2B2, together with CoCl2, an inhibitor of heme synthesis and an inducer of heme degradation, an excess of apo-P450 relative to the supply of the prosthetic heme was generated. In vitro incubation of heme with the liver homogenates and/or liver microsomes (present in the 640g pellet subfraction or reconstituted with the 104,000g supernatant subfraction) derived from these rats increased spectrally determined P450 and corresponding monooxygenase activities indicating structural and functional reconstitution of the holo-P450 enzymes.
Further evidence for heme-reconstitution of liver P450s has been obtained from the study of two classes of suicide substrates that N-alkylate their heme prosthetic group: a) Unsaturated drugs, such as AIA or norethindrone (NE) and b) 4-alkyl-dihydropyridines such as 4-ethyl-DDC. As discussed below, these drugs cause accumulation of apo-P450s in a heme-free form that can be reconstituted by exchangeable cellular heme or by exogenous heme.
Destruction of heme caused in vivo by compounds of both classes concerned not only the heme of the microsomal fraction (predominantly P450 heme), but also cytosolic heme (De Matteis, 1970; Abbritti and De Matteis, 1973). Heme added exogenously to microsomal incubations along with AIA (Unseld and De Matteis, 1978) or NE (Correia et al., 1981), or given in vivo to AIA- or NE-treated rats (Correia et al., 1979) was also destroyed. This suggested that the P450 apoprotein could act as the catalytic site for drug-induced destruction of exchangeable, non-committed pools of heme (Unseld and De Matteis, 1978; Correia et al., 1979; 1981), as follows (Fig. 4): 1) The heme of the susceptible P450 forms is first N-alkylated by a reactive metabolite of AIA, NE or 4-ethyl-DDC; 2) the N-alkylated heme leaves the apoprotein site; 3) fresh heme is taken up to reconstitute the holocytochrome. The newly incorporated heme is itself N-alkylated, thereby initiating a fresh N-alkylation/reconstitution cycle.
Fig. 4. The apoprotein of P450 as a catalytic center for conversion of exchangeable pools of heme to N-alkylated porphyrins.

4-Ethyl-DDC inactivates some P450 enzymes, releasing its 4-ethyl group in a reactive form that alkylates the heme prosthetic group at one of its pyrrole nitrogens. The alkylated product leaves the apoprotein site and reconstitution with fresh heme follows, leading to renewed heme alkylation. The main steps involved are discussed in the text and are based on data obtained in suspensions of isolated hepatocytes from rats induced with phenobarbital or β-naphthoflavone (De Matteis et al., 1986). A similar heme alkylation/reconstitution cycle has been described with unsaturated drugs such as 2-allyl-2-isopropylacetamide (Correia et al., 1979, 1991; De Matteis et al., 1986; Bornheim et al., 1986; Zgoda et al., 2002). Note that not all P450s inactivated by these drugs can be functionally restored: for example, reconstitution of 4-ethyl-DDC-inactivated CYP3A could not be demonstrated, possibly because the heme of this enzyme is broken down to fragments that bind the CYP3A protein at the active site (Correia et al., 1987).
More direct evidence for such a drug-induced (and apoprotein-directed) N-alkylation of exchangeable heme pools has been obtained from studies with hepatic microsomes or in primary rat hepatocyte cultures by employing heme analogs, such as deutero- and meso-heme. These analogs are sufficiently similar to the natural protoheme to enable reconstitution of enzymatically active holoP450 (Bornheim et al., 1984; 1986). Yet they are sufficiently different to allow a HPLC separation of the corresponding N-alkylated products (De Matteis et al., 1986). Thus the contribution of the endogenous and exogenous heme pools to drug-induced N-alkylation could be studied in the same experiment. N-alkylated derivatives of both types of heme could be demonstrated: That originating from the native endogenous protoheme predominated in the early stages of the experiment, that from the deutero- or mesoheme analog in the later stages. Both products exhibited chiral properties and a regio-isomeric composition that was influenced by the particular P450 isoform (CYP1A or CYP2B) predominating in the cell at the time of treatment (De Matteis et al., 1986). Similar P450 reconstitution in liver homogenates from AIA-treated rats by in vitro incubation with heme, has also been reported by Sardana et al. (1976) and by Bonkovsky et al. (1984). Consistent with such a heme-dependent reconstitution of liver P450 hemoproteins, Bonkovsky et al. (1991) have shown that intravenous heme injection increased antipyrine metabolism both in normal women and in women suffering from acute intermittent porphyria, a genetic condition characterized by a partial block in liver heme biosynthesis. These findings underscore the physiological relevance of this process.
Heme-mediated P450 reconstitution also reportedly occurs in extrahepatic tissues. Thus, Omiecinski et al. (1978, 1980) have described the activation of extrahepatic P450 monooxygenases by the addition of heme to brain, kidney, and testis tissue homogenates. An up to 70-fold stimulation of the metabolism of benzo[a]pyrene and similar substrates was observed after addition of heme to these homogenates, but similar heme addition to liver homogenates did not enhance the corresponding enzyme activities (Omiecinski et al., 1978, 1980). A similar heme-dependent stimulation was also found with placental preparations from human and other species (Namkung et al., 1983). Significant amounts of heme-reconstitutable CYP1A1 are also found in brain and kidney cytosol as detected both by immunoblotting analyses and EROD activity upon functional reconstitution with purified NADPH-cytochrome P450 reductase (Meyer et al., 2002).
Evidence for heme reconstitution of hemoproteins other than P450s has also been obtained. Such a dynamic reconstitution by heme is a property shared by most hemoproteins of the b-type i.e. those containing non-covalently bound protoheme. This reconstitution process has been employed to study (a) the factors which govern hemoprotein stability; (b) the role of heme in apoprotein folding; (c) the orientation of heme and heme analogs within the apoprotein; and (d) the mechanism of such a process i.e. whether purely chemical or chaperone-assisted.
2.2.2. Factors that govern heme affinity for the apoprotein and hemoprotein stability
The heme-holding/binding ability of b5 is found to depend mostly on the strong axial ligation provided by two of its histidines residues, His39 and His63. Even so, when wild-type b5 was incubated with apo-myoglobin there was a slow loss of heme to the apo-myoglobin. Two b5 proteins, wherein the His39 is mutated (His39Cys and His39Ser), exhibited much reduced stability compared to the wild-type enzyme (i.e. lost heme more easily when incubated with apo-myoglobin), and interestingly, were defective in heme reconstitution (Wang et al., 2003). Similarly, when wild-type sperm whale myoglobin was compared with two sperm whale myoglobin mutants, the stability (i.e. ability to retain heme) of the three holo-myoglobins was found to depend almost entirely on their affinity for heme (Hargrove and Olson, 1996). A more recent study of the interactions for binding heme in b-type hemoproteins including P450s, has reemphasized the importance of the proximal ligand(s), but additional “hot spots” for binding have been identified at contacts with the pyrrole rings and the propionate groups (Schneider et al., 2007). These data therefore suggest that the affinity of an apoprotein for heme determines both the stability of the corresponding holo-hemoprotein and probably also the efficiency of reconstitution by heme of a heme-free apoprotein.
2.2.3. The role of heme in apoprotein folding
Studies with the heme-binding domain of myoglobin suggested that although the native apo-myoglobin probably provides a scaffolding for heme recognition, heme serves as a template for final protein folding (Grandori et al., 2000). The same may be true of P450s, since the existence of a heme-denuded apoP450 has been documented by in vitro reconstitution experiments (discussed above), and this again argues for a properly folded apoprotein preassembled for heme recognition and binding.
2.2.4. Orientation of heme and heme analogs upon apoprotein uptake
Several NMR studies have shown that in the reconstitution of myoglobin, hemoglobin and a b-type hemoprotein obtained by heme removal from cytochrome c, heme and heme analogs could be incorporated in one of two possible orientations. These differed from each other by a 180° rotation with respect to the heme α, γ-meso-axis. Of the two possible orientations “ordered” as in native hemoglobin, or “disordered”, the one observed depended on the 2,4-substituents of the heme molecule, the axial ligands provided by the apoprotein and/or other factors (La Mar et al., 1978; Ishimori & Morishima, 1988; Tomlinson and Ferguson, 2000). Bornheim et al. (1986) have examined a range of heme isomers and analogs and demonstrated the importance of the 2,4-substituents of heme for a functional P450 reconstitution. However, it was unclear in which of the two possible orientations exogenous heme was taken up by the P450 apoprotein. De Matteis et al., 1986 reported that incubation of CYP2B in isolated hepatocytes with AIA and mesoheme resulted in chiral N-alkylated heme products: That from heme representing the endogenous heme pool, and that from mesoheme representing fresh heme uptake through apoprotein reconstitution. These products both showed a similar CD spectrum, with a band of ellipticity at the Soret maximum. This suggested that mesoheme was most likely incorporated in one orientation, possibly the same ordered orientation found for the prosthetic heme in the native protein (Ortiz de Montellano et al., 1983).
2.2.5. Mechanism of heme insertion in the P450 reconstitution process
The mechanism by which an apo-hemoprotein takes up new heme to reconstitute the holo-hemoprotein is not yet completely understood. Reconstitution at high efficiency has been observed in vitro by adding heme to purified apoprotein, prepared by removing heme from holoprotein with acidified acetone under non-denaturing conditions. A functional P450 enzyme was obtained with this approach for both P450cam (Yu and Gonsalus, 1974) and P450 BM3 (Modi et al., 1995), two soluble bacterial enzymes. Since no additional components were present in these experiments (except for very high cysteine concentrations), the uptake must have involved direct molecular recognition of the two reactants and was driven, presumably, by physicochemical forces alone. However, the process may be different in more complex biological systems where additional ligands for heme potentially exist and specific heme uptake by the apoprotein may need to be facilitated or directed in some way, thus ensuring an ordered heme orientation with respect to the essential active site cysteinyl residue.
Several studies have addressed the mechanism by which heme reconstitution of P450 occurs in biological systems and suggested a facilitatory role of cytosolic factors. A role for both reducing factors and cytosolic proteins has been reported. Thus, Sadano and Omura (1985) found that the in vitro transfer of labeled heme from cytosol to the microsomal P450 was stimulated by the inclusion of a NADPH-generating system in the incubation. Similarly Zgoda et al. (2002) showed that the direct addition of heme to liver microsomal suspensions was very inefficient at reconstituting heme-depleted CYP2B, particularly when the liver microsomes were first “washed” with 0.075% sodium cholate: The effectiveness of heme reconstitution could be significantly improved by the addition of a rat liver cytosolic fraction (that itself could be replaced by GSH). Conceivably, GSH could facilitate heme reconstitution by maintaining the P450 active site cysteine-thiolate reduced and competent in heme- ligation (Zgoda et al., 2002).
Dean et al. (1986) also described a potentiation of the heme-mediated increase in microsomal monooxygenase activity by lung, kidney and perfused rat liver cytosolic fractions. The cytosolic activating factor(s) postulated to facilitate such heme-apocytochrome interaction, was a non-dialyzable, and trypsin- and heat-inactivated protein or complex of proteins, eluting in the void volume from Sephadex G-150 columns. Bender et al. (1998) subsequently reported that soluble proteins such as Hsp90, heterocomplexed with other cochaperones, could facilitate the heme binding by nNOS under conditions of limited heme availability. This possibly occurred via Hsp90-mediated sustained opening of the heme binding cleft, leading to functional activation of the enzyme. On the other hand, although Hsp90 was excluded as a participant in CYP2B1 heme reconstitution evidence was obtained for the involvement of GRP94, the ER Hsp90 homolog by Zgoda and coworkers (2002). These authors also showed that GRP94 levels promptly rose in the rat liver after AIA-treament, possibly to maintain the heme-denuded apoprotein in a heme-receptive state in vivo.
In conclusion, these observations suggest that the heme-free apoprotein can be assembled, folded properly and maintained in a state capable of reconstitution with heme. Secondary heme depletion may occur through N-alkylation of P450 prosthetic heme, impairment of liver heme biosynthesis caused by genetic defects or chemical inhibitors and also – possibly, see above - HO-1 induction. The association of heme with the apo-hemoproteins of the b type, such as P450s, should be viewed as a reversible interaction that is governed by the concentration of the two reactants and by the affinity of each specific apoprotein for the heme prosthetic group. However an important additional role in heme uptake appears to be played by the cellular chaperones as well as cytosolic factors such as GSH.
Additional mechanisms for modulation of hemoproteins, that alter the expression or stability of the protein moieties are discussed in Section 3. Such mechanisms also influence heme-apoprotein interactions by controlling the concentrations of one of the two reactants, the apoprotein moiety. Were the apoprotein concentration to increase, everything else being equal (that is excluding changes in the heme-affinity of the accumulating apoprotein or compensatory alterations in the total heme concentration), heme-binding will increase and the reverse will be true should the apoprotein concentration decrease.
The reconstitution/dissociation mechanism, schematically represented by the following reversible equation,
has important implications for the regulation of cellular heme metabolism and may contribute to the functional loss of the P450 system when the overall cellular heme status declines, as discussed below.
2.3. The P450 prosthetic heme is exchangeable with other cellular heme pools
Further evidence for the dynamic state of P450 prosthetic heme is provided by the findings that the P450 prosthetic heme moiety exchanges with other cellular heme pools and is apparently in equilibrium with them. This may occur though a transitory conversion of the holoenzyme into the heme-free apoprotein, with release of heme, followed by a prompt re-uptake of fresh heme as follows:
(a) When a pulse of radioactive ALA was chased by two larger doses of unlabeled ALA given 1 and 4 h after the radioactive precursor, no significant decrease in the radioactivity associated with the heme isolated from the total liver homogenate was found (compared to similarly treated rats given no ALA chase); however the distribution of the heme radioactivity between the microsomal fraction and the cytosol was altered, with an apparent migration of heme counts from the former to the latter (Abbritti and De Matteis, 1973). This suggests that the newly synthesized heme from the ALA chase readily equilibrated with the prelabeled microsomal heme (largely P450 heme), effectively diluting out the radioactive heme from the microsomal pool. (b) Similarly, a transfer of radioactivity to P450 could be demonstrated from labeled hemoglobin heme in vivo (Wyman et al., 1986) or when a P450 enzyme containing prelabeled heme was mixed with another unlabeled P450 enzyme in vitro and the radioactivity was then determined in both after resolving them from each other (Sadano and Omura, 1983; 1985). (c) As discussed above, the incorporation of exogenously administered labeled heme into P450 in the presence of a suicide inactivator, consequent generation of an inactivator-modified labeled heme adduct, followed by repeated cycles of P450 heme reconstitution and destruction, also argues for freely exchangeable heme pools (Unseld and De Matteis, 1978; Correia et al., 1979; 1981); as does the secondary induction of ALAS1 seen with AIA and similar P450 suicide inactivators, a response compatible with “regulatory” cellular heme depletion (see sections 1.1.2 and 2.2.1 for further discussion of these aspects). (d) Finally, as discussed earlier (section 1.2.4) an equilibrium between exchangeable pools of heme and P450 heme is likely under conditions of accelerated heme turnover, whether this is due to species differences, nutritional factors, or HO-1 induction.
3. Heme regulation of P450 protein synthesis and disposal
3.1. Heme regulation of cellular proteins: Available paradigms for the P450s
In addition to serving as the prosthetic moiety of cellular hemoproteins and finely tuning the size of the hepatic “free” heme pool through regulation of the enzymes involved in its own synthesis and degradation, heme plays important regulatory roles in transcriptional, translational and posttranslational events (Furuyama et al., 2007; Shibahara, 2003; Ortiz de Montellano, 2009; Padmanaban et al., 1989; Zhu et al., 2002a). Abundant biological precedents of heme regulation of cellular proteins exist, but the mechanisms of such regulation vary considerably (Padmanaban et al., 1989; Furuyama et al., 2007). Thus heme can regulate the content and function of certain cellular proteins by controlling their synthesis at transcriptional, translational or posttranscriptional/posttranslational steps (maturation and/or degradation of the corresponding mRNAs and proteins) (Padmanaban et al., 1989; Furuyama et al., 2007; and references therein). While the list of proteins so regulated is vast and continuously expanding, only a few select examples will be discussed as plausible paradigms available for the heme regulation of P450 hemoproteins.
