


Abstract
RNA is known to be involved in several cellular processes; however, it is only active when it is folded into its correct 3D conformation. The folding, bending and twisting of an RNA molecule is dependent upon the multitude of canonical and non-canonical secondary structure motifs. These motifs contribute to the structural complexity of RNA but also serve important integral biological functions, such as serving as recognition and binding sites for other biomolecules or small ligands. One of the most prevalent types of RNA secondary structure motifs are single mismatches, which occur when two canonical pairs are separated by a single non-canonical pair. To determine sequence–structure relationships and to identify structural patterns, we have systematically located, annotated and compared all available occurrences of the 30 most frequently occurring single mismatch-nearest neighbor sequence combinations found in experimentally determined 3D structures of RNA-containing molecules deposited into the Protein Data Bank. Hydrogen bonding, stacking and interaction of nucleotide edges for the mismatched and nearest neighbor base pairs are described and compared, allowing for the identification of several structural patterns. Such a database and comparison will allow researchers to gain insight into the structural features of unstudied sequences and to quickly look-up studied sequences.
INTRODUCTION
RNA is known to perform a variety of biological functions and to be involved in several cellular processes; however, it is only active when in its correct 3D conformation. The structural complexity and wide repertoire of structural components of RNA allows this biomolecule to effectively carry out a multitude of key functions. RNA consists of canonical double helical regions, along with non-canonical regions, such as internal loops, bulges, hairpins and multi-branch loops, which have implications for folding and stability of the correct tertiary and quaternary structures. Often times, these motifs are important for a variety of biological functions, such as serving as binding sites for proteins (1–10), metals (11–13), small molecules (14–19), or other nucleic acids (20). The scaffold of RNA tertiary structure is a result of the secondary structural components, which introduce kinks and turns in the RNA structure while providing available hydrogen bond donor and/or acceptor sites allowing for intermolecular interactions. Therefore, an understanding of the 3D conformation of RNA secondary structure motifs will give insight into RNA function.
An understanding of the structural propensities of common RNA secondary structure motifs should improve the prediction of RNA structure, function and recognition (21). Much work has been done to improve the prediction of RNA secondary structure from sequence (22–31), and methods are being developed to predict RNA tertiary structure (32–39). While the methods of NMR, crystallography and cryo-electron microscopy provide definitive tertiary structure information, they are not capable of keeping pace with the discovery of new and interesting RNA sequences. However, these tools have revealed a wide range of base pairing geometries commonly found in RNA (40,41). These different geometries have been shown to contribute to the complexity of RNA tertiary structure (42,43). Therefore, an understanding of these base–base conformations may allow for further understanding and accuracy in the prediction of RNA secondary and tertiary structure. One possible approach to begin developing a method to predict tertiary structure of RNA is to identify structural patterns for a given motif by structurally characterizing each occurrence of that motif in available 3D structures. Such structures have been deposited into the Protein Data Bank (PDB) (44–48), a world-wide archive of structural data of biomolecules, which includes all RNA structures solved by NMR, crystallography and cryo-electron microscopy. Currently, there are over 1600 structures containing RNA in the PDB (44–48) (accessed on 12 August 2009).
The structural characterization and comparison of all structures containing a particular secondary structure motif is not a trivial task; however, several laboratories have made significant contributions to analyzing RNA motifs found in the structures deposited in the PDB (44–48). The Fox laboratory has developed an internet-based, interactive database of non-canonical base pairs found in known RNA structures (NCIR). It contains over 2000 non-canonical base pairs with descriptions of the associated structural properties, such as sequence context, sugar pucker and glycosidic bond orientation (49,50). The Olson laboratory has also developed a user friendly internet-based database [the RNA base-pair structure (BPS) database] of canonical and non-canonical base pairs found in determined RNA structures. It contains over 91 000 bp and approximately 4000 higher-order base interactions. The database provides representative figures of the observed spatial patterns and the annotation of the structural and chemical features for each base pair (51). The Gutell laboratory has contributed a significant amount of data by investigating the occurrence and diversity of various motifs (52–54). The laboratories of Leontis and Westhof have provided a standardized method for the naming and classification of the various orientations of RNA base pairs to allow for unambiguous communication (55–62). The Brenner and Holbrook laboratories have developed the Structural Classification of RNA (SCOR) database, which provides details about the 3D structure, function, tertiary interactions and phylogentic relationships of RNA secondary structure motifs (63–65). The Major laboratory has developed computational tools which are compliant with the RNA ontology (66) and are incorporated into the computer program, MC-Annotate, which is capable of interpreting and labeling RNA base pairs and base stacking interactions of a given 3D structure (67–69). The Major laboratory has also developed the computer program MC-Search, which determines the locations of user-defined structural motifs in RNA (69–71). These efforts have advanced the understanding of the structural details of RNA and have provided tools to analyze RNA tertiary structure. However, with the exception of the recent structural characterization of hairpin triloops (69), no effort has been put forth to systematically locate, annotate and compare occurrences of a particular RNA secondary structure motif.
This work is focused on systematically locating, annotating and comparing the most frequently occurring RNA single mismatches in nature. Single mismatches are known to be the most frequently occurring secondary structure motif in ribosomal RNA (72) and often times serve integral structural and/or functional roles (73–83). Using the computer search algorithm MC-Search, single mismatches have been located in the deposited structures found in the PDB. The structural characteristics of each occurrence were then objectively annotated using MC-Annotate. The resulting data for each located and annotated single mismatch were exported into Microsoft Excel to allow for the extraction of the most frequently occurring single mismatch-nearest neighbor sequence combinations (84). Hydrogen bonding, stacking and interaction of nucleotide edges for the mismatched and nearest neighbor base pairs are described and compared, allowing for the identification of several structural patterns. Such a database and comparison will allow researchers to gain insight into the structural features of unstudied sequences and quickly look-up studied sequences. It is important to distinguish this work from previous databases, such as the NCIR and BPS databases. Both the NCIR and BPS databases contain structure information about non-canonical pairs in all secondary structure motifs. This work focuses on non-canonical pairs in single mismatches exclusively, allowing for the identification of structural patterns specific to isolated non-canonical pairs.
MATERIALS AND METHODS
Creation of a 3D RNA structure database
To create a database of previously solved RNA 3D structures, the PDB was searched for molecules containing RNA using the Molecule/Chain Type (since changed to Macromolecule Type) query in the Advanced Search menu on the PDB website (44–48) and selecting the molecules to contain RNA. All query results were selected and downloaded as uncompressed, .pdb formatted files. This search was conducted on 12 August 2009 and, therefore, includes all RNA-containing structures deposited into the PDB up to this date. The search was not limited by experimental method or resolution, but the resulting data is limited by the quality of the data deposited into the PDB.
Single mismatch database
The programs MC-Search (69–71) and MC-Annotate (67–69) were utilized to create the single mismatch database, and it is important to note they were not modified from the version provided by the authors. MC-Search (version 0.5) (69–71) was used to locate all single mismatches in the 3D structure database. In order to search 3D structures to locate a secondary structure motif, MC-Search requires an input descriptor (Figure 1). In simple terms, the input descriptor defines the size and type of the secondary structure motifs of interest. In order to define a single mismatch, 6 nt are involved, the 2 nt in the mismatch and the 2 nt in each of the 2 bp on either side of the mismatch. The type of interaction between the 2 nt in each pair was defined in the input descriptor, thereby limiting the nearest neighbor pairs to canonical pairs and the mismatch pair to a non-canonical pair. The pairing relations for the MC-Search input descriptor are defined by Roman (85–87) and Arabic (88,89) numerals, which indicate the presence of two or three hydrogen bonds and bifurcated or single hydrogen bonds, respectively. For example, Roman numeral XX (85–87) represents an A-U base pair with two hydrogen bonds (from A-N6 to U-NH3 and A-NH6 to U-O4) between the Watson–Crick face of each base with a cis-glycosidic bond orientation. Other Roman numerals represent other pairs in a similar fashion (85–87). Arabic numeral 51 (88,89) represents an A-U base pair with one hydrogen bond (A-NH6 to U-O4) between the Watson–Crick face of each base with a trans-glycosidic bond orientation. Other Arabic numerals represent other pairs in a similar fashion (88,89).
Figure 1.

Single mismatch graph (top) and MC-Search input descriptor (bottom). The nucleotides are numbered A1 to A3 and B1 to B3 in the 5′ to 3′ direction. The ‘A’ and ‘B’ letter designations specify opposing RNA strands. The letter ‘N’ represents any nucleotide. The input descriptor identifies the canonical nearest neighbors by limiting the allowed pairing interactions to the canonical pairs defined by the Roman (85–87) and Arabic (88,89) numerals. Not all possible numerals for A–U, U–A, G–C, C–G, G–U and U–G pairs are shown here due to space limitations. The input descriptor identifies the mismatched nucleotides by allowing an interaction defined by no hydrogen bonds, while also prohibiting the canonical pairing interactions defined by the Roman and Arabic numerals.
For the nearest neighbor pair, any pair described by the Roman or Arabic numeral naming system of base pairs was allowed, thereby allowing most conformations of G-C, C-G, A-U, U-A, G-U and U-G pairs. Conversely, the mismatch nucleotides were defined as any pair not described by the Roman and Arabic numeral naming system of base pairs, thereby disallowing the pairs previously listed. Once the input descriptor contained this information, MC-Search was able to locate all of the single mismatches in the three dimensional RNA structural database. For each single mismatch located in this manner, the nucleotides involved in the single mismatch-nearest neighbor sequence combination were ‘clipped’ (i.e. all nucleotides not involved in the single mismatch or nearest neighbor were removed) and saved as a .pdb file to allow for quick annotation and a simple 3D graphic to be produced.
