

Abstract
Assessing the significance of novel genetic variants revealed by DNA sequencing is a major challenge to the integration of genomic techniques with medical practice. Many variants remain difficult to classify by traditional genetic methods. Computational methods have been developed that could contribute to classifying these variants, but they have not been properly validated and are generally not considered mature enough to be used effectively in a clinical setting. We developed a computational method for predicting the effects of missense variants detected in patients with hypertrophic cardiomyopathy (HCM). We used a curated clinical data set of 74 missense variants in six genes associated with HCM to train and validate an automated predictor. The predictor is based on support vector regression and uses phylogenetic and structural features specific to genes involved in HCM. Ten-fold cross validation estimated our predictor's sensitivity at 94% (95% confidence interval: 83%–98%) and specificity at 89% (95% confidence interval: 72%–100%). This corresponds to an odds ratio of 10 for a prediction of pathogenic (95% confidence interval: 4.0–infinity), or an odds ratio of 9.9 for a prediction of benign (95% confidence interval: 4.6–21). Coverage (proportion of variants for which a prediction was made) was 57% (95% confidence interval: 49%–64%). This performance exceeds that of existing methods that are not specifically designed for HCM. The accuracy of this predictor provides support for the clinical use of automated predictions alongside family segregation and population frequency data in the interpretation of new missense variants and suggests future development of similar tools for other diseases.
Introduction
DNA sequencing is quickly becoming the method of choice for clinical genetic diagnostics. The improvement in clinical sensitivity that sequencing provides over genotyping platforms is invaluable, especially in disorders that show locus and allelic heterogeneity. However, there are also important challenges presented by the use of DNA sequencing, including the difficulty of interpreting novel sequence variants. There is currently little standardization of variant classification in the genetics community. Most clinics use a combination of traditional genetic methods relying on segregation with the disease in families, frequency in controls, biochemical characterization, and evolutionary conservation at the variant position.1 This manual classification process is time consuming and requires significant expert knowledge. More frustratingly, it often fails to produce a classification at all: variants with incomplete or conflicting data are routinely classified as variants of unknown significance (VUSs), and no confident classification is reported to the patient or the referring physician. In some genes, these VUSs comprise as many as one-quarter to one-half of all reported variants.2 This problem is only getting worse. As next-generation sequencing technologies begin to enter widespread clinical use, the volume of novel variants is expected to expand by several orders of magnitude. The genetics community must therefore begin to develop robust automated methods to classify novel variants accurately.
There currently exist several computational tools for predicting the functional effects of genetic variants.3–5 However, these tools in general were not designed for clinical use, have not been rigorously tested on individual genes or diseases, and have not undergone any kind of validation against well-curated data sets. Therefore, the sensitivities and specificities of these predictors are in general ill-defined. This lack of proper validation has created the perception among medical professionals that automated predictors cannot be trusted.6 Consequently, although most geneticists are familiar with these tools, the predictions they produce are typically not formally included in clinical variant classification methods and are therefore not communicated to physicians via clinical reports. Several studies have attempted to address this problem by validating existing predictors against known disease-causing variants, largely arriving at the conclusion that these methods are not yet mature enough for clinical use.6–8
Variant classification pipelines that are considered mature enough for clinical use are generally designed from the ground up with clinical use in mind and are designed, demonstrated, and validated using variants classified according to clinical criteria. Examples of such pipelines include the classification procedure currently in use at the Laboratory for Molecular Medicine (LMM), a clinical diagnostic laboratory in the U.S., and the integrated evaluation of BRCA gene variants that developed from the work of Goldgar et al.9 However, fully automated computational predictors are not currently designed in this way. We therefore set out to test whether this methodology could successfully create an automated predictor that would be useful to medical professionals as a tool for classifying novel missense variants. We chose to target one specific disease and a limited number of genes in which disease-causing variants might be found so that we would be able to generate a high-quality set of manually classified missense variants to use as the gold standard for training and validating our predictions. We also hoped that focusing on a limited number of functionally related genes would allow us to identify common features of these genes and common mechanisms of disease in these genes, which would help us to make our predictor more accurate.
The disease we chose was hypertrophic cardiomyopathy (HCM [MIM 192600]), an autosomal dominant disease of the myocardium (heart muscle) with an incidence of roughly one in 500 individuals and a largely genetic basis.10 Variants in over 20 genes are associated with HCM, with over 900 unique variants reported in the literature, and sequencing of many of these genes can be ordered for clinical testing in CLIA-approved laboratories. The vast majority of pathogenic variants are found in eight genes that encode for units of the cardiac sarcomere, a contractile protein complex in the heart: β-cardiac myosin heavy chain (MYH7 [MIM 160760]), cardiac actin (ACTC1 [MIM 102540]), cardiac troponin T (TNNT2 [MIM 191045]), α-tropomyosin (TPM1 [MIM 191010]), cardiac troponin I (TNNI3 [MIM 191044]), cardiac myosin-binding protein C (MYBPC3 [MIM 600958]), and the myosin light chains (MYL2 [MIM 160781] and MYL3 [MIM 160790]). Sequencing of these genes yields a high number of novel variants, mainly because of the high prevalence of private familial variants. Roughly 50% of probands tested have a disease-causing variant in one of these genes, and approximately 80% of those are in MYH7 and MYBPC3 (H.L.R., unpublished data).11 Missense variants represent nearly all such variants detected in MYH7 and 35% of those in MYBPC3. Missense variants exerting dominant negative effects on the sarcomere structure represent the vast majority of all variants. The notable exception is MYBPC3, where missense variants constitute only 35% of all variants, the remainder being splice, nonsense, or frameshift variants leading to loss of function. At the time of this study, the LMM had identified over 700 variants in HCM-related genes over 5 years of testing, over half of which were novel at the time of reporting and over half of which were missense changes. We performed a systematic manual classification of these variants, producing a final data set of 74 missense variants with extremely confident manual classifications. Using these 74 variants as our gold standard, we then set out to develop and validate a computational method that could predict the pathogenicity of any variant in these six genes.