As discussed above (section 1.1.2), heme negatively regulates its own biosynthesis, by affecting the first and rate-limiting enzyme ALAS1 in the heme biosynthetic pathway, transcriptionally, posttranscriptionally and posttranslationally (Kikuchi & Hayaishi, 1981; Drew & Ades, 1986; Srivastava et al., 1989, 1990; Hamilton et al, 1991). Furthermore, as discussed above, heme is also known to directly induce its terminal catabolic enzyme, microsomal HO-1, primarily by increasing the rate of HO-1 gene transcription, and only slightly by stabilizing its mRNA turnover (section 1.2.3). As discussed above (section 1.2.3), this modulation of HO-1 gene transcription is finely regulated by heme through its control of the antagonistic interactions between Bach-1 and Nrf2 transcription factors for binding to the MARE of HO-1 gene promoter through their mutually exclusive competition for sMafs.
Very similar heme-Bach1-interactions with MARE-like transcriptional activator NF-E2/AP-1 (NA) site are also evident in the heme regulation of α- and β-globin genes in human erythroleukemia K562 and mouse erythroleukemia (MEL) cells (Tahara et al., 2004a, b). SA-mediated heme depletion of these cells enhances the inhibitory interactions of Bach1 with the NA site, an effect that is reversed by heme treatment. On the other hand, heme-binding of Bach1-HRMs in these cells also increased its nuclear export thereby synergizing the positive transcriptional regulation of both a- and b-globin genes via the NA-site. Additional mechanisms for heme-mediated induction of hemoglobin via extracellular signal-regulated kinase 1/2 (Erk1/2) (Woessman & Mivechi, 2001) and of fetal globin gene expression via soluble guanylate cyclase-protein kinase G (sGC-PKG) have also been reported in K562 cells (Ikuta et al., 2001). However, the precise significance of such positive heme-mediated transcriptional activation of globin genes is unclear, in view of the much more dramatic translational regulation of globin synthesis in reticulocytes via the heme-regulated inhibitor (HRI) eIF2α kinase (see below). Perhaps, as previously proposed (Furuyama et al., 2007), such heme-regulated transcriptional activation may provide some fine-tuning of globin expression at a late stage of differentiation while the erythroid cells are still nucleated. Similar heme-induced sGC-PKG-mediated transcriptional activation of neuroglobin, a hemoprotein that transports O2 to neuronal mitochondria (Burmester et al., 2000), has been reported (Zhu et al., 2002b).
Positive coordinate heme-protein regulation of genes (CYC1, COX4, COX6 and COX5A) respectively encoding the mitochondrial hemoproteins isocytochrome 1c, cytochrome c oxidase subunits IV, VI, and Va (a V isolog) occurs in Saccharomyces cerevisiae (Guarente & Mason, 1983; Pfeifer et al., 1987; Pinkham et al., 1987; Myers et al., 1987; Trawick et al., 1989). Such heme regulation of CYC1 and COXVI genes is exerted at the initiation of transcription, mediated by transcriptional factors (HAP1/2) and well-defined upstream activation sites within their 5′-flanking regions (Guarente & Mason, 1983; Pfeifer et al., 1987; Pinkham et al., 1987; Myers et al., 1987; Trawick et al., 1989). Heme activates Hap1 through well-defined HRMs (Pfeifer et al., 1989; Zhang & Guarente, 1995). Heme also exerts positive coordinate regulation of non-mitochondrial hemoproteins such as catalase by a similar mechanism that includes a transcriptional activator (HAP1) and an upstream activation site within the 5′-distal region of its gene (Hörtner et al. 1982; Winkler et al., 1988). The expression of non-heme proteins such as metallothionein (Alam & Smith, 1992) and Mn2+- superoxide dismutase (SOD2) (Pinkham et al., 1997) is apparently also regulated by heme at the initiation of transcription at a 5′ distal site, albeit by mechanisms different from that of HO-1 induction, in that different signals and/or transcriptional activators are involved (Alam & Smith, 1992; Pinkham et al., 1997; Ren & Smith, 1995).
Heme is required for both basal and dexamethasone (DEX)-inducible expression and function of TDO (Ren and Correia, 2000), a hepatic cytosolic enzyme induced via glucocorticoid (GC)-mediated transcriptional activation as well as by substrate-induced stabilization (Schutz et al., 1972; Schimke et al., 1965). Preliminary Northern analyses of hepatic TDO mRNA from rats acutely depleted of hepatic heme coupled with in vitro nuclear run-on assays had suggested that both basal and DEX-mediated transcriptional activation are heme-dependent (Ren and Correia, 2000), implying the existence of “heme responsive elements”. However, specific deletion analyses of both 1.95 kb and full length 15 kb-TDO promoter luciferase reporter plasmid revealed that other than the two GC-response elements (GREs) none of the potential TF-binding sites in the TDO promoter region were critical to heme-dependent modulation of DEX-mediated TDO gene expression (Liao & Correia, unpublished findings). Furthermore, qRT-PCR analyses also revealed no effect of acute heme depletion on hepatic TDO mRNA expression (Liao et al., 2007). Collectively, these findings indicated that heme had no significant role in TDO transcription. Indeed, more recent findings (Liao et al., 2007) reveal that heme-regulation of hepatic TDO expression largely occurs via HRI eIF2α kinase-mediated translational control very similar to that of P450 enzymes (see below).
Other examples of heme regulation of protein synthesis at the translational level also exist. Indeed, this is the major mechanism by which heme regulates hemoglobin synthesis in reticulocytes: Globin synthesis is controlled through the phosphorylation of the α-subunit of eukaryotic initiation factor 2 (eIF2α) by HRI eIF2α kinase (Chen, 2007). Heme inhibits HRI activity and consequently modulates the extent of eIF2α phosphorylation and the translational initiation of protein (globin) synthesis (Traugh, 1989; Chen, 2007). In reticulocyte lysates, heme is apparently required for the translation of most proteins (Traugh, 1989; Chen, 2007; and references therein). Impaired protein synthesis also occurs in freshly isolated hepatocytes depleted of their heme by the porphyrinogenic P450 suicide inhibitor AIA (Fagard & Guguen-Guillouzo, 1983). This impairment is dramatically reversed by heme (Fagard & Guguen-Guillouzo, 1983), thereby implicating a hemin-sensitive inhibitor of protein synthesis such as HRI. Low level translation of mRNA encoding catalase T and catalase A is also observed in cell free protein synthesis systems from heme-deficient S. cerevisiae, that is reversed by the addition of heme (Hamilton et al., 1982).
Heme also regulates cellular protein content posttranslationally. For instance, heme is required for the posttranslational processing of the precursor form of myeloperoxidase (MPX), a hemoprotein synthesized in the ER lumen, proteolytically processed to its mature functionally active species and subsequently packaged in azurophilic granules (Pinnix et al., 1994). Heme deficiency does not impair either MPX transcription or translation qualitatively or quantitatively, but it greatly retards MPX maturation to its fully processed functional state and its translocation from the ER, resulting in its ER-accumulation in SA-treated HL-60 cells. It also accelerates the degradation of the mature MPX species (Pinnix et al., 1994). All these effects are reversed by heme resupplementation, indicating their heme-dependence. The hemoproteins catalase (Price et al., 1962) and TDO also undergo rapid turnover on losing their heme. Thus TDO is rapidly degraded after hepatic heme depletion (Yao & Correia, 1991; Ren & Correia, 2000).
Another example of posttranslational regulation by heme is its role in microRNA processing through binding and promoting the dimerization of the RNA-binding protein DiGeorge critical region 8 (DGCR8). The heme-bound DGCR8 is active in the cleavage of primary microRNA transcripts to the mature active microRNA (Faller et al., 2007).
3.2.1. Heme regulation of hepatic P450s: Transcriptional activation
Cellular syntheses of the P450 heme and apoprotein moieties are tightly coordinated under normal conditions. This is in common with that of other hemoproteins such as mitochondrial cytochromes, hemoglobin, myoglobin (Graber & Woodworth, 1986), neuroglobin (Zhu et al., 2002b), and catalase, wherein heme has been established as a transcriptional activator (reviewed by Padmanaban et al., 1989). Similar heme-dependent transcriptional activation was proposed to be involved in the PB-mediated induction of rat liver P450s CYP2B1/2B2 (henceforward termed CYP2B because of the 97% identity in the coding regions, and the highly similar PB-induction profiles of the two isoforms) (Dwarki et al., 1987; Bhat & Padmanaban, 1988; Rangarajan & Padmanaban, 1989). PB-mediated CYP2B induction unequivocally involves transcriptional activation of both the protein and heme synthetic machinery (Waxman and Azaroff, 1992; De Matteis and Marks, 1996; and references therein). However, the role of heme as a specific activator of CYP2B transcription remained controversial for many years with one research group advocating a positive role for heme in CYP2B transcriptional activation (Dwarki et al., 1987; Rangarajan and Padmanaban, 1989). In their elucidation of this heme role, heme-mediated enhancement of CYP2B transcriptional activation was shown to occur both in vivo after heme administration to animals depleted of hepatic heme by treatment with 3-aminotriazole or CoCl2, and in vitro, after heme addition to cell-free nuclear extracts from CoCl2-treated rat livers (Dwarki et al., 1987; Sultana et al., 1997). The authors concluded that heme acted as a positive modulator of CYP2B gene transcription by binding to a purified nuclear 65-kDa protein among other nuclear transcription factor(s), and enhancing their interaction with specific regulatory elements within the CYP2B gene promoter (Ravishankar & Padmanaban, 1985; Venkateswar & Padmanaban, 1991; Sultana et al., 1997). This 65-kDa protein was proposed to mediate the requirement for heme in the basal transcription of CYP2B gene.
Other investigators who examined the effect of heme depletion on CYP2B mRNA expression in SA-treated rats or cultured hepatocytes found no impairment of PB-mediated CYPB transcriptional activation, thereby concluding that heme is not involved in CYP2B transcriptional activation (Hamilton et al., 1988; Srivastava et al., 1989; Sinclair et al., 1990). Interestingly however, one of these reports documented decreases in CYP2B protein in SA-treated hepatocytes (Sinclair et al., 1990; see below). Similarly, heme was also excluded as a transcriptional activator of CYP3A11 synthesis in mice treated with the heme synthesis inhibitor Pb-acetate. Although CYP3A11 transcription was indeed impaired in these Pb-acetate treated rats, it was refractory to heme (Jover et al., 1996). By contrast, a positive role for heme in PB-mediated CYP2A5 transcriptional activation was documented in a heme-deficient mouse model generated by genetic disruption of the heme-synthetic enzyme porphobilinogen deaminase (PBGD), that reduced hepatic PBGD activity to 31% of normal (Jover et al., 2000). However, neither the expression nor function of CYP2B10, the mouse liver CYP2B ortholog, was affected by heme deficiency in these mice (Jover et al., 2000). Assessment of hepatic heme deficiency status of these mice indicated that heme synthesis monitored by the rate of 14C-ALA incorporation into hepatic heme was reduced to just 46% of normal, while the TDO heme-saturation was reduced to the non-statistically significant value of 78% of normal (Lindberg et al., 1996). These indices revealed that the extent of hepatic heme depletion might have been insufficient to conclusively determine whether heme regulates CYP2B hemoprotein synthesis.
Differences in the extent of hepatic heme depletion and/or cytotoxicity induced by the chemical agent employed in each study could potentially rationalize the apparent discrepancies in the analyses of whether or not heme regulates PB-mediated CYP2B transcriptional activation. However, the evidence for a positive role for heme in the PB-induced CYP2B transcriptional activation (Rangarajan and Padmanaban, 1989) was based on in vitro findings obtained with a plasmid (pP450e179) containing just 360 nt of the CYP2B2 gene (−179/+181). This required reexamination because subsequent reports had established that the PB-responsive element/unit/enhancer module (PBRE/PBRU/PBREM) lay far upstream in the 5′ flanking promoter region (−2317/−2155 bp) of the rat, mouse and human CYP2B genes, and was critical for PB-responsiveness (Ramsden et al., 1993; Trottier et al., 1995; Park et al., 1996; Kim & Kemper, 1997; Kemper, 1998; Honkakoski & Negishi, 1998; Honkakoski et al., 1998a, 1998b; Stoltz et al., 1998; Kawamoto et al., 1999; Stoltz & Anderson, 1999). In the case of CYP2B10 this region was shown to bind the constitutive androstane receptor (CAR), an orphan nuclear receptor (Honkakoski & Negishi, 1998; Honkakoski et al., 1998a, 1998b; Kawamoto et al., 1999). Indeed, it is now known that PB-mediated activation of cytosolic CAR results in its translocation into the nucleus whereupon it heterodimerizes with another nuclear receptor, retinoid X receptor (RXR). The CAR-RXR heterodimeric complex then interacts with PBREM in the 5′-promoter region of CYP2B genes, thereby inducing their expression through enhanced transcriptional-translational activation (Kim et al., 2001; Williams et al., 2004; Timsit and Negishi, 2007). These collective findings, together with the demonstration that CYP2B is PB-inducible in transgenic mice bearing the full 19 kb of 5′-flank (hence containing the −2317/minus;2155 PBREM domain), but not the first 800 bp of the 5′-flank of a rat CYP2B2 transgene (Ramsden et al., 1993), underscores the fact that PBREM is indeed responsible for conferring PB-responsiveness. Thus, assessment of heme regulation of PB-induced CYP2B transcriptional activation would need to include this “PBREM”. Because of these combined issues, the particular controversy surrounding the specific agent used for hepatic heme depletion, its relative heme depleting potential and the possibly insufficient extent of hepatic heme depletion both in chemically treated or even PBGD-deficient mice, the issue of heme-regulation of CYP2B synthesis was revisited in a rat model of acute hepatic heme depletion (Han et al., 2005).
In this rat model, hepatic heme is acutely depleted after treatment with the P450 suicide inactivator, 4-ethyl-DDC (Litman and Correia, 1983, 1985; Ren and Correia, 2000). 4-Ethyl-DDC irreversibly destroys the heme of hepatic P450s 2C11, 2C6 and 3A, but not CYP2B1 (Lee et al., 1988; Sugiyama et al., 1989). Such CYP2C11 and CYP2C6 destruction through heme pyrrole N-ethylation generates N-ethylprotoporphyrins (NEPP), potent inhibitors of FC, the terminal heme synthetic enzyme. This dual DDEP effect on hepatic P450 heme destruction and the inhibition of heme synthesis results in profound hepatic heme depletion in rats, much more severe than that seen in PBGD-deficient mice (Jover et al., 2000) or after treatment with any other heme inhibitor. This was assessed by a <25% heme saturation of the cytosolic hemoprotein TDO (Litman & Correia, 1985), a reporter of the “free” heme pool (Bissell & Hammaker, 1977). While understandably, this extent of severe hepatic heme depletion may be incompatible with “normal” life, nevertheless, the 4-ethylDDC-treated rats survived for >32 h post-treatment (Litman & Correia, 1983). Furthermore, similar treatment of rats or mice with 4-alkyldihydropyridines such as DDC or 4-ethylDDC is known to markedly stimulate liver ALAS-1 activity and consequent porphyrin production/accumulation, and this is further augmented by coadministration of lipophilic drugs (De Matteis & Gibbs, 1972; De Matteis et al., 1973; Litman & Correia, 1983). These indices are fully consistent with the chemical induction of acute hepatic porphyria in these rats.
Similar acute hepatic heme depletion can be documented in cultured hepatocytes treated with 4-ethyl-DDC or N-methylprotoporphyrins (NMPP), the methyl analogs of NEPP that are even more potent FC inhibitors (De Matteis et al., 1980; Ortiz de Montellano et al., 1980, 1981), and inhibit 14C-ALA incorporation into heme to 15% of controls (Han et al., 2005). It is noteworthy that these cultured hepatocytes also sustained this severe heme depletion without any overt signs of cytotoxicity for >72 h (Han et al., 2005; Han & Correia, unpublished findings). In cultured hepatocytes, as in intact animal livers, 4-alkyldihydropyridines such as 4-ethylDDC also induced hepatic ALAS-1 mRNA with stimulation of its hepatic activity and consequent porphyrin accumulation (Marks et al., 1985; Mackie & Marks, 1989; Mackie et al., 1991; Brady & Lock, 1992). Similar stimulation of hepatic ALAS-1 activity and/or porphyrin accumulation was also observed after addition of NMPP or NEPP to the hepatocyte cultures (De Matteis & Marks, 1983; Brady & Lock, 1992). These models for acute hepatic heme depletion (Litman & Correia, 1985; Han et al., 2005) were therefore employed to reexamine the controversial role of heme in PB-mediated transcriptional activation of CYP2B in 4-ethyl-DDC- or NMPP-treated rat hepatocytes in primary monolayer culture (Han et al., 2005). These findings in both 4-ethyl-DDC- and NMPP-treated hepatocytes revealed that heme is most definitely required for PB-mediated induction of CYP2B protein (Han et al., 2005). However, qRT-PCR analyses of CYP2B1/2B2 mRNA expression in PB/4-ethyl-DDC-treated rat hepatocytes indicated no impairment of PB-mediated CYP2B mRNA induction by acute heme depletion (Han et al., 2005). These results essentially confirmed the previous findings in SA-treated rats and cultured rat hepatocytes (Sinclair et al., 1990; Srivastava et al., 1989). These findings once again indicated that although hepatic PB-inducible CYP2B content was dramatically decreased in these heme depleted hepatocytes, such regulation is not exerted at the level of either CYP2B1 or 2B2 mRNA induction, but rather occurs at a posttranscriptional/translational step (Han et al., 2005).
