

Abstract
Dihydrofolate reductase (DHFR) is a critical enzyme in folate metabolism and an important target of antineoplastic, antimicrobial, and antiinflammatory drugs. We describe three individuals from two families with a recessive inborn error of metabolism, characterized by megaloblastic anemia and/or pancytopenia, severe cerebral folate deficiency, and cerebral tetrahydrobiopterin deficiency due to a germline missense mutation in DHFR, resulting in profound enzyme deficiency. We show that cerebral folate levels, anemia, and pancytopenia of DHFR deficiency can be corrected by treatment with folinic acid. The characterization of this disorder provides evidence for the link between DHFR and metabolism of cerebral tetrahydrobiopterin, which is required for the formation of dopamine, serotonin, and norepinephrine and for the hydroxylation of aromatic amino acids. Moreover, this relationship provides insight into the role of folates in neurological conditions, including depression, Alzheimer disease, and Parkinson disease.
Main Text
Folates are critical cofactors for single-carbon metabolism in biological processes, including DNA synthesis, regulation of gene expression, and synthesis of amino acids, neurotransmitters, and myelin.1 Systemic folate deficiency, most frequently due to dietary insufficiency, manifests as biochemical, hematological and neurological disturbance. Six inherited disorders of folate transport and metabolism have been described (Table S1 available online). Of particular note, mutations in FOLR1 (MIM 136430), encoding folate receptor alpha, cause a brain specific folate-transport defect that manifests between 2 and 3 years of age and is characterized by irritability, slow head growth, psychomotor retardation, cerebellar ataxia, pyramidal tract signs, dyskinesia, and seizures.2 The only consistent biochemical abnormality observed in children affected with FOLR1 mutations is a low level of 5-methyltetrahydrofolate (5-MTHF) in the cerebrospinal fluid (CSF) with normal serum and erythrocyte folate levels. Cerebral folate deficiency (CFD) can also result from impaired transport of 5-MTHF across the blood-CSF barrier due to antibodies to folate receptors.3 Here, we describe the clinical features and molecular mechanism of an inborn error of folate metabolism characterized by infantile-onset megaloblastic anemia and/or pancytopenia, severe CFD, and moderate cerebral BH4 deficiency.
Our proband (family 1, II:6 in Figure 1A) is the fourth child of first-cousin British Pakistani parents (I-1 and I-2), born at full term after an uneventful pregnancy and with no neonatal problems. His weight and head circumference (HC) at birth were 2.83 kg (9th–25th centile) and 33 cm (2nd–9th centile), respectively. He presented at 4 mo of age with increasing pallor, poor feeding, and secondary microcephaly with an HC of 36.8 cm (< 0.4th centile). Investigations revealed severe anemia (Table 1), and his blood film showed a dual red cell population with oval macrocytes and microcytes and hypersegmented neutrophils (Figure S1A). Within 2 days, he was pancytopenic. His bone marrow demonstrated megaloblastic erythropoesis (Figure 1B). Serum folate, vitamin B12, and ferritin levels were normal. A working diagnosis of transcobalamin II deficiency (MIM 275350) was made, and hydroxocobalamin was started. After 1 week, he started having generalized tonic-clonic and right-sided focal seizures that were refractory to phenytoin, benzodiazepines, and pyridoxine. His hematological profile remained unresponsive to hydroxocobalamin. Partial seizure control was achieved with phenobarbitone and levetiracetam. His brain MRI showed severe cerebellar and cerebral atrophy (Figures 1C, 1D, and 1E). CSF neurotransmitters were analyzed by high-performance liquid chromatography with electrochemical and/or fluorescence detection4 and revealed markedly decreased CSF 5-MTHF, with low CSF BH4 and normal dihydrobiopterin (BH2) levels (Table 1). Metabolic investigations showed that there was no methylmalonic aciduria and that plasma homocysteine and phenylalanine levels were normal. Table S2 provides details of all other metabolic investigations that were performed.
Figure 1.
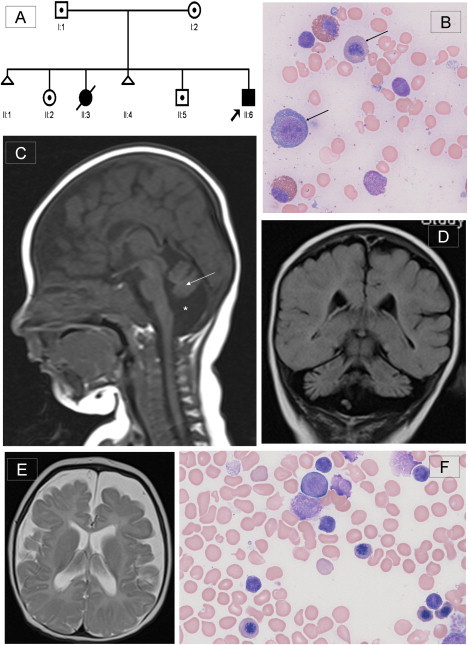
Pedigree of the Family, Bone Marrow Morphology, and Brain MRI of the Proband
(A) Pedigree of family of the proband (II:6). DNA from individuals II:2, II:5, and II:6 was used for the autozygosity mapping.
(B) Proband's pretreatment bone marrow aspirate with modified Wright's stain at 500× magnification demonstrating early and late megaloblasts (marked by arrows), consistent with megaloblastic erythropoesis. Giant metamyelocytes and excess siderocytes were seen, and megakaryocytes were reduced (not shown here).
(C) T1-weighted midline sagittal section of brain MRI at 4 months showing cerebellar vermian hypoplasia with atrophy of cerebellar hemispheres (marked by arrow) and surrounding enlarged CSF space (marked by ∗). The corpus callosum is thin, and cerebral atrophy can also be seen. Additionally, there was a chronic right sided subdural collection (not shown), possibly due to atrophic changes within the brain.
(D) TI-weighted coronal section showing severe cerebellar atrophy with prominence of the folia.
(E) T2-weighted axial section demonstrating prominence of the sulci and extensive extraaxial CSF space. The white matter is poorly myelinated. No abnormality of the basal ganglia is seen.
(F) Proband's posttreatment marrow aspirate, confirming return to normoblastic erythropoiesis.
Table 1.