Material and Methods
We created a computational method to predict the pathogenicity of a novel variant in any of the six genes we chose to screen for HCM mutations. Our method, like other existing methods12–16 and, particularly, the recently developed algorithm PolyPhen-2,17 integrates phylogenetic and structural information from several heterogeneous sources with a probabilistic classifier. However, unlike these methods, it exploits the narrow focus on six specific genes known to contain variants that cause the disease to improve the prediction strategy significantly. Also unlike these methods, it uses variants classified according to clinical criteria of pathogenicity to train the probabilistic classifier. The selection and classification of these variants, the features used for classification, and the training and validation of the classifier are all described below. This study was performed under an institutional-review-board-approved protocol through Partners Healthcare System.
Selection of Target Genes
HCM is caused primarily by variants in eight genes encoding protein subunits of the cardiac sarcomere. We initially attempted to use all eight genes to develop our predictor. However, after constructing our data set (see Manual Classification of HCM Variants below), we examined the distribution of variants and found that the final data set contained no variants in ACTC1 and only one in MYL3. We discarded these two genes and built our classifier around the remaining six (MYH7, TNNT2, TPM1, TNNI3, MYPC3, and MYL2).
Manual Classification of HCM Variants
We relied on LMM's standard variant-assessment pipeline to create our data set of manually classified variants. To ensure unbiased training and testing of our computational method, we excluded from manual classification information that was accessible to the method such as evolutionary conservation or structural data, even though this information is currently used in the pipeline. Each variant recieved a classification of pathogenic, likely pathogenic benign, likely benign, or VUS. The basic decision process we used is described below and shown in Figure 1.
Figure 1.
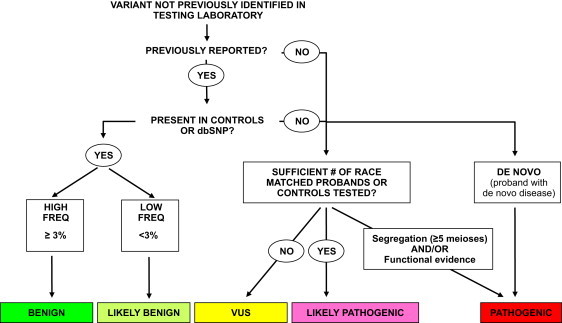
Process Used to Classify Variants at the LMM
This process is described in detail in Material and Methods. We treat the pathogenic, benign, and likely benign categories as high-confidence classifications for the purposes of training the automatic classifier.
Pathogenic
Variants with a minimum of five informative meioses supporting familial cosegregation with HCM, absent in healthy controls, and/or having strong functional data are classified as pathogenic. In HCM, informative meioses typically only include individuals who are positive for both phenotype and genotype. This level of stringency is required because of the highly variable expressivity and reduced penetrance, which make individuals without the phenotype largely uninformative, regardless of their genotype.
Likely Pathogenic
The minimum requirement to classify a variant as likely pathogenic is absence from race-matched controls or a large cohort of race-matched probands. The LMM has previously sequenced sarcomere genes in over 1000 HCM probands of European ancestry. Absence from this cohort was accepted in lieu of healthy control data because it serves to set a maximum population frequency of one per the total number of probands tested. Novel variants detected in minority populations are therefore often classified as VUSs because of the lack of control cohorts or large proband datasets.
Benign or Likely Benign
Variants that are frequent in the general population (at least 3%) are classified as benign. Variants present in controls at frequencies below 3% and without other suspicion for pathogenicity are classified as likely benign.
VUS
This class commonly includes variants for which there is insufficient evidence to classify the variant in any of the other four categories, or variants for which the evidence is conflicting.
Figure 2 shows the distribution of variants by the classification category in our database.
Figure 2.
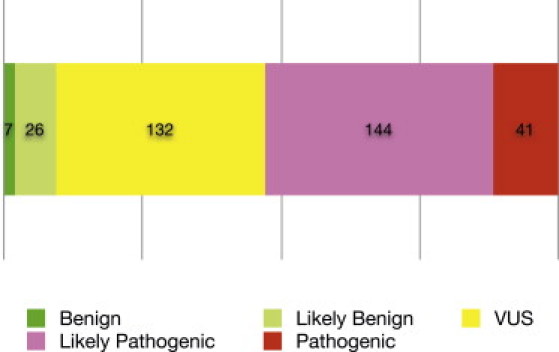
Distribution of Variant Pathogenicity
We categorized 350 missense variants in six genes according to the criteria described in Figure 1. The three categories pathogenic, benign, and likely benign were treated as high-confidence classifications and used as training data for our classifier (enumerated in Table S1).
After applying these criteria to the complete set of variants collected by LMM, we filtered the resulting data set to exclude unconfident predictions. We excluded variants in the likely-pathogenic category because we considered the classification for this category to be not stringent enough. We also excluded variants in the VUS category because this category carries no clinical or biological significance. This left us with 41 pathogenic variants, which we treated as truly pathogenic, and seven benign and 26 likely benign variants, all of which we treated as truly benign. These 74 variants became our gold standard for validation of our predictor. The complete list of 74 variants is shown in Table S1.