Once the results from the MC-Search and MC-Annotate scripts were tabulated, the results were searched for false-positives. A false-positive results, for example, when MC-Annotate does not annotate a G-C pair with a Roman or Arabic numeral. As a result, this G-C pair is considered a single mismatch. All G-C, C-G, A-U, U-A, G-U and U-G identified by the scripts as single mismatches were considered false positives and were removed from the database of true single mismatches.
Single mismatch annotation
The located single mismatches were structurally characterized by the program MC-Annotate (version 1.6.2) (67–69), which analyzes the atomic coordinates to determine the nucleotide interactions and classifies the type of base pairing. MC-Annotate utilizes four characterization parameters which include: (i) residue conformation, (ii) adjacent stackings, (iii) non-adjacent stackings and (iv) base-pairs. The residue conformation defines the sugar pucker as endo or exo and the glycosidic bond orientation as syn or anti. The adjacent and non-adjacent stackings define the relative orientation of each base, which are identified by MC-Annotate utilizing the method proposed by Gabb et al. (90). The nomenclature used to describe these orientations was proposed by Major and Thibault (91), which includes four base-stacking types: upward, downward, outward and inward. The nomenclature incorporated to illustrate the base pairing annotations is based on the Leontis and Westhof (56,57) classification scheme, which describes the interacting edges [i.e. the Watson–Crick (W), Hoogsteen (H) and Sugar (S) edges] of the two bases. This scheme has been further defined and described previously by Lemieux and Major (68). The resulting data for each located and annotated single mismatch were exported into Microsoft Excel.
Analysis of data and identification of structural patterns
Due to the excessive amount of data generated from the search and annotation (4899 single mismatches identified), the analysis of the data and the identification of structural patterns focused on the 30 most frequently occurring single mismatches in nature (84). To allow for the extraction of the most frequently occurring single mismatch-nearest neighbor sequence combinations (84) and further allow for the identification of structural patterns, the Leontis and Westhof (56,57) naming scheme was utilized when determining general structural trends and patterns because annotation is subject to interpretation and small geometrical variations (32), which could arise due to experimental conditions.
It is important to note some single mismatches have been excluded from the following analysis. In order to prevent over-counting and to simplify the analysis, ensembles of structures determined by NMR were excluded from the analysis. PDB structures consisting of a single averaged NMR structure, however, were included. Several clipped PDB files were not included in the analysis for various reasons (i.e. 13 single mismatch containing PDB files were not in the correct .pdb format, which prevented nucleotide annotation by MC-Annotate). These PDB files are denoted in Supplementary Table S1. Lastly, it is important to note the structural trends and patterns may be skewed due to repetitive representation of a molecule in the PDB. For example, the crystal structure of the large ribosomal subunit of Haloarcula marismortui has been solved unbound (PDB I.D. 1ffk) and bound (PDB I.D. 1n8r) to antibiotics.
RESULTS
3D RNA structure database
The PDB (44–48) search returned 1666 RNA-containing structures which were then used to create the 3D RNA structure database. A complete listing of the obtained structures can be found in the Supplementary Data (Supplementary Table S1).
Single mismatch structural database
Incorporation of a single mismatch-specific input descriptor into the MC-Search (69–71) program followed by a search of the structures contained in the 3D RNA structure database returned an extremely large dataset. Each of these 4899 identified single mismatches were structurally characterized using MC-Annotate. Of the 30 most frequently occurring single mismatches in a secondary structure database (84), 21 were located in the 3D structure database (Table 1 and Supplementary Table S2) and are the focus of the rest of this study. The nine frequently occurring single mismatch-nearest neighbor sequences (84) not found in the structural database were: 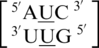 ,
, 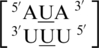 ,
, 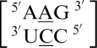 ,
, 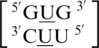 ,
, 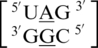 ,
,  ,
, 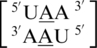 ,
,  and
and 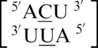 , with frequencies of 94, 62, 54, 43, 38, 38, 34, 34 and 34, respectively (84). For each of the remaining single mismatch-nearest neighbor combinations found in the top 30 (84), a wide variance in the number of times they were found in the structural database resulted (Table 1). Single mismatches were found in a wide repertoire of RNAs, including ribosomal RNAs (free and bound to antibiotics and proteins), riboswitches, tRNAs and viral RNAs.
, with frequencies of 94, 62, 54, 43, 38, 38, 34, 34 and 34, respectively (84). For each of the remaining single mismatch-nearest neighbor combinations found in the top 30 (84), a wide variance in the number of times they were found in the structural database resulted (Table 1). Single mismatches were found in a wide repertoire of RNAs, including ribosomal RNAs (free and bound to antibiotics and proteins), riboswitches, tRNAs and viral RNAs.
Table 1.
Summary of the structural orientation and interaction of the 30 frequently occurring single mismatchesa
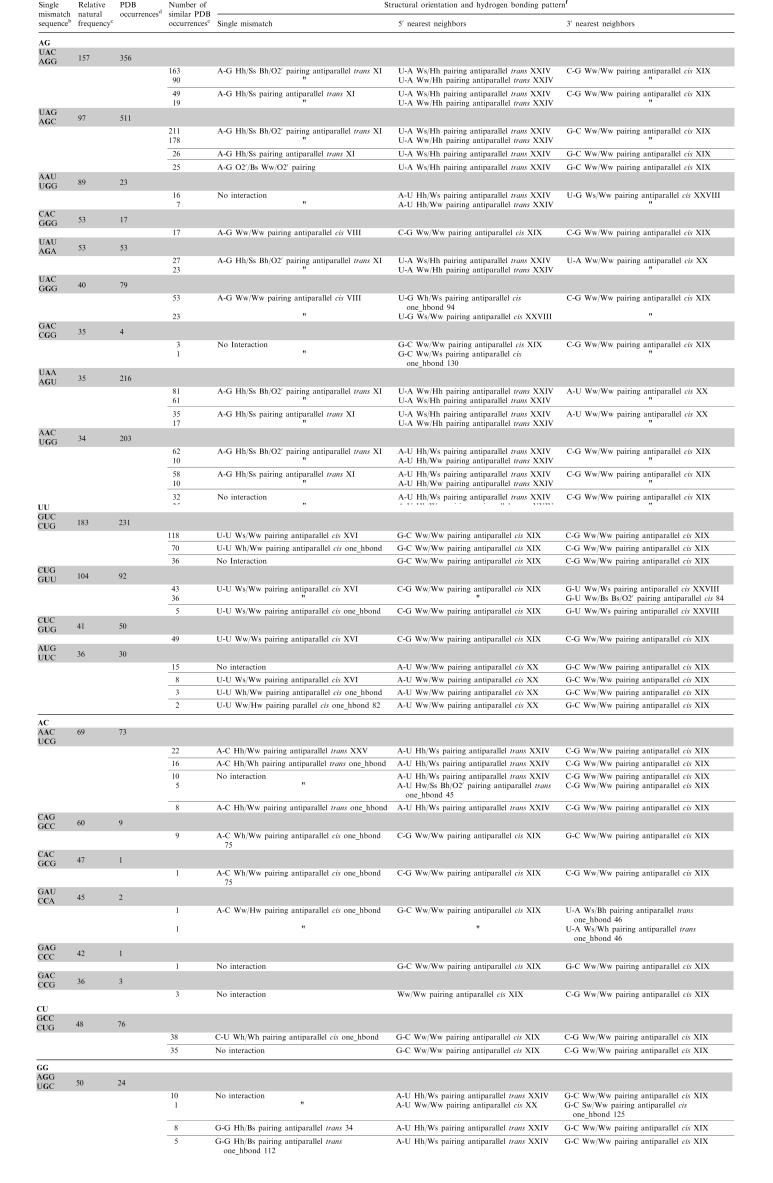 |
aAll possible orientations and hydrogen bonding patterns are not shown for each single mismatch-nearest neighbor combination. Only those representing at least 5% of total occurrences are included.
bFor each sequence, the top strand is written 5′–3′, and the bottom strand is written 3′–5′. Duplexes are written in alphabetical order by the loop nucleotide (A over G, not G over A). If the loop nucleotides are identical, then duplexes are written in alphabetical order by the nearest neighbors (CUG over GUU, not GUU over CUG).
cFrequency of occurrence in the database (84).
dNumber of times each single mismatch-nearest neighbor sequence combination was located in the three dimensional RNA structure database compiled from structures deposited into the PDB.
eNumber of occurrences in each subclass, which is determined among each sequence combination, considering four parameters: interacting edges for the single mismatch nucleotides and the nearest neighbor base pairs and hydrogen bond patterns for the single mismatch nucleotides and the nearest neighbor base pairs.
fAnnotated orientations and hydrogen bonding patterns of the single mismatch and 5′- and 3′-nearest neighbor nucleotides, which is described in ‘Materials and Methods’ section.
Due to the immense amount of data collected, a table summarizing the common structural characteristics for each single mismatch-nearest neighbor sequence combination in the top 30 (84) is provided in Table 1 and Supplementary Table S2. To determine structural classes, or specimens (69), among each sequence combination, four parameters were considered: interacting edges for both the single mismatch nucleotides and the nearest neighbor base pairs and hydrogen bond patterns for both the single mismatch nucleotides and the nearest neighbor base pairs. Interactions involving a mismatched nucleotide and a nearest neighbor nucleotide were only considered when occurring in >5% of the total population for each single mismatch-nearest neighbor sequence combination.