The Results of Hematological and CSF Investigations of the Proband
|
Pretreatment |
After Folinic Acid Treatment |
|||||
|---|---|---|---|---|---|---|
|
Investigation (Reference Range) |
At Presentation | 2 Days after Presentation | After Transfusion | After 1 Week of Treatment | After 1 Month of Treatment | After 4 Months of Treatment |
| Age in months | 3.5 | 3.5 | 4.5 | 5 | 6 | 9.5 |
| Haemoglobin (10.0–13.0 g/dl) |
5.2 ↓ | 3.7 ↓ | 8.2 ↓ | 8.6 ↓ | 10.6 | 12.7 |
| Red cell count (3.80–4.90 × 109/l) |
1.9 ↓ | 1.31 ↓ | 3.03 ↓ | 3.18 ↓ | 3.91 | 4.65 |
| Mean corpuscular volume (73–88 fl) | 85 | 82 | 81 | 84 | 82 | 86 |
| White cell count (6.0–17.0 × 109/l) |
8.4 | 2.3 ↓ | 3.6 ↓ | 5.8 ↓ | 9.2 | 8.0 |
| Neutrophils (1.00–6.00 × 109/l) |
2.7 | 0.95 ↓ | 1.76 ↓ | 2.47 | 2.63 | 2.78 |
| Lymphocytes (3.30–11.50 × 109/l) |
4.2 | 1.11 ↓ | 1.34 ↓ | 2.25 ↓ | 5.52 | 4.19 |
| Monocytes (0.20 −1.30 × 109/l) |
0.7 | 0.17 ↓ | 0.24 | 0.56 | 0.76 | 0.92 |
| Eosinophils (0.05–1.10 × 109/l) |
0.6 | 0.08 | NA | 0.55 | 0.31 | NA |
| Basophils (0.02–0.20 × 109/l) |
0.2 | 0 ↓ | 0 ↓ | 0.01 ↓ | 0.01 ↓ | 0.01 ↓ |
| Platelets (150–560 × 109/l) |
240 | 138 ↓ | 316 | 423 | 390 | 358 |
| CSF 5-MTHF (72–305 nmol/l) |
NA | NA | 9 ↓ | 98 | NA | 81 |
| CSF total neopterin (7–65 nmol/l) |
NA | NA | 36 | 20 | NA | NA |
| CSF tetrahydrobiopterin (∗nmol/l) |
NA | NA | 23 ↓ (27–105) |
13 ↓ (23–55) |
NA | NA |
| CSF dihydrobiopterin (0.4–13.9 nmol/l) |
NA | NA | 8.9 | 5.3 | NA | NA |
| CSF pyridoxal phosphate (∗nmol/l) |
NA | NA | 52 (44–89) |
51 (23 – 87) |
NA | NA |
| CSF homovanillic acid (∗nmol/l) |
NA | NA | 288 ↓ (324–1098) |
298 ↓ (362–955) |
NA | 330 (176–851) |
| CSF 5-hydroxyindoleacetic acid (∗nmol/l) | NA | NA | 198 ↓ (199–608) |
142 (63–503) |
NA | 165 (68–451) |
Reference ranges for investigations marked with ∗ are age dependent and are given with individual measurements. NA, not available; ↓, results below normal.
Oral folinic acid (30 mg daily) was initiated, after which anemia (Figure 1F and Figure S1B), seizure control, and CSF 5-MTHF level showed rapid and significant improvement, although the BH4 level dropped further (Table 1). Now, at the age of 19 months, the patient remains profoundly delayed with central hypotonia, poor head control, and inability to fix and follow. He has two to three short focal seizures daily, is fed via gastrostomy, and suffers from frequent lower respiratory tract infections.
An older sibling (II:3) died at 28 weeks of age with a history of anemia and intractable seizures of undefined cause. Postmortem examination showed a small brain, weighing 480 g (normal 615 g), with ventricular dilatation and white matter atrophy of the cerebrum. There was neuronal loss in the cerebellar folia with a reduction of both the internal and external granular cell layers. Neuronal and vascular calcifications were seen in the basal ganglia and subcortical white matter, respectively, with gliosis in the periventricular white matter and possible optic atrophy. The cause of death was Klebsiella aerogenes pneumonia. The proband's two remaining older siblings (II-2 and II-5) and parents (I-1 and I-2) are healthy.
CFD and megaloblastic anemia in the presence of normal serum folate excluded the possibility of defects in any of the known folate transporters.2,5 Absence of hyperhomocysteinemia indicated that the defect did not involve the methylation cycle but was most likely to be in the DNA-synthesis arm of folate metabolism. Ethical approval for the study was obtained from the University of Manchester (06138) and NHS ethics committees (06/Q1406/52). Informed consent was obtained from all participants or their parents.
We undertook autozygosity mapping and genotyped the proband (II:6) and two unaffected siblings (II:2 and II:5) by using the Genome-Wide Human SNP Array 6.0 (Affymetrix, Santa Clara, CA, USA) as described previously.6 Genotype calls were generated with the use of the Birdseed V2 algorithm within the same software, and the results were analyzed by AutoSNPa.7 Multiple homozygous regions, equivalent to approximately 180 Mb and unique to the affected individual, were identified (Table S3). This included a 3 Mb region at chromosome 5q14.1 flanked by rs4521453 and rs10059759, containing DHFR.
Primers for PCR and sequencing were designed with Primer3 on the basis of NCBI reference sequence NM_000791.3, covering the coding regions and intron-exon boundaries of exons 1 to 6 of the DHFR and ensuring no amplification of the pseudogenes. Primer sequences and PCR conditions are provided in Table S4. Sequence variants were named in accordance with HGV nomenclature, and NM_000791.3 was used as the reference sequence.
DNA sequence analysis of DHFR revealed a homozygous missense mutation, c.238C>T (p.Leu80Phe), in exon 3 (Figure 2A). The mutation segregated with the disorder in the family (Figure 2B) and, importantly, was homozygous in DNA extracted from a neonatal blood spot available from the affected deceased sibling (patient 2, II:3). The mutation was absent in 292 chromosomes from ethnically matched Pakistani controls (Figure 2C).
Figure 2.