3.2.2. Heme-mediated translational control of P450s via hepatic HRI
Indeed, along with marked (>80%) impairment of de novo CYP2B synthesis (determined by 35S-Met/Cys-incorporation into immunoprecipitable protein), acute hepatic heme depletion resulted in global suppression of de novo hepatic protein synthesis and enhanced eIF2α phosphorylation (Han et al., 2005). These effects were effectively reversed by heme, thereby suggesting that this impaired CYP2B induction resulted from blocked translational initiation stemming from the activation of a hepatic heme-sensitive eIF2α kinase. Four independent eIF2α kinases catalyze eIF2α phosphorylation in mammalian cells: dsRNA/interferon-inducible PKR [RNA-dependent protein kinase (EIF2AK2)], the ER-stress inducible PERK [PKR-like ER kinase (EIF2AK3)], amino acid deprivation- and UV-irradiation-inducible GCN2 [general control non-derepressible-2 (EIF2AK4)] and HRI [the heme-regulated inhibitor (EIF2AK1)] activated by heme-deficiency (Chen and London, 1995; Harding et al., 2003; Scheuner et al., 2001; 2006; Wek, 1994; Wek et al., 2006; Zhang et al., 2002a; 2002b). Of these, PKR could a priori be excluded as the relevant eIF2α kinase as the hepatocyte cultures were not exposed to dsRNA or interferons. Similarly, immunoblotting analyses of heme depleted hepatocytes revealed no corresponding activation via autophosphorylation of either PERK or GCN2, thereby also excluding these two eIF2α kinases in the enhanced eIF2α phosphorylation observed in heme depleted hepatocytes (Acharya et al., 2010). These findings implicated hepatic HRI in the heme-regulation of CYP2B induction.
Indeed in erythroid cells as discussed above, HRI is well recognized to critically regulate hemoglobin synthesis through coordinated synthesis of its heme and globin moieties (Chen, 2007). Although HRI eIF2α kinase was once viewed as erythroid-specific (Chen, 2000; Chen and London, 1995; Crosby et al., 1994; Pal et al., 1991), the presence of HRI mRNA in non-erythroid tissues, albeit at much lower concentrations than that in reticulocytes, and the isolation and characterization of a mouse liver HRI protein have been documented (Berlanga et al., 1998; Delaunay et al., 1977; Igarashi et al., 2004; Mellor et al., 1994). However, the apparent low abundance (per mg protein) of hepatic HRI relative to that of the erythroid HRI may have largely contributed to overlooking its hepatic relevance (Acharya et al., 2010). Given this relatively low abundance of hepatic HRI, its immunochemical detection within the enormous hepatocellular protein background required very high protein amounts and highly specific and sensitive detection systems that led to the documentation of a heme-sensitive 76 kDa protein as a bona fide HRI in cultured rat hepatocytes (Liao et al., 2007; Acharya et al., 2010). Accordingly, acute hepatic heme depletion autoactivates this eIF2α kinase via protein autophosphorylation to a 92 kDa species, resulting in enhanced eIF2α phosphorylation and global protein translational shutoff (Han et al., 2005; Liao et al., 2007; Acharya et al., 2010). Heme repletion functionally inhibits HRI and reverses these effects (Han et al., 2005; Liao et al., 2007; Acharya et al., 2010). Thus, the de novo syntheses of the PB-inducible CYP2B enzymes (Han et al., 2005; Acharya et al., 2010) and Dex-inducible TDO (Liao et al., 2007) were greatly impaired in heme-depleted rat hepatocytes in a heme-reversible manner. Direct proof of the functional involvement of hepatic HRI in the translational shutoff of PB-inducible CYP2B enzymes, was obtained in a HRI gene knockout mouse model [HRI(−/−)] (Han et al., 2001). In this model, genetic ablation of HRI was documented to abrogate the heme-deficiency induced impairment of hepatic PB-inducible CYP2B synthesis (Acharya et al., 2010), resulting in hepatic CYP2B protein accumulation even in the absence of heme. Essentially similar findings were obtained with CYP3A. Collectively, these findings indicate that heme regulates hepatic P450 synthesis via HRI-mediated translational control.
3.2.3. Hepatic HRI eIF2α kinase
Much that is known about the structure-function and heme-regulation of this enzyme has been derived from purified recombinant erythroid and/or mouse liver HRI proteins (Bauer et al., 2001; Chen et al., 1989; Chefalo et al., 1998; Chen, 2000; 2007; Rafie-Kolpin et al., 2000; 2003; Igarashi et al., 2004; 2008; Inuzuka et al., 2004; Miksanova et al., 2006). The predicted monomeric molecular mass of HRI is 72 kDa, with an apparent mass of 76–93.4 kDa after SDS-PAGE. Multi-angle light scattering analyses combined with size exclusion chromatography yielded a molecular weight for the native liver HRI protein ranging from 396 kDa to 420–447 kDa, thus revealing that in its native state the enzyme is a homohexameric protein (Miksanova et al., 2006). The higher apparent monomeric mass (≈ 92 kDa) corresponds to the hyperphosphorylated HRI species as documented by its reversal to the 76 kDa species upon calf-intestinal alkaline phosphatase treatment (Acharya et al., 2010). Interestingly, in their native state, the rat and human liver HRI proteins exist as the 76 kDa species that can be inhibited by heme. By contrast, certain mouse liver HRI proteins exist as the 92 kDa hyperphosphorylated species in their basal state that is apparently heme-insensitive (Acharya et al., 2010; Rafie-Kolpin et al., 2003).
The monomeric subunit contains 626 residues, and consists of an N-terminal domain (NTD; 1–160 amino acids), a kinase insertion domain (KID, 231–420 amino acids) that splits a relatively conserved eIF2a kinase domain into two regions, and a C-terminal domain (CTD, 540–626 amino acids) (Fig. 5A). Such a large insert in the kinase domain is a hallmark of all four eIF2α kinases (Rafie-Kolpin et al., 2000; Dar et al., 2005; Igarashi et al., 2004; 2008; Miksanova et al., 2006). The purified HRI enzyme is a hemoprotein that exhibits spectral characteristics typical of cytochrome b-type hemoproteins. Although the mouse liver enzyme contains two distinct “HRMs” one centered around Cys409-Pro410 in KID, and the other around Cys550-Pro551 in CTD, recent studies indicate that it binds just one heme moiety per subunit (i.e. 1:1 stoichiometry; Miksanova et al., 2006; Igarashi et al., 2008). In its inactive heme-inhibited state, His119/His120 in the NTD provides one axial ligand to the HRI heme-iron, while Cys409 in the KID provides the other. This dual heme-tethering of the NTD to the KID retains the subunit in an inactive basal state. The enzyme incurs multi-site phosphorylation on Thr483, Thr485 and possibly other Thr, Ser and Tyr residues. Thr485 in the activation loop is conserved in all other eIF2α kinases and its autophosphorylation is required for eIF2α kinase activity. Oligomerization of the enzyme requires the NTD, but the NTD-deleted HRI subunit still retains its eIF2α kinase activity.
Fig. 5. HRI eIF2α kinase.


A. An inactive, heme-bound HRI 76 kDa subunit is schematically shown. Heme tethers the regulatory N-terminal (NT) subunit via His119/His120 to Cys409 in the Cys-Pro HRM in the kinase insert (KI) domain. Mutation of either of these His residues results in “ligand switching” to the other (Igarashi et al., 2008). This large insert (spanning residues 231–420) in the kinase domain is a hallmark of all eIF2α kinases. The HRI protein undergoes phosphorylation at multiple sites, but the Thr residue corresponding to Thr485 in the HRI kinase K-II activation loop is conserved in all eIF2α kinases and incurs autophosphorylation. B. A plausible model for HRI activation and subsequent eIF2α phosphorylation is illustrated, based on that reported for PKR eIF2α kinase (Dar et al., 2005; Dey et al., 2005) and various reports in the HRI literature (Bauer et al. 2001; Miksanova et al., 2006; Igarashi et al., 2008). The HRI NT domain is essential for HRI-homodimerization/oligomerization. Heme depletion results in the loss of the HRI NT-heme tethering to the KI domain, resulting in homodimerization, trans-interdimer autophosphorylation, conformational changes leading to the active HRI homotrimeric/hexameric structure, eIF2α recognition and phosphorylation at Ser51.
Direct analogy to the PKR eIF2α kinase mechanism of autoactivation (Dar et al., 2005; Dey et al., 2005) suggests that on loss of HRI heme after hepatic heme depletion, the NTD may be released from its heme tether to the KID, thereby leading to the dimerization of NTDs from two different HRI subunits, trans-interdimer Thr485 autophosphorylation and subsequent oligomerization of the HRI protein into the activated hexameric species capable of recognizing and phosphorylating Ser51 of eIF2α (Fig. 5B). Such eIF2α phosphorylation stabilizes the GDP-eIF2-eIF2B complex, thereby sequestering eIF2B, the relatively less abundant and thus rate-limiting regulatory subcomplex and preventing eIF2B-catalyzed GTP-GDP exchange required for subsequent regeneration of GTP-eIF2 for fresh initiation cycles (Wek et al., 2006; Chen 2000; 2007; Kaufmam, 2004; Harding et al., 2003). Such functional HRI eIF2α kinase autoactivation following acute hepatic heme depletion results in global translational arrest of de novo synthesis of hepatic proteins, including that of P450 enzymes. However in this context, two issues are particularly noteworthy: (a) that not all hepatic proteins are translationally shut down by eIF2α kinase activation (Wek et al., 2006). Some including the ER-stress chaperones Grp78 and Grp94 are actually induced (Acharya et al., 2010), as is ALAS1 (see above); and (b) hepatic heme depletion need not be very severe for HRI activation and translational arrest. Thus, the extent of translational arrest depends on the relative level of activated HRI species that in turn determines how much eIF2αP is actually generated in the hepatocyte. Because not only is the cellular content of eIF2B normally limiting, comprising just 10–15% of the corresponding eIF2α content, but also its affinity for eIF2αP is relatively higher than for eIF2α, very little eIF2αP can efficiently sequester all of the cellular eIF2B thereby effectively shutting down translational initiation. Accordingly, Sinclair et al. (1990) reported that SA-mediated hepatic heme depletion, even though less severe than that caused by DDEP (as monitored by hepatic TDO heme saturation; Liu & Correia, unpublished observations), resulted in appreciable reduction of PB-inducible hepatic CYP2B protein. Thus, the extent of inhibited P450 translation is apparently proportional to the extent of hepatic heme depletion.
3.2.4. Hepatic HRI activation and implications for the acute hepatic porphyries
The finding that HRI exists in human hepatocytes in its 76 kDa, unphosphorylated, heme inhibitable latent form (Acharya et al., 2010), may be clinically relevant. This form is functionally activated in heme-deficient states, thus serving as a heme sensor to shut-off protein synthesis and conserve vital energy and nutrients as well as contain ER- and oxidative stress, events that enable the hepatocyte to withstand the stress until return to normalcy. This may explain why in the absence of lipophilic/porphyrinogenic agents, hepatic ALAS1 induction is not always observed under these conditions (Hamilton et al., 1988; Ryan & Ades, 1989). However, such otherwise protective heme-deficiency mediated hepatic HRI activation could be particularly treacherous in patients with genetically compromised heme synthesis that predisposes them to the acute hepatic porphyrias for two reasons. The first is the significant induction of HRI protein in hepatocytes following treatment with P450 inducers such as PB (Acharya et al., 2010). These drugs are clinically notorious for precipitating acute attacks of hepatic porphyria and are contraindicated in genetically susceptible individuals (Anderson et al., 2005). The basis of their porphyric action is that by inducing hepatic P450 proteins, the major consumers of hepatic heme, they aggravate the inherent hepatic heme deficiency (Anderson et al., 2005; De Matteis, 1978). The second is that in addition to exacerbating hepatic heme deficiency, P450 inducers also markedly increase hepatic HRI mRNA and both total and autoactivated/autophosphorylated hepatic HRI content (Acharya et al., 2010). Conceivably, this would enhance the translational arrest of key hepatic proteins such as the ER-bound drug-metabolizing P450s and the cytosolic TDO, a key rate-limiting enzyme in tryptophan catabolism that determines the CNS and ANS serotonergic tone (Han et al., 2005; Liao et al., 2007). Similar HRI activation in genetically predisposed individuals could lead to global translational arrest of physiologically relevant enzymes and proteins resulting in the severe and often fatal clinical symptomatology of the acute hepatic porphyrias.
3.3. Posttranslational heme-regulation of P450 repair and disposal
The factors that determine the “normal” turnover of hemoproteins associated with the physiological “wear and tear” encountered during their active catalytic life deserve some consideration. All hemoproteins capable of binding O2, including P450s, are at danger of spontaneous oxidative damage and inactivation, since their dioxygen-heme complexes can suffer abortive breakdown and generate O2− and H2O2, potentially capable of damaging the hemoprotein with heme-protein cross-linked adducts (Guengerich, 1978; Osawa & Korzekwa, 1991) and/or amino acid oxidation. Additional reactive species normally formed by P450 and peroxidases are the high-valent ferryl oxygen intermediates that may occasionally damage heme or its environment. These structural changes may provide the appropriate signal for apoprotein degradation and form the basis for “normal” hemoprotein turnover (Tierney et al., 1992). This concept is supported by the observation that binding of troleandomycin, a macrolide antibiotic CYP3A inhibitor that forms a stable metabolic intermediate complex with CYP3A heme, decreased the proteolytic degradation of CYP3A (Watkins et al., 1986), possibly by inhibiting the catalytic generation of oxidants that damage the protein. Similarly, conditional deletion of P450 reductase in vivo or chemical inhibition of P450 reductase in vitro also resulted in P450 3A stabilization (Zhukov & Ingelman-Sundberg, 1999; Goasduff & Cederbaum, 1999; Hendersen et al., 2003). P450 enzymes exhibit relatively constant rates of heme turnover (t1/2 ≈ 8–10 h) but differ markedly in their rates of protein turnover (t1/2 ≈ 7–37 h) (Correia, 2003; and references therein). The reasons for such marked differences are not yet completely understood. The stabilization of some P450 by an appropriate substrate could contribute to these differences, but also, as described below, the pathway of hemoprotein disposal utilized for their degradation.
The marked CYP2E1 stabilization observed in the presence of a substrate as well as the rapid CYP2E1 turnover observed in its absence have been recently rationalized on the basis of CYP2E1 and Hsp90 “client” protein-protein interactions, as treatment of cells with the Hsp90 inhibitor, radicicol, increased this CYP2E1 degradation (Morishima et al., 2005). CYP2E1 interactions with the Hsp90-system were proposed to stabilize the enzyme, and these interactions were promoted by its substrate ethanol.
Whether responsible for the normal base-line turnover of the hemoproteins, or consequent to the abrupt large scale damage caused by foreign chemicals, the mechanism of hemoprotein disposal appears to follow a number of distinct steps, which have been characterized by recent studies on P450 and NOS enzymes. Two distinct pathways have been identified, one involving ubiquitin-dependent 26S proteasomal disposal, the other relying on autophagic-lysosomal degradation (Correia & Liao, 2007; and references therein). Suicide inhibitors can inactivate P450s by forming adducts with the apoprotein or cross-linking of heme and/or its oxidative fragments to the protein moiety (Correia et al., 1987; Osawa & Pohl, 1989). The latter type of damage is irreversible and apparently plays a major role in proteolytic recognition, acting as a trigger for rapid proteolytic disposal of the damaged P450 via the ubiquitin-mediated 26S proteasomal degradation (Correia et al., 1987; Correia et al., 1992a; 1992b; Tierney et al., 1992; Korsmeyer et al., 1999; Wang et al., 1999).