DISCUSSION
A·G single mismatches
A·G single mismatches are the most frequently occurring single mismatch type found in the secondary structure database (84) when categorized by only the mismatched nucleotides. There are 10 A·G mismatch-nearest neighbor sequence combinations found in the 30 most frequently occurring single mismatches (84), and nine are represented in the RNA single mismatch structural database (Table 1 and Supplementary Table S2), with a total of 1462 occurrences. These nine can be divided into three groups based upon the geometric configuration of the mismatch nucleotides. The first group consists of the most common geometric orientation of the mismatched nucleotides, 5′(A)H/3′(G)S pairing, antiparallel, trans glycosidic bond conformation, with 83% of the total occurrences found with these characteristics (Figure 2). 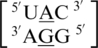 ,
, 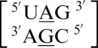 ,
, 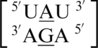 ,
, 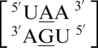 and
and 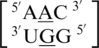 are the five sequence combinations with these geometric features, and, interestingly, they each contain a U-A or A-U base pair on the 5′ side of the A·G mismatch. Considering these five single mismatch-nearest neighbor sequence combinations, the most common base-pair orientation and hydrogen bonding pattern of the 5′ and 3′ nearest neighbors are 5′(U)W/3′(A)H pairing, antiparallel, trans XXIV and 5′W/3′W pairing, antiparallel, cis XIX, respectively. Although the orientation of the 5′ nearest neighbors are reversed for
are the five sequence combinations with these geometric features, and, interestingly, they each contain a U-A or A-U base pair on the 5′ side of the A·G mismatch. Considering these five single mismatch-nearest neighbor sequence combinations, the most common base-pair orientation and hydrogen bonding pattern of the 5′ and 3′ nearest neighbors are 5′(U)W/3′(A)H pairing, antiparallel, trans XXIV and 5′W/3′W pairing, antiparallel, cis XIX, respectively. Although the orientation of the 5′ nearest neighbors are reversed for 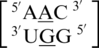 (A–U instead of U–A), the A–U pair still exhibits a 5′(U)W/3′(A)H pair. It is interesting to note the 5′ A–U or U–A nearest neighbor does not have the expected 5′W/3′W pairing. Perhaps this is due to the structural perturbation resulting from the accommodation of the A·G mismatch, a purine–purine mismatch. The helical geometry may be disrupted to accommodate this type of noncanonical base pair. However, it is unclear why the 3′ nearest neighbor is not similarly disrupted.
(A–U instead of U–A), the A–U pair still exhibits a 5′(U)W/3′(A)H pair. It is interesting to note the 5′ A–U or U–A nearest neighbor does not have the expected 5′W/3′W pairing. Perhaps this is due to the structural perturbation resulting from the accommodation of the A·G mismatch, a purine–purine mismatch. The helical geometry may be disrupted to accommodate this type of noncanonical base pair. However, it is unclear why the 3′ nearest neighbor is not similarly disrupted.
Figure 2.

Representation of an A·G mismatch in the 5′(A)H/3′(G)S pairing, antiparallel, trans orientation with XI hydrogen bonding pattern (PDB ID 1C04), which is the most common orientation and interaction determined for the most frequently occurring A·G mismatch-nearest neighbor combinations (84) that were also represented in the PDB.
The second group of A·G mismatches consist of mismatch nucleotides with 5′(A)W/3′(G)W pairing, antiparallel, cis orientation forming two hydrogen bonds in the VIII pattern. 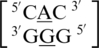 and
and 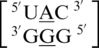 are the two sequence combinations with these geometric features. They have similar nearest neighbors, with 5′Y/3′G (where Y is a pyrimidine) and 5′C/3′G on the 5′ and 3′ side of the A·G single mismatch, respectively. The 5′ and 3′ nearest neighbors are both characterized as 5′W/3′W pairing, antiparallel, cis XIX.
are the two sequence combinations with these geometric features. They have similar nearest neighbors, with 5′Y/3′G (where Y is a pyrimidine) and 5′C/3′G on the 5′ and 3′ side of the A·G single mismatch, respectively. The 5′ and 3′ nearest neighbors are both characterized as 5′W/3′W pairing, antiparallel, cis XIX.
The third group of A·G mismatches consists of mismatch nucleotides which are annotated not to form any interactions with each other. 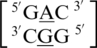 and
and 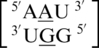 are the two sequence combinations with these geometric features. No interactions are found between the A·G mismatch nucleotides in
are the two sequence combinations with these geometric features. No interactions are found between the A·G mismatch nucleotides in 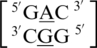 because the A is flipped out from the center of the helix and is interacting with the surrounding solvent. The nucleotides of the base pairing nearest neighbors for
because the A is flipped out from the center of the helix and is interacting with the surrounding solvent. The nucleotides of the base pairing nearest neighbors for 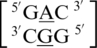 were most commonly annotated to both be in the 5′W/3′W pairing, antiparallel, cis orientation forming three hydrogen bonds in the XIX pattern (one of the four examples was annotated to form only one hydrogen bond in the 130 base-pairing pattern). Although
were most commonly annotated to both be in the 5′W/3′W pairing, antiparallel, cis orientation forming three hydrogen bonds in the XIX pattern (one of the four examples was annotated to form only one hydrogen bond in the 130 base-pairing pattern). Although 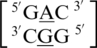 contains similar nearest neighbor sequence combinations and geometries as
contains similar nearest neighbor sequence combinations and geometries as 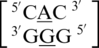 (discussed above in the second group), the geometry of the single mismatch is different.
(discussed above in the second group), the geometry of the single mismatch is different. 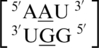 also is annotated not to have any interactions between the mismatched nucleotides; however, the geometries of the 5′ and 3′ nearest neighbors are the same as those in the first group discussed above, 5′(U)W/3′(A)H pairing, antiparallel, trans XXIV and 5′(U)W/3′(G)W pairing, antiparallel, cis XIX, respectively.
also is annotated not to have any interactions between the mismatched nucleotides; however, the geometries of the 5′ and 3′ nearest neighbors are the same as those in the first group discussed above, 5′(U)W/3′(A)H pairing, antiparallel, trans XXIV and 5′(U)W/3′(G)W pairing, antiparallel, cis XIX, respectively.
Inter- and intra-strand interactions involving a mismatched nucleotide and a nearest neighbor nucleotide were found to occur prevalently in eight of the nine A·G mismatch-nearest neighbor sequence combinations (data not shown). The sequence without these types of interactions is 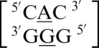 , and it is unclear why this A·G mismatch does not engage in these types of interactions. Characterizing the single mismatch-nearest neighbor sequences as
, and it is unclear why this A·G mismatch does not engage in these types of interactions. Characterizing the single mismatch-nearest neighbor sequences as 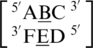 , all eight involved an inter-strand interaction between nucleotides A and E. The sequence combinations of
, all eight involved an inter-strand interaction between nucleotides A and E. The sequence combinations of 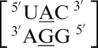 and
and 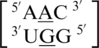 also formed an intra-strand interaction between nucleotides B and C through the O2P/Bh (i.e. one of the free oxygen atoms at the phosphorous between nucleotides B and C is the hydrogen bond acceptor which forms a bifurcated hydrogen bond with the two amino hydrogen atoms found on the Hoogsteen edge of the C) adjacent pairing with upward stacking. It is interesting to note, these two sequences only differ by the orientation of their 5′ nearest neighbor. The sequences
also formed an intra-strand interaction between nucleotides B and C through the O2P/Bh (i.e. one of the free oxygen atoms at the phosphorous between nucleotides B and C is the hydrogen bond acceptor which forms a bifurcated hydrogen bond with the two amino hydrogen atoms found on the Hoogsteen edge of the C) adjacent pairing with upward stacking. It is interesting to note, these two sequences only differ by the orientation of their 5′ nearest neighbor. The sequences  and
and 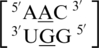 formed an intra-strand interaction between nucleotides F and E, and
formed an intra-strand interaction between nucleotides F and E, and 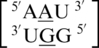 has an additional intra-strand interaction between nucleotides E and D. These types of interactions may contribute to single mismatch stability and are, therefore, important to understand and further study their effects.
has an additional intra-strand interaction between nucleotides E and D. These types of interactions may contribute to single mismatch stability and are, therefore, important to understand and further study their effects.