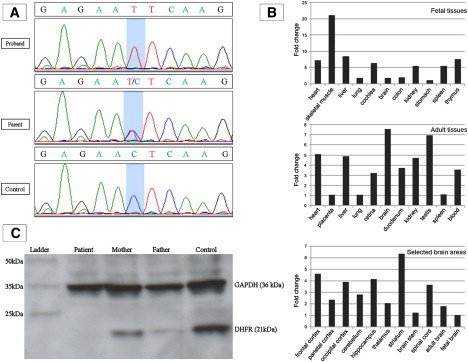
Results of Sequencing, Expression Analyses, Immunoblotting
(A) Sequence trace showing missense mutation c.238C>T in exon 3 of DHFR. This mutation was homozygous in the proband (II:6) and two other affected individuals. The mutation was heterozygous in I:1, I:2, II:2, and II:5 and in an unaffected sibling of patient 3. A normal sequence trace from an ethnically matched control is also shown.
(B) Expression of DHFR in different fetal tissues (top panel), adult tissues (middle panel), and selected brain areas (bottom panel). Relative expression levels are given as the fold change in comparison to the tissue or area with the lowest expression level. All fetal tissues are from 20- or 21-week-old embryos after gestation, except for cochlear RNA, which was isolated from an 8-week-old embryo.
(C) Immunoblot of protein from cellular and nuclear lysates from lymphoblast cell lines derived from the proband, his parents, and an unrelated control. Mouse monoclonal antibody to human GAPDH (36 kDa) was used to demonstrate equal protein loading. Polyclonal antibody to human DHFR (21 kDa) produced in rabbit was purchased from ProteinTech (Chicago, IL). DHFR was undetectable in the patient's sample and was reduced in both parents in comparison to the control. Of note, the expression level of DHFR in parents is slightly different even though they carry the same mutation, which is probably because DHFR is a dynamically expressed protein.
Another child (family 2) with CFD, megaloblastic anemia, and normal serum folate was ascertained. He is the first child born to first-cousin British Pakistani parents, delivered at full term with a birth HC of 33.5 cm (25th centile). He presented at 10 weeks of age with reduced oral intake, icterus, and hepatomegaly. His hemoglobin (Hb) level was 6.1 g/dl, with a mean corpuscular volume of 95 fl. His HC was 37 cm (0.4th–2ndcentile). Within 5 days, he developed leucopenia (5.4 × 109/l) and thrombocytopenia (71 × 109/l). A diagnosis of megaloblastic anemia of unknown cause was made, and folic acid was started. His anemia resolved and his neurodevelopment was satisfactory. Folic acid was stopped at 7.5 months of age, and within 6 weeks he presented in status epilepticus. His brain MRI demonstrated marked hypoplasia of the cerebellar vermis. At 11 months of age, his CSF 5-MTHF level was extremely low, at 3 nmol/l. Treatment 5 mg folinic acid twice daily was begun. Brain MRI, repeated at 23 months, showed atrophy of the cerebellar hemispheres, patchy high signal in the subcortical white matter, and delayed myelination. Currently, at 5 years of age, he is microcephalic (HC < 0.4th centile), with severe developmental delay and cerebellar ataxia. DHFR mutation analysis in this child, apparently unrelated to the proband from family 1, revealed the same homozygous missense change, p.Leu80Phe.
We screened 20 additional patients with CFD of unknown etiology: 19 without anemia and one with anemia but with a low serum folate. No variants in the DHFR coding sequence were identified in these patients. Human, mouse, chicken, zebrafish, and Drosophila DHFR protein sequences were aligned by ClustalW and demonstrated conservation of the leucine residue at position 80 across species to Drosophila (Figure S2).
We assessed the expression profile of DHFR in human tissues by quantitative real-time PCR analysis (qPCR), using SYBR Green and Taqman (both from Applied Biosystems, Foster City, CA, USA) techniques. Total RNA for qPCR analyses from different adult human brain areas was purchased from Stratagene (La Jolla, CA, USA), except for hippocampus, thalamus, and spinal cord total RNA, which was ordered from Biochain (Hayward, CA, USA). RNA was available from a human embryonic brain panel of forebrain, telencephalon, diencephalon, midbrain, hindbrain, and optic vesicles (MRC Newcastle Brain Tissue Resource, Newcastle, UK) from Carnegie stage 16 (week 6) and stage 21 (week 8) human embryos.6 Total RNA from different human adult and fetal tissues was purchased from Stratagene Europe (Amsterdam, The Netherlands), except for cochlear RNA, isolated from an 8-week-old embryo with the use of the NucleoSpin RNA II Kit (Macherey-Nagel, Düren, Germany). Whole-blood RNA was isolated from the proband, his parents, and three healthy control individuals with the use of the PAXgene Blood RNA Kit (QIAGEN, Venlo, The Netherlands). Whole-blood and cochlear RNA was treated with DNase I (Invitrogen, Leek, The Netherlands), and the concentration and purity were determined by optical densitometry.
SYBR Green-based QPCR expression analysis was performed on a 7500 Fast Real-Time PCR System with the use of Power SYBR Green PCR Master Mix (Applied Biosystems). Primers were designed with the use of the primer3 program, and GUSB and PPIB were used as reference genes (Table S5). Five micrograms of total RNA was transcribed into cDNA with the use of the iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA, USA), and cDNA was purified with the NucleoSpin Extract II Kit (Macherey-Nagel, Düren, Germany). QPCR quantifications were performed in duplicate on the equivalent of 12.5 ng total RNA input. Differences in expression of a gene of interest between two samples were calculated by the comparative Ct or 2ΔΔCt method.8
TaqMan-based QPCR analysis was performed on a StepOne Plus Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). Predesigned, validated primers and probe mix for DHFR were purchased from Applied Biosystems. 18S and HPRT1 were used as reference genes. Four hundred nanograms of RNA was transcribed to cDNA with the use of the high-capacity RNA-to-cDNA Kit (Applied Biosystems). qPCR quantifications were performed in three triplicate experiments with water controls. The results were analyzed by StepOne software (Applied Biosystems). Differences in expression of a gene of interest between two samples were calculated by the comparative 2ΔΔCt method.
SYBR Green and Taqman real-time PCR analyses demonstrated that DHFR is widely expressed in fetal and adult tissues, including throughout the fetal and adult brains and whole blood (Figure 2B). Of note, expression was higher in the adult brain than in the fetal brain. Levels of DHFR transcripts from lymphocytes from the proband and his heterozygous parents were not significantly different from each other (data not shown).