By contrast, as discussed above, the loss of prosthetic heme following P450 suicide inactivation may be repaired by reconstituting the holoenzyme with freshly supplied heme, provided heme is readily available and the apoprotein is still competent for heme reconstitution. On the other hand, covalent modification of the P450 heme can significantly activate P450 proteolytic disposal if heme availability is insufficient to repair the hemoprotein. This is the case of 4-ethyl-DDC-mediated suicide inactivation of CYP2C11 via N-ethylation of one or more of its pyrrole moieties (Lee et al. 1988; Sugiyama et al., 1989; Licad-Coles et al., 1997) and consequent inhibition of heme synthesis. In the absence of sufficient heme, the N-ethylated heme moiety of CYP2C11 is possibly not replaced, thereby resulting in a “heme-stripped” CYP2C11 protein. In vivo, under these circumstances, this protein is rapidly cleared via ubiquitin-dependent proteasomal degradation (Correia et al., 1992a; Halpert et al., 1994). Heme inhibits this CYP2C11 loss not only by functionally reconstituting any available heme-stripped CYP2C11 protein rendering it less proteolytically susceptible, but also by blocking the proteasomal degradation of any heme-stripped CYP2C11 protein that has incurred ubiquitination, and thus leading to the accumulation of the ubiquitinated CYP2C11 species. Because intact CYP2C11 is normally degraded via autophagic lysosomal degradation (Murray et al., 2002), prosthetic heme incorporation thus appears to control the route of CYP2C11 protein degradation and thus its ultimate cellular fate.
In summary, the damage caused by reactive metabolic drug intermediates to the P450 prosthetic heme moiety can, provided heme is available, often be repaired by fresh heme uptake and reconstitution of a functional holoenzyme. More extensive/irreversible structural damage, especially that arising from heme-protein cross linking, cannot be repaired and will signal apoprotein disposal via the ubiquitin-dependent 26S proteasomal pathway. Apoprotein degradation may also result from the normal “wear and tear” associated with the physiological catalytic activity and thus contribute towards the “normal” turnover of the hemoproteins. It remains to be determined whether the relative fast versus slow rates of P450 protein turnover are determined by their specific catalytic turnover rates; or whether the presence of yet to be characterized intrinsic structural determinants “degrons” commit a P450 enzyme to proteolytic degradation by the 26S proteasomal route, rather than the lysosomal route. Nevertheless, the heme-saturation of the enzyme and the binding of specific substrates control P450 structural and proteolytic stability and thus are important determinants of P450 lifetimes.
Conclusions
In addition to serving as the essential prosthetic moiety of P450 enzymes, responsible for their highly versatile catalytic function and consequently diverse biological roles, heme also controls P450 de novo protein synthesis, assembly, content, repair and disposal. Thus, heme exquisitely regulates every aspect from birth to death in the biological lives of these important endo- and xenobiotic metabolizing enzymes. Pathologies of heme metabolism can and do influence P450 function, and thus may result in clinically relevant drug-drug interactions, as those observed in patients with acute hepatic porphyria (Anderson et al., 2005).
Footnotes
Heme stands for iron-protoporphyrin IX, irrespective of the redox state of the iron. In in vitro studies, heme is usually added as hematin (the hydroxide derivative dissolved in NaOH) or as hemin (the hydrochloride salt dissolved in DMSO), but to avoid confusion, we have used the term heme throughout.
We regret our inability to cite every relevant literature article due to considerations of the overall length of this manuscript.
Supported by NIH grants DK26506 and GM44037 to MAC.
Contributor Information
Maria Almira Correia, Departments of Cellular & Molecular Pharmacology, Pharmaceutical Chemistry, Bioengineering & Therapeutic Sciences, and The Liver Center, University of California, San Francisco, CA 94158, USA.
Peter R. Sinclair, Veterans Administration, White River Junction, VT 05001, and Departments of Biochemistry and Pharmacology/Toxicology, Dartmouth Medical School, Hanover, NH03755, USA
Francesco De Matteis, School of Biological and Chemical Sciences, Birkbeck College, University of London, London WC1 7HX, UK.
References
- Abraham NG, Cao J, Sacerdoti D, Li X, Drummond G. Heme oxygenase: the key to renal function regulation. Am J Physiol Renal Physiol. 2009;297:F1137–52. doi: 10.1152/ajprenal.90449.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbritti G, De Matteis F. Effect of 3,5-Diethoxycarbonyl-1,4-dihydrocollidine on degradation of liver heme. Enzyme. 1973;16:196–202. doi: 10.1159/000459381. [DOI] [PubMed] [Google Scholar]
- Acharya P, Chen JJ, Correia MA. Hepatic heme-regulated inhibitor (HRI) eukaryotic initiation factor 2alpha kinase: a protagonist of heme-mediated translational control of CYP2B enzymes and a modulator of basal endoplasmic reticulum stress tone. Mol Pharmacol. 2010;77:575–592. doi: 10.1124/mol.109.061259. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Ades IZ. Heme production in animal tissues: The regulation of biogenesis of delta-aminolevulinate synthase. Int J Biochem. 1990;22:565–78. doi: 10.1016/0020-711x(90)90032-x. [DOI] [PubMed] [Google Scholar]
- Ajioka RS, Phillips JD, Kushner JP. Biosynthesis of heme in mammals. Biochim Biophys Acta. 2006;1763:723–36. doi: 10.1016/j.bbamcr.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Alam J, Smith A. Heme-hemopexin-mediated induction of metallothionein gene expression. J Biol Chem. 1992;267:16379–16384. [PubMed] [Google Scholar]
- Alterman L, Negishi M, Sabatini DD. Synthesis and insertion of cytochrome P-450 into endoplasmic reticulum membranes. Proc Natl Acad Sci U S A. 1980;77:965–969. doi: 10.1073/pnas.77.2.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KE. Effects of antihypertensive drugs on hepatic heme biosynthesis, and evaluation of ferrochelatase inhibitors to simplify testing of drugs for heme pathway induction. Biochim Biophys Acta. 1978;543:313–27. doi: 10.1016/0304-4165(78)90049-1. [DOI] [PubMed] [Google Scholar]
- Anderson KE, Bloomer JR, Bonkovsky HL, Kushner JP, Pierach CA, Pimstone NR, Desnick RJ. Recommendations for the diagnosis and treatment of the acute porphyries. Ann Intern Med. 2005;142:439–450. doi: 10.7326/0003-4819-142-6-200503150-00010. [DOI] [PubMed] [Google Scholar]
- Bakken AF, Thaler MM, Schmid R. Metabolic regulation of heme catabolism and bilirubin production. I. Hormonal control of hepatic heme oxygenase activity. J Clin Invest. 1972;51:530–536. doi: 10.1172/JCI106841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Nun S, Kreibich G, Adesnik M, Alterman L, Negishi M. Synthesis and insertion of cytochrome P-450 into endoplasmic reticulum membranes. Proc Natl Acad Sci USA. 1980;77:965–969. doi: 10.1073/pnas.77.2.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barañano DE, Rao M, Ferris CD, Snyder SH. Biliverdin reductase: a major physiologic cytoprotectant. Proc Natl Acad Sci USA. 2002;99:16093–16098. doi: 10.1073/pnas.252626999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer M, Bauer I. Heme Oxygenase-1: Redox Regulation and Role in the Hepatic Response to Oxidative Stress. Antioxid Redox Signal. 2004;4:749–758. doi: 10.1089/152308602760598891. [DOI] [PubMed] [Google Scholar]
- Bauer BN, Rafie-Kolpin M, Lu L, Han A, Chen JJ. Multiple autophosphorylation is essential for the formation of the active and stable homodimer of heme-regulated eIF2alpha kinase. Biochemistry. 2001;40:11543–11551. doi: 10.1021/bi010983s. [DOI] [PubMed] [Google Scholar]
- Bender AT, Silverstein AM, Demady DR, Kanelakis KC, Noguchi S, Pratt WB, Osawa Y. Nitric-oxide Synthase Is Regulated by the hsp90-based Chaperone System in Vivo. J Biol Chem. 1998;274:1472–1478. doi: 10.1074/jbc.274.3.1472. [DOI] [PubMed] [Google Scholar]
- Berlanga JJ, Herrero S, de Haro C. Characterization of the hemin-sensitive eukaryotic initiation factor 2alpha kinase from mouse nonerythroid cells. J Biol Chem. 1998;273:32340–32346. doi: 10.1074/jbc.273.48.32340. [DOI] [PubMed] [Google Scholar]
- Bhat GJ, Padmanaban G. Heme is a positive regulator of cytochrome P-450 gene transcription. Arch Biochem Biophys. 1988;264:584–590. doi: 10.1016/0003-9861(88)90324-4. [DOI] [PubMed] [Google Scholar]
- Bissell DM, Hammaker LE. Effect of endotoxin on tryptophan pyrrolase and delta-aminolaevulinate synthase: evidence for an endogenous regulatory haem fraction in rat liver. Biochem J. 1977;166:301–304. doi: 10.1042/bj1660301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonkovsky HL, Sinclair JF, Healey JF, Sinclair PR, Smith EL. Formation of cytochrome P-450 containing haem or cobalt-protoporphyrin in liver homogenates of rats treated with phenobarbital and allylisopropylacetamide. Biochem J. 1984;222:453–462. doi: 10.1042/bj2220453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonkovsky HL, Healey JF, Lourie AN, Gerron GG. Intravenous heme albumin in acute intermittent porphyria: evidence for repletion of hepatic hemoproteins and regulatory heme pools. Am J Gastroenterol. 1991;86:1050–1056. [PubMed] [Google Scholar]
- Borgese N, Meldolesi J. Localization and biosynthesis of NADH-cytochrome b5 reductase, an integral membrane protein in rat liver cells. I. Distribution of the enzyme activity in microsomes, mitochondria and Golgi complex. J Cell Biol. 1980;85:501–515. doi: 10.1083/jcb.85.3.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornheim LM, Correia MA, Smith KM. Functional reconstitution of rat liver cytochrome P-450 with mesohemin. Biochem Biophys Res Commun. 1984;121:95–101. doi: 10.1016/0006-291x(84)90692-2. [DOI] [PubMed] [Google Scholar]
- Bornheim LM, Parish DW, Smith KM, Litman DA, Correia MA. The influence of side chain modifications of the heme moiety on prosthetic acceptance and function of rat hepatic cytochrome P-450 and tryptophan pyrrolase. Arch Biochem Biophys. 1986;246:63–74. doi: 10.1016/0003-9861(86)90449-2. [DOI] [PubMed] [Google Scholar]
- Brady AM, Lock EA. Inhibition of ferrochelatase and accumulation of porphyrins in mouse hepatocyte cultures exposed to porphyrinogenic chemicals. Arch Toxicol. 1992;66:175–181. doi: 10.1007/BF01974011. [DOI] [PubMed] [Google Scholar]
- Burmester T, Weich B, Reinhardt S, Hankeln T. A vertebrate globin expressed in the brain. Nature. 2000;407:520–523. doi: 10.1038/35035093. [DOI] [PubMed] [Google Scholar]
- Camus-Randon AM, Raffalli F, Bereziat JC, McGregor D, Konstandi M, Lang MA. Liver injury and expression of cytochromes P450: evidence that regulation of CYP2A5 is different from that of other major xenobiotic metabolizing CYP enzymes. Toxicol Appl Pharmacol. 1996;138:140–148. doi: 10.1006/taap.1996.0107. [DOI] [PubMed] [Google Scholar]
- Cable EE, Pepe JA, Karamitsios NC, Lambrecht RW, Bonkovsky HL. Differential effects of metalloporphyrins on messenger RNA levels of delta-aminolevulinate synthase and heme oxygenase. Studies in cultured chick embryo liver cells. J Clin Invest. 1994;94:649–54. doi: 10.1172/JCI117381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cable EE, Miller TG, Isom HC. Regulation of heme metabolism in rat hepatocytes and hepatocyte cell lines: delta-aminolevulinic acid synthase and heme oxygenase are regulated by different heme-dependent mechanisms. Arch Biochem Biophys. 2000;384:280–95. doi: 10.1006/abbi.2000.2117. [DOI] [PubMed] [Google Scholar]
- Chefalo PJ, Oh J, Rafie-Kolpin M, Kan B, Chen JJ. Heme-regulated eIF-2alpha kinase purifies as a hemoprotein. Eur J Biochem. 1998;258:820–830. doi: 10.1046/j.1432-1327.1998.2580820.x. [DOI] [PubMed] [Google Scholar]
- Chen JJ, London IM. Regulation of protein synthesis by heme-regulated eIF-2 alpha kinase. Trends Biochem Sci. 1995;20:105–108. doi: 10.1016/s0968-0004(00)88975-6. [DOI] [PubMed] [Google Scholar]
- Chen JJ, Yang JM, Petryshyn R, Kosower N, London IM. Disulfide bond formation in the regulation of eIF-2 alpha kinase by heme. J Biol Chem. 1989;264:9559–9564. [PubMed] [Google Scholar]
- Chen JJ. Heme-regulated eIF-2a kinase. In: Sonenberg N, Hershey JWB, Mathews MB, editors. Translational Control of Gene Expression. Cold Springs Harbor: Cold Spring Harbor Laboratory Press; 2000. pp. 529–546. [Google Scholar]
- Chen JJ. Regulation of protein synthesis by the heme-regulated eIF2alpha kinase: relevance to anemias. Blood. 2007;109:2693–2699. doi: 10.1182/blood-2006-08-041830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Converso DP, Taillé C, Carreras MC, Jaitovich A, Poderoso JJ, Boczkowski J. HO-1 is located in liver mitochondria and modulates mitochondrial heme content and metabolism. FASEB J. 2006;20:1236–1238. doi: 10.1096/fj.05-4204fje. [DOI] [PubMed] [Google Scholar]
- Correia MA. Hepatic cytochrome P450 degradation: mechanistic diversity of the cellular sanitation brigade. Drug Metab Rev. 2003;35:107–143. doi: 10.1081/dmr-120023683. and references therein. [DOI] [PubMed] [Google Scholar]
- Correia MA, Meyer UA. Apocytochrome P-450: reconstitution of functional cytochromme with hemin in vitro. Proc Natl Acad Sci USA. 1975;72:400–404. doi: 10.1073/pnas.72.1.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia MA, Liao M. Cellular proteolytic systems in P450 degradation: evolutionary conservation from Saccharomyces cerevisiae to mammalian liver. Expert Opin Drug Metab Toxicol. 2007;3:33–49. doi: 10.1517/17425255.3.1.33. [DOI] [PubMed] [Google Scholar]
- Correia MA, Farrell GC, Schmid R, Ortiz de Montellano PR, Yost GS, Mico BA. Incorporation of exogenous heme into hepatic cytochrome P450 in vivo. J Biol Chem. 1979;254:15–17. [PubMed] [Google Scholar]
- Correia MA, Farrell GC, Olson S, Wong JS, Schmid R, Ortiz de Montellano PR, Beilan HS, Kunze KL, Mico BA. Cytochrome P-450 heme moiety. The specific target in drug-induced heme alkylation. J Biol Chem. 1981;256:5466–5470. [PubMed] [Google Scholar]