An interesting structural and thermodynamic comparison is found for the two mismatch-nearest neighbor sequence combinations of 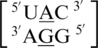 and
and 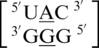 , which only differ by the identity of the 5′ nearest-neighbor, U-A versus U-G, respectively; however, they have experimental free energy values of −0.6 and 1.2 kcal/mol (84). There are 356 examples of
, which only differ by the identity of the 5′ nearest-neighbor, U-A versus U-G, respectively; however, they have experimental free energy values of −0.6 and 1.2 kcal/mol (84). There are 356 examples of 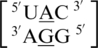 found in the structural database, and the 5′ nearest neighbor, A·G mismatch and the 3′ nearest neighbor nucleotides are annotated to have the following characteristics in 90% of these occurrences: 5′(U)W/3′(A)H pairing antiparallel trans XXIV (two hydrogen bonds), 5′(A)H/3′(G)S pairing antiparallel trans XI (two hydrogen bonds) and 5′(C)W/3′(G)W pairing antiparallel cis XIX (three hydrogen bonds), respectively. Additionally, this mismatch-nearest neighbor sequence generally forms intra- and inter-strand interactions, which are described above. There are 79 examples of
found in the structural database, and the 5′ nearest neighbor, A·G mismatch and the 3′ nearest neighbor nucleotides are annotated to have the following characteristics in 90% of these occurrences: 5′(U)W/3′(A)H pairing antiparallel trans XXIV (two hydrogen bonds), 5′(A)H/3′(G)S pairing antiparallel trans XI (two hydrogen bonds) and 5′(C)W/3′(G)W pairing antiparallel cis XIX (three hydrogen bonds), respectively. Additionally, this mismatch-nearest neighbor sequence generally forms intra- and inter-strand interactions, which are described above. There are 79 examples of 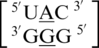 found in the structural database, and the 5′ nearest neighbor, A·G mismatch and the 3′ nearest neighbor nucleotides are annotated to have the following characteristics in 67% of these occurrences: 5′(U)W/3′(G)W pairing antiparallel cis one_hbond (one hydrogen bond), 5′(A)W/3′(G)W pairing antiparallel cis VII (two hydrogen bonds), and 5′(C)W/3′(G)W pairing antiparallel cis XIX (three hydrogen bonds), respectively. It is important to note another 29% of the occurrences of
found in the structural database, and the 5′ nearest neighbor, A·G mismatch and the 3′ nearest neighbor nucleotides are annotated to have the following characteristics in 67% of these occurrences: 5′(U)W/3′(G)W pairing antiparallel cis one_hbond (one hydrogen bond), 5′(A)W/3′(G)W pairing antiparallel cis VII (two hydrogen bonds), and 5′(C)W/3′(G)W pairing antiparallel cis XIX (three hydrogen bonds), respectively. It is important to note another 29% of the occurrences of 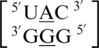 have similar structural characteristics and only differ by the hydrogen bonding pattern of the 5′ nearest neighbor, which is annotated to be XXVIII (two hydrogen bonds). However, this mismatch-nearest neighbor sequence is not annotated to engage in intra- and inter-strand interactions. Comparing the structural and interaction differences between these two mismatch-nearest neighbor sequences to the difference in free energy contribution of the respective single mismatches to duplex stability, it is unclear what the major contributing factor is that is resulting in such a large difference in thermodynamic stability. However, the additional stability of
have similar structural characteristics and only differ by the hydrogen bonding pattern of the 5′ nearest neighbor, which is annotated to be XXVIII (two hydrogen bonds). However, this mismatch-nearest neighbor sequence is not annotated to engage in intra- and inter-strand interactions. Comparing the structural and interaction differences between these two mismatch-nearest neighbor sequences to the difference in free energy contribution of the respective single mismatches to duplex stability, it is unclear what the major contributing factor is that is resulting in such a large difference in thermodynamic stability. However, the additional stability of 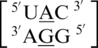 may partially be a result of the additional intra- and inter-strand hydrogen bonding.
may partially be a result of the additional intra- and inter-strand hydrogen bonding.
U·U single mismatches
There are seven U·U RNA single mismatch-nearest neighbor combinations found in the top 30 naturally occurring single mismatches (84), and four of these combinations, which include 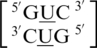 ,
, 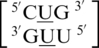 ,
, 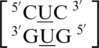 and
and 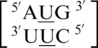 , are represented in the RNA single mismatch structural database with a total of 403 occurrences (Table 1). Comparing these sequence combinations, the most common orientation of mismatch and nearest neighbor nucleotides for each are similar. Most commonly, the U·U mismatch nucleotides adopt the 5′W/3′W pairing, antiparallel, cis conformation in 344 (85%) of the occurrences. When the U·U mismatches are found in this orientation, XVI and one_hbond (note this hydrogen bonding pattern has not been defined by an Arabic numeral in the literature) are the two hydrogen bonding patterns observed for 257 (75%) (Figure 3) and 87 (25%) of these occurrences, respectively. Also, when only considering this U·U conformation, 343 (∼100%) and 302 (88%) of the 5′ and 3′ nearest neighbor base pairs, respectively, are interacting in the 5′Ww/3′Ww pairing, antiparallel, cis XIX orientation. Interestingly, the 5′ nearest neighbors vary in sequence identity, including G-C, C-G and A-U, but they are all observed with the same type of orientation and interaction. The 3′ nearest neighbors also vary in sequence identity, including G-C, C-G and G-U; however, the 3′ nearest neighbor of the sequence combination
, are represented in the RNA single mismatch structural database with a total of 403 occurrences (Table 1). Comparing these sequence combinations, the most common orientation of mismatch and nearest neighbor nucleotides for each are similar. Most commonly, the U·U mismatch nucleotides adopt the 5′W/3′W pairing, antiparallel, cis conformation in 344 (85%) of the occurrences. When the U·U mismatches are found in this orientation, XVI and one_hbond (note this hydrogen bonding pattern has not been defined by an Arabic numeral in the literature) are the two hydrogen bonding patterns observed for 257 (75%) (Figure 3) and 87 (25%) of these occurrences, respectively. Also, when only considering this U·U conformation, 343 (∼100%) and 302 (88%) of the 5′ and 3′ nearest neighbor base pairs, respectively, are interacting in the 5′Ww/3′Ww pairing, antiparallel, cis XIX orientation. Interestingly, the 5′ nearest neighbors vary in sequence identity, including G-C, C-G and A-U, but they are all observed with the same type of orientation and interaction. The 3′ nearest neighbors also vary in sequence identity, including G-C, C-G and G-U; however, the 3′ nearest neighbor of the sequence combination 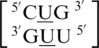 is observed to always have the same orientation but with the two different hydrogen bonding patterns of XIX (forming three hydrogen bonds) and XXVIII (forming two hydrogen bonds) for 40 (44%) and 50 (56%) of the occurrences, respectively.
is observed to always have the same orientation but with the two different hydrogen bonding patterns of XIX (forming three hydrogen bonds) and XXVIII (forming two hydrogen bonds) for 40 (44%) and 50 (56%) of the occurrences, respectively.
Figure 3.

Representation of a U·U mismatch in the 5′(U)W/3′(U)W pairing, antiparallel, cis orientation with XVI hydrogen bonding pattern (PDB ID 1FJG), which is the most common orientation and interaction determined for the most frequently occurring U·U mismatch-nearest neighbor combinations (84) that were also represented in the PDB.
It is interesting to note for these four U·U single mismatch-nearest neighbor sequence combinations, 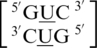 ,
, 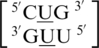 ,
, 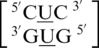 and
and 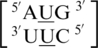 , there is at least one occurrence found for each where the U·U mismatch nucleotides are found to have no interaction with each other and are observed to be flipped-out from the center of the helix or to be positioned in such a way where hydrogen bonding is not possible through the 5′W/3′W paring type (data not shown). Furthermore, U·U mismatch nucleotides involved in the
, there is at least one occurrence found for each where the U·U mismatch nucleotides are found to have no interaction with each other and are observed to be flipped-out from the center of the helix or to be positioned in such a way where hydrogen bonding is not possible through the 5′W/3′W paring type (data not shown). Furthermore, U·U mismatch nucleotides involved in the 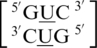 and
and 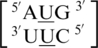 sequence combinations are annotated to have no interaction for 16 and 50% of the total hits of each, respectively. This may suggest U·U mismatches are dynamic and interact with the surrounding environment under certain conditions, such as what is observed for the
sequence combinations are annotated to have no interaction for 16 and 50% of the total hits of each, respectively. This may suggest U·U mismatches are dynamic and interact with the surrounding environment under certain conditions, such as what is observed for the 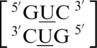 sequence combination, which is annotated and observed to be in a hydrogen bonded (one or two bonds formed), stacked conformation (Figure 4a) and a non-hydrogen bonded, unstacked conformation, where one of the U nucleotides involved in the single mismatch is flipped-out from the center of the helix and is interacting with surrounding solvent (Figure 4b) in 84 and 16% of the occurrences, respectively. However, it is further interesting to note both of these geometric orientations were annotated to have the same 5′W/3′W nearest neighbors; therefore, it appears the difference in spatial arrangement of the mismatched nucleotides does not affect that of the adjacent base pairs. This loop sequence was thermodynamically measured to contribute favorably to duplex stability (92), which may result from the ability of one of the loop nucleotides to rotate between two positions without distorting the geometrical orientation of the nearest neighbors.
sequence combination, which is annotated and observed to be in a hydrogen bonded (one or two bonds formed), stacked conformation (Figure 4a) and a non-hydrogen bonded, unstacked conformation, where one of the U nucleotides involved in the single mismatch is flipped-out from the center of the helix and is interacting with surrounding solvent (Figure 4b) in 84 and 16% of the occurrences, respectively. However, it is further interesting to note both of these geometric orientations were annotated to have the same 5′W/3′W nearest neighbors; therefore, it appears the difference in spatial arrangement of the mismatched nucleotides does not affect that of the adjacent base pairs. This loop sequence was thermodynamically measured to contribute favorably to duplex stability (92), which may result from the ability of one of the loop nucleotides to rotate between two positions without distorting the geometrical orientation of the nearest neighbors.
Figure 4.

Representation of 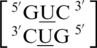 in the hydrogen bonded, stacked orientation (PDB ID 1O9M) (a) and in the non-hydrogen bonded, unstacked orientation (PDB ID 1O9M) (b).
in the hydrogen bonded, stacked orientation (PDB ID 1O9M) (a) and in the non-hydrogen bonded, unstacked orientation (PDB ID 1O9M) (b).
