

Abstract
Although there has been extensive research on various Cytochrome P450s, especially Cyt P450cam, there is much to be learned about the mechanism of how its functional unit, a heme b ligated by an axial cysteine, is finely tuned for catalysis by its second coordination sphere. Here we study how the hydrogen bonding network affects the proximal cysteine and the Fe-S(Cys) bond in ferric Cyt P450cam. This is accomplished using low-temperature magnetic circular dichroism (MCD) spectroscopy on wild-type (wt) Cyt P450cam, and on the mutants Q360P (pure ferric high-spin at low temperature) and L358P with which the “Cys pocket” has been altered (by removing amino acids involved in the hydrogen bonding network), and Y96W (pure ferric low-spin). The MCD spectrum of Q360P reveals fourteen electronic transitions between 15200 and 31050 cm-1. Variable-temperature variable-field (VTVH) saturation curves were used to determine the polarizations of these electronic transitions, with respect to in-plane (xy) and out-of-plane (z) polarization relative to the heme. The polarizations, oscillator strengths, and TD-DFT calculations were then used to assign the observed electronic transitions. In the lower energy region, prominent bands at 15909 and 16919 cm-1 correspond to porphyrin (P) → Fe charge transfer (CT) transitions. The band at 17881 cm-1 has distinct sulfur S(π)→ Fe CT contributions. The Q band is observed as a pseudo A-term (derivative shape) at 18604 and 19539 cm-1. In the case of the Soret band, the negative component of the expected pseudo A-term is split into two features due to mixing with another π → π* and potentially a P → Fe CT excited state. These features are observed at 23731, 24859, and 25618 cm-1. Most importantly, the broad, prominent band at 28570 cm-1 is assigned to the S(σ)→ Fe CT transition, whose intensity is generated through a multitude of CT transitions with strong iron character. For wt, Q360P, and L358P, this band occurs at 28724, 28570, and 28620 cm-1, respectively. The small shift of this feature upon altering the hydrogen bonds to the proximal cysteine indicates that the role of the Cys pocket is not primarily for electronic fine tuning of the sulfur donor strength, but is more for stabilizing the proximal thiolate against external reactants (NO, O2, H3O+), and for properly positioning cysteine to coordinate to the iron center. This aspect is discussed in detail.
Cytochrome P450s (Cyt P450s) are a major part of a super family of b-type heme-containing enzymes found in various forms of life, such as bacteria, yeast, insects, plants and mammals.(1) P450s generally activate dioxygen or hydrogen peroxide to oxidize a wide range of substrates. These enzymes catalyze diverse types of reactions including hydroxylation, epoxidation, dealkylation and heteroatom oxidations, making them an attractive class of enzymes for industrial applications in catalysis.(2-4) In humans, Cyt P450s are involved in hormone synthesis and metabolism, steroid biosynthesis, and the metabolism of small molecules for excretion (liver).(1, 5) Other members of this class are nitric oxide synthases, which facilitate the biosynthesis of nitric oxide (NO) in humans and mammals for signaling and immune defense.(6-8) Cyt P450s have also been implicated as activators of many chemical carcinogens, due to the ability of these enzymes to process a wide range of non-physiological substrates, including drugs.(9-13) Much research has been focused on the unique ability of Cyt P450s to activate and process such a wide variety of substrates.(1-3, 14-18) One important aspect in this regard is to establish how Cyt P450s control the mechanism(s) by which diatomic molecules are activated and react with substrates.
In general, the factors that control the reactivity of heme proteins include the nature of the axial ligand, the amino acid side chains near the active site (via bulk or charged residues), conformational changes of the heme upon binding diatomic molecules or substrates, and hydrogen bonding donors near the active site. For Cyt P450, several of the intermediates involved in the overall mechanism by which dioxygen is activated are reasonably established at this point; the reaction involves multiple intermediates, including Fe(III)-hydroperoxo (Compound 0), high-valent [Fe(IV)=O P•+] (Compound I; P = general porphyrin2- ligand), and [Fe(IV)=O P] (Compound II) species.(1, 3, 4) On the other hand, additional factors that control or fine-tune the reactivity of Cyt P450s are less well known. In particular, the role(s) of the proximal cysteine that coordinates to the heme iron is not well understood. This cysteine residue is strongly electron donating and presumably facilitates heterolysis of the peroxide that coordinates trans to the cysteine ligand,(19, 20) leading to the formation of Compound I. The importance of thiolate coordination in P450 catalysis is best exemplified by three findings: 1) P420, the inactivated form of P450, where the proximal Cys has been protonated yielding a neutral thiol ligand, usually shows no enzymatic activity;(21) 2) C357H, a mutant of P450cam (P450 that hydroxylates camphor) where the proximal cysteine is substituted by histidine, shows lack of catalytic activity for camphor hydroxylation as well as a reduced rate for heterolytic O-O bond cleavage with cumene hydroperoxide;(22) and 3) Mb H93C, a myoglobin mutant in which the proximal histidine is replaced by cysteine, exhibits enhanced heterolytic O-O bond cleavage of peroxide.(23) Furthermore, theoretical calculations on different Cyt P450s underline the importance of thiolate ligation for catalysis in this enzyme family.(24-34)
Surrounding the crucial proximal cysteine ligand is a hydrogen-bonding network. In Cyt P450cam from Pseudomonas putida, this hydrogen-bonding network, also known as the “Cys pocket”, is composed of Leu 358 (L358), Gly 359 (G359) and Gln 360 (Q360)(35). The Cys pocket stabilizes the heme-bound cysteine (C357) by three hydrogen bonds with the amide N-H groups of these amino acids, and one hydrogen bond from the side chain of glutamine to the carbonyl oxygen of Cys 357, as shown in Figure 1. It is important to note that the Cys pocket is not limited to P450cam, but is also found in other P450 enzymes such as P450terp, P450eryF, and P450BM-3,(36-38) suggesting that hydrogen bonds to the proximal cysteinate are key features of the Cyt P450 active site. These H-bonds have been implicated in controlling the function of the enzyme by decreasing the negative charge on the thiolate ligand, hence increasing the reduction potential of the iron center. For example, Cyt P450cam mutants L358P and Q360L, in which one of the conserved amide protons is removed, and Q360P, where the amide proton and the side chain hydrogen bonds are removed, show decreased reduction potentials in the presence of camphor of -170 (L358P), -180 (Q360L), and -205 mV (Q360P), respectively, compared to -134 mV for wild-type (wt) enzyme.(39) This idea is confirmed by Cyt P450 model complexes such as tetrapeptide heme,(40) arene-thiolate,(41, 42) and alkane-thiolate complexes,(43) all showing an increase in the heme redox potential when hydrogen bonding exists. Density functional theory (DFT) calculations (in particular on the latter model systems) show the roles of H-bonding for fine-tuning the Fe-S bond covalency, in agreement with XAS results.(44, 45)
Figure 1.
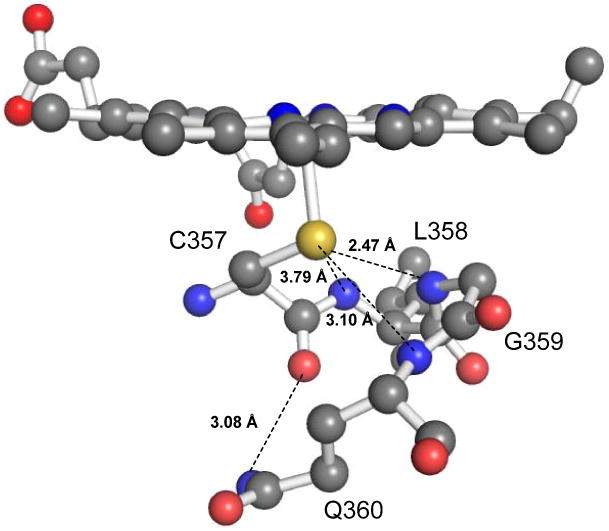
Proximal hydrogen bonding network of Cytochrome P450cam. The orange, yellow and blue balls represent the heme iron, the sulfur atom and the main chain amide nitrogen atoms of Leu358, Gly359, and Gln360, respectively. The dashed lines represent the NH⋯S hydrogen bonds in the Cys pocket. The image was generated using PyMOL from PDB code 2CPP.(35)
A complete understanding of the role of the Fe-S bond and the proximal hydrogen-bonding network in Cyt P450 catalysis necessitates careful analysis of the active site electronic structure. Changes in redox potential are very indirect measures of electronic structural changes, as these can relate to the stabilization/destabilization of the oxidized and/or reduced form. In this paper, we focus on determining how the Cys pocket affects the ferric form of the active site of Cyt P450s using Cyt P450cam as a model. Cyt P450cam exists as a six-coordinate, low-spin (ls, S = 1/2) ferric heme in its resting state at room temperature that becomes five-coordinate, high-spin (hs, S = 5/2) upon binding of camphor.(46, 47) A variety of spectroscopic techniques such as UV–vis,(39, 48, 49) electron paramagnetic resonance (EPR),(50-52) resonance Raman (rR)(39, 53), room temperature (RT) magnetic circular dichroism (MCD)(3, 48, 54) and X-ray absorption(44) spectroscopies have been used to investigate the ferric form of Cyt P450s. Despite these efforts, a detailed assignment of the electronic spectra of the catalytically active, high-spin ferric form of Cyt P450s has not yet been obtained. However, the assignment of the spectra, and in particular, the identification of S → Fe charge transfer (CT) transitions as sensitive probes for the electronic structure of the site, would be highly desirable.
This paper presents the first, detailed low-temperature MCD investigation on wt Cyt P450cam and specific variants in order to assign the electronic spectra of the hs ferric form of this protein. MCD is a powerful technique for investigating paramagnetic complexes.(55-57, 58, 59, 60) Compared to UV–vis absorption spectroscopy, MCD gives a better resolution of electronic transitions due to the fact that MCD signals have a sign. Second, unlike EPR spectroscopy, this technique is not restricted to non-integer spin systems. Third, the polarizations of the electronic transitions are accessible through detailed simulations of variable-temperature variable-field (VTVH) MCD saturation curves, even in samples of randomly oriented molecules. Recently, MCD spectra of the five-coordinate ferric high-spin complex [Fe(TPP)Cl] have been reported,(61) and it has been demonstrated that the nesting of the VTVH curves can be used to distinguish in-plane and out-of-plane polarized transitions (relative to the heme plane). In particular, this allows for the straightforward identification of CT transitions involving the axial ligands and the heme. Assignments were further confirmed by comparison to calculated (via TD-DFT) extinction coefficients and polarizations, and finally, compared to results from rR spectroscopy.(62) The methodology developed in ref. (61) is applied here to ferric wt Cyt P450cam and corresponding hs and ls variants. The goals of this study are twofold: first, to elucidate the complex electronic spectra of Cyt P450, and second, to unequivocally identify sulfur-to-iron CT transitions. The energies of these CT transitions are then compared for oxidized Cyt P450 wt and variants in which the hydrogen bonding network has been altered to reveal the effects of the proximal hydrogen bonding network on the Fe-S bond. Understanding the effect of the hydrogen bonding network on the donor strength of the thiolate ligand is crucial in determining whether the role of the Cys pocket is predominantly for stabilizing the thiolate, or whether this also serves to fine-tune the Fe-S bond strength for catalysis, for example in binding diatomic molecules and activating peroxides for heterolysis.
Experimental and Computational Methods
Expression and purification of P450cam and corresponding mutants
Wild-type Cyt P450cam, the ferric high-spin (hs) mutant Q360P, the ferric low-spin (ls) mutant Y96W, and L358P were prepared as previously described by Spolitak et al.(63)
UV–vis spectroscopy
Room temperature absorption spectra were recorded on Analytik Jena Specord S600 and Shimadzu 2501 spectrophotometers. The absorption spectra of wt Cyt P450cam, Q360P and L358P, and Y96W were taken in 50 mM potassium phosphate buffer (pH 7.4) containing 500 μM d-camphor. The same buffer systems were also utilized for MCD spectroscopy, except that 50% glycerol (v/v) was included.
MCD theoretical background
The theoretical background of MCD spectroscopy as it pertains to this work has been described in detail by Paulat and Lehnert.(61) Importantly, MCD intensity is proportional to the strength of the magnetic field, and to three contributions designated as the temperature-independent A- and B-terms, and the temperature-dependent C-term, which is specifically observed for paramagnetic systems.(55, 56, 64) The C-term intensity is inversely proportional to temperature, and therefore requires measurements at very low temperature (2-30 K). C-term intensity arises from a (spin) degenerate ground state that splits in the presence of a magnetic field due to the Zeeman effect. At low temperatures when kT is in the order of the Zeeman splitting, there is a greater population of the lower energy sublevels of the ground state than the sublevels of the higher energy levels. Further decreases in temperature or increases in magnetic field result in the complete depopulation of higher energy sublevels, which allows the C-term intensity to reach its maximum value, and hence saturate. The measurement of the temperature- and field-dependent C-term intensity is extremely useful because it contains the complete information of the ground state, including g values, zero field splitting (ZFS), and most importantly, polarizations of the electronic transitions in samples of randomly oriented molecules. The polarizations greatly help in making spectral assignments: as shown in ref. (61) for [Fe(TPP)Cl], the saturation curves are very sensitive to the polarization (either in-plane or out-of-plane relative to the heme) of the electronic transitions in metalloporphyrins. In this way, transitions involving axial ligands can be identified.