- Correia MA, Decker C, Sugiyama K, Caldera P, Bornheim L, Wrighton SA, Rettie AE, Trager WF. Degradation of rat hepatic cytochrome P-450 heme by 3,5-dicarbethoxy-2,6-dimethyl-4-ethyl-1,4-dihydropyridine to irreversibly bound protein adducts. Arch Biochem Biophys. 1987;258:436–451. doi: 10.1016/0003-9861(87)90365-1. [DOI] [PubMed] [Google Scholar]
- Correia MA, Yao K, Wrighton SA, Waxman DJ, Rettie AE. Differential apoprotein loss of rat liver cytochromes P450 after their inactivation by 3,5-dicarbethoxy-2,6-dimethyl-4-ethyl-1,4-dihydropyridine: a case for distinct proteolytic mechanisms? Arch Biochem Biophys. 1992a;294:493–503. doi: 10.1016/0003-9861(92)90716-a. [DOI] [PubMed] [Google Scholar]
- Correia MA, Davoll SH, Wrighton SA, Thomas PE. Degradation of rat liver cytochromes P450 3A after their inactivation by 3,5-dicarbethoxy-2,6-dimethyl-4-ethyl-1,4-dihydropyridine: characterization of the proteolytic system. Arch Biochem Biophys. 1992b;297:228–238. doi: 10.1016/0003-9861(92)90666-k. [DOI] [PubMed] [Google Scholar]
- Crosby JS, Lee K, London IM, Chen JJ. Erythroid expression of the heme-regulated eIF-2 alpha kinase. Mol Cell Biol. 1994;14:3906–3914. doi: 10.1128/mcb.14.6.3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dailey TA, Woodruff JH, Dailey HA. Examination of mitochondrial protein targeting of haem synthetic enzymes: in vivo identification of three functional haem-responsive motifs in 5-aminolaevulinate synthase. Biochem J. 2005;386:381–6. doi: 10.1042/BJ20040570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar AC, Dever TE, Sicheri F. Higher-order substrate recognition of eIF2alpha by the RNA-dependent protein kinase PKR. Cell. 2005;122:887–900. doi: 10.1016/j.cell.2005.06.044. [DOI] [PubMed] [Google Scholar]
- Dean PA, Rettie AE, Turnblom SM, Namkung MJ, Juchau MR. Cytosolic activation of hematin-dependent microsomal monooxygenase activity in the lung. Chem Biol Interact. 1986;58:79–94. doi: 10.1016/s0009-2797(86)80088-6. [DOI] [PubMed] [Google Scholar]
- Delaunay J, Ranu RS, Levin DH, Ernst V, London IM. Characterization of a rat liver factor that inhibits initiation of protein synthesis in rabbit reticulocyte lysates. Proc Natl Acad Sci USA. 1977;74:2264–2268. doi: 10.1073/pnas.74.6.2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Matteis F. Loss of Haem in rat liver caused by the porphyrogenic agent 2-allyl-2-isopropylacetamide. Biochem J. 1970;124:767–777. doi: 10.1042/bj1240767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Matteis F. Hepatic Porphyrias. Heme and Hemoproteins. In: De Matteis F, Aldridge WN, editors. Handbook Exp Pharmacol. Vol. 44. 1978. pp. 129–155. [Google Scholar]
- De Matteis F. Loss of microsomal components in drug-induced liver damage, in cholestasis and after administration of chemicals which stimulate heme catabolism. In: Schenkman JB, Kupfer D, editors. Hepatic Cytochrome P-450 Monoxygenase System International Encyclopedia of Pharmacol Therap, Section 108. Oxford: Pergamon Press; 1982. pp. 307–340. [Google Scholar]
- De Matteis F, Gibbs A. Stimulation of liver 5-aminolaevulinate synthetase by drugs and its relevance to drug-induced accumulation of cytochrome P-450. Studies with phenylbutazone and 3,5-diethoxycarbonyl-1,4-dihydrocollidine. Biochem J. 1972;126:1149–60. doi: 10.1042/bj1261149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Matteis F, Abbritti G, Gibbs AH. Decreased liver activity of porphyrin-metal chelatase in hepatic porphyria caused by 3,5-diethoxycarbonyl-1,4-dihydrocollidine. Studies in rats and mice. Biochem J. 1973;134:717–727. doi: 10.1042/bj1340717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Matteis F, Gibbs AH. Stimulation of the pathway of porphyrin synthesis in the liver of rats and mice by griseofulvin, 3,5-Diethoxycarbonyl-1,4-dihydrocollidine and related drugs: evidence for two basically different mechanisms. Biochem J. 1975;146:285–7. doi: 10.1042/bj1460285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Matteis F, Gibbs AH. The effect of cobaltous chloride on liver haem metabolism in the rat. Evidence for inhibition of haem synthesis and for increased haem degradation. Ann Clin Res. 1976;8:193–197. [PubMed] [Google Scholar]
- De Matteis F, Unseld A. Increased liver haem degradation caused by foreign chemicals: a comparison of the effects of 2-allyl-2-isopropylacetamide and cobaltous chloride. Biochem Soc Trans. 1976;4:205–209. doi: 10.1042/bst0040205. [DOI] [PubMed] [Google Scholar]
- De Matteis F, Gibbs AH, Smith AG. Inhibition of protohaem ferro-lyase by N-substituted porphyrins. Structural requirements for the inhibitory effect. Biochem J. 1980;189:645–648. doi: 10.1042/bj1890645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Matteis F, Ray DE. Studies on cerebellar haem metabolism in the rat in vivo. J Neurochem. 1982;39:551–6. doi: 10.1111/j.1471-4159.1982.tb03980.x. [DOI] [PubMed] [Google Scholar]
- De Matteis F, Marks GS. The effect of N-methylprotoporphyrin and succinyl-acetone on the regulation of heme biosynthesis in chicken hepatocytes in culture. FEBS Lett. 1983;159:127–31. doi: 10.1016/0014-5793(83)80430-x. [DOI] [PubMed] [Google Scholar]
- De Matteis F, Harvey C, Martin SR. N-alkylation of hexogenous heme analogues caused by drugs in isolated hepatocytes. Structural isomerism and chirality of the resulting porphyrins. Biochem J. 1986;238:263–268. doi: 10.1042/bj2380263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Matteis F, Marks GS. Cytochrome P450 and its interactions with the heme biosynthetic pathway. Can J Physiol Pharmacol. 1996;74:1–8. [PubMed] [Google Scholar]
- Dey M, Cao C, Dar AC, Tamura T, Ozato K, Sicheri F, Dever TE. Mechanistic link between PKR dimerization, autophosphorylation, and eIF2alpha substrate recognition. Cell. 2005;122:901–913. doi: 10.1016/j.cell.2005.06.041. [DOI] [PubMed] [Google Scholar]
- Dierks P. Molecular biology of eukaryotic 5-aminolevulinate synthase. In: Dailey HA, editor. Biosynthesis of heme and chlorophylls. McGraw-Hill; New York: 1990. pp. 201–234. [Google Scholar]
- Drew PD, Ades IZ. Regulation of production of embryonic chick liver delta-aminolevulinate synthase: effects of testosterone and of hemin on the mRNA of the enzyme. Biochem Biophys Res Commun. 1986;140:81–7. doi: 10.1016/0006-291x(86)91060-0. [DOI] [PubMed] [Google Scholar]
- Drummond GS, Rosenberg DW, Kappas A. Metal induction of haem oxygenase without concurrent degradation of cytochrome P-450. Protective effects of compound SKF 525A on the haem protein. Biochem J. 1982;202:59–66. doi: 10.1042/bj2020059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duez H, Staels B. Rev-erbα: a potential target for the treatment of circadian disorders. Heart Metab. 2008;44:21–24. [Google Scholar]
- Duvigneau JC, Piskernik C, Haindl S, Kloesch B, Hartl RT, Hüttemann M, Lee I, Ebel T, Moldzio R, Gemeiner M, Redl H, Kozlov AV. A novel endotoxin-induced pathway: upregulation of heme oxygenase 1, accumulation of free iron, and free iron-mediated mitochondrial dysfunction. Lab Invest. 2008;88:70–77. doi: 10.1038/labinvest.3700691. [DOI] [PubMed] [Google Scholar]
- Dwarki VJ, Francis VN, Bhat GJ, Padmanaban G. Regulation of cytochrome P-450 messenger RNA and apoprotein levels by heme. J Biol Chem. 1987;262:16958–16962. [PubMed] [Google Scholar]
- Elder GH. Porphyria cutanea tarda. Semin Liver Dis. 1998;18:67–75. doi: 10.1055/s-2007-1007142. [DOI] [PubMed] [Google Scholar]
- Fagard R, Guguen-Guillouzo C. The effect of hemin and of allyl isopropyl acetamide on protein synthesis in rat hepatocytes. Biochem Biophys Res Comm. 1983;114:612–619. doi: 10.1016/0006-291x(83)90824-0. [DOI] [PubMed] [Google Scholar]
- Faller M, Matsunaga M, Yin S, Loo JA, Guo F. Heme is involved in microRNA processing. Nat Struct Mol Biol. 2007;14:23–29. doi: 10.1038/nsmb1182. [DOI] [PubMed] [Google Scholar]
- Figueiredo RT, Fernandez PL, Mourao-Sa DS, Porto BN, Dutra FF, Alves LS, Oliveira MF, Oliveira PL, Graca-Souza AV, Bozza MT. Characterization of heme as activator of Toll-like receptor 4. J Biol Chem. 2007;282:20221–20229. doi: 10.1074/jbc.M610737200. [DOI] [PubMed] [Google Scholar]
- Fraser DJ, Podvinec M, Kaufmann MR, Meyer UA. Drugs mediate the transcriptional activation of the 5-aminolevulinic acid synthase (ALAS1) gene via the chicken xenobiotic-sensing nuclear receptor (CXR) J Biol Chem. 2002;277:34717–26. doi: 10.1074/jbc.M204699200. [DOI] [PubMed] [Google Scholar]
- Fraser DJ, Zumsteg A, Meyer UA. Nuclear receptors constitutive androstane receptor and pregnane X receptor activate a drug-responsive enhancer of the murine 5-aminolevulinic acid synthase gene. J Biol Chem. 2003;278:39392–401. doi: 10.1074/jbc.M306148200. [DOI] [PubMed] [Google Scholar]
- Furuyama K, Kaneko K, Vargas PD. Heme as a magnificent molecule with multiple missions: heme determines its own fate and governs cellular homeostasis. Tohoku J Exp Med. 2007;213:1–16. doi: 10.1620/tjem.213.1. [DOI] [PubMed] [Google Scholar]
- Gachon F, Olela FF, Schaad O, Descombes P, Schibler U. The circadian PAR-domain basic leucine zipper transcription factors DBP, TEF, and HLF modulate basal and inducible xenobiotic detoxification. Cell Metab. 2006;4:25–36. doi: 10.1016/j.cmet.2006.04.015. [DOI] [PubMed] [Google Scholar]
- Goasduff T, Cederbaum AI. NADPH-dependent microsomal electron transfer increases degradation of CYP2E1 by the proteasome complex: role of reactive oxygen species. Arch Biochem Biophys. 1999;370:258–270. doi: 10.1006/abbi.1999.1399. [DOI] [PubMed] [Google Scholar]
- Gozzelino R, Jeney V, Soares MP. Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol. 2010;50:323–54. doi: 10.1146/annurev.pharmtox.010909.105600. [DOI] [PubMed] [Google Scholar]
- Graber SG, Woodworth RC. Myoglobin expression in L6 muscle cells. Role of differentiation and heme. J Biol Chem. 1986;261:9150–9154. [PubMed] [Google Scholar]
- Grandori R, Schwarzinger S, Müller N. Cloning, overexpression and characterization of micro-myoglobin, a minimal heme-binding fragment. Europ J Biochem. 2000;267:1168–1172. doi: 10.1046/j.1432-1327.2000.01114.x. [DOI] [PubMed] [Google Scholar]
- Granick S, Urata G. Increase in activity of alpha-aminolevulinic acid synthetase in liver mitochondria induced by feeding of 3,5-dicarbethoxy-1,4-dihydrocollidine. J Biol Chem. 1963;238:821–827. and references therein. [PubMed] [Google Scholar]
- Granick S, Sinclair P, Sassa S, Grieninger G. Effects by heme, insulin, and serum albumin on heme and protein synthesis in chick embryo liver cells cultured in a chemically defined medium, and a spectrofluorometric assay for porphyrin composition. J Biol Chem. 1975;250:9215–25. [PubMed] [Google Scholar]
- Guarente L, Mason T. Heme regulates transcription of the CYC1 gene of S. cerevisiae via an upstream activation site. Cell. 1983;32:1279–1286. doi: 10.1016/0092-8674(83)90309-4. [DOI] [PubMed] [Google Scholar]
- Guengerich FP. Destruction of heme and hemoproteins mediated by liver microsomal reduced nicotinamide adenine dinucleotide phosphate-cytochrome P-450 reductase. Biochemistry. 1978;17:3633–3639. doi: 10.1021/bi00610a033. [DOI] [PubMed] [Google Scholar]
- Guengerich FP. Mechanisms of cytochrome P450 substrate oxidation: MiniReview. J Biochem Mol Toxicol. 2007;21:163–8. doi: 10.1002/jbt.20174. [DOI] [PubMed] [Google Scholar]
- Guéye PM, Glasser N, Férard G, Lessinger JM. Influence of human haptoglobin polymorphism on oxidative stress induced by free hemoglobin on red blood cells. Clin Chem Lab Med. 2006;44:542–547. doi: 10.1515/CCLM.2006.095. [DOI] [PubMed] [Google Scholar]
- Halpert JR, Guengerich FP, Bend JR, Correia MA. Selective inhibitors of cytochromes P450. Toxicol Appl Pharmacol. 1994;125:163–175. doi: 10.1006/taap.1994.1061. [DOI] [PubMed] [Google Scholar]
- Hamilton B, Hofbauer R, Ruis H. Translational control of catalase synthesis by hemin in the yeast Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 1982;79:7609–7613. doi: 10.1073/pnas.79.24.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton JW, Bement WJ, Sinclair PR, Sinclair JF, Wetterhahn KE. Expression of 5-aminolaevulinate synthase and cytochrome P-450 mRNAs in chicken embryo hepatocytes in vivo and in culture. Effect of porphyrinogenic drugs and haem. Biochem J. 1988;255:267–75. Erratum in: Biochem J 1989: 257:following 934. [PMC free article] [PubMed] [Google Scholar]
- Hamilton JW, Bement WJ, Sinclair PR, Sinclair JF, Alcedo JA, Wetterhahn KE. Heme regulates hepatic 5-aminolevulinate synthase mRNA expression by decreasing mRNA half-life and not by altering its rate of transcription. Arch Biochem Biophys. 1991;289:387–392. doi: 10.1016/0003-9861(91)90428-l. [DOI] [PubMed] [Google Scholar]
- Han AP, Fleming MD, Chen JJ. Heme-regulated eIF2alpha kinase modifies the phenotypic severity of murine models of erythropoietic protoporphyria and beta-thalassemia. J Clin Invest. 2005;115:1562–1570. doi: 10.1172/JCI24141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han AP, Yu C, Lu L, Fujiwara Y, Browne C, Chin G, Fleming M, Leboulch P, Orkin SH, Chen JJ. Heme-regulated eIF2alpha kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. Embo J. 2001;20:6909–6918. doi: 10.1093/emboj/20.23.6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han XM, Lee G, Hefner C, Maher JJ, Correia MA. Heme-reversible impairment of CYP2B1/2 induction in heme-depleted rat hepatocytes in primary culture: Translational control by a hepatic alpha-subunit of the eukaryotic initiation factor kinase? J Pharmacol Exp Ther. 2005;314:128–138. doi: 10.1124/jpet.105.084699. [DOI] [PubMed] [Google Scholar]
- Handschin C, Meyer UA. Induction of drug metabolism: the role of nuclear receptors. Pharmacol Rev. 2003;55:649–673. doi: 10.1124/pr.55.4.2. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- Hargrove MS, Olson JS. The Stability of Holomyoglobin Is Determined by Heme Affinity. Biochemistry. 1996;35:11310–11318. doi: 10.1021/bi9603736. [DOI] [PubMed] [Google Scholar]
- Henderson CJ, Otto DM, Carrie D, Magnuson MA, McLaren AW, Rosewell I, Wolf CR. Inactivation of the hepatic cytochrome P450 system by conditional deletion of hepatic cytochrome P450 reductase. J Biol Chem. 2003;278:13480–13486. doi: 10.1074/jbc.M212087200. [DOI] [PubMed] [Google Scholar]
- Heinemann IU, Jahn M, Jahn D. The biochemistry of heme biosynthesis. Arch Biochem Biophys. 2008;474:238–51. doi: 10.1016/j.abb.2008.02.015. [DOI] [PubMed] [Google Scholar]