A·C single mismatches
Six of the eight A·C RNA single mismatch-nearest neighbor sequence combinations of the 30 most frequently occurring single mismatches in nature (84) are found in the RNA single mismatch structural database compiled here (Table 1). Considering these six combinations, a total of 89 A·C RNA single mismatch occurrences are found in the database; however, 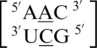 accounts for 73 (82%) of these hits, with all other combinations accounting for only 4% each, on average. The mismatched nucleotides of
accounts for 73 (82%) of these hits, with all other combinations accounting for only 4% each, on average. The mismatched nucleotides of 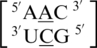 are most commonly observed in the 5′(A) H/3′(C) W pairing, antiparallel, trans orientation with the XXV (forming two hydrogen bonds) (Figure 5) or one_hbond hydrogen bonding pattern (each occurring ∼50% of the time). When A·C mismatches are found with this type of orientation and these interactions, the 5′ and 3′ nearest neighbors are always found in the 5′(A)Hh/3′(U)Ws pairing, antiparallel, trans XXIV and 5′Ww/3′Ww pairing, antiparallel, cis XIX orientation and interaction, respectively. Similar to A·G single mismatches, the 5′ nearest neighbor does not have the expected 5′W/3′W pairing. Contrary to A·G mismatches, A·C mismatches are not expected to disrupt the neighboring base pairs because this type of mismatch is comprised of one purine and pyrimidine base; therefore, it is similar in size to a canonical pair. This mismatch-nearest neighbor sequence combination was also found to engage in intra- and inter-strand interactions similar to what is observed for A·G mismatches. If the mismatch-nearest neighbor sequence is simply characterized as above, then inter- and intra-strand interactions are observed to form between nucleotides A and E and nucleotides B and C, respectively.
are most commonly observed in the 5′(A) H/3′(C) W pairing, antiparallel, trans orientation with the XXV (forming two hydrogen bonds) (Figure 5) or one_hbond hydrogen bonding pattern (each occurring ∼50% of the time). When A·C mismatches are found with this type of orientation and these interactions, the 5′ and 3′ nearest neighbors are always found in the 5′(A)Hh/3′(U)Ws pairing, antiparallel, trans XXIV and 5′Ww/3′Ww pairing, antiparallel, cis XIX orientation and interaction, respectively. Similar to A·G single mismatches, the 5′ nearest neighbor does not have the expected 5′W/3′W pairing. Contrary to A·G mismatches, A·C mismatches are not expected to disrupt the neighboring base pairs because this type of mismatch is comprised of one purine and pyrimidine base; therefore, it is similar in size to a canonical pair. This mismatch-nearest neighbor sequence combination was also found to engage in intra- and inter-strand interactions similar to what is observed for A·G mismatches. If the mismatch-nearest neighbor sequence is simply characterized as above, then inter- and intra-strand interactions are observed to form between nucleotides A and E and nucleotides B and C, respectively.
Figure 5.

Representation of an A·C mismatch in the 5′(A)H/3′(C)W pairing, antiparallel, trans orientation with XXV hydrogen bonding pattern (PDB ID 1FJG)), which is the most common orientation and interaction determined for the A·C mismatch-nearest neighbor combination of 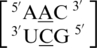 . This mismatch-nearest neighbor sequence combination is found in the 30 most frequently occurring single mismatches (84) and accounts for 80% of the total A·C mismatches found in this study.
. This mismatch-nearest neighbor sequence combination is found in the 30 most frequently occurring single mismatches (84) and accounts for 80% of the total A·C mismatches found in this study.
The remaining five A·C mismatch-nearest neighbor sequence combinations include 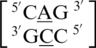 ,
, 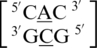 ,
, 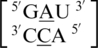 ,
, 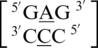 and
and 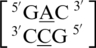 . These five can be divided into three groups based upon the geometric configuration of the mismatch nucleotides. The first group consists of the sequences
. These five can be divided into three groups based upon the geometric configuration of the mismatch nucleotides. The first group consists of the sequences 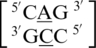 and
and 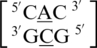 and the mismatched nucleotides are annotated with 5′(A)Wh/3′(C)Ww pairing, antiparallel, cis 75 (one hydrogen bond) geometric features. The second group consists of the sequences
and the mismatched nucleotides are annotated with 5′(A)Wh/3′(C)Ww pairing, antiparallel, cis 75 (one hydrogen bond) geometric features. The second group consists of the sequences 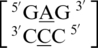 and
and 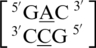 and are annotated to have no interaction. Interestingly, the first and second groups exhibit the same 5′ and 3′ nearest neighbor orientations and interactions. These nearest neighbors are annotated to both be in the 5′Ww/3′Ww pairing antiparallel cis orientation forming the canonical three hydrogen bonds in the XIX pattern. All four of these sequence combinations have G–C or C–G nearest neighbor base pairs at both the 5′ and 3′ side of the mismatch. Based upon the similarities in the type and orientation of the adjacent base pairs in these two groups, it is unclear why the A·C mismatched nucleotides are adopting different conformations.
and are annotated to have no interaction. Interestingly, the first and second groups exhibit the same 5′ and 3′ nearest neighbor orientations and interactions. These nearest neighbors are annotated to both be in the 5′Ww/3′Ww pairing antiparallel cis orientation forming the canonical three hydrogen bonds in the XIX pattern. All four of these sequence combinations have G–C or C–G nearest neighbor base pairs at both the 5′ and 3′ side of the mismatch. Based upon the similarities in the type and orientation of the adjacent base pairs in these two groups, it is unclear why the A·C mismatched nucleotides are adopting different conformations.
The third group only consists of the 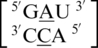 sequence combination, and the mismatched nucleotides are annotated to be in the 5′(A)Ww/3′(C)Hw pairing antiparallel cis, one_hbond orientation. The 5′ nearest neighbor of this mismatch-nearest neighbor sequence exhibits the same geometric orientation and hydrogen bonding pattern as the first and second group of A·C mismatches. However, the 3′ nearest neighbor is unique in identity and orientation when compared to these groups. The U-A base pair at this position is either annotated to be in the 5′(A)W/3′(C)Bh or 5′(A)W/3′(C)W pairing, antiparallel, trans orientation with the 46 (one hydrogen bond) hydrogen bonding pattern.
sequence combination, and the mismatched nucleotides are annotated to be in the 5′(A)Ww/3′(C)Hw pairing antiparallel cis, one_hbond orientation. The 5′ nearest neighbor of this mismatch-nearest neighbor sequence exhibits the same geometric orientation and hydrogen bonding pattern as the first and second group of A·C mismatches. However, the 3′ nearest neighbor is unique in identity and orientation when compared to these groups. The U-A base pair at this position is either annotated to be in the 5′(A)W/3′(C)Bh or 5′(A)W/3′(C)W pairing, antiparallel, trans orientation with the 46 (one hydrogen bond) hydrogen bonding pattern.
C·U single mismatches
C·U RNA single mismatches are the fourth most frequently occurring mismatch type, with three C·U mismatch-nearest neighbor sequences found in the 30 most frequently occurring single mismatches (84). Only one of these combinations is represented in the RNA single mismatch structure database presented here. There are 76 occurrences of 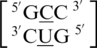 , and the C·U mismatch nucleotides are either in the 5′(C)W/3′(U)W pairing, antiparallel, cis one_hbond conformation (Figure 6) or the nucleotides are annotated to have no interaction. However, it is important to note the C·U mismatches annotated to have no interaction are also observed in the 5′(C)W/3′(U)W orientation. The 5′ and 3′ nearest neighbor base pairs are both in the 5′Ww/3′Ww pairing, antiparallel, cis XIX orientation.
, and the C·U mismatch nucleotides are either in the 5′(C)W/3′(U)W pairing, antiparallel, cis one_hbond conformation (Figure 6) or the nucleotides are annotated to have no interaction. However, it is important to note the C·U mismatches annotated to have no interaction are also observed in the 5′(C)W/3′(U)W orientation. The 5′ and 3′ nearest neighbor base pairs are both in the 5′Ww/3′Ww pairing, antiparallel, cis XIX orientation.
Figure 6.

Representation of a C·U mismatch in the 5′(C)W/3′(U)W pairing, antiparallel, cis orientation with one_hbond hydrogen bonding pattern (PDB ID 1FJG), which is the most common orientation and interaction determined for the most frequently occurring C·U mismatch-nearest neighbor combinations (84) that were also represented in the PDB.
A·A single mismatches
A·A RNA single mismatches are the fifth most frequently occurring mismatch type (84). Additionally, there is only one A·A mismatch-nearest neighbor sequence combination, 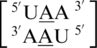 , found in the top 30, and it is not represented in the RNA 3D structure database. Therefore, this work does not contain structural information for this type of mismatch, but we are currently working to locate and annotate other A·A mismatch-nearest neighbor sequence combinations.
, found in the top 30, and it is not represented in the RNA 3D structure database. Therefore, this work does not contain structural information for this type of mismatch, but we are currently working to locate and annotate other A·A mismatch-nearest neighbor sequence combinations.
G·G single mismatches
G·G RNA single mismatches are the sixth most frequently occurring type of mismatch in nature (84). There is only one example of this mismatch type in the top 30 single mismatches, 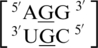 , and it is represented in the database presented here with 24 occurrences. The G·G mismatch nucleotides are either annotated to have no interaction (Figure 7) or in the 5′H/3′Bs pairing, antiparallel, trans conformation. When the nucleotides are interacting, the two hydrogen bond patterns annotated are 34 (bifurcated hydrogen bond) or 112 (one hydrogen bond). However, the G·G mismatches annotated to have no interaction are also observed in the 5′H/3′S orientation. Interestingly, regardless of the orientation and interaction of the mismatched nuceotides, the 5′ and 3′ nearest neighbor base pairs are always found in the 5′Hh/Ws3′ pairing, antiparallel, trans XXIV and 5′Ww/3′Ww pairing, antiparallel, cis XIX conformations, respectively. Once again, it is interesting to note the 5′ nearest neighbor does not form the canonical 5′W/3′W pairing type.
, and it is represented in the database presented here with 24 occurrences. The G·G mismatch nucleotides are either annotated to have no interaction (Figure 7) or in the 5′H/3′Bs pairing, antiparallel, trans conformation. When the nucleotides are interacting, the two hydrogen bond patterns annotated are 34 (bifurcated hydrogen bond) or 112 (one hydrogen bond). However, the G·G mismatches annotated to have no interaction are also observed in the 5′H/3′S orientation. Interestingly, regardless of the orientation and interaction of the mismatched nuceotides, the 5′ and 3′ nearest neighbor base pairs are always found in the 5′Hh/Ws3′ pairing, antiparallel, trans XXIV and 5′Ww/3′Ww pairing, antiparallel, cis XIX conformations, respectively. Once again, it is interesting to note the 5′ nearest neighbor does not form the canonical 5′W/3′W pairing type.