MCD spectroscopy
The proteins in 50mM potassium phosphate buffer (pH 7.4) containing 500 μM d-camphor with 50% glycerol added (used as a glassing agent) were placed between two quartz plate windows in a custom-made MCD holder. The samples were flash frozen in liquid nitrogen until a clear glass formed. Two sets of concentrations were used for each protein sample to probe the Soret and Q-regions. The sample concentrations for wt (20 and 170 μM), Q360P (26 and 140 μM), Y96W (8 and 180 μM), and L358P (15 and 165 μM) allowed for maximum signal-to-noise ratios in these two regions.
The MCD setup employs an OXFORD SM4000 cryostat and a JASCO J-815 CD spectrometer. The SM4000 cryostat consists of a liquid helium-cooled superconducting magnet providing horizontal magnetic fields of 0-7 Tesla. The J-815 spectrometer uses a gaseous nitrogen-cooled xenon lamp and a detector system consisting of two interchangeable photomultiplier tubes in the UV-vis and NIR range. The samples were loaded into a 1.5-300 K variable temperature insert (VTI), which offers optical access to the sample via four optical windows made from Suprasil B quartz. The MCD spectra were measured in [θ] = mdeg and manually converted to Δε [M-1 cm-1 T-1] using the conversion factor Δε = θ / (32980·c·d·B), where c is the concentration, B is the magnetic field, and d is the path length. The product c·d can be substituted by AMCD/εUV-vis, where A is the absorbance of the sample measured by the CD spectrometer. Complete spectra were recorded at different temperatures (2, 5, 10, 20, and 100 K) and magnetic fields (0-7 T) to obtain the VTVH data for all unambiguous MCD bands. This was done by varying the field at a specific temperature.
Gaussian Deconvolution of Spectra
Gaussian deconvolution of the UV-Vis and MCD C-term spectra was performed using the program PeakFit (version v4.12). The smallest number of Gaussian functions necessary was used for the deconvolution analysis, and the quality of the individual fits was monitored using χ2. Bandwidths and band centers of the Gaussian curves for each band were allowed to vary 2 % between the UV-vis and MCD C-term spectra to account for the different experimental conditions employed. The observed band broadening between Soret and CT bands was roughly the same (± 40 cm-1).
Raman spectroscopy
The resonance Raman data were obtained on 100 μM solutions of wt Cyt P450cam using 351 nm, 413 nm, and 457 nm excitation from a SpectraPhysics BeamLok 2060-RS Kr+-Ar+ laser. The excitation beam (20 mW) was focused onto 200 μL of the sample in an EPR tube contained in an EPR coldfinger dewar filled with liquid N2 to prevent thermal degradation. The scattered photons were dispersed by an Acton two-stage TriVista 555 monochromator, and detected by a liquid N2-cooled Princeton Instruments Spec-10:400B/LN CCD camera. A typical resolution in these experiments was 0.5 cm-1.
Density Functional Calculations
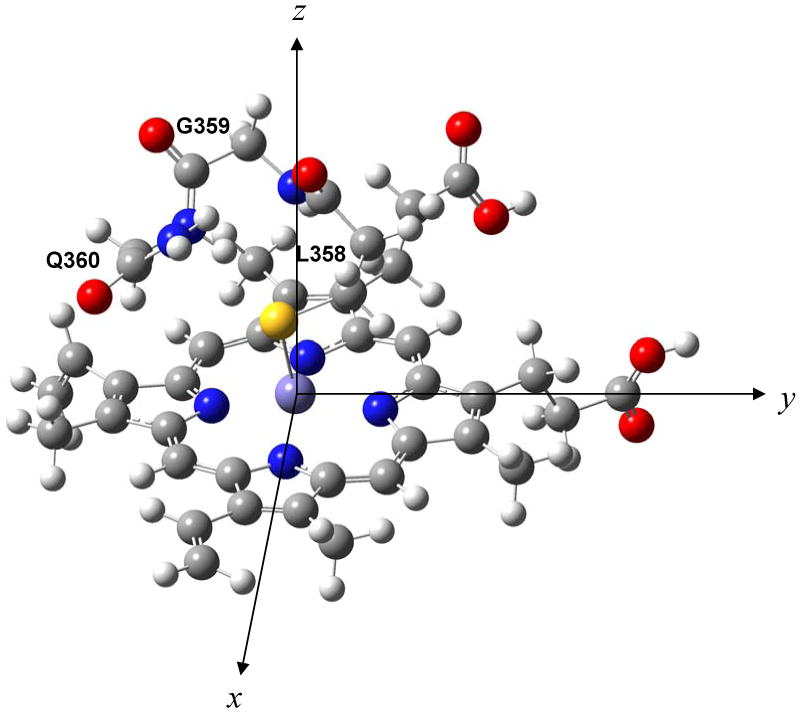
The coordinates of the high-resolution crystal structure of camphor-bound ferric Cyt P450cam (PDB code: 2CPP)(35) were obtained from the Protein Data Bank (PDB), and the coordinates of the heme b and the Cys pocket were identified and isolated. The structure of the active site was simplified by removing the side chains of the amino acids of the Cys pocket. Hydrogen atoms were added to the structure and optimized applying the B3LYP hybrid functional(65, 66) and the LanL2DZ basis set,(67-69) using the Gaussian 03 program package.(70) The resulting active site model is shown in Scheme 1. The theoretical absorption spectrum of hs ferric wt Cyt P450cam was generated from this structure using time-dependent density functional theory (TD-DFT) calculations(71-73) with the BP86 functional(74, 75) and the TZVP basis set,(76, 77) as implemented in the program ORCA.(78) Gradient-corrected functionals have previously been successfully applied in TD-DFT calculations.(71-73) The number of excited states for the TD-DFT calculations was set to 260, delivering electronic transitions in the 6122 to 34592 cm-1 range. The most important orbitals for analyzing the UV-vis spectra are the HOMO ± 13. The applied coordinate system is shown in Scheme 1, where the z-axis points approximately along the Fe-S bond.
Scheme 1.
Fitting of the MCD VTVH Data
The VTVH data of the different C-term bands observed in the MCD spectra of hs Q360P were fitted according to a method by Neese and Solomon.(79) The analysis includes the relative MCD intensities, effective transition dipole moment products, ZFS parameters and g values, and Boltzmann populations over the magnetically split sublevels of the ground state. The zero field splitting (D = 3.8 cm-1) and degree of rhombicity (E/D = 0.087) obtained experimentally for wt hs ferric Cyt P450cam(50) were incorporated in the fit to reliably determine the relative polarizations of the electronic transitions. The individual polarizations of the observed electronic transitions were calculated from the effective transition dipole moment products, obtained from fitting the VTVH saturation curves.(79)
Results
A. Ferric Cyt P450 Electronic Structure
The coordinates of the active site of wild type (wt) hs Cyt P450cam were obtained from the crystal structure of the enzyme in the presence of camphor (PDB code: 2CPP). In order to obtain a reliable description of the electronic structure, it is imperative to utilize the actual coordinates from the enzyme that show the correct conformation of heme b, and orientation of the sulfur in C357 relative to iron. For the TD-DFT calculations, the model was further simplified by truncating the side chains of the amino acids of the Cys pocket surrounding C357 as shown in Scheme 1. This simplification does not affect the calculated absorption spectrum (test calculations were performed for the 0 to 34592 cm-1 energy range), but it greatly simplifies the analysis of TD-DFT results. Scheme 1 shows the coordinate system applied here for the following discussion of molecular orbitals (MOs) and electronic transitions. According to the applied coordinate system, the x and y axes are located along the Fe-N bonds of the porphyrin ligand, while the z axis is approximately aligned with the Fe-S bond.
In order to evaluate the TD-DFT results, it is important to first consider the MO diagram of Cyt P450, obtained for the model system shown in Scheme 1 using BP86/TZVP. In five-coordinate, ferric Cyt P450 the iron center is high-spin (hs, S = 5/2), and therefore, all α-d orbitals are fully occupied, whereas the β-d orbitals are empty (in a spin-unrestricted scheme).2 Because of this, ligand → metal charge transfer (CT) transitions are limited to the β-spin MOs, which are therefore analyzed as described next. The fact that all β-d orbitals are accessible to CT transitions leads to a multitude of porphyrin (P) → Fe(III) and sulfur (S) → Fe(III) CT transitions, and hence, to very complicated electronic spectra in hs ferric hemes.(61) Figure 2 shows the MO diagram of Cyt P450cam obtained here. Contour plots of important β-MOs are depicted in Figure 3, and charge contributions of the MOs are listed in Table 1. Heme b core orbitals are labeled according to the MO diagram of free porphine2- shown in Figure S1 (for example, A2u<81>).(61) Coordination of iron(III) to the heme is mostly mediated by the β-dx2-y2 orbital of Fe, which forms a strong σ bond with the pyrrole nitrogens of the porphyrin. The corresponding antibonding combination, dx2-y2_B1g<80>(β<222>), has 61 % dx2-y2 and 32 % heme b contribution. The Eg<82/83> LUMO of the free porphyrin ligand (cf. Figure S1) is mixed with the dxz and dyz orbitals of iron, generating the MOs β<219> and β<220> that show 15 % and 12 % contributions from dxz and dyz, respectively. The dxy orbital is non-bonding and corresponds to the lowest unoccupied β molecular orbital (β-LUMO) of the complex, β<216>. The Fe-S(cys) π-bond is mediated by dxz of iron and S(px) of sulfur. The bonding and antibonding MOs, β<211> (9 % Fe and 36 % S contribution) and β<218> (55 % Fe and 19 % S admixture), respectively, indicate a quite strong interaction. The Fe-S(Cys) σ-bond is mediated by the dz2 orbital of iron and S(pz). The corresponding bonding combination, (β<201>) has 15 % Fe and 38 % S character, whereas the antibonding combination (β<221>) shows 56% Fe, 17% S, and 14% porphyrin contributions. The porphyrin A2u<81> orbital (the HOMO of the free porphyrin ligand) also exhibits some dz2 and S(pz) admixture as noted before.(61) The other important porphyrin π-orbital, A1u<79>, is non-bonding with respect to the iron d-orbitals and constitutes the β-HOMO-1 (β<214>) of the complex. Conversely, the β-HOMO contains neither contributions from the porphyrin ring nor iron, but is instead located on the amino acid side chains (sc) of the Cys pocket (cp) (π/σsc(cp), β<215>).
Figure 2.

Molecular orbital diagram (BP86/TZVP) of the Cyt P450cam model system shown in Scheme 1, as taken from the crystal structure (PDB code: 2CPP). The S(px), S(py), and S(pz) orbitals pertain to the 3p orbitals of sulfur. The π/σsc(cp) refers to a MO formed from π and σ bonding side chain (sc) orbitals from the Cys pocket (cp). The labels of the porphyrin core orbitals refer to free porphine, see Figure S1 in the Supporting Information. Molecular orbital labels a_b indicate that orbital a interacts with b and that a has a larger contribution in the resulting MO.
Figure 3.

Contour plots of important β molecular orbitals of the Cyt P450cam model system shown in Scheme 1 (BP86/TZVP). For labels, see Figure 2.
Table 1. Charge Contributions to the β spin Molecular Orbitals of the Cyt P450cam active site model (cf. Scheme 1) calculated with BP86/TZVP.