There is a possibility that the manual method of variant classification may have selected variants resulting in the most severe phenotypes, such as those seen in early-onset cases, which may reduce the utility of our classifier for less severe variants. To investigate this possibility, we used the age at which an individual was tested as a proxy for age at onset. The distribution of ages of all probands tested is roughly trimodal with clear peaks at and 15 years of age and a broad distribution centered around 50 years of age (Figure S1). The distribution of pathogenic variants in this population follows a similar distribution with pathogenic variants detected across a wide range of age groups tested. If we were indeed selecting for only the most severe, early-onset phenotypes, we would expect pathogenic variants to be overrepresented in newborns and teenagers and to be absent in late-onset cases. This does not appear to be the case, and we are confident that our training set does not only consist of pathogenic variants that lead to high penetrance, early-onset disease.
Predictive Features
We used four features in the final predictor. These features are described below.
PolyPhen-2 Prediction
Our first feature was a prediction made by the existing method PolyPhen-2.17 PolyPhen-2's predictions integrate several sources of phylogenetic and structural information using machine learning. Its output represents a general-purpose prediction made without knowledge of the specific disease under consideration. The PolyPhen-2 software reports a score ranging from 0 (neutral) to 1 (damaging), which represents the confidence of its internal classifier. We used this integrated score as a single feature in our predictor.
MrBayes Substitution Rate Score
Our second feature was the rate of evolution for each site in each gene. We computed this using the Markov chain Monte Carlo algorithm in the MrBayes software package.18 This score took several days of computer time to calculate for all six genes and would not have been feasible to calculate for a genome-wide data set.
Examples of the MrBayes instruction files we used are available as Figure S2. We used a function that infers site-specific evolution rates and includes them in the program's output. MrBayes reports the rate at positions with insufficient alignment depth as 1.000, so all scores of exactly 1.000 were treated as missing data. We normalized this rate so that the mean rate for each gene was 1.000.
Coiled-Coil Score
Our next two features took advantage of specific properties of the six target genes. Four of the six target proteins had significant coiled-coil regions: MYH7, TNNI3, TNNT2, and TPM1. We used the COILS2 software to predict the tendencies of the wild-type and mutant sequences to form coiled coils.19,20 Variants that significantly change the coiled-coil tendency of the sequence are likely to interfere with protein function.
For each of the four proteins, we downloaded annotations from SMART to determine the locations of coiled-coil regions.21 For any variant in a coiled-coil region, we ran COILS2 on both the wild-type and variant sequences of the coiled-coil region that contained the variant. COILS2 outputs a score indicating coiled-coil tendency for each residue in the input sequence with each score depending on the entire sequence. The feature we used in the final predictor was the magnitude of the largest single-residue change.
Protein Structure Comparison Score
Four of the six target proteins are contractile proteins studied in multiple conformations (MYH7 and MYL2 in ATP, ADP, and nucleotide-free states; TNNI3 and TNNT2 in Ca2+-activated and Ca2+-free states). For these four proteins, we measured the motion of each residue between the two conformations. Highly mobile residues were considered functionally important to the conformational change, whereas highly immobile residues were considered structurally important. Intermediately mobile residues were scored as unimportant. We measured the size of each residue's motion by comparing the displacement of the residue to the expected probability distribution of displacements under random thermal motion.
We used two sets of structures to compute this score. One was a set of six structures of a three-chain scallop myosin complex, consisting of the myosin heavy chain (corresponding to MYH7 in human heart muscle) and the two myosin light chains (corresponding to MYL2 and MYL3 in human heart muscle).22,23 One of these structures was not bound to a nucleotide (PDB ID 1KK7), two were bound to ADP analogs (PDB ID 1KK8 and 1B7T), and three were bound to ATP analogs (PDB ID 1KQM, 1KWO, and 1L2O). The other set of structures was a pair of structures of a three-chain chicken troponin complex, consisting of troponin I (corresponding to TNNI3 in human heart muscle), troponin T (corresponding to TNNT2 in human heart muscle), and troponin C (corresponding to TNNC1 in human heart muscle).24 One of these structures was activated by calcium ions (PDB ID 1YTZ), and the other had no calcium bound to it (PDB ID 1YV0).
We performed pairwise comparisons between structures that represented the same molecule in different biological states. Pairs of structures that represented the same biological state (such as 1KK8 and 1B7T, which both represent the ADP-bound state of myosin) were excluded under the assumption that differences between these structures would represent differences in the experimental preparation rather than a meaningful conformational change. We aligned each pair of structures with LovoAlign and measured the displacement between the α carbons of corresponding residues.25
The variance in the position of an atom in a crystal structure is given by , where B is the crystallographic temperature factor for the atom. We computed this variance for the α carbon of each residue, estimating B as the average of the reported temperature factor for that atom across the two crystal structures. We used Student's t test to compare the squared displacement of the atom with its expected variance. This produced a p value for the observed squared displacement, with numbers close to 0 representing motion much smaller than expected, numbers close to 1 representing motion much larger than expected, and numbers close to 0.5 representing the expected amount of motion. Finally, scores below 0.5 were subtracted from 1, so that a higher score would consistently represent a more important residue.