Figure 7.

Representation of a G·G mismatch annotated as having no interaction (PDB ID 2QAM), which is the most common orientation and interaction determined for the most frequently occurring G·G mismatch-nearest neighbor combination, 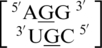 (84) that was also represented in the PDB.
(84) that was also represented in the PDB.
C·C single mismatches
C·C RNA single mismatches are the least frequently occurring mismatch type, and there are no C·C mismatch-nearest neighbor combinations found in the top 30 frequently occurring singe mismatches (84). Therefore, this work does not contain structural information for this type of mismatch, but we are currently working to locate and annotate C·C mismatch-nearest neighbor sequence combinations.
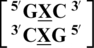 Nearest neighbor comparison
Nearest neighbor comparison
There are four examples in the top 30 of the nearest neighbor combination 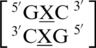 , where X is any nucleotide, and all are represented here, which include
, where X is any nucleotide, and all are represented here, which include 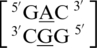 ,
, 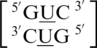 ,
, 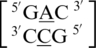 and
and 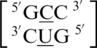 . It is important to note all three possible types of mismatches are present in this group: R·Y, R·R and Y·Y, when A and G are categorized as purines (R) and C and U are categorized as pyrimidines (Y). R·Y mismatches are similar in size to a canonical base pair since they are comprised of one purine and one pyrimidine; therefore, R·Y single mismatches are not likely disrupting the duplex backbone. R·R and Y·Y single mismatches are likely to disrupt the duplex backbone by causing the backbone to bulge-out or –in, respectively, to accommodate the mismatched nucleotides. Conversely, regardless of the mismatch type for these four sequence combinations, the 5′ and 3′ nearest neighbors are both in the 5′W/3′W pairing, antiparallel, cis XIX conformation in ∼99% of the occurrences.
. It is important to note all three possible types of mismatches are present in this group: R·Y, R·R and Y·Y, when A and G are categorized as purines (R) and C and U are categorized as pyrimidines (Y). R·Y mismatches are similar in size to a canonical base pair since they are comprised of one purine and one pyrimidine; therefore, R·Y single mismatches are not likely disrupting the duplex backbone. R·R and Y·Y single mismatches are likely to disrupt the duplex backbone by causing the backbone to bulge-out or –in, respectively, to accommodate the mismatched nucleotides. Conversely, regardless of the mismatch type for these four sequence combinations, the 5′ and 3′ nearest neighbors are both in the 5′W/3′W pairing, antiparallel, cis XIX conformation in ∼99% of the occurrences.
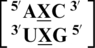 Nearest neighbor comparison
Nearest neighbor comparison
There are three examples in the top 30 of the nearest neighbor combination 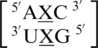 , but only two are represented in the RNA structural database,
, but only two are represented in the RNA structural database, 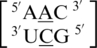 and
and 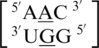 . It is important to note the difference of mismatch type, R·Y versus R·R, for reasons stated in the previous section in regards to the size of the nucleotides comprising the mismatched base pair and the hypothesized effect on the backbone. Interestingly, the 5′ and 3′ nearest neighbors are most commonly found in the 5′H/3′W pairing, antiparallel, trans XXIV and 5′Ww/3′Ww pairing, antiparallel, cis XIX conformations, respectively.
. It is important to note the difference of mismatch type, R·Y versus R·R, for reasons stated in the previous section in regards to the size of the nucleotides comprising the mismatched base pair and the hypothesized effect on the backbone. Interestingly, the 5′ and 3′ nearest neighbors are most commonly found in the 5′H/3′W pairing, antiparallel, trans XXIV and 5′Ww/3′Ww pairing, antiparallel, cis XIX conformations, respectively.
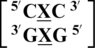 Nearest neighbor comparison
Nearest neighbor comparison
There are three examples in the top 30 of the nearest neighbor combination 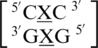 , which are all represented in the structural database and include
, which are all represented in the structural database and include 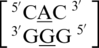 ,
, 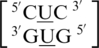 and
and 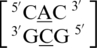 . Similar to the previous nearest neighbor sequence combinations, both the 5′ and 3′ nearest neighbors are found in the 5′Ww/3′Ww pairing, antiparallel, cis XIX conformation, in ∼100% of the occurrences. It is interesting to note the 3′ nearest neighbor for
. Similar to the previous nearest neighbor sequence combinations, both the 5′ and 3′ nearest neighbors are found in the 5′Ww/3′Ww pairing, antiparallel, cis XIX conformation, in ∼100% of the occurrences. It is interesting to note the 3′ nearest neighbor for 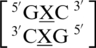 ,
, 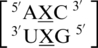 , and
, and 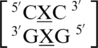 is C-G, and the orientation and interaction of this base pair is found to be the same for each, regardless of the identities of 5′ nearest neighbor base pair and the mismatch nucleotides.
is C-G, and the orientation and interaction of this base pair is found to be the same for each, regardless of the identities of 5′ nearest neighbor base pair and the mismatch nucleotides.
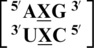 Nearest neighbor comparison
Nearest neighbor comparison
There are three examples in the top 30 of the nearest neighbor combination 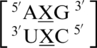 , but only two are found in the structural database,
, but only two are found in the structural database, 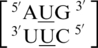 and
and 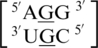 . The 5′ nearest neighbor conformation is different for each sequence combination. However, the 3′ nearest neighbor is identical in 98% of the total occurrences and is found to be 5′Ww/Ww3′ pairing, antiparallel, cis XIX, which is the same orientation and hydrogen bond pattern found in the above nearest neighbor comparisons.
. The 5′ nearest neighbor conformation is different for each sequence combination. However, the 3′ nearest neighbor is identical in 98% of the total occurrences and is found to be 5′Ww/Ww3′ pairing, antiparallel, cis XIX, which is the same orientation and hydrogen bond pattern found in the above nearest neighbor comparisons.
In conclusion, the PDB is a rich source of structural information, and this work has undertaken the task of systematically locating, annotating and comparing the most frequently occurring RNA single mismatches in nature. The 2046 single mismatches presented here (Table 1 and Supplementary Table S2) account for only 42% of the total number of single mismatches found in the available PDB structures. Therefore, this study only begins to investigate the available data, and we are currently looking at and comparing the remaining single mismatches to identify more structural patterns.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institute of General Medical Sciences (R15GM085699 to B.M.Z.). Monsanto Scholars Graduate Fellowship and the Saint Louis University Graduate School Dissertation Fellowship to A.R.D. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors would like to thank Francois Major for providing them with executable versions of MC-Search and MC-Annotate and for providing assistance with the software in the context of the work described here. The authors would also like to thank Pamela Vanegas for assistance in refining the search script.