| No. | Labela | Energy/Hartree | Fe, d | S, p | Porphyrin |
|---|---|---|---|---|---|
| β<228> | πsc(cp) + π/σsc(por) | -0.03146 | 0 | 0 | 2 |
| β<227> | πsc(por)+B1u | -0.03898 | 1 | 0 | 34 |
| β<226> | π/σsc(por) | -0.04348 | 0 | 0 | 2 |
| β<225> | πsc(por) | -0.04681 | 0 | 0 | 0 |
| β<224> | πsc(por) | -0.04704 | 0 | 0 | 0 |
| β<223> | B1u<84> | -0.06886 | 2 | 0 | 72 |
| β<222> | dx2-y2_ B1g<80> | -0.08264 | 61 | 0 | 32 |
| β<221> | dz2_S(pz)+A2u<81> | -0.10842 | 56 | 17 | 14 |
| β<220> | Eg<82/83>_dyz | -0.11245 | 12 | 0 | 77 |
| β<219> | Eg<82/83>_dxz+S(px) | -0.11598 | 15 | 1 | 71 |
| β<218> | dxz_S(px) | -0.13665 | 55 | 19 | 18 |
| β<217> | dyz | -0.15322 | 68 | 1 | 22 |
| β<216> | dxy (LUMO) | -0.15777 | 93 | 0 | 4 |
| β<215> | π/σsc(cp) (HOMO) | -0.18567 | 0 | 0 | 0 |
| β<214> | A1u<79> | -0.19331 | 0 | 0 | 89 |
| β<213> | π/σsc(cp) | -0.19560 | 0 | 1 | 1 |
| β<212> | A2u<81>_dz2 + S(px) | -0.20275 | 4 | 7 | 82 |
| β<211> | S(px)_dxz | -0.20470 | 9 | 36 | 39 |
| β<210> | πsc(cp) | -0.21646 | 1 | 2 | 1 |
| β<209> | πsc(cp) | -0.21693 | 1 | 1 | 1 |
| β<208> | B2u<74>+S(px) | -0.22086 | 1 | 2 | 51 |
| β<207> | π/σsc(por)+πpor | -0.22569 | 3 | 3 | 31 |
| β<206> | A1u(dis)+S(py) + πsc(cp,por) | -0.22943 | 4 | 6 | 46 |
| β<205> | πsc(cp) + πpor+S(px)+σsc(por) | -0.23415 | 3 | 10 | 22 |
| β<204> | πsc(cp) + πpor + dz2 | -0.23693 | 8 | 5 | 27 |
| β<203> | πpor+πsc(por) | -0.24000 | 3 | 2 | 47 |
| β<202> | πpor+π/σsc(por) | -0.24530 | 3 | 1 | 44 |
| β<201> | S(pz)_dz2 | -0.24839 | 15 | 38 | 1 |
| β<200> | π/σsc(cp) | -0.24944 | 1 | 3 | 2 |
| β<199> | πsc(por) | -0.25893 | 1 | 0 | 11 |
| β<198> | Eg<59/60> | -0.26373 | 1 | 1 | 80 |
| β<197> | Eg<59/60> | -0.26706 | 0 | 0 | 90 |
| β<196> | Eu(σ) | -0.28341 | 6 | 0 | 89 |
| β<195> | A1u(dis)+πsc(por) | -0.28439 | 2 | 0 | 52 |
| β<194> | πsc(por) | -0.28804 | 0 | 0 | 5 |
| β<193> | Eu(σ)_dx2y2 | -0.29459 | 10 | 2 | 62 |
| β<192> | σpor_dx2-y2 | -0.29840 | 11 | 0 | 60 |
| β<191> | π/σsc(por)+πpor | -0.30347 | 0 | 0 | 24 |
| β<190> | π/σsc(por)+πpor + S(py) | -0.30480 | 2 | 2 | 27 |
| β<189> | B2u(dis)+S(py) + π/σsc(cp) | -0.30572 | 3 | 8 | 38 |
| β<188> | B1u+πsc(por) + S(py) | -0.31095 | 1 | 2 | 46 |
| β<187> | B1u+πsc(por,cp) + S(py) | -0.31186 | 1 | 5 | 42 |
| β<186> | πsc(cp)+S(px/y)_dz2 | -0.31519 | 2 | 6 | 3 |
Abbreviations: sc – side chain, cp – Cys pocket, por – porphyrin, dis – distorted, S(px/y) – electron densities are in between the x and y axis. S(px), S(py) and S(pz) are the sulfur 3p orbitals.
Based on the MO diagram in Figure 2, all the pertinent electronic transitions identified from TD-DFT can be classified. The transitions are subdivided into inner porphyrin (π → π*) and porphyrin to iron CT (P → Fe CT) transitions, which are mainly in-plane (xy) polarized, and sulfur to iron CT (S → Fe CT) transitions, which are out-of-plane (z) polarized. In addition, there are other transitions that give rise to out-of-plane polarization such as the Cys pocket (cp) to porphyrin or iron transitions (cp → P/Fe), which, however, are overlap forbidden, and hence, should not contribute to the spectral intensity and the observed polarizations. The d-d transitions will not be considered because they are spin forbidden due to the 6A1 ground-state of hs Fe(III). The following one-electron transitions are relevant for the analysis of the electronic spectra:
-
Inner porphyrin transitions (cf. Scheme 2; xy-polarized)
π → π* (0): A1u, A2u<79,81> → Eg<82,83> (Soret and Q-band)
π → π* (1): A2u<72> → Eg<82,83>
π → π* (2): B2u<74> → Eg<82,83>
π → π* (3): πpor → Eg<82,83> (πpor = low-symmetry porphyrin π orbitals)
-
Porphyrin to iron CT transitions (P → Fe CT; cf. Scheme 2; xy-polarized)
: A1u <79> → dxz
: A1u <79> → dyz
: A1u <79> → dx2-y2
: B2u <74> → dxz
: B2u <74> → dyz
: Eu(σ) → dxz (z-polarized)
: Eu(σ) → dxy (xy-polarized)
: Eu(σ) → dyz (z-polarized)
CT(4): Eg<59/60> → dxz
CT(5): A2u<81>_dz2 + S(px) → dz2_S(pz)+A2u<81> (z-polarized)
-
Sulfur to iron CT transitions (S → Fe CT; cf. Scheme 2, z-polarized)
CT(S,π): S(px) → dxz
CT(S,σ): S(pz) → dz2
Scheme 2.

As mentioned above, some of the MOs involved in these transitions are highly mixed. Correspondingly, certain P → Fe CTs may involve contributions from sulfur or Cys pocket orbitals. These contributions are noted accordingly. A complete list of these transitions and the assignments of experimentally observed bands are presented in Table S2.
B. Spectroscopic results
In the resting state, wt Cyt P450s contain a six-coordinate (6C), low-spin (S = 1/2) ferric heme, which, in general, becomes five-coordinate (5C), high-spin (S = 5/2) upon binding of the substrate (camphor for Cyt P450cam).(46, 47) In the case of Cyt P450cam, cooling this 5C species to liquid helium temperature for spectroscopic measurements results in a mixture of hs and ls ferric species due to water coordination at the distal site of the iron center. This is clearly evident from the EPR spectra of wt ferric Cyt P450cam, which show both rhombic hs signals at g = 7.78, 3.95, and 1.76 and ls signals at g = 2.41, 2.23, and 1.97.(39) The same situation is also encountered in the low-temperature MCD spectra of ferric wt Cyt P450cam. In order to analyze the MCD data of wt enzyme, the spectra have to be deconvoluted into the contributions from the hs and ls forms. In order to achieve this, we have first investigated the variants, Q360P and Y96W, because these yield pure hs and ls spectra, even at low temperature. Q360P is a Cys pocket derivative where glutamine on the proximal side of the heme is replaced by proline at position 360. This replacement removes two hydrogen bonds: one between the amide group of Q360 and the sulfur of C357, and one between the side chain of Q360 and the backbone carbonyl of C357 (cf. Figure 1). This variant can be considered to be in a pure hs ferric state, even at low temperatures (15 K), as evidenced by EPR spectroscopy, where g values of 7.82, 3.82, and 1.75 are observed.(39) Another active site variant of P450cam, Y96W, has tyrosine on the distal side of the heme replaced by tryptophan at position 96. Y96 is positioned at the end of the B′ helix near the presumed channel where substrates enter the active site. Tyrosine forms a hydrogen bond with its phenol group to the carbonyl oxygen of camphor. Mutation of Y96 to tryptophan retains a six-coordinate, ls ferric heme species, as observed by low-temperature EPR spectroscopy,(80) potentially because the tryptophan indole fills the active site preventing camphor from binding, thereby leaving water coordinated to the heme. Finally, the L358P variant has been studied, and it forms a mixture of hs and ls Fe(III) species at low temperature as observed from EPR.(39) This Cys pocket mutant has one less hydrogen bond to the sulfur, relative to wt P450cam. Comparison of S → Fe CT transitions for wt (4 H-bonds), L358P (3-H bonds) and Q360P (2 H-bonds) therefore allows for the systematic study of the effect(s) of hydrogen bonding (located on the proximal side of the heme) on the strength of the Fe-S bond, as manifested by the energies of the S → Fe CT transitions.
Identification of the S → Fe CT is best achieved in the ferric hs state, since the electron configuration of this state with all five d-orbitals half occupied allows for the maximum number of CT transitions to be observed (cf. ref (61)). Moreover, the hs state is more relevant for the catalytic mechanism of Cyt P450s. The electronic spectra of Q360P (pure hs mutant) are therefore presented first, and then compared to the hs signals in the mixed hs/ls spectra of wt enzyme and L358P. The identified transitions from the MCD spectra of Q360P are assigned using VTVH measurements and TD-DFT calculations.
1. UV–vis spectroscopy
In order to understand the UV–vis absorption features of metalloporphyrins, a brief explanation of Gouterman's four-orbital model is necessary (see also ref. (61)).(81-83) In this model, the two highest occupied molecular orbitals (HOMOs), A1u<79> and A2u<81> (in D4h symmetry), and the two-fold degenerate lowest unoccupied molecular orbital (LUMO), Eg<82/82>, of heme are considered. The excited states resulting from the π → π* transitions, A1u → Eg and A2u → Eg, have the same symmetry (A1u × Eg = A2u × Eg = Eu), and show strong configuration interaction. For the resulting higher energy transition, the individual transition dipole moments add up leading to the intense Soret band, whereas they cancel for the lower energy transition, the Q band. The Q band further “steals” ∼10 % of the intensity from the Soret band through vibronic coupling, producing the vibronic Qv band as evident from resonance Raman spectroscopy.(62, 84, 85) Qv is found ∼1300 cm-1 to higher frequency relative to Q.(81)
The UV-vis spectra of ferric hs (Q360P) and ls (Y96W) Cyt P450cam in the presence of d-camphor at room temperature are shown in Figure 4. The spectrum of Q360P shows a rather broad Soret band at 391 nm, and weak intensity features in the Q-region at 514, 540, and 642 nm. The absorption spectrum of Q360P is very similar to that of wt enzyme, indicating that the resting state of ferric Cyt P450cam is also high-spin at room temperature, as has been previously established.(46, 47) Compared to Q360P, the spectrum of Y96W displays a sharper Soret band at 417 nm with a high-energy shoulder at 355 nm, and more defined bands at 536 and 570 nm in the Q-region. The extinction coefficient of the Soret band of Y96W is distinctively larger than that of Q360P (cf. Figure 4), which is a result of the smaller bandwidth in the ls form and is usually seen with ls P450s. The spectrum of Y96W is very similar to wt P450cam in the absence of d-camphor, presumably because the substrate does not have proper access to the active site.
Figure 4.

Room temperature absorption spectra of Q360P, Y96W, and wt Cyt P450cam in 50 mM potassium phosphate buffer (pH 7.4) containing 500 μM d-camphor.
2. Magnetic Circular Dichroism (MCD) spectroscopy
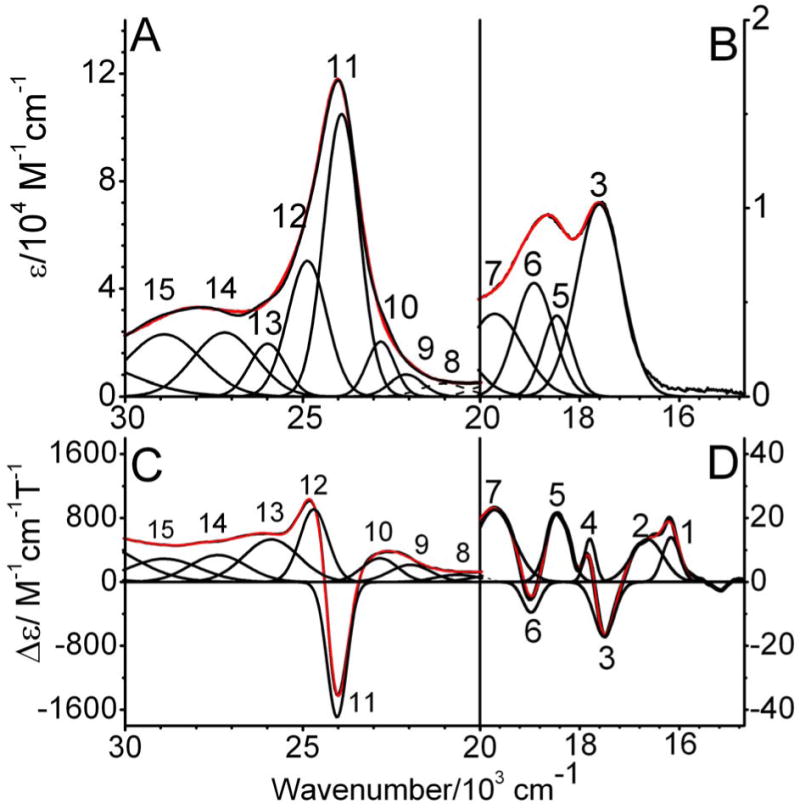
The low temperature MCD C-term spectra of Q360P, Y96W, and wt Cyt P450cam are presented in Figures 5-7, respectively. The C-term spectrum of hs Q360P shows complex features in the 15200-31050 cm-1 region as shown in Figure 5. The correlated Gaussian fit of the MCD C-term and UV–vis spectra of Q360P reveals fourteen electronic transitions that would have been impossible to identify from the UV–vis spectrum by itself, as evident in Figure 5. The Q region shows distinguishable features at 15222, 15909, 16919, 17881, and 18604 cm-1, and a higher energy feature at 19539 cm-1 that tails into the low energy region of the Soret band (features 1-6 in Figure 5 and Table 2). To lower energy of the Soret region three low intensity features are observed at 20539, 21807, and 22685 cm-1 (bands 7-9). The Soret band at 391 nm (from absorption) is deconvoluted into three surprisingly weak MCD features at 23731, 24859, and 25618 cm-1 (bands 10-12), which show a -Δε/+Δε/-Δε pattern similar to that observed for [Fe(TPP)Cl].(61) Since the Soret band, corresponding to an Eu excited state in D4h symmetry, should give rise to a derivative-shaped C-term feature,(86, 87) the observation of three components is indicative of selective mixing of the negative component of the Soret band with either a porphyrin π → π* or a P → Fe CT transition, as observed first for [Fe(TPP)Cl].(61) Finally, a very prominent peak at 28570 cm-1 (band 13) observed to higher energy of the Soret band is identified in the MCD spectrum, whereas this feature is hidden in the high-energy tail of the Soret band in the UV–vis spectrum. This band is relatively weak in absorption but strong in MCD, indicating significant metal contribution, and hence, is assigned to a charge-transfer transition. This feature had previously been observed for hs ferric wt P450cam via low temperature MCD,(48, 54) but was not further considered. The electronic transition energies along with molar intensities in the UV–vis and MCD C-term spectra of Q360P are presented in Table 2.