We performed an immunoblot on fibroblasts from a control line and identified very faint bands, even with high-protein loading, showing that DHFR is expressed relatively poorly in the fibroblasts (data not shown). Therefore, we undertook immunoblot analysis of cellular and nuclear lysates from EBV-immortalized lymphoblastoid cells, which revealed that DHFR was undetectable in the proband and was present at lower levels in his parents compared to controls (Figure 2C).
DHFR activity in EBV-immortalized lymphoblastoid cells was measured by the formation of tetrahydrofolate from dihydrofolate. A DHFR activity kit (Sigma-Aldrich, St. Louis, MO, USA) was used for the assay. Lymphoblastoid cells were incubated with 300 μM NADPH and 250 μM DHF for 30 min. Samples were stabilized with 10 mM mercaptoethanol and frozen immediately. Lysates were prepared from cells with the use of three freeze-thaw cycles. After centrifugation, the supernatant was acidified with 10 mM formic acid, and an internal standard ([13C5]-5-methenyltetrahydrofolate) was added. Proteins were removed by a molecular weight cut-off filter (Millipore, molecular weight < 10,000). Tetrahydrofolate concentration was determined by liquid chromatography-tandem mass spectrometry (LC-MS/MS).9
DHFR activity in the proband's immortalized lymphoblasts was about 100-fold lower compared to controls, confirming the diagnosis of DHFR deficiency (Table 2). We were unable to determine whether the remaining activity in the proband's sample was residual DHFR activity or background activity due to unexpected processes influencing the signal in LC-MS/MS that we considered as folates. The obligate heterozygote parents showed an intermediate reduced DHFR activity compared to controls.
Table 2.
The Results of Measurement of DHFR Activity and Folate Vitamers in the Proband on Folinic Acid Therapy and in His Heterozygous Parents
| Assay (Units) | Reference range | Patient | Mother | Father | Control 1 | Control 2 |
|---|---|---|---|---|---|---|
| DHFR activity (nmol THF/hr/mg protein) | NA | 1.5 ↓↓ | 59.7 ↓ | 52.9 ↓ | 154.7 | 149.6 |
| MTHF (nmol/l) | 95–470 | 169 | 117 | 43 ↓ | NA | NA |
| NonmethylTHF (nmol/l) measured as THF + methylene-THF + methenyl-THF + formyl-THF |
0–67 | 77 ↑ | 9.8 | 45 | NA | NA |
| Dihydrofolate + folic acid (nmol/l) | 0 | 31 ↑↑ | 0 | 0 | NA | NA |
NA, not available; THF, tetrahydrofolate; ↑, values above the reference range; ↓, values below the reference range. Double arrows indicate values that are severely abnormal.
The effect of the DHFR mutation on the concentrations of folate vitamers in erythrocytes in vivo was studied with the use of a previously described method.9 Methylfolate and nonmethylfolate metabolites were determined by LC-MS/MS, which distinguishes 5-MTHF, folic acid (sum of folic acid and dihydrofolate, which converts to folic acid during assay), nonmethyltetrahydrofolates, and unsubstituted tetrahydrofolate. This showed an accumulation of folic acid and dihydrofolate in the proband, but not in his heterozygous parents (Table 2).
To explore the mechanism of loss of enzyme activity, a homology model of the p.Leu80Phe variant was constructed with the MOE molecular modeling program, version 2009.10 (Chemical Computing Group, Montreal, Canada). Energy minimizations, homology models, and protein-geometry analysis were performed with the compute module with the use of the CHARMm22 force field and a distance-dependent dielectric (ɛ = 1) as implemented in MOE.
The wild-type crystallographic structure of human DHFR (Protein Data Bank [PDB] coordinates 2W3M) was used for all calculations. The crystallographic waters along with the NADPH and folate were removed from the structure. Hydrogens were added at the normal ionization state of amino acids at pH 7.0, and atomic partial charges were fixed to the CHARMm22 atom types. To eliminate potential steric clashes, the backbone atoms of the protein were tethered to their initial positions by application of a tethering force constant of 1 kcal·mol−1, and the initial structure was minimized with the use of a conjugate gradient minimization until a convergence of 0.001 kcal·mol−1 was attained (Figure 3B).
Figure 3.
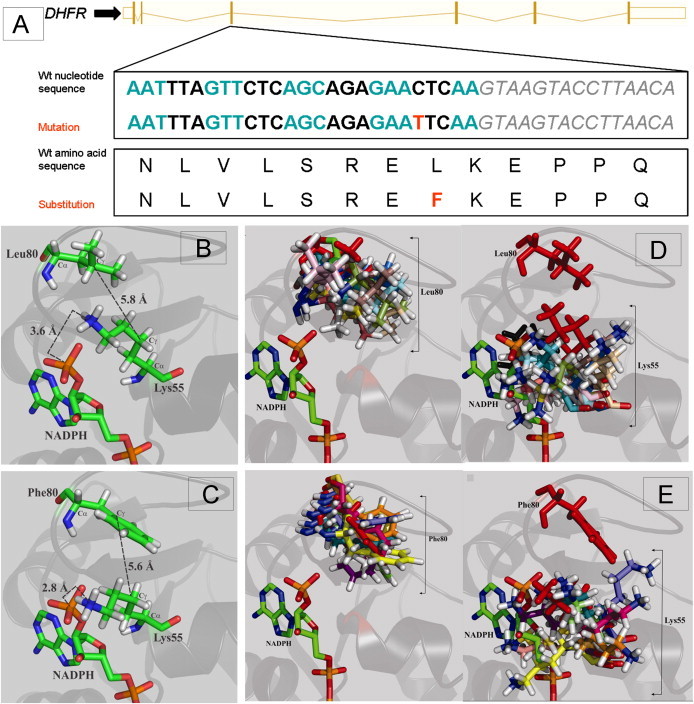
Results of Protein Modeling
(A) Schematic representation of the wild-type nucleotide and amino acid sequence of DHFR, with alternate codons in blue and black and the intron 3 sequence in gray italics. The mutation in exon 3 and resultant amino acid substitution are given in red.