REFERENCES
- 1.Mao H, White SA, Williamson JR. A novel loop-loop recognition motif in the yeast ribosomal protein L30 autoregulatory RNA complex. Nat. Struct. Biol. 1999;6:1139–1147. doi: 10.1038/70081. [DOI] [PubMed] [Google Scholar]
- 2.Lee J-H, Culver G, Carpenter S, Dobbs D. Analysis of the EIAV rev-responsive element (RRE) reveals a conserved RNA motif required for high affinity rev binding in bond HIV-1 and EIAV. PLoS ONE. 2008;3:e2272. doi: 10.1371/journal.pone.0002272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones S, Daley DTA, Luscombe NM, Berman HM, Thornton JM. Protein-RNA interactions: a structural analysis. Nucleic Acids Res. 2001;29:943–954. doi: 10.1093/nar/29.4.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beuth B, García-Mayoral MF, Taylor IA, Ramos A. Scaffold-independent analysis of RNA-protein interactions: the nova-1 KH3-RNA complex. J. Am. Chem. Soc. 2007;129:10205–10210. doi: 10.1021/ja072365q. [DOI] [PubMed] [Google Scholar]
- 5.Messias AC, Sattler M. Structural basis of single-stranded RNA recognition. Acc. Chem. Res. 2004;37:279–287. doi: 10.1021/ar030034m. [DOI] [PubMed] [Google Scholar]
- 6.Hall KB. RNA-protein interactions. Curr. Opin. Struct. Biol. 2002;12:283–288. doi: 10.1016/s0959-440x(02)00323-8. [DOI] [PubMed] [Google Scholar]
- 7.Hori T, Taguchi Y, Uesugi S, Kurihara Y. The RNA ligands for mouse proline-rich RNA-binding protein (mouse Prrp) contain two consensus sequences in separate loop structure. Nucleic Acids Res. 2005;33:190–200. doi: 10.1093/nar/gki153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dubey AK, Baker CS, Romeo T, Babitzke P. RNA sequence and secondary structure participate in high-affinity CsrA-RNA interaction. RNA. 2005;11:1579–1587. doi: 10.1261/rna.2990205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nagai K. RNA-protein complexes. Curr. Opin. Struct. Biol. 1996;6:53–61. doi: 10.1016/s0959-440x(96)80095-9. [DOI] [PubMed] [Google Scholar]
- 10.Steitz TA. RNA Recognition by Proteins. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1999. [Google Scholar]
- 11.Huppler A, Nikstad LJ, Allmann AM, Brow DA, Butcher SE. Metal binding and base ionization in the U6 RNA intramolecular step-loop structure. Nat. Struct. Biol. 2002;9:431–435. doi: 10.1038/nsb800. [DOI] [PubMed] [Google Scholar]
- 12.Grilley D, Misra V, Caliskan G, Draper DE. Importance of partially unfolded conformations for Mg2+-induced folding of RNA tertiary structure: structural models and free energies of Mg2+ interactions. Biochemistry. 2007;46:10266–10278. doi: 10.1021/bi062284r. [DOI] [PubMed] [Google Scholar]
- 13.Casiano-Negroni A, Sun X, Al-Hashimi HM. Probing Na+-induced changes in the HIV-1 TAR conformational dynamics using NMR residual dipolar couplings: new insights into the role of counterions and electrostatic interactions in adaptive recognition. Biochemistry. 2007;46:6525–6535. doi: 10.1021/bi700335n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Donarski J, Shammas C, Banks R, Ramesh V. NMR and molecular modelling studies of the binding of amicetin antibiotic to conserved secondar structural motifs of 23S ribosomal RNA. J. Antibiot. 2006;59:177–183. doi: 10.1038/ja.2006.25. [DOI] [PubMed] [Google Scholar]
- 15.Liu X, Thomas JR, Hergenrother PJ. Deoxystreptamine dimers bind to RNA hairpin loops. J. Am. Chem. Soc. 2004;126:9196–9197. doi: 10.1021/ja048936l. [DOI] [PubMed] [Google Scholar]
- 16.Chushak Y, Stone MO. In silico selection of RNA aptamers. Nucleic Acids Res. 2009;37:e87. doi: 10.1093/nar/gkp408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meyer ST, Hergenrother PJ. Small molecular ligands for bulged RNA secondary structures. Org. Lett. 2009;11:4052–4055. doi: 10.1021/ol901478x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Childs-Disney JL, Wu M, Pushechnikov A, Aminova O, Disney MD. A small molecule microarray platform to select RNA internal loop-ligand interactions. ACS Chem. Biol. 2007;2:745–754. doi: 10.1021/cb700174r. [DOI] [PubMed] [Google Scholar]
- 19.Gallego J, Varani G. Targeting RNA with small-molecule drugs: Therapeutic promise and chemical challenges. Accounts Chem. Res. 2001;34:836–843. doi: 10.1021/ar000118k. [DOI] [PubMed] [Google Scholar]
- 20.Chang K-Y, Tinoco I., Jr The structure of an RNA “kissing” hairpin complex of the HIV TAG hairpin loop and its complement. J. Mol. Biol. 1997;269:52–66. doi: 10.1006/jmbi.1997.1021. [DOI] [PubMed] [Google Scholar]
- 21.Shankar N, Kennedy SD, Chen G, Krugh TR, Turner DH. The NMR structure of an internal loop from 23S ribosomal RNA differs from its structure in crystals of 50S ribosomal subunits. Biochemistry. 2006;45:11776–11789. doi: 10.1021/bi0605787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu ZJ, Turner DH, Mathews DH. A set of nearest neighbor parameters for predicting the enthalpy change of RNA secondary structure formation. Nucleic Acids Res. 2006;34:4912–4924. doi: 10.1093/nar/gkl472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mathews DH, Disney MD, Childs JC, Schroeder SJ, Zuker M, Turner DH. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl Acad. Sci., USA. 2004;101:7287–7292. doi: 10.1073/pnas.0401799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mathews DH, Sabina J, Zuker M, Turner DH. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- 25.Hofacker IL. Vienna RNA secondary structure server. Nucleic Acids Res. 2003;31:3429–3431. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu ZJ, Gloor JW, Mathews DH. Improved RNA secondary structure prediction by maximizing expected pair accuracy. RNA. 2009;15:1805–1813. doi: 10.1261/rna.1643609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andronescu M, Condon A, Hoos HH, Mathews DH, Murphy KP. Efficient parameter estimation for RNA secondary structure prediction. Bioinformatics. 2007;23:i19–i28. doi: 10.1093/bioinformatics/btm223. [DOI] [PubMed] [Google Scholar]
- 29.Do CB, Woods DA, Batzoglou S. CONTRAfold: RNA secondary structure prediction without physics-based models. Bioinformatics. 2006;22:e90–e98. doi: 10.1093/bioinformatics/btl246. [DOI] [PubMed] [Google Scholar]
- 30.Hamada M, Kiryu H, Sato K, Mituyama T, Asai K. Prediction of RNA secondary structure using generalized centroid estimators. Bioinformatics. 2009;25:465–473. doi: 10.1093/bioinformatics/btn601. [DOI] [PubMed] [Google Scholar]
- 31.Dowell RD, Eddy SR. Evaluation of several lightweight stochastic context-free grammars for RNA secondary structure prediction. BMC Bioinformatics. 2004;5:71–84. doi: 10.1186/1471-2105-5-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parisien M, Cruz JA, Westhof É, Major F. New metrics for comparing and assessing discrepancies between RNA 3D structures and models. RNA. 2009;15:1875–1885. doi: 10.1261/rna.1700409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Das R, Baker D. Automated de novo prediction of native-like RNA tertiary structures. Proc. Natl Acad. Sci. 2007;104:114664–114669. doi: 10.1073/pnas.0703836104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ding F, Sharma S, Chalasani P, Demidov VV, Broude NE, Dokholyan NV. Ab initio RNA folding by discrete molecular dynamics: from structure prediction to folding mechanisms. RNA. 2008;14:1164–1173. doi: 10.1261/rna.894608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jonikas MA, Radmer RJ, Laederach A, Das R, Pearlman S, Herschlag D, Altman RB. Coarse-grained modeling of large RNA molecules with knowledge-based potentials and structural filters. RNA. 2009;15:189–199. doi: 10.1261/rna.1270809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martinez HM, Maizel JV, Shapiro BA. RNA2D3D: a program for generating, viewing, and comparing three-dimensional models of RNA. J. Biomol. Struct. Dyn. 2008;25:669–683. doi: 10.1080/07391102.2008.10531240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Massire C, Westhof E. MANIP: an interactive tool for modelling RNA. J. Mol. Graphics Modell. 1998;16:197–205. doi: 10.1016/s1093-3263(98)80004-1. 255–257. [DOI] [PubMed] [Google Scholar]
- 38.Michel F, Westhof E. Modeling of the three-dimensional architecture of group I catalytic introns based on comparative sequence analysis. J. Mol. Biol. 1990;216:585–610. doi: 10.1016/0022-2836(90)90386-Z. [DOI] [PubMed] [Google Scholar]
- 39.Parisien M, Major F. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature. 2008;452:51–55. doi: 10.1038/nature06684. [DOI] [PubMed] [Google Scholar]
- 40.Batey RT, Rambo RP, Lucast L, Rha B, Doudna JA. Tertiary motifs in RNA structure and folding. Angew. Chem., Int. Ed. 1999;38:2326–2343. doi: 10.1002/(sici)1521-3773(19990816)38:16<2326::aid-anie2326>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 41.Westhof E, Fritsch V. RNA folding: beyond Watson-Crick pairs. Structure with Folding & Design. 2000;8:R55–R65. doi: 10.1016/s0969-2126(00)00112-x. [DOI] [PubMed] [Google Scholar]
- 42.Ferré-D'Amare AR, a DJA. RNA folds: insights from recent crystal structures. Annu. Rev. Biophys. Biophys. Chem. 1999;28:57–73. doi: 10.1146/annurev.biophys.28.1.57. [DOI] [PubMed] [Google Scholar]
- 43.Hermann TaPDJ. Stitching together RNA tertiary architectures. J. Mol. Biol. 1999;294:829–849. doi: 10.1006/jmbi.1999.3312. [DOI] [PubMed] [Google Scholar]