The human genes were aligned to the structures with BLAST. Each residue in the human sequence was scored the same as the residue it aligned to. Residues that did not align to the structures were not given a score. Only 84 human residues failed to align to the structures, which represent 3.2% of all positions in the four proteins to which we applied this score.
Multiple Sequence Alignments
Both PolyPhen-2 and the MrBayes score described above use comparative sequence analysis as a source of phylogenetic information. These methods take as input aligned sequences of multiple homologous proteins, and their predictive values critically depend on the quality of the multiple sequence alignments used. Existing computational methods, including PolyPhen-2 and SIFT, rely on automated pipelines to construct multiple sequence alignments.12,13,17 We used the standard automated alignment pipeline provided by PolyPhen-2 but, because we only needed to construct six alignments, we were able to inspect and adjust each alignment manually.
We noticed in our manual inspection that some of the automated alignments were of very poor quality. The worst alignments were for the two genes, MYBPC3 and MYH7, that were most highly represented in our data set. These genes have numerous homologs at the domain level, arising from the multiple immunoglobulin domains of MYBPC3 and the highly conserved myosin motor domain of MYH7, and the multiple sequence alignments produced with automatically selected homologs are therefore of poor quality. We created new alignments for MYBPC3 and MYH7 by manually removing problematic sequences from the automatically generated alignments. This approach allowed us to tune the alignments manually while still taking advantage of PolyPhen-2's automatic filtering of poor alignments and incorrect sequences. The alignments were very deep to begin with, allowing us to remove a large number of sequences without having the alignments become too shallow to use.
The sequences we removed from the alignments were those that appeared to have only domain-level homology to the target sequences and/or did not appear to have a sufficiently similar function to the target sequences. In other words, we attempted to create an alignment for MYBPC3 that consisted only of forms of myosin-binding protein C from various tissues and organisms and an alignment for MYH7 that consisted only of forms of myosin heavy chain from various tissues and organisms. The resulting alignments were used as input to the PolyPhen-2 classifier and to MrBayes. The sequences used are listed in Tables S3–S6, and the resulting alignments are shown in Figure S3.
Training and Validation
We trained the classifier on the manually curated set of 74 missense variants in six genes. For each variant in the training set, we computed the four features described above (PolyPhen-2 prediction, MrBayes substitution rate score, coiled-coil score, and protein structure comparison score). The values of each feature for each variant can be found in Table S2. The training algorithm (Figure S4) aims to maximize accuracy of classification while keeping the required level of coverage. To avoid overfitting, the training algorithm uses 10-fold cross validation (Figure S5). This method splits the training data into ten parts (six parts of seven samples, four parts of eight samples), trains the classifier on nine training parts, and tests it on the remaining testing part. It then repeats the split-train-test procedure ten times, each time with a different part of the data used for testing. In order to account for the different results that would be produced by using different random divisions of the data in this process, we ran 1000 iterations of 10-fold cross validation, using a different random division of the data each time. We also tested the final classifier using a leave-one-out cross-validation strategy. The classifier assigns a prediction of pathogenic, benign, or no call to each variant. The no call prediction is given to variants the classifier cannot predict confidently. This category is included so that we can improve the accuracy (fraction of variants predicted correctly) of our confident predictions by sacrificing coverage (fraction of variants predicted to be either pathogenic or benign).2
Feature Selection
To verify that each of these four features made an important contribution, we constructed four incomplete classifiers, each one missing one of the four features. We performed validation on each of these classifiers as described above, and performed a random permutation test to show that the complete classifier had higher accuracy than each of the incomplete classifiers. We performed 106 permutations, so that the minimum p value we could find was . Out of our four features, only the PolyPhen-2 score had a one-sided p value greater than this minimum, with ; the other three features all had one-sided p values of less than . We also performed the same test to establish that using manual alignments instead of automatic alignments improved the score and found that it did with a one-sided p value of less than . Figure 5 shows the distributions of accuracies for each set of features in 1000 runs of cross validation.
Figure 5.
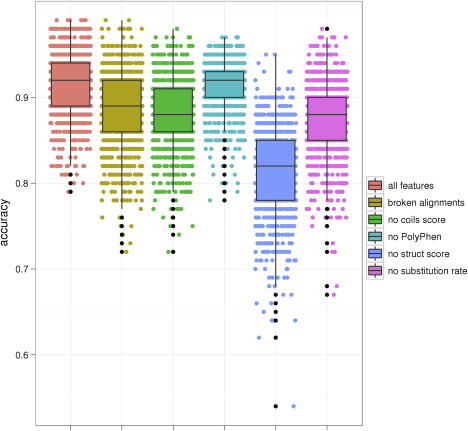
Feature Selection Experiment
Each column shows the distribution of accuracies in 1000 runs of cross validation for a classifier built with a different set of features: “all features” represents the final four-feature classifier with manual alignments, “broken alignments” represents the four-feature classifier without automatic alignments, and each of the other four columns represents a three-feature classifier missing the specified feature. Box plots show lower and upper quartiles (50% confidence intervals), and whiskers show 1.5 IQR ranges. The addition of each feature appears to improve the classifier, which is confirmed by a permutation test.
In addition to the four features in our final classifier, we also tried replacing PolyPhen-2 with the similar tools SIFT and PANTHER.12,13,26,27 We found that each performed comparably to PolyPhen-2, though the classifier with PolyPhen-2 performed very slightly better than either, again with one-sided p values less than . Interestingly, though PolyPhen-2, SIFT, and PANTHER were each far more informative individually than any other single feature, each made by far the least individual contribution to the full four-feature classifier that included it. Evidently, the other three features together contain enough information to make the PolyPhen-2, SIFT, or PANTHER score largely redundant.