- Hockin LJ, Paine AJ. The role of 5-aminolaevulinate synthase, haem oxygenase and ligand formation in the mechanism of maintenance of cytochrome P-450 concentration in hepatocyte culture. Biochem J. 1983;210:855–857. doi: 10.1042/bj2100855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honkakoski P, Moore R, Washburn KA, Negishi M. Activation by diverse xenochemicals of the 51-base pair phenobarbital-responsive enhancer module in the CYP2B10 gene. Mol Pharmacol. 1998;53:597–601. doi: 10.1124/mol.53.4.597. [DOI] [PubMed] [Google Scholar]
- Honkakoski P, Negishi M. Protein serine/threonine phosphatase inhibitors suppress phenobarbital-induced Cyp2b10 gene transcription in mouse primary hepatocytes. Biochem J. 1998;330:889–895. doi: 10.1042/bj3300889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honkakoski P, Zelko I, Sueyoshi T, Negishi M. The nuclear orphan receptor CAR-retinoid X receptor heterodimer activates the phenobarbital-responsive enhancer module of the CYP2B gene. Mol Cell Biol. 1998;18:5652–5658. doi: 10.1128/mcb.18.10.5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hörtner H, Ammerer G, Hartter E, Hamilton B, Rytka J, Bilinski T, Ruis H. Regulation of synthesis of catalases and iso-1-cytochrome c in Saccharomyces cerevisiae by glucose, oxygen and heme. Eur J Biochem. 1982;128:179–184. doi: 10.1111/j.1432-1033.1982.tb06949.x. [DOI] [PubMed] [Google Scholar]
- Igarashi J, Sato A, Kitagawa T, Yoshimura T, Yamauchi S, Sagami I, Shimizu T. Activation of heme-regulated eukaryotic initiation factor 2alpha kinase by nitric oxide is induced by the formation of a five-coordinate NO-heme complex: optical absorption, electron spin resonance, and resonance raman spectral studies. J Biol Chem. 2004;279:15752–15762. doi: 10.1074/jbc.M310273200. [DOI] [PubMed] [Google Scholar]
- Igarashi J, Murase M, Iizuka A, Pichierri F, Martinkova M, Shimizu T. Elucidation of the heme binding site of heme-regulated eukaryotic initiation factor 2alpha kinase and the role of the regulatory motif in heme sensing by spectroscopic and catalytic studies of mutant proteins. J Biol Chem. 2008;283:18782–18791. doi: 10.1074/jbc.M801400200. [DOI] [PubMed] [Google Scholar]
- Ikuta T, Ausenda S, Cappellini MD. Mechanism for fetal globin gene expression: role of the soluble guanylate cyclase-cGMP-dependent protein kinase pathway. Proc Natl Acad Sci U S A. 2001;98:1847–1852. doi: 10.1073/pnas.041599798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inuzuka T, Yun BG, Ishikawa H, Takahashi S, Hori H, Matts RL, Ishimori K, Morishima I. Identification of crucial histidines for heme binding in the N-terminal domain of the heme-regulated eIF2alpha kinase. J Biol Chem. 2004;279:6778–6782. doi: 10.1074/jbc.C300464200. [DOI] [PubMed] [Google Scholar]
- Ishimori K, Morishima I. Study of the specific heme orientation in reconstituted hemoglobins. Biochemistry. 1988;27:4747–4753. doi: 10.1021/bi00413a025. [DOI] [PubMed] [Google Scholar]
- Jacobs JM, Sinclair PR, Bement WJ, Lambrecht RW, Sinclair JF, Goldstein JA. Oxidation of uroporphyrinogen by methylcholanthrene-induced cytochrome P-450. Essential role of cytochrome P-450d. Biochem J. 1989;258:247–53. doi: 10.1042/bj2580247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jover R, Hoffmann K, Meyer UA. Induction of 5-aminolevulinate synthase by drugs is independent of increased apocytochrome P450 synthesis. Biochem Biophys Res Commun. 1996;226:152–7. doi: 10.1006/bbrc.1996.1325. [DOI] [PubMed] [Google Scholar]
- Jover R, Lindberg RL, Meyer UA. Role of heme in cytochrome P450 transcription and function in mice treated with lead acetate. Mol Pharmacol. 1996;50:474–481. [PubMed] [Google Scholar]
- Jover R, Hoffmann F, Scheffler-Koch V, Lindberg RL. Limited heme synthesis in porphobilinogen deaminase-deficient mice impairs transcriptional activation of specific cytochrome P450 genes by phenobarbital. Eur J Biochem. 2000;267:7128–37. doi: 10.1046/j.1432-1327.2000.01815.x. [DOI] [PubMed] [Google Scholar]
- Kaasik K, Lee CC. Reciprocal regulation of haem biosynthesis and the circadian clock in mammals. Nature. 2004;430:467–471. doi: 10.1038/nature02724. [DOI] [PubMed] [Google Scholar]
- Kanno Y, Otsuka S, Hiromasa T, Nakahama T, Inouye Y. Diurnal difference in CAR mRNA expression. Nucl Recept. 2004;2:6. doi: 10.1186/1478-1336-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman RJ. Regulation of mRNA translation by protein folding in the endoplasmic reticulum. Trends Biochem Sci. 2004;29:152–158. doi: 10.1016/j.tibs.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Kawamoto T, Sueyoshi T, Zelko I, Moore R, Washburn K, Negishi M. Phenobarbital-responsive nuclear translocation of the receptor CAR in induction of the CYP2B gene. Mol Cell Biol. 1999;19:6318–6322. doi: 10.1128/mcb.19.9.6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemper B. Regulation of cytochrome P450 gene transcription by phenobarbital. Prog Nucleic Acid Res Mol Biol. 1998;61:23–64. [PubMed] [Google Scholar]
- Kim J, Kemper B. Phenobarbital alters protein binding to the CYP2B1/2 phenobarbital-responsive unit in native chromatin. J Biol Chem. 1997;272:29423–29425. doi: 10.1074/jbc.272.47.29423. [DOI] [PubMed] [Google Scholar]
- Kim YM, Bergonia HA, Müller C, Pitt BR, Watkins WD, Jack R, Lancaster JR., Jr Loss and degradation of enzyme-bound heme Induced by cellular nitric oxide synthesis. J Biol Chem. 1995;270:5710–5713. doi: 10.1074/jbc.270.11.5710. [DOI] [PubMed] [Google Scholar]
- Kim J, Min G, Kemper B. Chromatin assembly enhances binding to the CYP2B1 phenobarbital-responsive unit (PBRU) of nuclear factor-1, which binds simultaneously with constitutive androstane receptor (CAR)/retinoid X receptor (RXR) and enhances CAR/RXR-mediated activation of the PBRU. J Biol Chem. 2001;276:7559–7567. doi: 10.1074/jbc.M008090200. [DOI] [PubMed] [Google Scholar]
- Kimura I, Nakayama Y, Yamauchi H, Konishi M, Miyake A, Mori M, Ohta M, Itoh N, Fujimoto M. Neurotrophic activity of neudesin, a novel extracellular heme-binding protein, is dependent on the binding of heme to its cytochrome b5-like heme/steroid-binding domain. J Biol Chem. 2008;283:4323–4331. doi: 10.1074/jbc.M706679200. [DOI] [PubMed] [Google Scholar]
- Kikuchi G, Hayashi N. Regulation by heme of synthesis and intracellular translocation of delta-aminolevulinate synthase in the liver. Mol Cell Biochem. 1981;37:27–41. doi: 10.1007/BF02355885. [DOI] [PubMed] [Google Scholar]
- Kolluri S, Sadlon TJ, May BK, Bonkovsky HL. Haem repression of the housekeeping 5-aminolaevulinic acid synthase gene in the hepatoma cell line LMH. Biochem J. 2005;392:173–80. doi: 10.1042/BJ20050354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsmeyer KK, Davoll S, Figueiredo-Pereira ME, Correia MA. Proteolytic degradation of heme-modified hepatic cytochromes P450: a role for phosphorylation, ubiquitination and the 26S proteasome? Arch Biochem Biophys. 1999;365:31–44. doi: 10.1006/abbi.1999.1138. [DOI] [PubMed] [Google Scholar]
- La Mar GN, Budd DL, Viscio DB, Smith KM, Langry KC. Proton nuclear magnetic resonance characterization of heme disorder in hemoproteins. Proc Natl Acad Sci USA. 1978;75:5755–5759. doi: 10.1073/pnas.75.12.5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlois MR, Delanghe JR. Biological and clinical significance of haptoglobin polymorphism in humans. Clinical Chemistry. 1996;42:1589–1600. [PubMed] [Google Scholar]
- Lathrop JT, Timko MP. Regulation by heme of mitochondrial protein transport through a conserved amino acid motif. Science. 1993;259:522–5. doi: 10.1126/science.8424176. [DOI] [PubMed] [Google Scholar]
- Layer G, Reichelt J, Jahn D, Heinz DW. Structure and function of enzymes in heme biosynthesis. Protein Sci. 2010;19:1137–1161. doi: 10.1002/pro.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarow P, deDuve C. The synthesis and turnover of rat liver peroxisomes: IV. Biochemical pathway of catalase synthesis. J Cell Biol. 1973;59:491–506. doi: 10.1083/jcb.59.2.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Jacobsen NE, Ortiz de Montellano PR. 4-alkyl radical extrusion in the cytochrome P-450-catalyzed oxidation of 4-alkyl-1,4-dihydropyridines. Biochemistry. 1988;27:7703–7710. doi: 10.1021/bi00420a020. [DOI] [PubMed] [Google Scholar]
- Levi F, Schibler U. Circadian rhythms: mechanisms and therapeutic implications. Annu Rev Pharmacol Toxicol. 2007;47:593–628. doi: 10.1146/annurev.pharmtox.47.120505.105208. [DOI] [PubMed] [Google Scholar]
- Liao M, Pabarcus MK, Wang Y, Hefner C, Maltby DA, Medzihradszky KF, Salas-Castillo SP, Yan J, Maher JJ, Correia MA. Impaired dexamethasone-mediated induction of tryptophan 2,3-dioxygenase in heme-deficient rat hepatocytes: translational control by a hepatic eIF2alpha kinase, the heme-regulated inhibitor. J Pharmacol Exp Ther. 2007;323:979–989. doi: 10.1124/jpet.107.124602. [DOI] [PubMed] [Google Scholar]
- Licad-Coles E, He K, Yin H, Correia MA. Cytochrome P450 2C11: Escherichia coli expression, purification, functional characterization, and mechanism-based inactivation of the enzyme. Arch Biochem Biophys. 1997;338:35–42. doi: 10.1006/abbi.1996.9795. [DOI] [PubMed] [Google Scholar]
- Lindberg RL, Porcher C, Grandchamp B, Ledermann B, Burki K, Brandner S, Aguzzi A, Meyer UA. Porphobilinogen deaminase deficiency in mice causes a neuropathy resembling that of human hepatic porphyria. Nat Genet. 1996;12:195–199. doi: 10.1038/ng0296-195. [DOI] [PubMed] [Google Scholar]
- Litman DA, Correia MA. L-tryptophan: a common denominator of biochemical and neurological events of acute hepatic porphyria? Science. 1983;222:1031–1033. doi: 10.1126/science.6648517. [DOI] [PubMed] [Google Scholar]
- Litman DA, Correia MA. Elevated brain tryptophan and enhanced 5-hydroxytryptamine turnover in acute hepatic heme deficiency: Clinical implications. J Pharmacol Exp Ther. 1985;232:337–345. [PubMed] [Google Scholar]
- Mackie JE, Marks GS. Synergistic induction of delta-aminolevulinic acid synthase activity by N-ethylprotoporphyrin IX and 3,5-diethoxycarbonyl-1,4-dihydro-2,6-dimethyl-4-isobutylpyridine. Biochem Pharmacol. 1989;38:2169–2173. doi: 10.1016/0006-2952(89)90072-5. [DOI] [PubMed] [Google Scholar]
- Mackie JE, Back DW, Hamilton JW, Marks GS. Elevation of delta-aminolevulinic acid synthase and cytochrome PB1 P450 messenger RNA levels by dihydropyridines, dihydroquinolines, sydnones, and N-ethylprotoporphyrin IX. Biochem Pharmacol. 1991;42:475–483. doi: 10.1016/0006-2952(91)90308-r. [DOI] [PubMed] [Google Scholar]
- Maines MD, Kappas A. Cobalt stimulation of heme degradation in the liver. Dissociation of microsomal oxidation of heme from cytochrome P-450. J Biol Chem. 1975;250:4171–7. [PubMed] [Google Scholar]
- Maines MD, Trakshel GM, Kutty RK. Characterization of two constitutive forms of rat liver microsomal heme oxygenase. Only one molecular species of the enzyme is inducible. J Biol Chem. 1986;261:411–419. [PubMed] [Google Scholar]
- Maines MD. Heme Oxygenase, Clinical Applications and Functions. CRC Press; Boca Raton: 1992. [Google Scholar]
- Mancuso C, Barone E. The heme oxygenase/biliverdin reductase pathway in drug research and development. Curr Drug Metab. 2009;10:579–94. doi: 10.2174/138920009789375405. [DOI] [PubMed] [Google Scholar]
- Marks GS, Goldman DR, McCluskey SA, Sutherland EP, Lyon ME. The effects of dihydropyridine calcium antagonists on heme biosynthesis in chick embryo liver cell culture. Can J Physiol Pharmacol. 1986;64:438–443. doi: 10.1139/y86-070. [DOI] [PubMed] [Google Scholar]
- Maxwell JD, Meyer UA. Effect of lead on hepatic delta-aminolaevulinic acid synthetase activity in the rat: a model for drug sensitivity in intermittent acute porphyria. Eur J Clin Invest. 1976;6:373–9. doi: 10.1111/j.1365-2362.1976.tb00531.x. [DOI] [PubMed] [Google Scholar]
- May BK, Borthwick IA, Srivastava G, Pirola BA, Elliott WH. Control of 5-aminolevulinate synthase in animals. Curr Top Cell Regul. 1986;28:233–62. doi: 10.1016/b978-0-12-152828-7.50008-1. [DOI] [PubMed] [Google Scholar]
- Mellor H, Flowers KM, Kimball SR, Jefferson LS. Cloning and characterization of cDNA encoding rat hemin-sensitive initiation factor-2 alpha (eIF-2 alpha) kinase. Evidence for multitissue expression. J Biol Chem. 1994;269:10201–10204. [PubMed] [Google Scholar]
- Mense SM, Zhang L. Heme: a versatile signaling molecule controlling the activities of diverse regulators ranging from transcription factors to MAP kinases. Cell Res. 2006;16:681–692. doi: 10.1038/sj.cr.7310086. [DOI] [PubMed] [Google Scholar]
- Meyer UA. Endo-xenobiotic crosstalk and the regulation of cytochromes P450. Drug Metab Rev. 2007;39:639–646. doi: 10.1080/03602530701498737. [DOI] [PubMed] [Google Scholar]
- Meyer UA, Marver HS. Chemically-induced porphyria. Increased microsomal heme turnover after treatment with allylisopropylacetamide. Science. 1971;171:64–66. doi: 10.1126/science.171.3966.64. [DOI] [PubMed] [Google Scholar]
- Meyer RP, Podvinec M, Meyer UA. Cytochrome P450 CYP1A1 accumulates in the cytosol of kidney and brain and Is activated by heme. Mol Pharmacol. 2002;62:1061–1067. doi: 10.1124/mol.62.5.1061. [DOI] [PubMed] [Google Scholar]
- Miksanova M, Igarashi J, Minami M, Sagami I, Yamauchi S, Kurokawa H, Shimizu T. Characterization of heme-regulated eIF2alpha kinase: roles of the N-terminal domain in the oligomeric state, heme binding, catalysis, and inhibition. Biochemistry. 2006;45:9894–9905. doi: 10.1021/bi060556k. [DOI] [PubMed] [Google Scholar]
- Modi S, Primrose WU, Lian L-Y, Roberts GCK. Effect of replacement of ferriprotoporphyrin IX in the haem domain of cytochrome P-450 BM-3 on substrate binding and catalytic activity. Biochem J. 1995;310:939–943. doi: 10.1042/bj3100939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monier P, Van Luc G, Kreibich G, Sabatini DD, Adesnik M. Signals for the incorporation and orientation of cytochrome P450 in the endoplasmic reticulum membrane. J Cell Biol. 1998;107:457–470. doi: 10.1083/jcb.107.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan WT, Muster P, Tatum F, Kao SM, Alam J, Smith A. Identification of the histidine residues of hemopexin that coordinate with heme-iron and of a receptor-binding region. J Biol Chem. 1993;268:6256–6262. [PubMed] [Google Scholar]