- 44.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berman H, Henrick K, Nakamura H, Markley JL. The worldwide Protein Data Bank (wwPDB): ensuring a single, uniform archive of PDB data. Nucleic Acids Res. 2007;35:D301–D303. doi: 10.1093/nar/gkl971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Westbrook J, Feng Z, Chen L, Huanwang Y, Berman HM. The Protein Data Bank and structural genomics. Nucleic Acids Res. 2003;31:489–491. doi: 10.1093/nar/gkg068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Westbrook J, Feng Z, Jain S, Bhat TN, Thanki N, Ravichandran V, Gilliland GL, Bluhm W, Weissig H, Greer DS, et al. The Protein Data Bank: unifying the archive. Nucleic Acids Res. 2002;30:245–248. doi: 10.1093/nar/30.1.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deshpande N, Addess KJ, Bluhm WF, Merino-Ott JC, Townsend-Merino W, Zhang Q, Knezevich C, Xie L, Chen L, Feng Z, et al. The RCSB Protein Data Bank: a redesigned query system and relational database based on the mmCIF schema. Nucleic Acids Res. 2005;33:D233–D237. doi: 10.1093/nar/gki057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nagaswamy U, Voss N, Zhang ZD, Fox GE. Database of non-canonical base pairs found in known RNA structures. Nucleic Acids Res. 2000;28:375–376. doi: 10.1093/nar/28.1.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nagaswamy U, Larios-Sanz M, Hury J, Collins S, Zhang ZD, Zhao Q, Fox GE. NCIR: A database of non-canonical interactions in known RNA structures. Nucleic Acids Res. 2002;30:395–397. doi: 10.1093/nar/30.1.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xin Y, Olson WK. BPS: a database of RNA base-pair structures. Nucleic Acids Res. 2009;37:D38–D88. doi: 10.1093/nar/gkn676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schnare MN, Damberger SH, Gray MW, Gutell RR. Comprehensive comparison of structural characteristics in eukaryotic cytoplasmic large subunit (23 S-like) ribosomal RNA. J. Mol. Biol. 1996;256:701–719. doi: 10.1006/jmbi.1996.0119. [DOI] [PubMed] [Google Scholar]
- 53.Gautheret D, Konings D, Gutell RR. A major family of motifs involving G.A mismatches in ribosomal RNA. J. Mol. Biol. 1994;242:1–8. doi: 10.1006/jmbi.1994.1552. [DOI] [PubMed] [Google Scholar]
- 54.Gautheret D, Konings D, Gutell RR. GU base-pairing motifs in ribosomal-RNA. RNA. 1995;1:807–814. [PMC free article] [PubMed] [Google Scholar]
- 55.Leontis NB, Westhof E. Conserved geometrical base-pairing patterns in RNA. Q. Rev. Biophys. 1998;31:399–455. doi: 10.1017/s0033583599003479. [DOI] [PubMed] [Google Scholar]
- 56.Leontis NB, Westhof E. Geometric nomenclature and classification of RNA base pairs. RNA. 2001;7:499–512. doi: 10.1017/s1355838201002515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leontis NB, Westhof E. Survey and summary: the non-Watson-Crick pairs and their associated isostericity matrices. Nucleic Acids Res. 2002;30:3497–3531. doi: 10.1093/nar/gkf481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lescoute A, Westhof E. The interaction networks of structured RNAs. Nucleic Acids Res. 2006;34:6587–6604. doi: 10.1093/nar/gkl963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Leontis NB, Lescoute A, Westhof E. The building blocks and motifs of RNA architecture. Curr. Opin. Struct. Biol. 2006;16:279–287. doi: 10.1016/j.sbi.2006.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lescoute A, Leonteis NB, Massire C, Westhof E. Recurrent structural RNA motifs. isostericity matrices and sequence alignments. Nucleic Acids Res. 2005;33:2395–2409. doi: 10.1093/nar/gki535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leontis NB, Westhof E. Analysis of RNA motifs. Curr. Opin. Struct. Biol. 2003;13:300–308. doi: 10.1016/s0959-440x(03)00076-9. [DOI] [PubMed] [Google Scholar]
- 62.Leontis NB, Westhof E. The annotation of RNA motifs. Comparative Funct Genomics. 2002;3:518–524. doi: 10.1002/cfg.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Klosterman PS, Tamura M, Holbrook SR, Brenner SE. SCOR: a structural classification of RNA database. Nucleic Acids Res. 2002;30:392–394. doi: 10.1093/nar/30.1.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Klosterman PS, Hendrix DK, Tamura M, Holbrook SR, Brenner SE. Three-dimensional motifs from the SCOR, structural classification of RNA database: extruded strands, base triples, tetraloops and U-turns. Nucleic Acids Res. 2004;32:2342–2352. doi: 10.1093/nar/gkh537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tamura M, Hendrix DK, Klosterman PS, Schimmelman NRB, Brenner SE, Holbrook SR. SCOR: structrual classification of RNA, version 2.0. Nucleic Acids Res. 2004;32:D182–D184. doi: 10.1093/nar/gkh080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Leontis NB, Altman RB, Berman HM, Brenner SE, Brown JW, Engelke DR, Harvey SC, Holbrook SR, Jossinet F, Lewis SE, et al. The RNA Ontology Consortium: an open invitation to the RNA community. RNA. 2006;12:533–541. doi: 10.1261/rna.2343206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gendron P, Lemieux S, Major F. Quantitative analysis of nucleic acid three-dimensional structures. J. Mol. Biol. 2001;308:919–936. doi: 10.1006/jmbi.2001.4626. [DOI] [PubMed] [Google Scholar]
- 68.Lemieux S, Major F. RNA canonical and non-canonical base-pairing types: a recognition method and complete repertoire. Nucleic Acids Res. 2002;30:4250–4263. doi: 10.1093/nar/gkf540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lisi V, Major F. A comparative analysis of the triloops in all high-resolution RNA structures reveals sequence-structure relationships. RNA. 2007;13:1537–1545. doi: 10.1261/rna.597507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hoffmann B, Mitchell GT, Gendron P, Major F, Anderson AA, Collins RA, Legault P. NMR structure of the active conformation of the Varkud satellite ribozyme cleavage site. Proc. Natl Acad. Sci. USA. 2003;100:7003–7008. doi: 10.1073/pnas.0832440100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Olivier C, Poirier G, Gendron P, Boisgontier A, Major F, Chartrand P. Identification of a conserved RNA motif essential for She2p recognition and mRNA localization to the yeast bud. Mol. Cell. Biol. 2005;25:4752–4766. doi: 10.1128/MCB.25.11.4752-4766.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Peritz AE, Kierzek R, Sugimoto N, Turner DH. Thermodynamic study of internal loops in oligoribonucleotides: Symmetric loops are more stable than asymmetric loops. Biochemistry. 1991;30:6428–6436. doi: 10.1021/bi00240a013. [DOI] [PubMed] [Google Scholar]
- 73.Calin-Jageman I, Nicholson AW. Mutational analysis of an RNA internal loop as a reactivity epitope for Escherichia coli ribonuclease III substrates. Biochemistry. 2003;42:5025–5034. doi: 10.1021/bi030004r. [DOI] [PubMed] [Google Scholar]
- 74.Saito H, Richardson CC. Processing of mRNA by ribonuclease III regulates expression of gene 1.2 of bacteriophage T7. Cell. 1981;27:533–542. doi: 10.1016/0092-8674(81)90395-0. [DOI] [PubMed] [Google Scholar]
- 75.Du T, Zamore PD. MicroPrimer: the biogenesis and function of microRNA. Development. 2005;132:4645–4652. doi: 10.1242/dev.02070. [DOI] [PubMed] [Google Scholar]
- 76.Bae SH, Cheong HK, Lee JH, Cheong C, Kainosho M, Choi BS. Structural features of an influenza virus promoter and their implications for viral RNA synthesis. Proc. Natl Acad. Sci. USA. 2001;98:10602–10607. doi: 10.1073/pnas.191268798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huthoff H, Berkhout B. Multiple secondary structure rearrangements during HIV-1 RNA dimerization. Biochemistry. 2002;41:10439–10445. doi: 10.1021/bi025993n. [DOI] [PubMed] [Google Scholar]
- 78.Schüler M, Connell SR, Lescoute A, Giesebrecht J, Dabrowski M, Schroeer B, Mielke T, Penczek PA, Westhof E, Spahn CMT. Structure of the ribosome-bound cricket paralysis virus IRES RNA. Nat. Struct. Mol. Biol. 2006;13:1092–1096. doi: 10.1038/nsmb1177. [DOI] [PubMed] [Google Scholar]
- 79.Wientges J, Putz J, Giege R, Florentz C, Schwienhorst A. Selection of viral RNA-derived tRNA-like structures with improved valylation activities. Biochemistry. 2000;39:6207–6218. doi: 10.1021/bi992852l. [DOI] [PubMed] [Google Scholar]
- 80.Thunder C, Witwer C, Hofacker IL, Stadler PF. Conserved RNA secondary structures in Flaviviridae genomes. J. Gen. Virol. 2004;85:1113–1124. doi: 10.1099/vir.0.19462-0. [DOI] [PubMed] [Google Scholar]
- 81.Shi P-Y, Brinton MA, Veal JM, Zhong YY, Wilson WD. Evidence for the existence of a pseudoknot structure at the 3' terminus of the Flavivirus genomic RNA. Biochemistry. 1996;35:4222–4230. doi: 10.1021/bi952398v. [DOI] [PubMed] [Google Scholar]
- 82.Everett CM, Wood NW. Trinucleotide repeats and neurodegenerative disease. Brain. 2004;127:2385–2405. doi: 10.1093/brain/awh278. [DOI] [PubMed] [Google Scholar]
- 83.Ranum LPW, Day JW. Myotonic dystrophy: RNA pathogenesis comes into focus. Amer. J. Hum. Gen. 2004;74:793–804. doi: 10.1086/383590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Davis AR, Znosko BM. Thermodynamic characterization of single mismatches found in naturally occurring RNA. Biochemistry. 2007;46:13425–13436. doi: 10.1021/bi701311c. [DOI] [PubMed] [Google Scholar]
- 85.Donohue J, Trueblood KN. Base-pairing in DNA. J. Mol. Biol. 1960;2:363–371. doi: 10.1016/s0022-2836(60)80047-2. [DOI] [PubMed] [Google Scholar]
- 86.Donohue J. Hydrogen-bonded helical configurations of polynucleotides. Proc. Natl Acad. Sci. USA. 1956;42:60–65. doi: 10.1073/pnas.42.2.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Saenger W. Principles of Nucleic Acid Structure. NY: Springer-Verlag New York, Inc.; 1984. [Google Scholar]
- 88.Gautheret D, Gutell RR. Inferring the conformation of RNA base pairs and triples from patterns of sequence variation. Nucleic Acids Res. 1997;25:1559–1564. doi: 10.1093/nar/25.8.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lemieux S, Chartrand P, Cedergren R, Major F. Modeling active RNA structures using the intersection of conformational space: application to the lead-activated ribozyme. RNA. 1998;4:739–749. doi: 10.1017/s1355838298971266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gabb HA, Sanghani SR, Rober CH, Prevost C. Finding and visualizing nucleic acid base stacking. J. Mol. Graphics Modell. 1996;14:23–24. doi: 10.1016/0263-7855(95)00086-0. [DOI] [PubMed] [Google Scholar]
- 91.Major F, Thibault P. RNA tertiary structure prediction. In: Lengauer T, editor. Bioinformatics: From Genentics to Therapies. Germany: Wiley-VCH, Weinheim; 2007. pp. 491–539. [Google Scholar]
- 92.Kierzek R, Burkard ME, Turner DH. Thermodynamics of single mismatches in RNA duplexes. Biochemistry. 1999;38:14214–14223. doi: 10.1021/bi991186l. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.