We also investigated the effect each feature had on coverage. This was of particular concern for the structure pair score and the coiled-coil score, each of which is missing entirely from several genes and regions, which could reduce the predictor's ability to make confident classifications in these regions. We found that both the structure pair score and the coiled-coil score actually increase the coverage, whereas neither of the other features has a significant effect. This suggests that it is rare for a variant that could be scored confidently with the PolyPhen and substitution-rate scores to be demoted to no call because it is missing one or both of the other features. In other words, the coiled-coil and structure pair scores tend to increase confidence where they are present rather than decreasing it where they are absent.
Results
The Prediction Method
We created an automated method to predict the pathogenicity of missense variants in six genes known to contain variants that cause HCM. In designing this predictor, we set out to take advantage of the fact that we were focusing on a small set of functionally related genes to improve our predictions. We identified two ways to accomplish this: first, by exploiting unique structural and biochemical properties of the six target genes and, second, by applying more rigorous methods that would be difficult to implement for large numbers of genes. With these principles in mind, we developed a total of three predictive features, which we used in conjunction with the existing PolyPhen-2 classifier.17 Two of these features reflect specific structural properties of sarcomeric proteins. One scores the effect of amino acid change on coiled-coil regions, whereas the other scores the importance of the mutated residue to functionally important conformational transitions in ATP and Ca2+-binding domains. The remaining feature is an estimated rate of evolution at the variant position. This feature was extremely time consuming to compute and would not have been feasible to apply to a genome-wide data set. It also was computed from manually adjusted multiple sequence alignments of homologous sequences, which required human intervention to produce. These same manually adjusted alignments were also used as input to PolyPhen-2, improving its performance. We combined these three features and the PolyPhen-2 score using machine learning with our set of 74 manually classified variants as a training set. The complete method is presented graphically in Figure 3.
Figure 3.
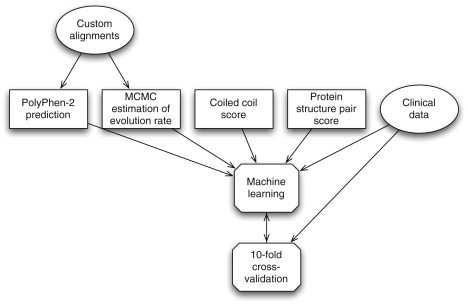
The Automated Prediction Process
For each variant, we computed four features and combined them by a machine-learning classifier. We trained this classifier on the high-confidence variants classified with clinical data and validated the classifier against the same data using 10-fold cross validation.
We also experimented with a small number of alternative features. The most notable among these were a different estimate of the rate of evolution computed with a genomic alignment of 46 vertebrate species, and several of the individual phylogenetic scores used as predictive features in PolyPhen-2. Addition of these features did not improve the performance of the predictor.
Validation of the Method against Manually Classified Variants
Given the small size of our gold standard data set (74 variants), the choice of training and validation method was important. Because we had so few variants, it was not feasible for us to use the simplest validation method of splitting the data set in half and using one half for training and the other for testing. Instead, we applied 10-fold cross validation, which is the accepted procedure in such cases (see Material and Methods). We ran this validation process a total of 1000 times to obtain median results and confidence intervals. Figure 4 shows the results of this validation for six different classifiers at different levels of coverage and accuracy. We used the bottom row, highlighted in red, as our final classifier. The method predicts each variant as pathogenic, benign, or no call, where the no call result means that the predictor is not sufficiently confident to permit a prediction. The median accuracy for covered variants for the most accurate classifier (the fraction of correct predictions out of all pathogenic and benign predictions when no call results are disregarded) was 92%, with a 95% confidence interval of 83%–98% (Figure 5). The median coverage (the fraction of variants that were predicted as either pathogenic or benign), was 57%, with a 95% confidence interval of 49%–64%; in other words, the median classifier reported no call for 43% of variants. The median sensitivity for covered variants (the fraction of variants manually classified as pathogenic that were predicted as pathogenic, excluding those predicted as no call) was 94%, with a 95% confidence interval of 83%–98%. The median specificity for covered variants (the fraction of variants manually classified as benign that were predicted as benign, excluding those predicted as no call) was 89%, with a 95% confidence interval of 83%–98%. The median odds ratio for a prediction of pathogenic (the odds of a pathogenic variant being classified as pathogenic divided by the odds of a benign variant being classified as pathogenic) was 10, with a 95% confidence interval of 4.0–infinity (no upper bound could be set because more than 5% of trials had no false positives). The median odds ratio for a prediction of benign (the odds of a benign variant being classified as benign divided by the odds of a pathogenic variant being classified as benign) was 9.9, with a 95% confidence interval of 4.6–21. Leave-one-out cross validation also resulted in highly similar estimates of all these quantities.
Figure 4.
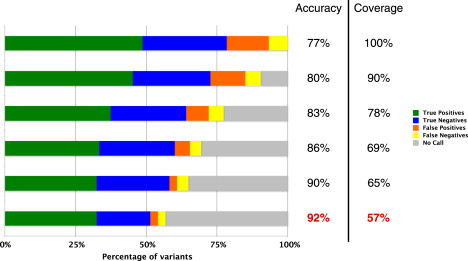
Results of Cross Validation
Rows contain median 10-fold cross-validation results for the gold standard data set at different levels of coverage. Horizontal bars correspond to different levels of coverage and median validation coverage and accuracy levels are indicated. “True Positives” are variants that were manually classified as pathogenic and that our method predicted as pathogenic. “True Negatives” are variants that were manually classified as benign or likely benign and that our method predicted as benign. “False Positives” are variants that were manually classified as benign” or likely benign but that our method predicted as pathogenic. “False Negatives” are variants that were manually classified as pathogenic but that our method predicted as benign. “Uncovered” are variants without a prediction (no call). The bottom-most coverage level, indicated in red, was used for our final predictor.