- Morishima Y, Peng HM, Lin HL, Hollenberg PF, Sunahara RK, Osawa Y, Pratt WB. Regulation of cytochrome P450 2E1 by heat shock protein 90-dependent stabilization and CHIP-dependent proteasomal degradation. Biochemistry. 2005;44:16333–16340. doi: 10.1021/bi0515570. [DOI] [PubMed] [Google Scholar]
- Morse D, Lin L, Choi AM, Ryter SW. Heme oxygenase-1, a critical arbitrator of cell death pathways in lung injury and disease. Free Radic Biol Med. 2009;47:1–12. doi: 10.1016/j.freeradbiomed.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray BP, Zgoda VG, Correia MA. Native CYP2C11: heterologous expression in Saccharomyces cerevisiae reveals a role for vacuolar proteases rather than the proteasome system in the degradation of this endoplasmic reticulum protein. Mol Pharmacol. 2002;61:1146–1153. doi: 10.1124/mol.61.5.1146. [DOI] [PubMed] [Google Scholar]
- Myers AM, Crivellone MD, Koerner TJ, Tzagoloff A. Characterization of the yeast HEM2 gene and transcriptional regulation of COX5 and COR1 by heme. J Biol Chem. 1987;262:16822–16829. [PubMed] [Google Scholar]
- Namkung MJ, Faustman-Watts E, Juchau MR. Hematin-mediated increases of benzo(a)pyrene mono-oxygenation in maternal, fetal and placental tissues of inducible and non-inducible mouse strains. Dev Pharmacol Ther. 1983;6:199–206. doi: 10.1159/000457295. [DOI] [PubMed] [Google Scholar]
- Negishi M, Fujii-Kuriyama Y, Tashiro Y, Imai Y. Site of biosynthesis of cytochrome P-450 in hepatocytes of phenobarbital treated rats. Biochem Biophys Res Commun. 1976;71:1153–1160. doi: 10.1016/0006-291x(76)90774-9. [DOI] [PubMed] [Google Scholar]
- Negishi M, Kreibich G. Coordinated polypeptide synthesis and insertion of protoheme in cytochrome P-450 during development of endoplasmic reticulum membranes. J Biol Chem. 1978;253:4791–4797. [PubMed] [Google Scholar]
- Nichols KD, Kirby GM. Expression of cytochrome P450 2A5 in a glucose-6-phosphate dehydrogenase-deficient mouse model of oxidative stress. Biochem Pharmacol. 2008;75:1230–1239. doi: 10.1016/j.bcp.2007.10.032. [DOI] [PubMed] [Google Scholar]
- Ogawa K, Sun J, Taketani S, Nakajima O, Nishitani C, Sassa S, Hayashi N, Yamamoto M, Shibahara S, Fujita H, Igarashi K. Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. Embo J. 2001;20:2835–2843. doi: 10.1093/emboj/20.11.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Frey AB, Guenthner TM, Oesch F, Sabatini DD, Kreibich G. Studies on the biosynthesis of microsomal membrane proteins: site of synthesis and mode of insertion of cytochrome b5, cytochrome b5 reductase, cytochrome P-450 reductase and epoxide hydrolase. Eur J Biochem. 1982;122:393–402. doi: 10.1111/j.1432-1033.1982.tb05894.x. [DOI] [PubMed] [Google Scholar]
- Okano S, Zhou L, Kusaka T, Shibata K, Shimizu K, Gao X, Kikuchi Y, Togashi Y, Hosoya T, Takahashi S, Nakajima O, Yamamoto M. Indispensable function for embryogenesis, expression and regulation of the nonspecific form of the 5-aminolevulinate synthase gene in mouse. Genes Cells. 2010;15:77–89. doi: 10.1111/j.1365-2443.2009.01366.x. [DOI] [PubMed] [Google Scholar]
- Omiecinski CJ, Bond JA, Juchau MR. Stimulation by hematin of monooxygenase activity in extra-hepatic tissues from rats, rabbits and chickens. Biochem Biophys Res Commun. 1978;83:1004–1011. doi: 10.1016/0006-291x(78)91495-x. [DOI] [PubMed] [Google Scholar]
- Omiecinski CJ, Namkung MJ, Juchau MR. Mechanistic aspects of the hematin-mediated increases in brain monooxygenase activities. Mol Pharmacol. 1980;17:225–232. [PubMed] [Google Scholar]
- Onisawa J, Labbe RF. Effects of diethyl-1, 4-dihydro-2, 4,6-trimethylpyridine-3,5-dicarboxylate on the metabolism of porphyrins and iron. J Biol Chem. 1963;238:724–727. [PubMed] [Google Scholar]
- Ortiz de Montellano PR, Mico BA, Yost GS. Suicidal inactivation of cytochrome P-450. Formation of a heme-substrate covalent adduct. Biochem Biophys Res Commun. 1978;83:132–137. doi: 10.1016/0006-291x(78)90407-2. [DOI] [PubMed] [Google Scholar]
- Ortiz de Montellano PR, Kunze KL, Cole SP, Marks GS. Inhibition of hepatic ferrochelatase by the four isomers of N-methylprotoporphyrin IX. Biochem Biophys Res Commun. 1980;97:1436–1442. doi: 10.1016/s0006-291x(80)80026-x. [DOI] [PubMed] [Google Scholar]
- Ortiz de Montellano PR, Kunze KL, Cole SP, Marks GS. Differential inhibition of hepatic ferrochelatase by the isomers of N-ethylprotoporphyrin IX. Biochem Biophys Res Commun. 1981;103:581–586. doi: 10.1016/0006-291x(81)90491-5. [DOI] [PubMed] [Google Scholar]
- Ortiz de Montellano PR, Kunze KL, Beilan HS. Chiral orientation of prosthetic heme in the cytochrome P-450 active site. J Biol Chem. 1983;258:45–47. [PubMed] [Google Scholar]
- Ortiz de Montellano PR, De Vos JJ. Substrate oxidation by cytochrome P450 enzymes. In: Ortiz de Montellano PR, editor. Cytochrome P450. Structure, Mechanism, and Biochemistry. Vol. 2004. Plenum Press; New York: 2004. pp. 183–246. and references therein. [Google Scholar]
- Ortiz de Montellano PR. Wiley Encyclopedia of Chemical Biology. John Wiley & Sons, Inc; 2009. Hemes in Biology; pp. 240–249. [Google Scholar]
- Osawa Y, Pohl LR. Covalent bonding of the prosthetic heme to protein: a potential mechanism for the suicide inactivation or activation of hemoproteins. Chem Res Toxicol. 1989;2:131–141. doi: 10.1021/tx00009a001. [DOI] [PubMed] [Google Scholar]
- Osawa Y, Korzekwa K. Oxidative modification by low levels of HOOH can transform myoglobin to an oxidase. Proc Natl Acad Sci USA. 1991;88:7081–7085. doi: 10.1073/pnas.88.16.7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanaban G, Venkateswar V, Rangarajan PN. Haem as a multifunctional regulator. Trends Biochem Sci. 1989;14:492–496. doi: 10.1016/0968-0004(89)90182-5. [DOI] [PubMed] [Google Scholar]
- Pal JK, Chen JJ, London IM. Tissue distribution and immunoreactivity of heme-regulated eIF-2 alpha kinase determined by monoclonal antibodies. Biochemistry. 1991;30:2555–2562. doi: 10.1021/bi00223a037. [DOI] [PubMed] [Google Scholar]
- Park Y, Li H, Kemper B. Phenobarbital induction mediated by a distal CYP2B2 sequence in rat liver transiently transfected in situ. J Biol Chem. 1996;271:23725–23728. doi: 10.1074/jbc.271.39.23725. [DOI] [PubMed] [Google Scholar]
- Paschos GK, Baggs JE, Hogenesch JB, FitzGerald GA. The role of clock genes in pharmacology. Annu Rev Pharmacol Toxicol. 50:187–214. doi: 10.1146/annurev.pharmtox.010909.105621. [DOI] [PubMed] [Google Scholar]
- Peterson SJ, Frishman WH, Abraham NG. Targeting heme oxygenase: therapeutic implications for diseases of the cardiovascular system. Cardiol Rev. 2009;17:99–111. doi: 10.1097/CRD.0b013e31819d813a. [DOI] [PubMed] [Google Scholar]
- Pfeifer K, Arcangioli B, Guarente L. Yeast HAP1 activator competes with the factor RC2 for binding to the upstream activation site UAS1 of the CYC1 gene. Cell. 1987;49:9–18. doi: 10.1016/0092-8674(87)90750-1. [DOI] [PubMed] [Google Scholar]
- Pinkham JL, Olesen JT, Guarente LP. Sequence and nuclear localization of the Saccharomyces cerevisiae HAP2 protein, a transcriptional activator. Mol Cell Biol. 1987;7:578–585. doi: 10.1128/mcb.7.2.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkham JL, Wang ZD, Alsina J. Heme regulates SOD2 transcription by activation and repression in Saccharomyces cerevisiae. Current Genetics. 1997;31:281–291. doi: 10.1007/s002940050207. [DOI] [PubMed] [Google Scholar]
- Pinnix IB, Guzman GS, Bonkovsky HL, Zaki SR, Kinkade JM., Jr The post-translational processing of myeloperoxidase is regulated by the availability of heme. Arch Biochem Biophys. 1994;312:447–458. doi: 10.1006/abbi.1994.1331. [DOI] [PubMed] [Google Scholar]
- Podvinec M, Handschin C, Looser R, Meyer UA. Identification of the xenosensors regulating human 5-aminolevulinate synthase. Proc Natl Acad Sci U S A. 2004;101:9127–9132. doi: 10.1073/pnas.0401845101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponka P. Cell biology of heme. Am J Med Sci. 1999;318:241–56. doi: 10.1097/00000441-199910000-00004. [DOI] [PubMed] [Google Scholar]
- Price VE, Sterling WR, Tarantola VR, Hartley RW, Jr, Rechcigl M., Jr The kinetics of catalase synthesis and destruction in vivo. J Biol Chem. 1962;237:3468–3475. [PubMed] [Google Scholar]
- Rafie-Kolpin M, Chefalo PJ, Hussain Z, Hahn J, Uma S, Matts RL, Chen JJ. Two heme-binding domains of heme-regulated eukaryotic initiation factor-2alpha kinase. N terminus and kinase insertion. J Biol Chem. 2000;275:5171–5178. doi: 10.1074/jbc.275.7.5171. [DOI] [PubMed] [Google Scholar]
- Rafie-Kolpin M, Han AP, Chen JJ. Autophosphorylation of threonine 485 in the activation loop is essential for attaining eIF2alpha kinase activity of HRI. Biochemistry. 2003;42:6536–6544. doi: 10.1021/bi034005v. [DOI] [PubMed] [Google Scholar]
- Raghuram S, Stayrook KR, Huang P, Rogers PM, Nosie AK, McClure DB, Burris LL, Khorasanizadeh S, Burris TP, Rastinejad F. Identification of heme as the ligand for the orphan nuclear receptors REV-ERBalpha and REV-ERBbeta. Nat Struct Mol Biol. 2007;14:1207–1213. doi: 10.1038/nsmb1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsden R, Sommer KM, Omiecinski CJ. Phenobarbital induction and tissue-specific expression of the rat CYP2B2 gene in transgenic mice. J Biol Chem. 1993;268:21722–21726. [PubMed] [Google Scholar]
- Rangarajan PN, Padmanaban G. Regulation of cytochrome P-450b/e gene expression by a heme- and phenobarbitone-modulated transcription factor. Proc Natl Acad Sci USA. 1989;86:3963–3967. doi: 10.1073/pnas.86.11.3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravishankar H, Padmanaban G. Regulation of cytochrome P-450 gene expression. Studies with a cloned probe. J Biol Chem. 1985;260:1588–1592. [PubMed] [Google Scholar]
- Regan RF, Chen J, Benvenisti-Zarom L. Heme oxygenase-2 gene deletion attenuates oxidative stress in neurons exposed to extracellular hemin. BMC Neuroscience. 2004;5:34. doi: 10.1186/1471-2202-5-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y, Smith A. Mechanism of metallothionein gene regulation by heme-hemopexin. Roles of protein kinase C, reactive oxygen species, and cis-acting elements. J Biol Chem. 1995;270:23988–23995. doi: 10.1074/jbc.270.41.23988. [DOI] [PubMed] [Google Scholar]
- Ren S, Correia MA. Heme: a regulator of rat hepatic tryptophan 2,3-dioxygenase? Arch Biochem Biophys. 2000;377:195–203. doi: 10.1006/abbi.2000.1755. [DOI] [PubMed] [Google Scholar]
- Rotenberg MO, Maines MD. Isolation, Characterization, and Expression in Escherichia coli of a cDNA Encoding Rat Heme Oxygenase-2. J Biol Chem. 1990;265:7501–7506. [PubMed] [Google Scholar]
- Ryan G, Ades IZ. Coordinate elevations of liver delta-aminolevulinate synthase and cytochrome P-450 RNA by phenobarbital in chicken embryos: the effects of heme. Int J Biochem. 1989;21:1025–31. doi: 10.1016/0020-711x(89)90235-8. [DOI] [PubMed] [Google Scholar]
- Sadano H, Omura T. Reversible transfer of heme between different molecular species of microsome-bound cytochrome P-450 in rat liver. Biochem Biophys Res Commun. 1983;116:1013–1019. doi: 10.1016/s0006-291x(83)80243-5. [DOI] [PubMed] [Google Scholar]
- Sadano H, Omura T. Incorporation of heme to microsomal cytochrome P450 in the absence of protein biosynthesis. J Biochem. 1985;98:1321–1331. doi: 10.1093/oxfordjournals.jbchem.a135399. [DOI] [PubMed] [Google Scholar]
- Sakaguchi M, Mibara K, Sato R. A short amino-terminal segment of microsomal cytochrome P-450 functions both as an insertion signal and as a stop-transfer sequence. EMBO J. 1987;6:2425–2431. doi: 10.1002/j.1460-2075.1987.tb02521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardana MK, Rajamanickam C, Padmanaban G. Differential role of heme in the synthesis of mitochondrial and microsomal hemoprotein. In: Doss M, editor. Porphyrins in Human Diseases; Proc. 1st Int. Porphyrin Meeting Freiburg; 1975; Basel: Karber; 1976. pp. 62–70. [Google Scholar]
- Satoh T, Satoh H, Iwahara S, Hrkal Z, Peyton DH, Muller-Eberhard U. Roles of heme iron-coordinating histidine residues of human hemopexin expressed in baculovirus-infected insect cells. Proc Natl Acad Sci U S A. 1994;91:8423–8427. doi: 10.1073/pnas.91.18.8423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuner D, Patel R, Wang F, Lee K, Kumar K, Wu J, Nilsson A, Karin M, Kaufman RJ. Double-stranded RNA-dependent protein kinase phosphorylation of the alpha-subunit of eukaryotic translation initiation factor 2 mediates apoptosis. J Biol Chem. 2006;281:21458–21468. doi: 10.1074/jbc.M603784200. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell. 2001;7:1165–1176. doi: 10.1016/s1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- Schimke RT, Sweeney EW, Berlin CM. The Roles of synthesis and degradation in the control of rat liver tryptophan pyrrolase. J Biol Chem. 1965;240:322–331. [PubMed] [Google Scholar]
- Schneider S, Marles-Wright J, Sharp KH, Paoli M. Diversity and conservation of interactions for binding heme in b-type heme proteins. Nat Prod Rep. 2007;24:621–630. doi: 10.1039/b604186h. [DOI] [PubMed] [Google Scholar]
- Schuller DJ, Wilks A, Ortiz de Montellano PR, Poulos TL. Crystal structure of human heme oxygenase-1. Nat Struct Biol. 1999;6:860–867. doi: 10.1038/12319. [DOI] [PubMed] [Google Scholar]
- Schutz G, Chow E, Feigelson P. Regulatory properties of hepatic tryptophan oxygenase. J Biol Chem. 1972;247:5333–5337. [PubMed] [Google Scholar]
- Sheftel AD, Kim SF, Ponka P. Non-heme induction of heme oxygenase-1 does not alter cellular iron metabolism. J Biol Chem. 2007;282:10480–10486. doi: 10.1074/jbc.M700240200. [DOI] [PubMed] [Google Scholar]
- Shibahara S, Muller R, Taguchi H, Yoshida T. Cloning and expression of cDNA for rat heme oxygenase. Proc Natl Acad Sci U S A. 1985;82:7865–7869. doi: 10.1073/pnas.82.23.7865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibahara S. The heme oxygenase dilemma in cellular homeostasis: New insights for the feedback regulation of heme catabolism. Tohoku J Exp Med. 2003;200:167–186. doi: 10.1620/tjem.200.167. [DOI] [PubMed] [Google Scholar]
- Sinclair PR, Granick S. Uroporphyrin formation induced by chlorinated hydrocarbons (lindane, polychlorinated biphenyls, tetrachlorodibenzo-p-dioxin). Requirement for endogenous iron, protein synthesis and drug-metabolizing activity. Biochem Biophys Res Commun. 1974;61:124–133. doi: 10.1016/0006-291x(74)90543-9. [DOI] [PubMed] [Google Scholar]