Figure 5.

Electronic spectra of ferric hs Q360P in 50 mM potassium phosphate buffer (pH 7.4) with 500 μM d-camphor. Top: UV-Vis absorption spectrum measured at room temperature. Bottom: MCD C-term spectrum measured at 5 K in phosphate buffer with 50 % (v/v) glycerol added. The colored lines represent a correlated Gaussian fit of these data (Table 2), where the predominant polarizations are color-coded (xy-polarizations in blue, z-polarizations in green, and undetermined polarizations in gray). The recorded spectra are red.
Figure 7.

The MCD spectra of wt Cyt P450cam (black), Q360P (green), and Y96W (blue) using the same conditions as in Figure 5. The red spectrum is the best fit to the wt MCD spectrum obtained by adding 86% Q360P and 14% Y96W.
Table 2. Correlated Fit of the UV–vis Absorption and MCD spectra of Cyt P450cam Q360P.
| No. | MCD | UV–vis | Calculated (average) | Assignmenta | Polarization from MCD | |||
|---|---|---|---|---|---|---|---|---|
| Energy, cm-1 | Δε | Energy, cm-1 | ε | |||||
| 1 | 15222 | 8 | 15223 | 3512 | NA | |||
| 2 | 15909 | 31 | 15790 | 4160 | 12480 |
|
x,y | |
| 3 | 16919 | -23 | 16800 | 5300 | 14721 | , A2u<81> → dxz | x,y | |
| 4 | 17881 | -32 | 17700 | 5402 | 16786 | CT(S,π) P → Fe CT | x,y,z | |
| 5 | 18604 | -47 | 18480 | 8707 | 19582 | Q-band | x,y | |
| 6 | 19539 | 37 | 19410 | 10786 | 19789 | x,y | ||
| 7 | 20539 | 83 | 20539 | 13578 | 25761 | π → π*(1), A2u<81> → dx2y2 | x,y | |
| 8 | 21807 | 112 | 21807 | 15193 | 27291 | , π → π* (3) | ||
| 9 | 22685 | 28 | 22607 | 14777 | 28089 | π → π* (3) | ||
| 10 | 23731 | -111 | 23558 | 28506 | 28367 | Soret, π → π* (3), πpor +πsc(por) → dz2 | x,y | |
| 11 | 24859 | 141 | 25013 | 78151 | 30636 | Soret, , πpor +/σsc(por) → dz2, σpor_dx2-y2 → dxy | x,y | |
| 12 | 25618 | -97 | 26492 | 52849 | 31099 | Soret, , CT(S,σ) | x,y | |
| 13 | 28570 | -179 | 28438 | 44742 | 31335 | CT(S,σ), , , , , π/σsc(por)+πpor → dxy | x,y,z | |
| 14 | 31042 | 47 | 31042 | 20066 | NA | |||
Note that σpor and πpor are distorted MOs that can only be classified as σ and π bonding porphyrin (por) orbitals, respectively. In some transitions, the propionate side chains (sc) of heme b are involved.
Unlike hs Q360P, the ls Y96W mutant exhibits less low-energy ligand to metal CT transitions, because the t2g type orbitals of Fe(III) are almost fully occupied. This limits the MCD C-term features in Y96W to mostly porphyrin π → π* transitions as observed before for model complexes.(81) More importantly, polarizations of the electronic transitions are not attainable in the ferric ls state. Hence, a detailed assignment of the low-temperature MCD spectra of Y96W, shown in Figure 6, has not been attempted. Nevertheless, the MCD C-term data of this species are important in order to analyze the MCD data of wt Cyt P450cam (vide infra). The molar MCD intensities of the Soret bands are in general one order of magnitude larger in ferric ls compared to ferric hs complexes, and the same is observed for Q360P and Y96W. This surprising result can be explained with differences in spin orbit coupling (SOC), as discussed by Paulat and Lehnert.(61) The difference in intensity between ferric hs and ls complexes has also previously been noted by Cheesman et al.(88) A correlated fit of the MCD and UV–vis spectra of the low-spin ferric complex Y96W is shown in Figure 6, and details of the fit are presented in Table 3.
Figure 6.
Electronic spectra of ferric ls Y96W under the same conditions as in Figure 5. Top: UV-Vis absorption spectrum measured at room temperature. Bottom: MCD C-term spectrum measured at 5 K in phosphate buffer with 50 % (v/v) glycerol added.
Table 3. Correlated Fit of the UV–vis Absorption and MCD spectra of Cyt P450 Y96W.
| No. | MCD | UV–vis | ||
|---|---|---|---|---|
| Energy, cm-1 | Δε | Energy, cm-1 | ε | |
| 1 | 16168 | 14 | - | - |
| 2 | 16643 | 14 | - | - |
| 3 | 17507 | -17 | 17605 | 10208 |
| 4 | 17797 | 14 | - | - |
| 5 | 18441 | 22 | 18455 | 4280 |
| 6 | 18975 | -10 | 18914 | 6017 |
| 7 | 19706 | 23 | 19701 | 4377 |
| 8 | 20877 | 21 | 21034 | 4813 |
| 9 | 21967 | 216 | 22081 | 8076 |
| 10 | 22821 | 297 | 22800 | 20266 |
| 11 | 24029 | -1696 | 23900 | 105000 |
| 12 | 24688 | 908 | 24871 | 50284 |
| 13 | 25887 | 532 | 25990 | 19504 |
| 14 | 27384 | 337 | 27200 | 23658 |
| 15 | 28906 | 291 | 28900 | 22986 |
In the case of wt Cyt P450cam, a mixture of high- and low-spin complexes is observed at low temperature. Knowledge of the pure hs and ls spectra from Q360P and Y96W allows one (a) to assign the observed MCD bands in wt enzyme to either the hs or ls form, and (b) to calculate the hs to ls ratio. For the wt sample presented in this paper, contributions of 86 % hs and 14% ls were estimated at 5 K using 3 T (Tesla) data as shown in Figure 7 (note that this ratio varies somewhat between samples – perhaps because of variation in the freezing conditions). The MCD C-term spectrum of wt Cyt P450cam reveals fifteen electronic transitions (Figure 8, bottom). In the low-energy Q region, three distinguishable weak bands at 15202, 15953, and 18077 cm-1 (bands 1-3) are observed, which are clearly separated from a group of five more intense, electronic transitions at 19441, 20194, 20921, 22219, and 23150 cm-1 (bands 4-8). The MCD features in this energy region (19000-23200 cm-1) are similar to those of Q360P, and hence, belong to the hs form. The Soret region displays four sharp peaks at 24101, 24618, 25132, and 25727 cm-1 (bands 9-12) that are more intense than in Q360P, and are closer in energy to those of Y96W, indicating the presence of the ls ferric form at low temperature, in agreement with EPR.(39) Curiously, the peak at 25132 cm-1 is not present in the hs or ls spectra. Hence, the nature of this peculiar band remains unknown. To higher energy, a weaker band at 26969 cm-1 (band 13) is observed, close to the prominent band at 28274 cm-1. This weak band may correspond to band 14 from the ls form based on relative energies. The broad intense band 14 of wt enzyme is very similar to band 13 of hs Q360P, but is absent in ls Y96W, and hence, clearly belongs to the hs complex (see Section C). Finally, the feature at 30980 cm-1 (band 15) in wt Cyt P450cam is similar to band 14 of Q360P (Figure 8, top). Table 4 lists the electronic transition energies and molar intensities for wt P450cam. A comparison of the MCD spectra of Q360P and wt Cyt P450cam is shown in Figure 8.
Figure 8.

MCD C-term spectra of ferric hs Q360P (top) and wt P450cam (bottom) in 50 M potassium phosphate buffer (pH 7.4) containing 500 μM d-camphor at 5 K with 50 % (v/v) glycerol. The black lines represent a correlated Gaussian fit of these data (cf. Tables 2 and 4). The asterisk on the band numbers in wt Cyt P450 indicates contributions from the ls form.
Table 4. Correlated Fit of the UV–vis Absorption and MCD spectra of wt Cyt P450cam.
| No. | MCD | UV–vis | ||
|---|---|---|---|---|
| Energy, cm-1 | Δε | Energy, cm-1 | ε | |
| 1 | 15202 | 7 | 15465 | 3129 |
| 2 | 15953 | 29 | 17234 | 5087 |
| 3 | 18077 | -42 | 18480 | 5780 |
| 4 | 19441 | 32 | 19367 | 7545 |
| 5 | 20196 | 48 | 20268 | 7983 |
| 6 | 20921 | 80 | 21451 | 9258 |
| 7 | 22219 | 135 | 22556 | 12106 |
| 8 | 23150 | 64 | 23240 | 12964 |
| 9 | 24101 | -259 | 23900 | 25612 |
| 10 | 24618 | 174 | 24650 | 48417 |
| 11 | 25132 | -124 | 25330 | 48492 |
| 12 | 25727 | 46 | 26000 | 47614 |
| 13 | 26969 | 68 | 26900 | 44028 |
| 14 | 28724 | -206 | 28500 | 36871 |
| 15 | 30980 | 43 | 30815 | 19189 |
3. Variable Temperature Variable Field (VTVH) measurements
VTVH saturation curves for hs Q360P were determined for all clearly identified transitions in order to provide insight into the relative polarizations of these features; in particular, with respect to in-plane (xy) and out-of-plane (z) polarized transitions. In-plane polarized transitions include porphyrin π → π* and porphyrin-to-iron CT transitions, while out-of-plane polarized transitions mostly correspond to sulfur-to-iron CT transitions. In the case of the C4v symmetric model complex [Fe(TPP)Cl], which shows axial zero field splitting (E/D = 0), clear differences in the VTVH curves for xy-, and z-polarized transitions have been observed.(61) In the case of Cyt P450cam that contains a heme b, an additional complication is the generally lower symmetry of the heme active site, as is evident from the distinct degree of rhombicity (E/D = 0.087) in the EPR spectra.(50) To understand how such a rhombic distortion affects the properties of the VTVH curves, calibration curves with different contributions of x, y and z polarizations were calculated as shown in Figure S2. A nesting (“N”) behavior is observed for x-polarized, and in particular, for y-polarized transitions. On the other hand, overlaying saturation isotherms (“O” behavior) are observed for z-polarized transitions and transitions with equal contributions of x and z polarization.
VTVH curves were generated for ten of the fourteen clearly identified bands in the MCD spectrum of hs Q360P, whereas the intensities of the remaining four bands 1, 9, 10, and 12 were either too weak or too convoluted (due to strongly overlapping features) to obtain reliable isotherms. Bands 2 and 3 show “N” behavior, characteristic of xy-polarization (cf. Figures 9 and S3). Bands 4 and 5 are strongly y-polarized (cf. Figures S3 and 10), which leads to very distinguishable saturation curves. Bands 6 and 7 are again in-plane polarized (Figure S3). As shown in Figure 5, bands 8-12 show relatively strong overlap, which renders it difficult to obtain unambiguous VTVH curves for these features. Bands 8 and 11 display VTVH data that cannot be simulated well, resulting in unreliable fits with ∼50% error margins (Figure S3). Since these bands occur in the Soret region, these transitions are most likely xy-polarized with major contributions from π → π* transitions. The expected polarizations of these transitions compare well with the TD-DFT calculations (vide infra). Finally, the prominent band at 28570 cm-1 (band 13) shows “O” saturation behavior (Figure 11) with ∼40% z-polarization, indicative of a significant contribution of sulfur-to-iron CT transitions. A summary of the polarizations of all clearly identified transitions is presented in Table 2.
Figure 9.
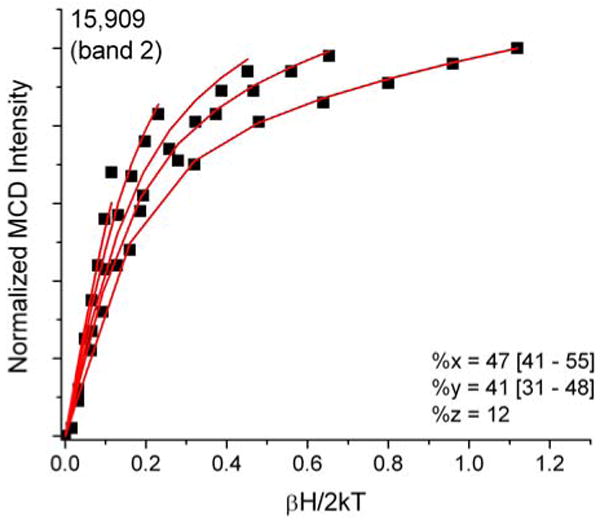
Ferric hs Q360P MCD C-term saturation magnetization curves (VTVH) for band 2 at 15909 cm-1 (red lines: fit; black dots: experimental data points) showing 88% xy-polarization. Buffer conditions were the same as in Figure 5.