Comparison with General-Purpose Methods
Because our predictor bases its predictions in part on predictions of the existing general-purpose method PolyPhen-2, we investigated whether our predictor was a significant improvement over the PolyPhen-2 predictor without our modifications and other general-purpose methods. In order to investigate this, we tested PolyPhen-2, SIFT, and PANTHER on the same data set. We applied the same 10-fold cross-validation method with each of these three scores as the only predictive feature. We found that all three general-purpose scores had comparable performance on this data set: PolyPhen-2's median cross-validation accuracy was 70% (95% confidence interval: 60%–77%), SIFT's was 74% (95% confidence interval: 64%–83%), and PANTHER's was 68% (95% confidence interval: 56%–79%). All of these estimates are much lower than the accuracies reported for these methods, which may reflect features of this data set. Our specialized predictor, on the other hand, had a median accuracy of 92% (95% confidence interval: 83%–98%), as reported above. A permutation test showed that all three general-purpose predictors performed worse than our specialized predictor, with one-sided p values of less than .
Predictions for Variants without Confident Classifications
The ultimate goal of our predictor is to provide accurate predictions for variants that are not confidently classified by manual methods. This will not be possible if there is some systematic biological difference between the confident and unconfident classifications, such as a difference in penetrance, severity, or mechanism of disease. To determine whether this is the case, we applied our method to a low-confidence data set, the set of missense variants that did not meet the confidence criteria to be manually classified as truly pathogenic or benign (Figure 6). Of the missense variants manually classified as likely pathogenic, 80% of those for which a prediction was made were predicted as pathogenic. This is consistent with the expectation that most of these variants that were classified as likely pathogenic are indeed pathogenic. It is also consistent with the expectation that the fraction of variants predicted as pathogenic in this set is lower than the fraction of variants manually classified as confidently pathogenic. Among variants manually classified as VUS, 70% of those for which a prediction was made were predicted to be pathogenic. Because these variants have been identified in individuals diagnosed with HCM, there is a higher a priori likelihood that they are indeed pathogenic, although we have no way of knowing what the true fraction should be. The fraction of variants predicted to be pathogenic remains lower in the VUS set than in the likely-pathogenic set, which is consistent with what would be expected.
Figure 6.
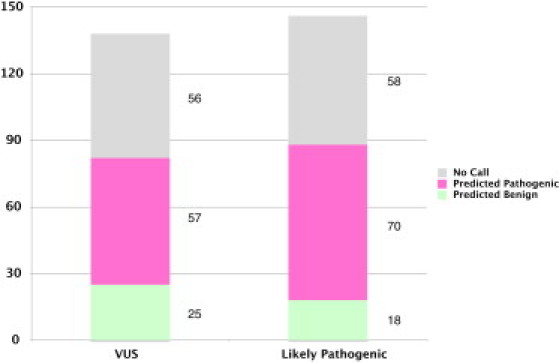
Results for Low-Confidence Data Set
Columns indicate, for each class of variants, the number of predictions in predicted categories produced by the final classifier.
We also used the low-confidence data set to generate an independent estimate of the predictor's coverage. We found that the predictor made a prediction for 60% of low-confidence variants, which is well within the confidence interval of 49%–64% for the estimated coverage on the gold standard variants.
Discussion
We developed and clinically validated an automated method to predict the pathogenic effect of missense variants that might cause HCM. Unlike current commonly used methods, our predictor has been validated against high-confidence manually curated data. This enabled us to estimate its specificity and sensitivity for the specific task of predicting HCM mutations, which will allow its predictions to be incorporated into clinical reports to health care professionals as one piece of evidence supporting a variant classification. Although this tool adds little for variants whose clinical significance is already supported by strong genetic and/or functional data, it will add value for those variants that had little or no prospect of ever being supported by solid family studies or large scale healthy control studies. Importantly, our classifier is particularly helpful for variants identified in minority populations, where healthy control cohorts, one of the pillars of traditional variant classification, are typically unavailable.
To maintain high accuracy, it was necessary to sacrifice coverage, i.e., the proportion of variants for which a prediction is made.2 As shown in Figure 4, an increase in coverage is accompanied by a rapid decline in accuracy. A method attempting to predict every variant as either pathogenic or benign could not achieve levels of accuracy acceptable for clinical use. We estimated the coverage of our predictor at 57%, with a 95% confidence interval of 49%–64%. We believe this level of coverage is still above the threshold of clinical usefulness. For comparison, note that out of 350 LMM missense variants in the six target genes, only 74 met the criteria for high-confidence manual classification, giving the manual classification process a coverage of only 21%. Note also that our method covers a different set of variants than the manual classification process, including 59% of the variants that the manual classification classifies as VUS.
The most important limitation of our automated prediction method stems from the size of the training data set. In general, training on small data sets may lead to overfitting of automated classifiers. An overfit classifier may be highly accurate on the training data but much less accurate on new data. We applied several safeguards against overfitting during training and validation. These included limiting the number of features in the classifier, using only features that we expected a priori to be informative, and performing cross validation to calibrate parameters and estimate accuracy. In this way, we hope we have avoided excessive overfitting in our final predictor.