(B) Normal orientation of Leu80 in the loop region of the crystal structure of wild-type DHFR (PDB coordinates: 2W3M). Leu80 is near the binding site of the adenine portion of NADPH and lies in close proximity to Lys55, a critical residue for cofactor binding and specificity. More specifically, the positively charged amine moiety of Lys55 lies within 3.6 Å of the 2′-phosphate group of NADPH (nitrogen to phosphorus distance), allowing for the formation of favorable ionic interactions that help secure and position the NADPH cofactor within the active site. Leu80 appears to provide the requisite sterics and hydrophobicity for optimal packing of the Lys55 amine relative to NADPH (Leu80-Cγ to Lys55-Cγ = 5.8 Å).
(C) The homology model of the Leu80Phe variant based on wild-type crystal structure of DHFR, demonstrating that the introduction of a phenylalanine residue at position 80 induces the Lys55 ɛ-nitrogen to be shifted to within 2.8 Å of the 2′-phosphorus of NADPH, thus resulting in a significant steric clash that would disrupt cofactor binding for the Leu80Phe mutant relative to wild-type DHFR. The Lys55-Cγ to Phe80-Cγ distance was found to be 5.6 Å, which is slightly shorter than the 5.8 Å found for wild-type DHFR, suggestive of the introduction of a weak π-cation interaction between the Lys55 ɛ-amine and the Phe80 phenyl ring. The formation of a new π-cation interaction in the Leu80Phe mutant would weaken the ionic interaction of Lys55 with NADPH.
(D) Overlay of simulated-annealing-derived conformations of wild-type DHFR, illustrating the degree of conformational sampling for the side chains of residues Leu80 (left) and Lys55 (right). The positions of the Leu80 and Lys55 residues in the energy-minimized X-ray structure (PDB 1W3M) are shown in red.
(E) Overlay of simulated-annealing-derived conformations for the mutant Leu80Phe variant DHFR, illustrating the degree of conformational sampling for the side chains of the mutational position Phe80 (left) and the Lys55 residue (right). The positions of the Phe80 and Lys55 residues in the energy-minimized homology model are shown in red. The wild-type Leu80 adopted a broader range of energy-minimized conformations than the mutant Phe80. This appears to be due to the bulk of the phenylalanine residue, which constrains its potential for conformational rearrangement. The average Lys55-Cα to residue 80-Cα was 11.0 ± 1.8 Å for the Phe80 variant, relative to 9.0 ± 0.7 Å for the wild-type. Similarly, the average Lys55-Cγ to Phe80-Cγ distance was increased to 7.9 ± 1.6 Å, relative to 6.3 ± 1.5 Å in the wild-type. Possibly, the shift in the backbone destabilizes the enzyme structure. In certain instances, the energy-minimized conformations adopted by the bulky Phe80 displace the Lys55 ɛ-amine into the NADPH binding pocket, lending support to the homology-modeling result in which a steric clash would hinder NADPH binding. Nonetheless, other instances place Phe80 farther away from the NADPH binding region, where it pulls the Lys55 ɛ-amine along with it. As a result, the ionic interaction between the Lys55 ɛ-amine and the NADPH phosphorus would be disrupted. In contrast, the poses adopted by Leu80 in the wild-type result in maintenance of more constant positioning of Lys55, thus generally maintaining the integrity of NADPH positioning. Overall, the poses obtained after molecular dynamics simulated annealing suggest that a potential destabilization of the protein and/or a disruption of NADPH binding could result from the Leu80Phe mutation in DHFR.
To construct a homology model of the p.Leu80Phe variant, the wild-type human DHFR template structure (PDB 2W3M) was modified to include the mutation. After removal of water and ligands and the addition of hydrogens and charges as for the wild-type, ten intermediate models were constructed and scored with the use of a generalized Born/volume integral internal fitness function within the MOE homology modeler.10 The model with the best internal fitness score was selected and subjected to a conjugate gradient minimization with a tethering force as described above, until a convergence of 0.001 kcal·mol−1 was attained. The structures for the wild-type and the p.Leu80Phe mutant of DHFR revealed that introduction of a phenylalanine induces a shift in the position of the critical lysine 55 residue, predicting a significant steric clash and disrupting cofactor binding for the p.Leu80Phe mutant relative to wild-type DHFR (Figure 3C).
Homology modeling holds the inherent disadvantage of exploring only the local conformational space, thus potentially remaining within a local energy minimum that does not represent the lowest energy structure. Therefore, we performed a molecular dynamics simulated annealing, which allowed the backbone and side-chains to sample the accessible conformational space much more widely for comparison of the effects of the variant on the structural integrity and dynamic behavior of DHFR. All molecular dynamics simulated annealing calculations were performed with the InsightII package, version 2000.1 (Accelrys, San Diego, CA, USA). The Biopolymer, Analysis, and Decipher modules were used for analysis of molecular dynamics simulated annealing trajectories. Simulated annealing calculations were performed with the Discover module employing the constant valence force field (CVFF), and calculations were run on an Origin 2000 Silicon Graphics Fuel Server. For the minimized structures of wild-type and mutant p.Leu80Phe DHFR, atomic potentials were fixed to the default CVFF atom types. The wild-type and mutant p.Leu80Phe DHFR were then subjected to a molecular dynamics simulated annealing protocol according to the following iterative procedure: The structure was heated to 1000 K over 5 ps, equilibrated at 1000 K for 3 ps, then cooled down to 300 K in 5 ps, with the use of an exponential rate constant (timtmp) of 0.7 ps for both the heating and cooling steps. The time step of the molecular dynamics simulations was set to 1 fs, and a distance-dependent dielectric constant of 1 was applied to the system. During the simulated annealing protocol, the backbone atoms of the protein were tethered to their initial positions by applying a force constant of 1200 kcal·mol−1·Å2. This force constant sufficiently restricts the protein backbone to limit local unfolding of secondary structural elements while allowing for subtle backbone movements that permit broad side-chain conformation exploration. After each simulated annealing cycle, the resulting cooled structures were subjected to a final energy minimization step involving 1000 steps of steepest descent followed by conjugate gradient minimization until a convergence of 0.01 kcal·mol−1·Å2 was reached. This cycle was repeated nine times for a total simulation time of 130 ps, yielding ten minimized structures for the wild-type DHFR. The protocol was repeated for mutant p.Leu80Phe, resulting in ten minimized conformations for the p.Leu80Phe mutant. This analysis suggested that the p.Leu80Phe mutation could result in potential destabilization of the protein and/or a disruption of NADPH binding (Figures 3D and 3E).