Figure 10.
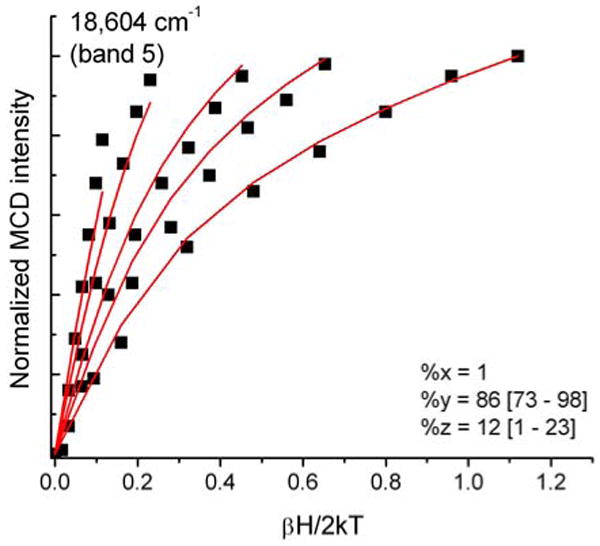
Ferric hs Q360P MCD C-term saturation magnetization curves (VTVH) for band 5 at 18604 cm-1 (red lines: fit; black dots: experimental data points) showing 87% xy-polarization. Buffer conditions were the same as in Figure 5.
Figure 11.
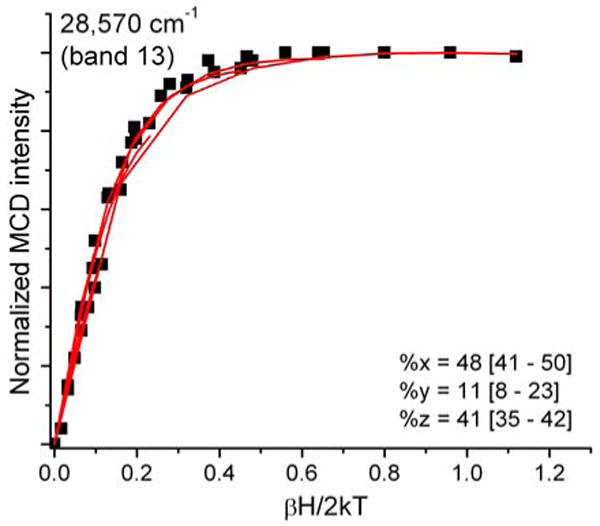
Ferric hs Q360P MCD C-term saturation magnetization curves (VTVH) for band 13 at 28570 cm-1 (red lines: fit; black dots: experimental data points) showing 41% z-polarization. Buffer conditions were the same as in Figure 5.
C. Assignments using TD-DFT calculations
Unlike the four-fold symmetric tetraphenylporphyrin2- ligand used previously in the MCD-spectroscopic investigations on [Fe(TPP)Cl],(61) heme b in Cyt P450cam has only twofold symmetry, which results from the presence of two propionate substituents on the porphyrin core (taking into account the additional vinyl and methyl groups, the symmetry reduces to C1). This is important to note because it affects the degeneracy of Eg-symmetric molecular orbitals, and correspondingly, the degeneracies, energies, and intensities of the transitions involving these orbitals (cf. Scheme 2). In the following, Figure 5 and Table S2 are used as reference.
On the basis of TD-DFT calculated polarizations, oscillator strengths, and in a few cases, relative energies compared to experiment, most bands in the MCD spectrum of Q360P could be assigned. Band 1 (15222 cm-1) has neither polarization data nor literature precedence available to unequivocally assign it to a specific transition. Paulat and Lehnert(61) assigned similar weak, low-energy features at 11584 and 13542 cm-1 in [Fe(TPP)Cl] to the spin-forbidden 6A1 → 4T1 + 4T2 ligand field transition, which is usually observed in the 10000-15000 cm-1 range for hs Fe(III). Band 1 is therefore tentatively assigned to this ligand field transition. To higher energy, the MCD spectrum of Q360P shows two features (bands 2 and 3) that form a pseudo-A term signal. The observation of a pseudo-A term usually indicates the presence of a degenerate excited state that is split into two components by low-symmetry distortions.(79, 86, 87) In the case of [Fe(TPP)Cl], a similar feature is observed at 15000 and 15600 cm-1, and has been assigned to the two components of the A1u → dxz, dyz transition (CT(1)).(61) We assign band 2 (15909 cm-1), which is purely xy-polarized, to the analogous . Interestingly, however, the corresponding transition occurs ∼3500 cm-1 to lower energy in Cyt P450cam, which is due to the large splitting between the dxz and dyz orbitals as shown in Figure 2. This is in agreement with the observed rhombicity of the ferric heme in Cyt P450s as evident from previously reported EPR studies.(39) The large splitting between dxz and dyz relates to the fact that (a) dxz forms a strong π-bond with S(px) of cysteine (major contribution; cf. MOs<211> and <218> in Figure 3), and (b) the low symmetry of the heme, which leads to anisotropic π-bonding of the heme with dxz and dyz. Comparison of the calculated and observed polarizations of band 2 also indicates that this feature must have other contributions besides . Because of the low energy of dyz, the other pseudo-A component, band 3 (16919 cm-1), cannot correspond to the transition. From the TD-DFT calculations, band 3 could either be identified with a calculated band at 14507 cm-1, corresponding to the A2u<81> → dxz transition (95 % xy-polarized), or a calculated feature at 14934 cm-1, which is of character (77 % xy-polarized). The sign change between bands 2 and 3 is best rationalized when band 3 corresponds to , which would allow for strong excited state SOC, in agreement with the observed pseudo A-term signal. Bands 2 and 3 also show minor contributions from the π → π*(0) transition (Q band), similar to what has been observed for the CT(1) transition in [Fe(TPP)Cl].(61)
Based on relative oscillator strengths and polarizations, band 4 at 17881 cm-1 corresponds to calculated features at 16206, 16372, and 17779 cm-1, which have significant CT(S,π) contributions, and hence, are largely z-polarized in the calculation. Experimentally, an increase in z-polarization is indeed observed for band 4 compared to bands 2 and 3, but the effect is less pronounced, indicating that band 4 is more dominated by xy-polarized transitions than predicted by TD-DFT. We assign this band to a mixture of P → Fe CT plus CT(S,π), consistent with the MCD data of hs [Fe(TPP)Cl](61), where CT(Cl,π)) is observed at 16444 cm-1. Other z-polarized contributions to band 4 are predicted to be of π/σsc(cp) → π/σpor and πsc(cp) → Fe(d) character, which, however, are overlap forbidden and hence, should not contribute to the observed intensities (oscillator strengths) and polarizations.
At higher energy, bands 5 (18604 cm-1) and 6 (19539 cm-1) give rise to another pseudo-A term signal. These features are assigned to the two components of the Q band. Compared to the TD-DFT calculations, band 5 is identified with the calculated features at 19407, 19552, and 19786 cm-1, and band 6 corresponds to the calculated transition at 19789 cm-1, which are all dominated by π → π* (0) in agreement with the assignment of these features to the Q band. Room temperature MCD spectra of hs ferric Cyt P450cam show a derivative-shaped band at ∼540 nm, almost exactly at the center of band 5, which further supports this assignment.(3, 48, 54) Interestingly, band 5 shows a quite large contribution of z-polarized intensity, which is similar to band 4, and likely arises from admixture of S(π) → Fe CT character. Note that the Q band is hardly detectable in the UV–vis or MCD spectra of the more symmetric model complex [Fe(TPP)Cl]. In Cyt P450cam, the increased intensity of the Q band might be due to the lower symmetry of heme b, which could counteract the intensity quenching of Q due to the cancellation of the transition dipole moments of the underlying A1u <79> → Eg and A2u <81> → Eg excitations, according to Gouterman's four orbital model (see also ref. 61).(81) Based on this assignment, the position of the two components of Qv are estimated at 19900 and 20800 cm-1 (in the region of band 7); however, these features are not observed experimentally.
The next three features, bands 7 (20539 cm-1), 8 (21807 cm-1) and 9 (22685 cm-1) are more difficult to assign, largely because the polarizations of bands 8 and 9 are not well defined. Here, the oscillator strengths and the energy sequence of these transitions were used as a guide for comparison with the TD-DFT results. Band 7 corresponds to two possible xy-polarized transitions calculated at 25225 cm-1 (34 % π → π* (1) character) and 26297 cm-1 (56% P → Fe CT transitions, Table S2), or a superposition of both of them. A similar situation is encountered for band 8, which is assigned to features calculated at 27088 cm-1 and 27493 cm-1 (π → π* (3)). The weakest among the three bands, band 9, is assigned to the transition calculated at 28089 cm-1, which has π → π* (3) character. In all three cases, TD-DFT predicts these states to be in-plane polarized, consistent with the type of transitions involved. The fact that these features are calculated about 6000 cm-1 too high in energy seems surprising, but is in agreement with the deviation observed for Soret band.
The Soret region shows three features, bands 10 (23731 cm-1), 11 (24859 cm-1) and 12 (25618 cm-1). Theoretical analysis shows that the Soret band, which corresponds to an Eu excited state (in ideal D4h symmetry), should exhibit a pseudo-A type signal in the MCD spectra.(86, 87) Previous work on the hs ferric model complex [Fe(TPP)Cl] has shown that the observed three Soret components result from selective mixing of the negative component of the Soret band with a porphyrin π → π* excited state, in this case of A2u<72> → Eg<82/83> character (π → π*(1)).(61) This leads to a splitting of the negative Soret component into two features. Interestingly, a similar splitting pattern is observed for Cyt P450cam in this study. Although experimental polarizations are not available for bands 10 and 12, and band 11 has large errors in its experimental polarizations, it can be expected that these features are in-plane polarized. Based on the TD-DFT calculations, band 10 is assigned to a feature calculated at 28367 cm-1, which has π → π* (0) and π → π* (3) character and 25 % contribution from other P → Fe CT transitions. Band 11 is likely comprised of two transitions predicted at 30608 and 30663 cm-1 that are very close in energy: both show quite large and other P → Fe CT contributions arising from porphyrin σ and π distorted MOs. Band 12 can also be identified with two possible transitions from TD-DFT, calculated at 31088 and 31110 cm-1, respectively. Both show contributions from .3
Importantly, band 13 at 28570 cm-1 shows very different VTVH behavior, and corresponds to a transition that is ∼40 % z-polarized. This band is very broad (full width at half height: 2080 cm-1), and therefore, likely corresponds to more than one transition. The weak absorption and strong MCD intensity of this feature indicates a large metal contribution, and hence, this feature is clearly a CT transition. From the TD-DFT calculations, thirteen features in the 31300-32700 cm-1 range are identified that could potentially contribute to band 13, and hence, a detailed assignment of this feature is not possible. According to TD-DFT, the in-plane intensity of this feature likely arises from , , , and contributions, whereas the out-of-plane polarized intensity is attributed to CT(S,σ). The latter is present in five of the thirteen transitions, although the percent contributions of CT(S,σ) are small, and hence, likely underestimated by TD-DFT. The occurrence of the S(σ) → Fe CT transition to higher energy of the Soret band at 28570 cm-1 parallels the observation of a Cl(π, σ) → Fe CT transition at a very similar spectral position (26181 cm-1) for [Fe(TPP)Cl],(61) which strongly supports our assignment of band 13. Lastly, band 14 (31042 cm-1) is located in an energy range beyond the 260 states calculated here. All assignments discussed in this section are summarized in Table S2.
D. The S(σ) → Fe CT transition
Besides the detailed assignment of the optical spectra of ferric hs Cyt P450cam, the identification of a CT band with strong S(σ) → Fe CT character (CT(S,σ)) is one of the major findings of this study. This band is observed to higher energy of the Soret band, and although relatively weak in absorption, is easily identified via low-temperature MCD spectroscopy. The position of this CT band is in agreement with the z-polarized single crystal absorption spectrum of Cyt P450cam showing a single peak at 30960 cm-1.(89) This assignment is further supported by resonance Raman spectroscopy: here, a strong enhancement of the ν(Fe-S) stretch at 351 cm-1 is observed upon excitation at 363 nm (= 27550 cm-1; see ref (39)) in the region of band 13, compared to spectra taken at 413 and 457 nm excitation as shown in Figure S4. From the literature, resonance Raman excitation profiles show two peaks split by ∼3200 cm-1 that are associated with the S → Fe CT.(90) The high-energy component occurs at ∼30960 cm-1 while the low-energy component is found at ∼28000 cm-1, the splitting of which is associated with a distinct lowering of the symmetry that is associated with the rhombic character of Cyt P450cam.
In summary, the prominent S(σ) → Fe MCD feature identified in this study allows us to use the energy of this band to monitor the effects of the proximal hydrogen bonding network on the Fe-S bond. This feature is only observed for the high-spin form of ferric P450cam (Figures 8 and S5), while this feature is absent in the ls ferric form (Y96W, Figure 6).