It is important to point out that this method may not accurately predict the effect of those missense variants that exert their effect partially or fully though affecting mRNA splicing. This is true for all currently available tools of this kind, including PolyPhen-2, SIFT, and others. For example, the MYBPC3 Glu258Lys variant was confidently manually classified as pathogenic but was incorrectly classified as benign in several runs of cross-validation (though not in the final predictor). Many MYBPC3 variants affect splicing, and there is evidence that the Glu258Lys variant causes disease via this mechanism. The underlying cDNA alteration is c.772>A, which affects the last base of exon 6. This position is known to be part of the splice consensus and five different splice predictors (SpliceSiteFinder-like, MatEntScan, NNSPLICE, GeneSplicer, and Human Splice Finder; see Figure S6) predict an impact on splicing. This is supported by evidence showing that this may result in skipping of exon 6.28,29 Therefore, the conservation of the nucleotide and not the amino acid at this position is essential, possibly explaining a misprediction by our predictor. This is a limitation of this method and clearly lends itself to future improvement and generation of tools that incorporate a splice assessment.
It is also important to point out that clinical laboratories are typically aware of this limitation. Novel variant assessment is a lengthy and complex process that relies on a large collection of different computer tools in combination with traditional genetic evidence such as familial segregation with disease and absence from race-matched healthy controls. All evidence is taken into account to synthesize a final probability for pathogenicity. In our laboratory, a splice assessment is performed for every variant, regardless of whether it changes an amino acid or not, and a benign prediction by this predictor would not lead to a final classification of benign, particularly not for genes for which pathogenic splice variants are known to be common.
This example illustrates that this predictor or any other predictor developed with this methodology should not be used as a sole foundation for a diagnosis but rather be used in combination with other lines of evidence in agreement with recommendations from the American College of Medical Genetics and the International Agency for Research on Cancer.1,2 We envision future development of a single probabilistic classifier that would automatically combine heterogeneous factors such as familial segregation, frequency in controls, functional evidence, and computational predictions following early work in this area.
Conclusion
We have addressed the problems that prevent automated predictors from being widely used in genomic medicine by developing a custom-tailored predictor specifically designed for clinical use. Our analysis suggests several important considerations that can increase the accuracy of computational methods. Manual adjustment of multiple sequence alignments and time-consuming computational methods of molecular evolution are feasible when focusing on a small set of genes and may improve predictions that use comparative sequence analysis. Exploitation of specific structural properties of proteins also becomes feasible when focusing on a specific disease. Most importantly, a highly accurate manually curated data set is necessary to train and validate an accurate predictor, and this level of validation enables clinical laboratories to include it as part of their variant assessment processes. Where previous studies have concluded that existing tools are not mature enough for clinical use, we believe that our tool is ready for clinical use now, in combination with other sources of information. Our collaborating clinical laboratory, the LMM, has already begun to use our predictor as a source of information about HCM variants, and we look forward to helping additional laboratories do the same. Our study focused on HCM, but we believe that our approach is general and that analogous methods can be constructed for many other diseases where genetic testing is an important part of the diagnosis. In the future, we expect to work with additional laboratories and on additional diseases to expand the use of automated predictors in genomic medicine and simplify the problem of interpreting novel variants.
Acknowledgments
This research was supported by grants from the National Institutes of Health (R01 GM078598, T32 GM008313). We would like to thank Steve DePalma, Dan Hermann, and Jon and Christine Seidman for their help with the CardioGenomics database and insights into HCM.
Contributor Information
Birgit H. Funke, Email: bfunke@partners.org.
Shamil R. Sunyaev, Email: ssunyaev@rics.bwh.harvard.edu.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
PolyPhen-HCM method, http://genetics.bwh.harvard.edu/hcm
References
- 1.Richards C.S., Bale S., Bellissimo D.B., Das S., Grody W.W., Hegde M.R., Lyon E., Ward B.E., Molecular Subcommittee of the ACMG Laboratory Quality Assurance Committee ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet. Med. 2008;10:294–300. doi: 10.1097/GIM.0b013e31816b5cae. [DOI] [PubMed] [Google Scholar]
- 2.Plon S.E., Eccles D.M., Easton D., Foulkes W.D., Genuardi M., Greenblatt M.S., Hogervorst F.B., Hoogerbrugge N., Spurdle A.B., Tavtigian S.V., IARC Unclassified Genetic Variants Working Group Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum. Mutat. 2008;29:1282–1291. doi: 10.1002/humu.20880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ng P.C., Henikoff S. Predicting the effects of amino acid substitutions on protein function. Annu. Rev. Genomics Hum. Genet. 2006;7:61–80. doi: 10.1146/annurev.genom.7.080505.115630. [DOI] [PubMed] [Google Scholar]