- Sinclair PR, Bement WJ, Gorman N, Liem HH, Wolkoff AW, Muller-Eberhard U. Effect of serum proteins on haem uptake and metabolism in primary cultures of liver cells. Biochem J. 1988;256:159–65. doi: 10.1042/bj2560159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair PR, Schuetz EG, Bement WJ, Haugen SA, Sinclair JF, May BK, Li D, Guzelian PS. Role of heme in phenobarbital induction of cytochromes P450 and 5-aminolevulinate synthase in cultured rat hepatocytes maintained on an extracellular matrix. Arch Biochem Biophys. 1990;282:386–392. doi: 10.1016/0003-9861(90)90133-j. [DOI] [PubMed] [Google Scholar]
- Sinclair PR, Gorman N, Dalton T, Walton HS, Bement WJ, Sinclair JF, Smith AG, Nebert DW. Uroporphyria produced in mice by iron and 5-aminolaevulinic acid does not occur in Cyp1a2(-/-) null mutant mice. Biochem J. 1998;330:149–53. doi: 10.1042/bj3300149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AG, Francis JE. Genetic variation of iron-induced uroporphyria in mice. Biochem J. 1993;291:29–35. doi: 10.1042/bj2910029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AG, Clothier B, Carthew P, Childs NL, Sinclair PR, Nebert DW, Dalton TP. Protection of the Cyp1a2(-/-) null mouse against uroporphyria and hepatic injury following exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Appl Pharmacol. 2001;173:89–98. doi: 10.1006/taap.2001.9167. [DOI] [PubMed] [Google Scholar]
- Srivastava G, Borthwick IA, Maguire DJ, Elferink CJ, Bawden MJ, Mercer JF, May BK. Regulation of 5-aminolevulinate synthase mRNA in different rat tissues. J Biol Chem. 1988;263:5202–9. [PubMed] [Google Scholar]
- Srivastava G, Bawden MJ, Hansen AJ, May BK. Heme may not be a positive regulator of cytochrome-P450 gene expression. Eur J Biochem. 1989;178:689–692. doi: 10.1111/j.1432-1033.1989.tb14499.x. [DOI] [PubMed] [Google Scholar]
- Srivastava G, Hansen AJ, Bawden MJ, May BK. Hemin administration to rats reduces levels of hepatic mRNAs for phenobarbitone-inducible enzymes. Mol Pharmacol. 1990;38:486–493. [PubMed] [Google Scholar]
- Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235:1043–1046. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- Stoltz C, Vachon MH, Trottier E, Dubois S, Paquet Y, Anderson A. The CYP2B2 phenobarbital response unit contains an accessory factor element and a putative glucocorticoid response element essential for conferring maximal phenobarbital responsiveness. J Biol Chem. 1998;273:8528–8536. doi: 10.1074/jbc.273.14.8528. [DOI] [PubMed] [Google Scholar]
- Stoltz C, Anderson A. Positive regulation of the rat CYP2B2 phenobarbital response unit by the nuclear receptor hexamer half-site. nuclear factor 1 complex. Biochem Pharmacol. 1999;57:1073–1076. doi: 10.1016/s0006-2952(98)00367-0. [DOI] [PubMed] [Google Scholar]
- Sugiyama K, Yao K, Rettie AE, Correia MA. Inactivation of rat hepatic cytochrome P-450 isozymes by 3,5-dicarbethoxy-2,6-dimethyl-4-ethyl-1,4-dihydropyridine. Chem Res Toxicol. 1989;2:400–410. doi: 10.1021/tx00012a008. [DOI] [PubMed] [Google Scholar]
- Sultana S, Nirodi CS, Ram N, Prabhu L, Padmanaban G. A 65-kDa protein mediates the positive role of heme in regulating the transcription of CYP2B1/B2 gene in rat liver. J Biol Chem. 1997;272:8895–8900. doi: 10.1074/jbc.272.14.8895. [DOI] [PubMed] [Google Scholar]
- Sun J, Hoshino H, Takaku K, Nakajima O, Muto A, Suzuki H, Tashiro S, Takahashi S, Shibahara S, Alam J, Taketo MM, Yamamoto M, Igarashi K. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J. 2002;21:5216–24. doi: 10.1093/emboj/cdf516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahara T, Sun J, Nakanishi K, Yamamoto M, Mori H, Saito T, Fujita H, Igarashi K, Taketani S. Heme positively regulates the expression of beta-globin at the locus control region via the transcriptional factor Bach1 in erythroid cells. J Biol Chem. 2004;279:5480–5487. doi: 10.1074/jbc.M302733200. [DOI] [PubMed] [Google Scholar]
- Tahara T, Sun J, Igarashi K, Taketani S. Heme-dependent up-regulation of the alpha-globin gene expression by transcriptional repressor Bach1 in erythroid cells. Biochem Biophys Res Commun. 2004;324:77–85. doi: 10.1016/j.bbrc.2004.09.022. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Kinoshita J-Y, Kuroda R, Ito A. Integration of cytochrome b5 into endoplasmic reticulum membrane: Participation of carboxy-terminal portion of the transmembrane domain. J Biochem. 2003;133:247–251. doi: 10.1093/jb/mvg034. [DOI] [PubMed] [Google Scholar]
- Tenhunen R, Marver H, Schmid R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc Natl Acad Sci USA. 1968;61:748–755. doi: 10.1073/pnas.61.2.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenhunen R, Marver HS, Schmid R. Microsomal heme oxygenase, characterization of the enzyme. J Biol Chem. 1969;244:6388–6394. [PubMed] [Google Scholar]
- Tephly TR, Gibbs AH, De Matteis F. Studies on the mechanism of experimental porphyria produced by 3,5-diethoxycarbonyl-1,4-dihydrocollidine: role of a porphyrin-like inhibitor of protohaem ferro-lyase. Biochem J. 1979;180:241–244. doi: 10.1042/bj1800241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tierney DJ, Haas AL, Koop DR. Degradation of cytochrome P450 2E1: Selective loss after labilization of the enzyme. Arch Biochem Biophys. 1992;293:9–16. doi: 10.1016/0003-9861(92)90358-4. [DOI] [PubMed] [Google Scholar]
- Timsit YE, Negishi M. CAR and PXR: the xenobiotic-sensing receptors. Steroids. 2007;72:231–246. doi: 10.1016/j.steroids.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolosano E, Altruda F. Hemopexin: Structure, Function, and Regulation. DNA and Cell Biology. 2002;21:297–306. doi: 10.1089/104454902753759717. [DOI] [PubMed] [Google Scholar]
- Tomlinson EJ, Ferguson SJ. Conversion of a c type cytochrome to a b type that spontaneously forms in vitro from apoprotein and heme: Implications for c type cytochrome biogenesis and folding. Proc Natl Acad Sci USA. 2000;97:5156–5160. doi: 10.1073/pnas.090089397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traugh JA. Heme regulation of hemoglobin synthesis. Sem Hematol. 1989;26:54–62. [PubMed] [Google Scholar]
- Trawick JD, Wright RM, Poyton RO. Transcription of yeast COX6, the gene for cytochrome c oxidase subunit VI, is dependent on heme and on the HAP2 gene. J Biol Chem. 1989;264:7005–7008. [PubMed] [Google Scholar]
- Trottier E, Belzil A, Stoltz C, Anderson A. Localization of a phenobarbital-responsive element (PBRE) in the 5′-flanking region of the rat CYP2B2 gene. Gene. 1995;158:263–268. doi: 10.1016/0378-1119(94)00916-g. [DOI] [PubMed] [Google Scholar]
- Unseld A, De Matteis F. Destruction of endogenous and exogenous haem by 2-allyl-2-isopropylacetamide: role of the liver cytochrome P450 which is inducible by phenobarbitone. Int J Biochem. 1978;9:865–869. doi: 10.1016/0020-711x(78)90061-7. [DOI] [PubMed] [Google Scholar]
- Urquhart AJ, Elder GH, Roberts AG, Lambrecht RW, Sinclair PR, Bement WJ, Gorman N, Sinclair JA. Uroporphyria produced in mice by 20-methylcholanthrene and 5-aminolaevulinic acid. Biochem J. 1988;253:357–62. doi: 10.1042/bj2530357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkateswar V, Padmanaban G. Involvement of heme in the transcriptional activation of CYPIIB1/B2 gene by phenobarbitone in rat liver--studies with succinylacetone. Arch Biochem Biophys. 1991;290:167–172. doi: 10.1016/0003-9861(91)90603-g. [DOI] [PubMed] [Google Scholar]
- Wang J, Ortiz de Montellano PR. The Binding Sites on Human Heme Oxygenase-1 for Cytochrome P450 Reductase and Biliverdin Reductase. J Biol Chem. 2003;278:20069–20076. doi: 10.1074/jbc.M300989200. [DOI] [PubMed] [Google Scholar]
- Wang HF, Figueiredo Pereira ME, Correia MA. Cytochrome P450 3A degradation in isolated rat hepatocytes: 26S proteasome inhibitors as probes. Arch Biochem Biophys. 1999;365:45–53. doi: 10.1006/abbi.1999.1139. [DOI] [PubMed] [Google Scholar]
- Wang WH, Lu JX, Yao P, Xie Y, Huang ZX. The distinct heme coordination environments and heme-binding stabilities of His39Ser and His39Cys mutants of cytochrome b5. Protein Engineering. 2003;16:1047–1054. doi: 10.1093/protein/gzg134. [DOI] [PubMed] [Google Scholar]
- Watkins PB, Wrighton SA, Schuetz EG, Maurel P, Guzelian PS. Macrolide antibiotics inhibit the degradation of the glucocorticoid-responsive cytochrome P-450 in rat hepatocytes in vivo and in primary monolayer culture. J Biol Chem. 1986;261:6264–6271. [PubMed] [Google Scholar]
- Waxman DJ, Azaroff L. Phenobarbital induction of cytochrome P-450 gene expression. Biochem J. 1992;281:577–592. doi: 10.1042/bj2810577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wek RC. eIF-2 kinases: regulators of general and gene-specific translation initiation. Trends Biochem Sci. 1994;19:491–496. doi: 10.1016/0968-0004(94)90136-8. [DOI] [PubMed] [Google Scholar]
- Wek RC, Jiang HY, Anthony TG. Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans. 2006;34:7–11. doi: 10.1042/BST20060007. [DOI] [PubMed] [Google Scholar]
- Whiting MJ, Granick S. Delta-Aminolevulinic acid synthase from chick embryo liver mitochondria. I. Purification and some properties. J Biol Chem. 1976;251:1340–6. [PubMed] [Google Scholar]
- Wilks A, Ortiz de Montellano PR. Rat liver heme oxygenase. High level expression of a truncated soluble form and nature of the meso-hydroxylating species. J Biol Chem. 1993;268:22357–22362. [PubMed] [Google Scholar]
- Wilks A, Black SM, Miller WL, Ortiz de Montellano PR. Expression and Characterization of Truncated Human Heme Oxygenase (hHO-1) and a Fusion Protein of hHO-1 with Human Cytochrome P450 Reductase. Biochemistry. 1995;34:4421–4427. doi: 10.1021/bi00013a034. [DOI] [PubMed] [Google Scholar]
- Williams SN, Dunham E, Bradfield CA. Induction of cytochrome P450 enzymes. In: Ortiz de Montellano PR, editor. Cytochrome P450. Structure, Mechanism, and Biochemistry. Vol. 2004. Plenum Press; New York: 2005. pp. 323–346. and references therein. [Google Scholar]
- Winkler H, Adam G, Mattes E, Schanz M, Hartig A, Ruis H. Co-ordinate control of synthesis of mitochondrial and non-mitochondrial hemoproteins: a binding site for the HAP1 (CYP1) protein in the UAS region of the yeast catalase T gene (CTT1) EMBO J. 1988;7:1799–1804. doi: 10.1002/j.1460-2075.1988.tb03011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woessmann W, Mivechi NF. Role of ERK activation in growth and erythroid differentiation of K562 cells. Exp Cell Res. 2001;264:193–200. doi: 10.1006/excr.2000.5124. [DOI] [PubMed] [Google Scholar]
- Wyman JF, Gollan JL, Settle W, Farell GC, Correia MA. Incorporation of haemoglobin haem into the rat hepatic haemoprotein tryptophan pyrrolase and cytochrome P-450. Biochem J. 1986;238:837–846. doi: 10.1042/bj2380837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Hayashi N, Kikuchi G. Evidence for the transcriptional inhibition by heme of the synthesis of delta-aminolevulinate synthase in rat liver. Biochem Biophys Res Commun. 1982;105:985–90. doi: 10.1016/0006-291x(82)91067-1. [DOI] [PubMed] [Google Scholar]
- Yao K, Correia MA. Enhanced proteolytic loss of rat hepatic tryptophan dioxygenase (TO) after drug-induced heme depletion. FASEB J. 1991;5:A1541. [Google Scholar]
- Yin L, Wu N, Curtin JC, Qatanani M, Szwergold NR, Reid RA, Waitt GM, Parks DJ, Pearce KH, Wisely GB, Lazar MA. Rev-erbalpha, a heme sensor that coordinates metabolic and circadian pathways. Science. 2007;318:1786–1789. doi: 10.1126/science.1150179. [DOI] [PubMed] [Google Scholar]
- Yu CA, Gunsalus IC. Cytochrome P450cam. III. Removal and replacement of ferriprotoporphyrin IX. J Biol Chem. 1974;249:107–110. [PubMed] [Google Scholar]
- Zgoda VG, Arison B, Mkrtchian S, Ingelman-Sundberg M, Correia MA. Hemin-mediated restoration of allylisopropylacetamide-inactivated CYP2B1: a role for glutathione and GRP94in the heme–protein assembly. Arch Biochem Biophys. 2002;408:58–68. doi: 10.1016/s0003-9861(02)00489-7. [DOI] [PubMed] [Google Scholar]
- Zhang L, Guarente L. Heme binds to a short sequence that serves a regulatory function in diverse proteins. Embo J. 1995;14:313–320. doi: 10.1002/j.1460-2075.1995.tb07005.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, McGrath B, Li S, Frank A, Zambito F, Reinert J, Gannon M, Ma K, McNaughton K, Cavener DR. The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol Cell Biol. 2002a;22:3864–3874. doi: 10.1128/MCB.22.11.3864-3874.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, McGrath BC, Reinert J, Olsen DS, Lei L, Gill S, Wek SA, Vattem KM, Wek RC, Kimball SR, Jefferson LS, Cavener DR. The GCN2 eIF2alpha kinase is required for adaptation to amino acid deprivation in mice. Mol Cell Biol. 2002b;22:6681–6688. doi: 10.1128/MCB.22.19.6681-6688.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YK, Yeager RL, Klaassen CD. Circadian expression profiles of drug-processing genes and transcription factors in mouse liver. Drug Metab Dispos. 2009;37:106–115. doi: 10.1124/dmd.108.024174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Shan Y, Lambrecht RW, Donohue SE, Bonkovsky HL. Differential regulation of human ALAS1 mRNA and protein levels by heme and cobalt protoporphyrin. Mol Cell Biochem. 2008;319:153–61. doi: 10.1007/s11010-008-9888-0. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Sun Y, Jin K, Greenberg DA. Hemin induces neuroglobin expression in neural cells. Blood. 2002b;100:2494–2498. doi: 10.1182/blood-2002-01-0280. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Silverman RB. Revisiting heme mechanisms. A perspective on the mechanisms of nitric oxide synthase (NOS), heme oxygenase (HO), and cytochrome P450s (CYP450s) Biochemistry. 2008;47:2231–2243. doi: 10.1021/bi7023817. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Hon T, Ye W, Zhang L. Heme deficiency interferes with the Ras-mitogen-activated protein kinase signaling pathway and expression of a subset of neuronal genes. Cell Growth Differ. 2002a;13:431–439. [PubMed] [Google Scholar]
- Zhukov A, Ingelman-Sundberg M. Relationship between cytochrome P450 catalytic cycling and stability: fast degradation of ethanol-inducible cytochrome P450 2E1 (CYP2E1) in hepatoma cells is abolished by inactivation of its electron donor NADPH-cytochrome P450 reductase. Biochem J. 1999;340:453–458. [PMC free article] [PubMed] [Google Scholar]