Compared to wt Cyt P450cam, the mutant L358P lacks one amide hydrogen bond to the proximal cysteine ligand (Cys357), because proline does not have an amide hydrogen atom. In Q360P, one amide hydrogen bond and in addition, the hydrogen bond from the Q side chain to the carbonyl group of Cys357 is missing. Removal of a hydrogen bond potentially increases the thiolate donor strength, and thus, the covalency of the Fe-S bond. This should be evident from changes in the S(σ) → Fe CT band. Figure 12 shows a superposition of CT(S,σ) for all three enzyme variants investigated here. Importantly, transition energies of 28724 (wt), 28620 (L358P), and 28570 cm-1 (Q360P) are observed, which indicates only a small change in the Fe-S bond upon removal of the hydrogen bonds. This aspect is further evaluated in the Discussion.
Figure 12.

MCD C-term spectra of wt Cyt P450cam, L358P, and Q360P under the same conditions as in Figure 5. These spectra focus on the S → Fe CT transition for each enzyme.
Discussion
Central to the active site of Cytochrome P450s is the thiolate ligand (Cys357 in Pseudomonas putida Cyt P450cam) coordinated to the iron of heme b. This thiolate ligand is strongly electron donating and is assumed to provide the “push” effect that facilitates heterolytic O-O bond cleavage, generating the high-valent [Fe(IV)=O P•+] species (Compound I),(91) which is the active oxidant for substrate hydroxylation. Although the overall features of the mechanism by which Cyt P450s activate dioxygen are reasonably well-established,(2, 3) the details of how Cys357 fine-tunes the enzyme active site, and hence, its reactivity are not well understood. One factor that potentially controls the reactivity is the hydrogen bonding network from the polypeptide amides that surround the thiolate ligand (the Cys pocket; cf. Figure 1). These hydrogen bonds are proposed to directly influence the properties of the Fe-S bond. In this work, Cyt P450cam and three representative variants are used to systematically study the effect of the hydrogen bonding network on the Fe-S bond strength. This includes L358P and Q360P, where one and two hydrogen bonds are removed from the Cys pocket. Low-temperature MCD spectroscopy on the high-spin (hs) ferric forms of these proteins is then use to monitor changes in the Fe-S bond via the energy of S → Fe CT transitions as the number of N-H⋯S hydrogen bonds is varied.
Importantly, studies on the heme redox potentials of wt Cyt P450cam and the “Cys” pocket variants mentioned above show a decrease in reduction potential along the series: -134 mV (wt), -170 (L358P), -180 (Q360L), and -205 mV (Q360P).(39) These results demonstrate a clear effect of the Cys pocket on the properties of the Cyt P450 active site. However, the redox potential of a metal site is a complex property that depends on the stabilization and/or destabilization of the oxidized and/or reduced forms of the complex. The observed changes could therefore relate to changes in (b) heme conformation, (c) charge distribution in the active site, or (d) axial ligand properties in either the reduced or oxidized form (or both).(92) Hence, it is not clear from redox potentials how exactly the hydrogen bonding network affects the Fe-S bond in the oxidized and reduced forms of Cyt P450s. On the other hand, spectroscopic results can contribute to a better understanding of these effects. In this work, we focus on the properties of the Fe-S bond in the (hs) ferric form of the enzyme. Electrochemical studies on Cyt P450 model complexes further support the above findings. For example, the redox potentials of the hs complexes [Fe(III)(OEP)(SPh)], [Fe(III)(OEP)(S-H1)] and [Fe(III)(OEP)(S-H2)] (OEP = octaethylporphyrin; S-H1 = S-2-(CF3CONH)C6H4; S-H2 = S-2,6-(CF3CONH)2C6H3), which have zero, one, and two strong NH-S hydrogen bonds, are -680 mV, -520 mV, and -350 mV (vs SCE), respectively.(41) In addition, the Fe-S bond lengths in these ferric compounds are 2.299, 2.327, and 2.356 Å, respectively, indicating that hydrogen bonding reduces the donor strength of the thiolate, and in this way, makes thiolate a weaker ligand. Similar trends in redox potential and Fe-S bond distance are observed for other arene-thiolate model complexes(42) and tetrapeptide-heme complexes.(40) Importantly, however, the observed changes are much larger for the model systems than for Cyt P450cam, indicating that the H-bonds in the latter are distinctively weaker.
Different spectroscopic methods have been applied to ferric Cyt P450cam and corresponding variants to directly detect the effect of H-bonding on the Fe-S bond. One indirect measure of Fe-S bond strength is the frequency of the Fe-S stretch, ν(Fe-S), which can be monitored via resonance Raman spectroscopy. A previous study by Yoshioka et al.(39) shows ν(Fe-S) for ferric wt Cyt P450cam, L358P, and Q360L at 351 cm-1, but at 347 cm-1 in Q360P. These frequencies strongly suggest that the Fe-S bond strength is unaltered upon removal of one (L358P) and two (Q360P) hydrogen bonds from the Cys pocket. Since vibrational frequencies are also influenced by mode mixing and effective masses, as has been recently demonstrated for six-coordinate ferric heme-nitrosyl model complexes [Fe(P)(SR)(NO)],(93) a more reliable comparison would be based on Fe-S force constants. However, such force constants are not available. Second, ligand K-edge X-ray absorption spectroscopy (XAS) can be applied, which directly probes ligand-metal covalency through the pre-edge feature resulting from the 1s (sulfur) to 3d (metal) transition. The effect of hydrogen bonding on the Fe-S bond covalency has been investigated for the above mentioned model complexes [Fe(OEP)(S-H2)], [Fe(OEP)(S-H1)], and [Fe(OEP)(SPh)].(45) Here, the total covalency decreased from 49 (L = SPh), 41 (L = S-H1), to 31 % (L = H2), and this was related to a weakening of the Fe(dxz)-S(Px) π bond covalency. XAS data have recently been obtained on hs and ls wt Cyt P450cam by the same group,(44) showing a total covalency of 41 % (hs) and 69 % (ls), respectively, indicating that [Fe(OEP)(S-H1) is the most appropriate model for wt hs ferric Cyt P450cam.
Another possibility to access changes in Fe-S covalency is via the energy and intensity of the S → Fe CT transitions, which are best observed by low-temperature MCD spectroscopy. However, application of this method is hampered by the fact that (a) low-temperature MCD spectra for ferric wt Cyt P450cam and Cys pocket mutants have not been reported, and (b) assignment of these spectra constitutes a significant challenge. In this study, we demonstrate how low-temperature MCD spectroscopy is applied to obtain assignments of the optical spectra of hs ferric Cyt P450cam, including identification of S → Fe CT bands. Since ferric wt Cyt P450cam exists as a mixture of hs and ls at low temperature, the pure hs mutant Q360P is used to assign the optical spectra of the hs form. The spectra of hs ferric hemes are generally complicated due to the hs d5 configuration of iron, which allows for a multitude of heme/axial ligand → Fe(III) CT transitions. The assignments of the observed features for Q360P are based on (a) the experimentally determined polarizations of the transitions obtained from VTVH measurements, (b) oscillator strengths and UV–vis/MCD intensity ratios, (c) the energy sequence and polarizations of the transition as determined from TD-DFT calculations (BP86/TZVP) on the active site model of Cyt P450cam shown in Scheme 1, and (d) comparison to the assignments for ferric hs [Fe(TPP)Cl] determined previously.(61) It is noted that while TD-DFT calculations aid in assigning the experimentally observed bands, there is a limit to the accuracy of the TD-DFT results. For instance, heme π → π* transitions are greatly overestimated in energy; for example, the Soret band is predicted to be ∼6000 cm-1 higher in energy compared to the experimental value (Figure S7). Hence, absolute TD-DFT energies cannot be used for spectral assignments. Three major types of electronic transitions are observed in the optical spectra of hs ferric hemes: porphyrin π → π* transitions (xy-polarized), porphyrin to Fe CT (P → Fe) transitions (almost all xy-polarized), and axial ligand (sulfur) to iron CT (S → Fe) transitions (z-polarized) (cf. Scheme 2). Many of these transitions involve orbitals that are strongly mixed, due to the low symmetry of heme b, which is an additional complication. Peculiarities of the low-temperature MCD spectra of Q360P that are addressed in this study include (a) the low MCD intensity of the Soret band, (b) the position of the Q and Qv bands, (c) the nature of the intense, broad band to higher energy of the Soret band, and (d) the location of the P → Fe and S → Fe CT transitions.
Table 5 shows a comparison of the MCD spectral features and assignments of the ferric hs complexes [Fe(TPP)Cl] and Cyt P450cam (Q360P). The Soret band of hs Q360P is split into three components that give rise to a -Δε/+Δε/-Δε pattern similar to that observed for [Fe(TPP)Cl].(61) The three component feature is a result of selective mixing of the negative component of the Soret band with another porphyrin (π → π*) or Fe → P CT excited state, as discussed before.(61) This seems to be a characteristic feature of hs ferric hemes. Interestingly, the intensity of the Soret band in Q360P (and hs Cyt P450cam in general) is quite weak (cf. Figures 5 and 8): in Q360P the positive Soret band is 141 mol-1 cm-1 T-1 compared to 479 mol-1 cm-1 T-1 in ferric hs [Fe(TPP)Cl].(61) The lowering of the Soret band intensity in Cyt P450cam is first of all a result of the redistribution of its intensity among bands 7 – 12, in agreement with the TD-DFT results. Another effect of the low symmetry of heme b is an increase in the energy splitting between the two components of the Eg<82/83> LUMO (775 cm-1 from the BP86/TZVP calculations), which reduces spin-orbit coupling matrix elements between the corresponding Eu (in D4h) excited state components.(61) This lowers the MCD C-term intensity that depends on excited state spin-orbit coupling. The presence of the axial thiolate further induces electronic anisotropy as is evident from the splitting of the (β) dxz and dyz orbitals, which is 3640 cm-1. In comparison, the higher (four-fold) symmetry of [Fe(TPP)Cl] is reflected by a smaller splitting of the Eg<82/83> (420 cm-1) and (β)dxz and dyz orbitals (770 cm-1; as obtained from B3LYP/LanL2DZ*).(61) Correspondingly, the EPR spectra of [Fe(TPP)Cl] and Q360P show axial and rhombic character, respectively.(39) The Q and Qv excited states of porphyrins have Eu symmetry (in D4h), similar to the Soret band, and hence, give rise to weak pseudo-A signals in the low temperature MCD spectra of metalloporphyrins. For Q360P, the bands at 18604 cm-1 (band 5) and 19539 cm-1 (band 6) are assigned to the two components of Q, based on the energies of the features relative to the Soret band, their expected in-plane polarization, and the observed pseudo-A signal of bands 5 and 6. In addition, the high temperature MCD spectra of wt ferric Cyt P450cam show a derivative band with a zero crossing at ∼540 nm (18520 cm-1),(3, 48, 54) which is very close in energy to band 5. Interestingly, the Qv band is not observed, but can be estimated to occur ∼1300 cm-1 to higher energy of Q;(81) in this case at ∼19900/20800 cm-1. Interestingly, in the case of the hs model complex [Fe(TPP)Cl], the room-temperature MCD spectra show the Qv band as a weak feature at 20202 cm-1, but Q is not observed.(61) We believe that the appearance of the Q band in the low temperature MCD spectrum of Q360P is a result of the low symmetry of the complex, causing incomplete cancellation of the individual transition dipole moments of the A1u → Eg and A2u → Eg components of Q (cf. ref. (61)).
Table 5. Comparison of transition energies and assignments of ferric Cyt P450cam (Q360P) and [Fe(TPP)Cl](61).
| Q360P | [Fe(TPP)Cl] | ||||
|---|---|---|---|---|---|
| Energy, cm-1 | Δε | Assignment | Energy, cm-1 | Δε | Assignment |
| 15222 | 8 | ||||
| 15909 | 31 | CT(1) | 15000 | 24 | CT(1), π → π* (0) |
| 16919 | -23 | CT(2), P → Fe CT | 15600 | -13 | CT(1), π → π* (0) |
| 16444 | -28 | CT(Cl,π) | |||
| 17881 | -32 | CT(S,π) | 17700 | -38 | CT(Cl,π) |
| 18336 | -18 | P → Fe CT | |||
| 18604 | -47 | Q band | |||
| 19539 | 37 | ||||
| 20150 | -22 | Qv | |||
| 20539 | 83 | π → π* (1), P → Fe CT | |||
| 21807 | 112 | CT(3), π → π* (3) | |||
| 22685 | 28 | π → π* (3) | 22232 | -101 | Soret |
| 23731 | -111 | Soret | 23266 | -226 | Soret |
| 24859 | 141 | 24269 | 479 | Soret | |
| 25618 | -97 | Soret, CT(3), CT(S,σ) | |||
| 28570 | -179 | CT(S,σ), P → Fe CT | 26181 | 214 | CT(Cl,σ,π), P → Fe CT |
| 31042 | 47 | ||||
| 26472 | -162 | π → π* (0), π → π* (2), P → Fe CT | |||
| 28098 | -48 | π → π* (2) | |||
| 29819 | 5 | π → π* (2) | |||
The energy sequence of CT transitions is quite similar in hs [Fe(TPP)Cl] and Cyt P450cam as shown in Table 5. Both complexes show a low energy pseudo-A signal at 15000-16000 cm-1 of P(π) → dxz,dyz character, followed by axial ligand(π) → dxz,dyz CT, mixed with inner porphyrin transitions. The latter transitions are located in the lower energy region of the optical spectra (16444 cm-1 for [Fe(TPP)Cl] and 17888 cm-1 for Q360P). Importantly, to higher energy of the Soret band, both systems show a strongly out-of-plane polarized feature that corresponds to a Cl(σ,π) → Fe (26181 cm-1) in the model complex and a S(σ) → Fe (28570 cm-1) CT transition in Cyt P450cam. This band shows remarkably large MCD intensity in Cyt P450cam, indicating that this feature has significant metal contribution, and hence, corresponds to a CT transition. Based on the TD-DFT results, other significant contributions to this feature are of P → Fe CT (in particular, , , , and ) character.