- 4.Thusberg J., Vihinen M. Pathogenic or not? And if so, then how? Studying the effects of missense mutations using bioinformatics methods. Hum. Mutat. 2009;30:703–714. doi: 10.1002/humu.20938. [DOI] [PubMed] [Google Scholar]
- 5.Jordan D.M., Ramensky V.E., Sunyaev S.R. Human allelic variation: perspective from protein function, structure, and evolution. Curr. Opin. Struct. Biol. 2010;20:342–350. doi: 10.1016/j.sbi.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tchernitchko D., Goossens M., Wajcman H. In silico prediction of the deleterious effect of a mutation: proceed with caution in clinical genetics. Clin. Chem. 2004;50:1974–1978. doi: 10.1373/clinchem.2004.036053. [DOI] [PubMed] [Google Scholar]
- 7.Dorfman R., Nalpathamkalam T., Taylor C., Gonska T., Keenan K., Yuan X.W., Corey M., Tsui L.C., Zielenski J., Durie P. Do common in silico tools predict the clinical consequences of amino-acid substitutions in the CFTR gene? Clin. Genet. 2010;77:464–473. doi: 10.1111/j.1399-0004.2009.01351.x. [DOI] [PubMed] [Google Scholar]
- 8.Tavtigian S.V., Greenblatt M.S., Lesueur F., Byrnes G.B., IARC Unclassified Genetic Variants Working Group In silico analysis of missense substitutions using sequence-alignment based methods. Hum. Mutat. 2008;29:1327–1336. doi: 10.1002/humu.20892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldgar D.E., Easton D.F., Deffenbaugh A.M., Monteiro A.N.A., Tavtigian S.V., Couch F.J., Breast Cancer Information Core (BIC) Steering Committee Integrated evaluation of DNA sequence variants of unknown clinical significance: application to BRCA1 and BRCA2. Am. J. Hum. Genet. 2004;75:535–544. doi: 10.1086/424388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang L., Seidman J.G., Seidman C.E. Narrative review: harnessing molecular genetics for the diagnosis and management of hypertrophic cardiomyopathy. Ann. Intern. Med. 2010;152:513–520, W181. doi: 10.1059/0003-4819-152-8-201004200-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richard P., Charron P., Carrier L., Ledeuil C., Cheav T., Pichereau C., Benaiche A., Isnard R., Dubourg O., Burban M., EUROGENE Heart Failure Project Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 2003;107:2227–2232. doi: 10.1161/01.CIR.0000066323.15244.54. [DOI] [PubMed] [Google Scholar]
- 12.Ng P.C., Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ng P.C., Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bromberg Y., Rost B. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 2007;35:3823–3835. doi: 10.1093/nar/gkm238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yue P., Moult J. Identification and analysis of deleterious human SNPs. J. Mol. Biol. 2006;356:1263–1274. doi: 10.1016/j.jmb.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 16.Yue P., Melamud E., Moult J. SNPs3D: candidate gene and SNP selection for association studies. BMC Bioinformatics. 2006;7:166. doi: 10.1186/1471-2105-7-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ronquist F., Huelsenbeck J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 19.Lupas A., van Dyke M., Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- 20.Lupas A. Prediction and analysis of coiled-coil structures. Methods Enzymol. 1996;266:513–525. doi: 10.1016/s0076-6879(96)66032-7. [DOI] [PubMed] [Google Scholar]
- 21.Letunic I., Doerks T., Bork P. SMART 6: recent updates and new developments. Nucleic Acids Res. 2009;37(Database issue):D229–D232. doi: 10.1093/nar/gkn808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Houdusse A., Kalabokis V.N., Himmel D., Szent-Györgyi A.G., Cohen C. Atomic structure of scallop myosin subfragment S1 complexed with MgADP: a novel conformation of the myosin head. Cell. 1999;97:459–470. doi: 10.1016/s0092-8674(00)80756-4. [DOI] [PubMed] [Google Scholar]
- 23.Himmel D.M., Gourinath S., Reshetnikova L., Shen Y., Szent-Györgyi A.G., Cohen C. Crystallographic findings on the internally uncoupled and near-rigor states of myosin: further insights into the mechanics of the motor. Proc. Natl. Acad. Sci. USA. 2002;99:12645–12650. doi: 10.1073/pnas.202476799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vinogradova M.V., Stone D.B., Malanina G.G., Karatzaferi C., Cooke R., Mendelson R.A., Fletterick R.J. Ca(2+)-regulated structural changes in troponin. Proc. Natl. Acad. Sci. USA. 2005;102:5038–5043. doi: 10.1073/pnas.0408882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martínez L., Andreani R., Martínez J.M. Convergent algorithms for protein structural alignment. BMC Bioinformatics. 2007;8:306. doi: 10.1186/1471-2105-8-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomas P.D., Campbell M.J., Kejariwal A., Mi H., Karlak B., Daverman R., Diemer K., Muruganujan A., Narechania A. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003;13:2129–2141. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomas P.D., Kejariwal A., Guo N., Mi H., Campbell M.J., Muruganujan A., Lazareva-Ulitsky B. Applications for protein sequence-function evolution data: mRNA/protein expression analysis and coding SNP scoring tools. Nucleic Acids Res. 2006;34(Web Server issue) doi: 10.1093/nar/gkl229. W645–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andersen P.S., Havndrup O., Bundgaard H., Larsen L.A., Vuust J., Pedersen A.K., Kjeldsen K., Christiansen M. Genetic and phenotypic characterization of mutations in myosin-binding protein C (MYBPC3) in 81 families with familial hypertrophic cardiomyopathy: total or partial haploinsufficiency. Eur. J. Hum. Genet. 2004;12:673–677. doi: 10.1038/sj.ejhg.5201190. [DOI] [PubMed] [Google Scholar]
- 29.Marston S., Copeland O., Jacques A., Livesey K., Tsang V., McKenna W.J., Jalilzadeh S., Carballo S., Redwood C., Watkins H. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ. Res. 2009;105:219–222. doi: 10.1161/CIRCRESAHA.109.202440. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.