DHFR catalyzes the NADPH-dependent reduction of dihydrofolate to tetrahydrofolate, an essential step in the synthesis of precursors of DNA, including glycine and purines, and the conversion of deoxyuridine monophosphate to deoxythymidine monophosphate.11 It is also the only enzyme that reduces folic acid, a synthetic vitamin not found in nature, to dihydrofolate. DHFR is a key enzyme in all prokaryotes and eukaryotes, being present in all dividing cells. Cell lines or animal models deficient in DHFR are considered not viable without supplemented glycine, purines, and thymidine, indicating its critical role in cellular function. Variants in DHFR, including a 19 base pair deletion in intron 1, have been studied in numerous disorders, including breast cancer,12 neural-tube defects,13 and preterm delivery.14 However, no molecularly confirmed cases of DHFR deficiency have been described to date. In 1967, Walters reported a child with megaloblastic anemia and proposed that he may have DHFR deficiency (T.R. Walters, 1967, Midwest Society for Pediatric Research, abstract). Skin fibroblasts from this child were later demonstrated to have normal enzyme activity.15 In 1976, Tauro et al. reported two neonates with congenital deficiency of DHFR.16 However, subsequent analysis revealed that the first child had methionine synthase reductase deficiency (MIM 602568),17 and the second patient was shown to have transcobalamin II deficiency (MIM 275350).18
The phenotype of the patients we describe with biallelic mutations in DHFR is distinct from patients with the other recognized disorders of folate metabolism or transport. Megaloblastic anemia was the presenting feature in our patients with DHFR deficiency, which resolved completely with folic acid or folinic acid replacement. In contrast, CSF 5-MTHF levels normalized and seizures responded better to folinic acid. The neurodevelopmental improvement was less marked than that reported in patients with FOLR1 mutations and cerebral folate receptor antibodies.2,3 The presence of cerebral and cerebellar atrophy also distinguishes CFD due to DHFR deficiency from other forms of CFD. Reduced or delayed myelination in the brain may also be a feature of the condition, but further identification of patients is required to confirm this, and studies should be undertaken to understand the mechanism in the context of normal homocysteine levels. Of note, none of our patients had a neural-tube defect. The biochemical profile of DHFR deficiency is unique, given that serum folate and homocysteine levels were normal in the affected individuals. The normal serum homocysteine levels may be explained by way of the fact that DHFR deficiency does not directly alter 5-MTHF levels and inhibits only the generation of tetrahydrofolate from dihydrofolate or folic acid. The folate vitamer profile of the proband demonstrated an accumulation of dihydrofolate and folic acid and nonmethyltetrahydrofolate within erythrocytes, consistent with the observed enzyme defect. In the assay, dihydrofolate is converted to folic acid, and therefore the two compounds are measured together. Of note, the patient's analyzed blood samples were obtained during folinic acid treatment. The reduction of folinic acid to tetrahydrofolate is DHFR independent, and thus the accumulation of dihydrofolate/folic acid is unlikely to be due to the ongoing therapy.19
Cerebral BH4 deficiency was noted in the proband. It was not corrected by folinic acid and could explain some of the residual neurological symptoms. The absence of hyperphenylalaninemia in our patients indicates that BH4 deficiency was primarily cerebral and was not due to any of the known causes of systemic BH4 deficiency. BH4 is a cofactor for phenylalanine-4-hydroxylase, tyrosine-3-hydroxylase, and tryptophan-5-hydroxylase, and it is required for the production of monoamines, including dopamine, serotonin, and norepinephrine.20 Of note, in the proband, CSF homovanillic acid (a major catecholamine metabolite) and 5-hydroxyindoleacetic acid (a serotonin metabolite) levels were slightly low and remained in the low-normal range after folinic acid treatment.
5-MTHF prevents peroxynitrite-induced BH4 oxidation in blood vessels.21 Thus, the low cerebral BH4 level in our patient could indicate a similar role for 5-MTHF in the CSF or brain. An alternative explanation could be derived from another recognized role of DHFR, whereby with NADPH it reduces BH2 to BH4 for pterin salvage22 and plays an important role in the coupling of endothelial nitric oxide synthase by maintaining the BH4:BH2 ratio.23 Our findings support a similar role for DHFR in the brain. Notably, the neurological phenotypes of DHFR and dihydrobiopterin reductase (DHPR—another key enzyme in pterin salvage) deficiency (MIM 261630) overlap considerably.24 In addition to developmental delay, DHPR deficiency results in early brain atrophy and intracranial perivascular calcifications. Interestingly, DHPR has a role in maintaining tetrahydrofolate in the reduced form and patients with DHPR deficiency also develop CFD.25
DHFR deficiency provides further evidence of the link between folates and pterin metabolism in the brain. Low folate levels are a risk factor for dementia, depression, and poor cognitive function in the elderly, and folic acid supplementation can improve symptoms of depression, Alzheimer disease, and Parkinson disease in patients on L-DOPA.26 BH4 levels in CSF also correlate with red cell folate in depression.27 Our study supports further evaluation of the role of folates in cerebral pterin metabolism.
Expression of DHFR is markedly upregulated during the S-phase of the cell cycle.28 In rapidly dividing cells, DFHR inhibition negatively affects DNA synthesis, leading to cell death, and therefore inhibition of DHFR is a key therapeutic target. The antibiotic trimethoprim and the antiprotozoal pyrimethamine both inhibit DHFR. Interestingly, hyperphenylalaninemia and hematological and neurological disturbances are well-recognized side effects of pyrimethamine.19,20 The anticancer and antiinflammatory drug methotrexate binds to DHFR more potently than folate and inhibits thymidine production. However, resistance to methotrexate can arise as the result of amplification and somatic mutations of DHFR.29,30 The use of methotrexate during organogenesis is associated with congenital defects, including intrauterine growth retardation, decreased calvarial ossification, dysmorphic features, and limb abnormalities.31 Likewise, methotrexate toxicity causes a range of symptoms, including hepatotoxicity, renal failure, myelosuppression, acute lung injury, seizures, motor dysfunction, and encephalopathy. The clinical features of both methotrexate embryopathy and toxicity are distinct from those present in our patients. Additionally, in contrast with DHFR deficiency, methotrexate therapy is associated with an increase in homocysteine levels.32 The biochemical and phenotypic differences between DHFR deficiency and methotrexate therapy may be due to off-target effects of methotrexate that require further characterization.