The most apparent difference between the MCD spectra of ls Y96W and hs Q360P is that the former has a Soret MCD C-term intensity that is ∼10 times larger (-1076 vs -111 M-1 cm-1 T-1, respectively). This finding reflects differences in spin orbit coupling (SOC), where the ls form gains additional C-term intensity from SOC between the ground state and low-lying ligand field excited states, whereas in the hs form, no such low-lying states are available (see ref (61)). Hence, in the latter case, all MCD intensity results from excited state SOC. This increase in Soret intensity in ls ferric metalloporphyrins is a general effect that has been observed in many cases, including horse heart cytochrome c,(94) the metmyoglobin-CN- complex,(94) cytochrome c oxidase,(95) and the cytochrome c oxidase-CN- complex.(95) Most importantly, the broad feature (band 13) assigned to a CT(S,σ) transition in hs Cyt P450cam is absent in the ls form.
The energies of the S(σ) → Fe CT bands observed at 28724, 28620, and 28570 cm-1 for wt, L358P, and Q360P Cyt P450cam, respectively (Figure 12), have implications for the effect of H-bonding on the Fe-S bond. In going from wt to L358P and Q360P, one hydrogen bond to sulfur is removed. In addition, Q360P is also lacking an additional hydrogen bond to the cysteine backbone. The removal of hydrogen bond(s) from the thiolate group of cysteine increases the electron density on sulfur, and thus, the sulfur donor strength. This, in turn, affects the strength of the Fe-S bond. DFT calculations on six-coordinate ferric nitrosyl model complexes [Fe(P)(SR)(NO)] support this idea, and show a correlated increase in the Fe-S force constant and a decrease in Fe-S bond distance upon stepwise removal of strong NH-S(thiolate) hydrogen bonds in model complexes.(93) The reason for this is an increase of S(pz) in energy as H-bonds are removed, leading to an increase of the Fe-S covalency, and hence, bond strength. This effect is quite pronounced in the model complexes (see above).(45) There is little doubt that L358, G359, and Q360 form amide hydrogen bonds to the sulfur donor atom of C357 in Cyt P450cam; the real question is how strong these interactions are, and how strongly they influence the Fe-S bond strength. One way to detect this change is the energy of the S → Fe CT transition: as the S(pz) orbital increases in energy, the energetic separation between the corresponding bonding and antibonding combinations of S(pz) and dz2 of iron(III) decreases and this shifts the corresponding S(σ) → Fe CT transition to lower energy. This notion is consistent with the trend observed in going from wt to L358P and to Q360P. However, this effect is quite small, in agreement with the small changes in Fe-S vibrational energies observed from Raman spectroscopy.(39) One could argue that the small changes are due to (a) rearrangement of the Cys pocket to strengthen the remaining H-bonds, or (b) incorporation of water in the Cys pocket in the mutants. However, the crystal structure of ferrous L358P indicates that this is likely not the case (Figure S6).(96)
Compared to the model complex data, there are two possible conclusions that can be drawn from this: (a) this could indicate that the amide N-H⋯S hydrogen bonds in the enzyme are generally weak, or (b) this could mean that hydrogen bonds from L358 and Q360 are weak, whereas that from G359 could then be much stronger. From crystallography,(96) the G359 H-bond is indeed clearly shorter than the other two, so it is possible that G359 provides the H-bond that is most significant for the Fe-S bond strength. This point requires further study. Our work therefore implies, based on the small changes in the S(σ) → Fe CT transition energy between wt, L358P, and Q360P, and corresponding small changes in ν(Fe-S), that the roles of the hydrogen bonds from L358 and Q360 are not primarily for fine-tuning the sulfur donor strength. In contrast, we believe that the primary role of these H-bonds is for the stabilization of the thiolate ligand to prevent protonation (or reaction with diatomics) that would lead to Cyt P420 formation, and for positioning the cysteinate for proper coordination to the iron center. Consistent with this, note that the removal of the amide proton in variant Q360P resulted in partial conversion of Cyt P450 to the P420 form, as evidenced by the large fraction of a 422 nm species in the ferrous-CO form of the enzyme prior to purification.(39) In this respect, it should also be noted that too strong N-H⋯S hydrogen bonds would be of disadvantage for Cyt P450 catalysis. In fact, weak H-bonds, leading to a strong Fe-S bond, are of key importance for efficient O-O bond cleavage and compound I formation, according to the push effect.(19) This finding disagrees with several hypotheses that point out the NH-S hydrogen bonds promote both the stabilization of the thiolate ligand and control the redox potential of the heme.(97, 98) In addition, the hydrogen bonds could be involved in structural changes upon putidaredoxin binding during P450cam catalysis. Based on these results, the observed changes in redox potential between wt and the Cys pocket mutants then likely relate to a destabilization of the ferrous form of the protein as a major source for the observed changes, and/or to structural changes in the heme conformation in the mutants.
Summary and Conclusions
Magnetic circular dichroism measurements were performed on wt Cyt P450cam, and the variants Q360P (pure hs; two hydrogen bonds to cysteine removed), L358P (one hydrogen bond to cysteine removed), and Y96W (pure ls). The low temperature MCD spectrum of Q360P, coupled to VTVH measurements and TD-DFT calculations, allowed for the assignment of the optical spectra of hs Cyt P450cam. This allowed the identification of the inner porphyrin (π → π*), P → Fe CT and S → Fe CT transitions. From these data, the S(σ) → Fe CT band (CT(S,σ)) was identified at 28570 cm-1, close to the corresponding CT(Cl,σ)) in hs [Fe(TPP)Cl].(61) The CT(S,σ) energy serves as a marker for the Fe-S interaction in Cyt P450s. This prominent band occurs at 28570, 28620, and 28724 cm-1 in Q360P, L358P, and wt, respectively. Importantly, the small change in the S(σ) → Fe CT transition energy implies that the amide H-bonds from L358 and Q360 to the proximal thiolate are weak. Hence, the role of these H-bonds lies less in fine-tuning of the Fe-S bond strength, but mostly in stabilizing the thiolate donor to prevent protonation (or reaction with diatomics), and hence, P420 formation, as well as in positioning the sulfur atom relative to heme. The remaining G359 H-bond is therefore potentially responsible for fine-turning the Fe-S interaction. Note that weak H-bonds, leading to strong Fe-S interactions, are advantageous for the catalytic role of Cys357 in the formation of Compound I. Hence, application of weak H-bonds constitutes the best compromise between stabilization/positioning of the proximal Cys and strong S → Fe donation advantageous for catalysis.
Supplementary Material
Acknowledgments
Professor F. Neese (University of Bonn) is acknowledged for help with the large scale TD-DFT calculations.
Abbreviations
- Cyt P450cam
Cytochrome P450 enzyme with camphor as the natural substrate
- wt
wild-type
- C357
cysteine residue at position 357
- L358
leucine residue at position 358
- G359
glycine residue at position 359
- Q360
glutamine residue at position 360
- “Cys pocket”
amino acids L358, G359, and Q360 that form a hydrogen bonding network to C357
- L358P
variant of wild-type where proline is substituted for leucine at position 358
- Q360P
variant of wild-type where proline is substituted for glutamine at position 360 (also the pure high-spin variant)
- Y96W
variant of wild-type where tryptophan is substituted for tyrosine at position 96 (also the pure low-spin variant)
- MCD
magnetic circular dichroism
- TD-DFT
time-dependent density functional theory
- VTVH
variable-temperature variable-field
- CT
charge-transfer
- hs
high-spin
- ls
low-spin
- PDB
protein data bank
Footnotes
This work is supported by the National Science Foundation (NSF # CHE-0846235) to N.L. and the National Institutes of Health (NIH # GM20877) to D. P. B.
In the spin-unrestricted scheme, two sets of molecular orbitals (MOs) are generated: one for the spin-up (α) and one for the spin-down (β) electrons. In the case of hs Fe(III), the d-electron configuration is therefore [α–d]5. Hence, the α-d orbitals are not involved in ligand to metal charge transfer (CT) transitions as they are fully occupied; instead, α–MOs only involve inner porphyrin transitions. On the other hand, the β-d orbitals are unoccupied and hence, allow for CT transitions into all of these orbitals, including porphyrin to metal and axial ligand to metal transitions.
Interestingly, the TD-DFT calculations seem to indicate that bands 10-12 only have 7, 9 and 2 % π → π* (0) character, which is unexpected for the Soret band. However, one should note that these low values are a clear underestimate. Since the obtained MOs from the DFT calculations are strongly mixed and complex, many other single excitations that contribute to these excited states also exhibit some degree of π → π* (0) excited state character, which somewhat complicates the analysis. This is reflected by the oscillator strength of band 11 (0.76), which corresponds well to the sum of oscillator strengths of the associated TD-DFT features at 30608 and 30663 cm-1, which is ∼0.5.