In summary, we have described an inborn error of metabolism due to germline mutations in DHFR and characterized by megaloblastic anemia and/or pancytopenia in the presence of normal serum folate, severe CFD, moderate cerebral BH4 deficiency, and cerebral and cerebellar atrophy. The phenotype of our patients is consistent with the known role of DHFR in actively dividing cells. Our work shows that DHFR-deficient cell lines can be viable and that an external source of folate may counter some of the effects resulting from loss of DHFR activity. The developing fetus gets its supply of 5-MTHF from the mother and thus can survive even in presence of DHFR deficiency. Notably, the half life of folates in the body is approximately 90 days,33 which coincides with onset of symptoms in all three patients. Although subtle neurodevelopmental delay can be missed, all three affected children probably had normal development until the age of 3 months. This raises the possibility of better outcome with active perinatal management or earlier diagnosis and treatment. The effect of differences in time of presentation and phenotype due to folate fortification across the world will be interesting. Our study indicates functional overlap between DHFR and BH4 metabolism in the brain, possibly at the level of DHPR, and supports further evaluation of the role of folates in cerebral pterin metabolism.
Acknowledgments
We thank the families for their help with the study. The support of the NIHR Manchester Biomedical Research Centre and Natural Sciences and Engineering Research Council of Canada is acknowledged. We are also thankful for the expert help of William Fergusson during cell culture.
Contributor Information
Simon Jones, Email: simon.jones@cmft.nhs.uk.
William G. Newman, Email: william.newman@manchester.ac.uk.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
AutoSNPa, http://dna.leeds.ac.uk/autosnpa
Primer3, http://frodo.wi.mit.edu/cgi-bin/primer3/primer3.cgi
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
- 1.Djukic A. Folate-Responsive Neurologic Diseases. Pediatr. Neurol. 2007;37:387–397. doi: 10.1016/j.pediatrneurol.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 2.Steinfeld R., Grapp M., Kraetzner R., Dreha-Kulaczewski S., Helms G., Dechent P., Wevers R., Grosso S., Gärtner J. Folate Receptor Alpha Defect Causes Cerebral Folate Transport Deficiency: A Treatable Neurodegenerative Disorder Associated with Disturbed Myelin Metabolism. Am. J. Hum. Genet. 2009;85:354–363. doi: 10.1016/j.ajhg.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramaekers V.T., Rothenberg S.P., Sequeira J.M., Opladen T., Blau N., Quadros E.V., Selhub J. Autoantibodies to Folate Receptors in the Cerebral Folate Deficiency Syndrome. N. Engl. J. Med. 2005;352:1985–1991. doi: 10.1056/NEJMoa043160. [DOI] [PubMed] [Google Scholar]
- 4.Hyland K., Surtees R.A.H., Heales S.J.R., Bowron A., Howells D.W., Smith I. Cerebrospinal fluid concentrations of pterins and metabolites of serotonin and dopamine in a pediatric reference population. Pediatr. Res. 1993;34:10–14. doi: 10.1203/00006450-199307000-00003. [DOI] [PubMed] [Google Scholar]
- 5.Qiu A., Jansen M., Sakaris A., Min S.H., Chattopadhyay S., Tsai E., Sandoval C., Zhao R., Akabas M.H., Goldman I.D. Identification of an Intestinal Folate Transporter and the Molecular Basis for Hereditary Folate Malabsorption. Cell. 2006;127:917–928. doi: 10.1016/j.cell.2006.09.041. [DOI] [PubMed] [Google Scholar]
- 6.Daly S.B., Urquhart J.E., Hilton E., McKenzie E.A., Kammerer R.A., Lewis M., Kerr B., Stuart H., Donnai D., Long D.A. Mutations in HPSE2 Cause Urofacial Syndrome. Am. J. Hum. Genet. 2010;86:963–969. doi: 10.1016/j.ajhg.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carr I.M., Flintoff K.J., Taylor G.R., Markham A.F., Bonthron D.T. Interactive visual analysis of SNP data for rapid autozygosity mapping in consanguineous families. Hum. Mutat. 2006;27:1041–1046. doi: 10.1002/humu.20383. [DOI] [PubMed] [Google Scholar]
- 8.Pfaffl M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith D.E.C., Kok R.M., Teerlink T., Jakobs C., Smulders Y.M. Quantitative determination of erythrocyte folate vitamer distribution by liquid chromatography-tandem mass spectrometry. Clin. Chem. Lab. Med. 2006;44:450–459. doi: 10.1515/CCLM.2006.085. [DOI] [PubMed] [Google Scholar]
- 10.Labute P. The generalized Born/volume integral implicit solvent model: Estimation of the free energy of hydration using London dispersion instead of atomic surface area. J. Comput. Chem. 2008;29:1693–1698. doi: 10.1002/jcc.20933. [DOI] [PubMed] [Google Scholar]
- 11.Trimble J.J., Murthy S.C., Bakker A., Grassmann R., Desrosiers R.C. A gene for dihydrofolate reductase in a herpesvirus. Science. 1988;239:1145–1147. doi: 10.1126/science.2830673. [DOI] [PubMed] [Google Scholar]
- 12.Xu X., Gammon M., Wetmur J., Rao M., Gaudet M., Teitelbaum S., Britton J., Neugut A., Santella R., Chen J. A functional 19-base pair deletion polymorphism of dihydrofolate reductase (DHFR) and risk of breast cancer in multivitamin users. Am. J. Clin. Nutr. 2007;85:1098–1102. doi: 10.1093/ajcn/85.4.1098. [DOI] [PubMed] [Google Scholar]
- 13.van der Linden I.J.M., Afman L.A., Heil S.G., Blom H.J. Genetic variation in genes of folate metabolism and neural-tube defect risk. Proc. Nutr. Soc. 2006;65:204–215. doi: 10.1079/pns2006495. [DOI] [PubMed] [Google Scholar]
- 14.Johnson W.G., Scholl T.O., Spychala J.R., Buyske S., Stenroos E.S., Chen X. Common dihydrofolate reductase 19-base pair deletion allele: a novel risk factor for preterm delivery. Am. J. Clin. Nutr. 2005;81:664–668. doi: 10.1093/ajcn/81.3.664. [DOI] [PubMed] [Google Scholar]
- 15.Erbe R.E. Inborn errors of folate metabolism. N. Engl. J. Med. 1975;293:753–757. doi: 10.1056/NEJM197510092931506. [DOI] [PubMed] [Google Scholar]
- 16.Tauro G.P., Danks D.M., Rowe P.B., Van der Weyden M.B., Schwarz M.A., Collins V.L., Neal B.W. Dihydrofolate Reductase Deficiency Causing Megaloblastic Anemia in Two Families. N. Engl. J. Med. 1976;294:466–470. doi: 10.1056/NEJM197602262940903. [DOI] [PubMed] [Google Scholar]
- 17.Rosenblatt D.S., Fenton W.A. Inherited disorders of folate and cobalamin transport and metabolism. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., editors. The Metabolic and Molecular Basis of Inherited Diseases. McGraw-Hill; New York: 2001. pp. 3903–3904. [Google Scholar]
- 18.Hoffbrand A., Tripp E., Jackson B., Luck W., Frater-Schröder M. Hereditary Abnormal Transcobalamin II Previously Diagnosed as Congenital Dihydrofolate Reductase Deficiency. N. Engl. J. Med. 1984;310:789–790. doi: 10.1056/nejm198403223101217. [DOI] [PubMed] [Google Scholar]
- 19.Padmanabhan S., Tripathi D.N., Vikram A., Ramarao P., Jena G.B. Methotrexate-induced cytotoxicity and genotoxicity in germ cells of mice: Intervention of folic and folinic acid. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2009;673:43–52. doi: 10.1016/j.mrgentox.2008.11.011. [DOI] [PubMed] [Google Scholar]
- 20.Thony B., Auerbach G., Blau N. Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem. J. 2000;347:1–16. [PMC free article] [PubMed] [Google Scholar]
- 21.Antoniades C., Shirodaria C., Warrick N., Cai S., de Bono J., Lee J., Leeson P., Neubauer S., Ratnatunga C., Pillai R. 5-Methyltetrahydrofolate Rapidly Improves Endothelial Function and Decreases Superoxide Production in Human Vessels: Effects on Vascular Tetrahydrobiopterin Availability and Endothelial Nitric Oxide Synthase Coupling. Circulation. 2006;114:1193–1201. doi: 10.1161/CIRCULATIONAHA.106.612325. [DOI] [PubMed] [Google Scholar]
- 22.Nichol C.A., Lee C.L., Edelstein M.P., Chao J.Y., Duch D.S. Biosynthesis of tetrahydrobiopterin by de novo and salvage pathways in adrenal medulla extracts, mammalian cell cultures, and rat brain in vivo. Proc. Natl. Acad. Sci. USA. 1983;80:1546–1550. doi: 10.1073/pnas.80.6.1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crabtree M.J., Tatham A.L., Hale A.B., Alp N.J., Channon K.M. Critical Role for Tetrahydrobiopterin Recycling by Dihydrofolate Reductase in Regulation of Endothelial Nitric-oxide Synthase Coupling. J. Biol. Chem. 2009;284:28128–28136. doi: 10.1074/jbc.M109.041483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ponzone A., Spada M., Ferraris S., Dianzani I., de Sanctis L. Dihydropteridine Reductase Defciency in Man: From Biology to Treatment. Med. Res. Rev. 2004;24:127–150. doi: 10.1002/med.10055. [DOI] [PubMed] [Google Scholar]
- 25.Pollock R.J., Kaufman S. Dihydropteridine Reductase May Function In Tetrahydrofolate Metabolism. J. Neurochem. 1978;31:115–123. doi: 10.1111/j.1471-4159.1978.tb12439.x. [DOI] [PubMed] [Google Scholar]
- 26.Stanger O., Fowler B., Piertzik K., Huemer M., Haschke-Becher E., Semmler A., Lorenzl S., Linnebank M. Homocysteine, folate and vitamin B12 in neuropsychiatric diseases: review and treatment recommendations. Expert Rev. Neurother. 2009;9:1393–1412. doi: 10.1586/ern.09.75. [DOI] [PubMed] [Google Scholar]
- 27.Bottiglieri T., Hyland K., Laundy M., Godfrey P., Carney M.W.P., Toone B.K., Reynolds E.H. Folate deficiency, biopterin and monoamine metabolism in depression. Psychol. Med. 1992;22:871–876. doi: 10.1017/s0033291700038447. [DOI] [PubMed] [Google Scholar]
- 28.Bjarnason G.A., Jordan R.C.K., Wood P.A., Li Q., Lincoln D.W., Sothern R.B., Hrushesky W.J.M., Ben-David Y. Circadian Expression of Clock Genes in Human Oral Mucosa and Skin: Association with Specific Cell-Cycle Phases. Am. J. Pathol. 2001;158:1793–1801. doi: 10.1016/S0002-9440(10)64135-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goker E., Waltham M., Kheradpour A., Trippett T., Mazumdar M., Elisseyeff Y., Schnieders B., Steinherz P., Tan C., Berman E. Amplification of the dihydrofolate reductase gene is a mechanism of acquired resistance to methotrexate in patients with acute lymphoblastic leukemia and is correlated with p53 gene mutations. Blood. 1995;86:677–684. [PubMed] [Google Scholar]
- 30.Gorlick R., Goker E., Trippett T., Waltham M., Banerjee D., Bertino J.R. Intrinsic and acquired resistance to methotrexate in acute leukemia. N. Engl. J. Med. 1996;335:1041–1048. doi: 10.1056/NEJM199610033351408. [DOI] [PubMed] [Google Scholar]
- 31.Bawle E.V., Conard J.V., Weiss L. Adult and two children with fetal methotrexate syndrome. Teratology. 1998;57:51–55. doi: 10.1002/(SICI)1096-9926(199802)57:2<51::AID-TERA2>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 32.van Ede A.E., Laan R.F.J.M., Blom H.J., Boers G.H.J., Haagsma C.J., Thomas C.M.G., de Boo T.M., van de Putte L.B.A. Homocysteine and folate status in methotrexate-treated patients with rheumatoid arthritis. Rheumatology. 2002;41:658–665. doi: 10.1093/rheumatology/41.6.658. [DOI] [PubMed] [Google Scholar]
- 33.Gregory J.F., III, Quinlivan E.P. In vivo kinetics of folate metabolism. Annu. Rev. Nutr. 2002;22:199–220. doi: 10.1146/annurev.nutr.22.120701.083554. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.