Supporting Information: Cartesian coordinates of the applied active site model of Cyt P450cam, detailed assignments of the electronic spectra of Q360P, selected VTVH curves, VTVH calibration curves, the resonance Raman spectra of wt enzyme, the MCD spectrum of L358P, the crystal structure of ferrous L358P, and the theoretical (TD-DFT) absorption spectrum are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Ortiz de Montellano PR. Cytochrome P450: Structure, Mechanism, and Biochemistry. 3rd. Kluver Academic/Plenum Publishers; New York: 2005. [Google Scholar]
- 2.Meunier B, De Visser SP, Shaik S. Mechanism of oxidation reactions catalyzed by Cytochrome P450 enzymes. Chem Rev. 2004;104:3947–3980. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- 3.Dawson JH, Sono M. Cytochrome P-450 and chloroperoxidase: thiolate-ligated heme enzymes. Spectroscopic determination of their active-site structures and mechanistic implications of thiolate ligation. Chem Rev. 1987;87:1255–1276. [Google Scholar]
- 4.Denisov IG, Makris TM, Sligar SG, Schlichting I. Structure and chemistry of Cytochrome P450. Chem Rev. 2005;105:2253–2278. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 5.Sato R, Omura T. Cytochrome P-450. Academic; New York: 1978. [Google Scholar]
- 6.White KA, Marletta MA. Nitric oxide synthase is a cytochrome P-450 type hemoprotein. Biochemistry. 1992;31:6627–6631. doi: 10.1021/bi00144a001. [DOI] [PubMed] [Google Scholar]
- 7.Stuehr DJ. Structure-Function Aspects in the Nitric Oxide Synthases. Annu Rev Pharmacol Toxicol. 1997;37:339–359. doi: 10.1146/annurev.pharmtox.37.1.339. [DOI] [PubMed] [Google Scholar]
- 8.Bredt DS, Snyder SH. Nitric Oxide: A Physiologic Messenger Molecule. Annu Rev Biochem. 1994;63:175–195. doi: 10.1146/annurev.bi.63.070194.001135. [DOI] [PubMed] [Google Scholar]
- 9.Coon MJ, Vatsis KP. Polycyclic hydrocarbons and cancer. Vol. 1. Academic; New York: 1978. [Google Scholar]
- 10.Lewis DFV. Physical methods in the study of the active site geometry of cytochromes P450. Drug Metab Rev. 1986;17:1. doi: 10.3109/03602538608998285. [DOI] [PubMed] [Google Scholar]
- 11.Vermeulen NPE. Prediction of drug metabolism: the case of cytochrome P450 2D6. Curr Top Med Chem. 2003;3:1227–1239. doi: 10.2174/1568026033451998. [DOI] [PubMed] [Google Scholar]
- 12.Durr UHN, Waskell L, Ayyalusamy R. The cytochromes P450 and b5 and their reductases - promising targes for structural studies by advanced solid-state NMR spectroscopy. Biochem Biophys Acta. 2007;1768:3235–3259. doi: 10.1016/j.bbamem.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 13.Guengerich FP. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem Res Toxicol. 2001;14:611–650. doi: 10.1021/tx0002583. [DOI] [PubMed] [Google Scholar]
- 14.Meunier B, Bernadou J. Active iron-oxo and iron-peroxo species in cytochromes P450 and peroxidases; oxo-hydroxo tautomerism with water-soluble metalloporphyrins. Struct Bonding. 2000;97:1–35. [Google Scholar]
- 15.Ortiz de Montellano PR, De Voss JJ. Oxidizing species in the mechanism of cytochrome P450. Nat Prod Rep. 2002;19:477–493. doi: 10.1039/b101297p. [DOI] [PubMed] [Google Scholar]
- 16.Yoshizawa K. Theoretical study on kinetic isotope effects in the C--H bond activation of alkanes by iron-oxo complexes. Coord Chem Rev. 2002;226:251–259. [Google Scholar]
- 17.Kamachi T, Yoshizawa K. A theoretical study on the mechanism of camphor hydroxylation by compound I of Cytochrome P450. J Am Chem Soc. 2003;125:4652–4661. doi: 10.1021/ja0208862. [DOI] [PubMed] [Google Scholar]
- 18.Mayer JM. Biomimetic Oxidations Catalyzed by Transition Metal Complexes. Imperial College Press; London: 2000. [Google Scholar]
- 19.Dawson JH, Holm RH, Trudell JR, Barth G, Linder RE, Bunnenberg E, Djerassi C, Tang SC. Magnetic circular dichroism studies. 43. Oxidized cytochrome P-450. Magnetic circular dichroism evidence for thiolate ligation in the substrate-bound form. Implications for the catalytic mechanism. J Am Chem Soc. 1976;98:3707–3709. doi: 10.1021/ja00428a054. [DOI] [PubMed] [Google Scholar]
- 20.Dawson JH. Probing structure-function relations in heme-containing oxygenases and peroxidases. Science. 1988;240:433–439. doi: 10.1126/science.3358128. [DOI] [PubMed] [Google Scholar]
- 21.Sabat J, Stuehr DJ, Yeh SR, Rousseau DL. Characterization of the proximal ligand in P420 form of inducible nitric oxide synthase. J Am Chem Soc. 2009;131:12186–12192. doi: 10.1021/ja901016a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoshioka S, Takahashi S, Hori H, Ishimori K, Morishima I. Proximal cysteine residue is essential for the enzymatic activities of cytochrome P450cam. Eur J Biochem. 2001;268:252–259. doi: 10.1046/j.1432-1033.2001.01872.x. [DOI] [PubMed] [Google Scholar]
- 23.Matsui TNS, Ishimori K, Watanabe Y, Morishima I. Preparation and reactions of myoglobin mutants bearing both proximal cysteine ligand and hydrophobic distal cavity: protein models for the active site of P450. Biochemistry. 1996;35:13118–13124. doi: 10.1021/bi960459z. [DOI] [PubMed] [Google Scholar]
- 24.Harris DL, Loew GH. Proton-assisted pathway to formation of the catalytically active, ferryl species of P450eryF. J Am Chem Soc. 1996;118:6377–6387. doi: 10.1021/ja954101m. [DOI] [PubMed] [Google Scholar]
- 25.Harris DL, Loew GH. Theoretical investigation of the proton assisted pathway to formation of cytochrome P450 compound I. J Am Chem Soc. 1998;120:8941–8948. [Google Scholar]
- 26.Green MT. Roles of the axial ligand in determining the spin state of resting cytochrome P450. J Am Chem Soc. 1998;120:10772–10773. [Google Scholar]
- 27.Green MT. Evidence for sulfur-based radicals in thiolate compound I intermediates. J Am Chem Soc. 1999;121:7939–7340. [Google Scholar]
- 28.De Visser SP, Ogliaro F, Harris N, Shaik S. Multi-state epoxidation of ethene by Cytochrome P450: a Quantum Chemical Study. J Am Chem Soc. 2001;123:3037–3047. doi: 10.1021/ja003544+. [DOI] [PubMed] [Google Scholar]
- 29.Altun A, Kumar D, Neese F, Thiel W. Multireference Ab initio quantum mechanics/molecular mechanics study on intermediates in the catalytic cycle of Cytochrome P450cam. J Phys Chem A. 2008;112:12904–12910. doi: 10.1021/jp802092w. [DOI] [PubMed] [Google Scholar]
- 30.Schoneboom JC, Neese F, Thiel W. Toward Identification of the Compound I Reactive Intermediate in Cytochrome P450 Chemistry: A QM/MM Study of Its EPR and Mössbauer Parameters. J Am Chem Soc. 2005;127:5840–5853. doi: 10.1021/ja0424732. [DOI] [PubMed] [Google Scholar]
- 31.Ogliaro F, Harris N, Cohen S, Filatov M, de Visser SP, Shaik S. A model “rebound” mechanism of hydroxylation by cytochrome P450: stepwise and effectively concerted pathways, and their reactivity patterns. J Am Chem Soc. 2000;122:8977–8989. [Google Scholar]
- 32.Kumar D, de Visser SP, Shaik S. How does product isotope effect prove the operation of a two-state “rebound” mechanism in C-H hydroxylation by Cytochrome P450? J Am Chem Soc. 2003;125:13024–13025. doi: 10.1021/ja036906x. [DOI] [PubMed] [Google Scholar]
- 33.Filatov M, Harris N, Shaik S. On the “rebound” mechanism of alkane hydroxylation by cytochrome P450: Electronic structure of the intermediate and the electron transfer character in the rebound step. Angew Chem Int Ed. 1999;38:3510–3512. doi: 10.1002/(sici)1521-3773(19991203)38:23<3510::aid-anie3510>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 34.de Visser SP, Ogliaro F, Sharma PK, Shaik S. Hydrogen Bonding Modulates the Selectivity of Enzymatic Oxidation by P450: Chameleon Oxidant Behavior by Compound I. Angew Chem Int Ed. 2002;41:1947–1951. [PubMed] [Google Scholar]
- 35.Poulos TL, Finzel BC, Howard AJ. High-resolution crystal structure of cytochrome P450cam. J Mol Biol. 1987;195:687–700. doi: 10.1016/0022-2836(87)90190-2. [DOI] [PubMed] [Google Scholar]
- 36.Cupp-Vickery J, Poulos TL. Structure of cytochrome P450 eryF: an enzyme involved in erythromycin biosynthesis. Nat Struct Biol. 1995;2:144–153. doi: 10.1038/nsb0295-144. [DOI] [PubMed] [Google Scholar]
- 37.Ravichandran KG, Boddupalli SS, Hasemann CA, Peterson J, Deisenhofer J. Crystal structure of hemoprotein domain of P450BM-3, a prototype for microsomal P450's. Science. 1993;261:731–736. doi: 10.1126/science.8342039. [DOI] [PubMed] [Google Scholar]
- 38.Hasemann CA, Ravichandran KG, Boddupalli SS, Peterson J, Deisenhofer J. Structure and function of cytochromes P450: a comparative analysis of three crystal structures. Structure. 1995;3:41–62. doi: 10.1016/s0969-2126(01)00134-4. [DOI] [PubMed] [Google Scholar]
- 39.Yoshioka S, Tosha T, Takahashi S, Ishimori K, Hori H, Morishima I. Roles of the Proximal Hydrogen Bonding Network in Cytochrome P450cam-Catalyzed Oxygenation. J Am Chem Soc. 2002;124:14571–14579. doi: 10.1021/ja0265409. [DOI] [PubMed] [Google Scholar]
- 40.Ueno T, Nishikawa N, Moriyama S, Adachi S, Lee K, Okamura T, Ueyama N, Nakamura A. Role of the invariant peptide fragment forming NH⋯S hydrogen bonds in the active site of cytochrome P-450 and chloroperoxidase: Synthesis and properties of Cys-containing peptide Fe(III) and Ga(III) (octaethylporphinato) complexes as models. Inorg Chem. 1999;38:1199–1210. doi: 10.1021/ic980710u. [DOI] [PubMed] [Google Scholar]
- 41.Ueyama N, Nishikawa N, Yamada Y, Okamura T, Nakamura A. Cytochrome P-450 model (porphinato)(thiolato)iron(III) complexes with single and double NH⋯S hydrogen bonds at the thiolate site. J Am Chem Soc. 1996;118:12826–12827. [Google Scholar]
- 42.Ueyama N, Nishikawa N, Yamada Y, Okamura T, Oka S, Sakurai H, Nakamura A. Synthesis and Properties of Octaethylporphinato(arenethiolato)iron(III) Complexes with Intramolecular NH⋯S Hydrogen Bond: Chemical Function of the Hydrogen Bond. Inorg Chem. 1998;37:2415–2421. [Google Scholar]
- 43.Suzuki N, Higuchi T, Urano Y, Kikuchi K, Uekusa H, Ohashi Y, Uchida T, Kitagawa T, Nigano T. Novel Iron Porphyrin-Alkanethiolate Complex with Intramolecular NH--S Hydrogen Bond: Synthesis, Spectroscopy, and Reactivity. J Am Chem Soc. 1999;121:11571–11572. [Google Scholar]
- 44.Dey A, Jiang Y, Ortiz de Montellano PR, Hodgson KO, Hedman B, Solomon EI. S K-edge XAS and DFT calculations on cytochrome P450: covalent and ionic contributions to the cysteine-Fe bond and their contribution to reactivity. J Am Chem Soc. 2009;131:7869–7878. doi: 10.1021/ja901868q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dey A, Okamura T, Ueyama N, Hedman B, Hodgson KO, Solomon EI. Sulfur K-Edge XAS and DFT Calculations on P450 Model Complexes: Effects of Hydrogen Bonding on Electronic Structure and Redox Potentials. J Am Chem Soc. 2005;127:12046–12053. doi: 10.1021/ja0519031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sligar SG. Coupling of spin, substrate, and redox equilibriums in cytochrome P450. Biochemistry. 1976;15:5399–5406. doi: 10.1021/bi00669a029. [DOI] [PubMed] [Google Scholar]
- 47.Raag R, Poulos TL. The structural basis for substrate-induced changes in redox potential and spin equilibrium in cytochrome P-450CAM. Biochemistry. 1989;28:917–922. doi: 10.1021/bi00428a077. [DOI] [PubMed] [Google Scholar]
- 48.Shimizu T, Iizuka T, Shimada H, Ishimura Y, Nozawa T, Hatano M. Magnetic circular dichroism studies of Cytochrome P450cam characterization of axial ligands of ferric and ferrous low-spin complexes. Biochem Biophys Acta. 1981;670:341–354. doi: 10.1016/0005-2795(81)90106-9. [DOI] [PubMed] [Google Scholar]
- 49.Dawson JH, Andersson LA, Sono M. Spectroscopic investigations of ferric Cytochrome P450cam ligand complexes. J Biol Chem. 1982;257:3606–3617. [PubMed] [Google Scholar]
- 50.Tsai R, Yu CA, Gunsalus IC, Peisach J, Blumberg W, Orme-Johnson WH, Beinert H. Spin-state changes in Cytochrome P-450cam on binding of specific substrates. PNAS. 1970;66:1157–1163. doi: 10.1073/pnas.66.4.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lipscomb JD. Electron paramagnetic resonance detectable states of cytochrome P-450cam. Biochemistry. 1980;19:3590–3599. doi: 10.1021/bi00556a027. [DOI] [PubMed] [Google Scholar]
- 52.Masuya F, Tsubaki M, Makino R, Hori H. EPR studies on the photoproducts of ferric cytochrome P450cam (CYP101) nitrosyl complexes: Effects of camphor and its analogues on ligandbound structures. J Biochem. 1994;116:1146–1152. doi: 10.1093/oxfordjournals.jbchem.a124641. [DOI] [PubMed] [Google Scholar]
- 53.Champion PM, Stallard BR, Wagner GC, Gunsalus IC. Resonance Raman detection of an Fe-S bond in Cyt P450cam. J Am Chem Soc. 1982;104:5469–5472. [Google Scholar]
- 54.Vickery LE, Salmon A, Sauer K. Magnetic circular dichroism studies on microsomal aryl hydrocarbon hydroxylase: comparison of cytochrome b5 and Cytochrome P450cam. Biochim Biophys Acta. 1975;386:87–98. doi: 10.1016/0005-2795(75)90249-4. [DOI] [PubMed] [Google Scholar]
- 55.Stephens PJ. Magnetic Circular Dichroism. Annu Rev Phys Chem. 1974;25:201–232. [Google Scholar]
- 56.Stephens PJ. Magnetic Circular Dichroism. Adv Chem Phys. 1976;35:197–264. [Google Scholar]
- 57.Landrum GA, Ekberg CA, Whittaker JW. A ligand field model for MCD spectra of biological cupric complexes. Biophys J. 1995;69:674–689. doi: 10.1016/S0006-3495(95)79943-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kirk ML, Peariso K. Recent applications of MCD spectroscopy to metalloenzymes. Curr Opin Chem Biol. 2003;7:220–227. doi: 10.1016/s1367-5931(03)00034-6. [DOI] [PubMed] [Google Scholar]
- 59.McMaster J, Carducci MD, Yi-Shan Y, Solomon EI, Enermark JH. Electronic Spectral Studies of Molybdenyl Complexes. 2. MCD Spectroscopy of [MoOS4]- Centers. Inorg Chem. 2001;40:687–702. doi: 10.1021/ic0005846. [DOI] [PubMed] [Google Scholar]
- 60.Decker A, Rohde JU, Klinker EJ, Wong SD, Que L, Jr, Solomon EI. Spectroscopic and Quantum Chemical Studies on Low-Spin FeIVO Complexes: Fe–O Bonding and Its Contributions to Reactivity. J Am Chem Soc. 2007;129:15983–15996. doi: 10.1021/ja074900s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Paulat F, Lehnert N. Detailed Assignment of the Magnetic Circular Dichroism and UV-Vis Spectra of Five-Coordinate High-Spin Ferric [Fe(TPP)(Cl)] Inorg Chem. 2008;47:4963–4976. doi: 10.1021/ic8002838. [DOI] [PubMed] [Google Scholar]
- 62.Paulat F, Praneeth VKK, Näther C, Lehnert N. Quantum Chemistry-Based Analysis of the Vibrational Spectra of Five-Coordinate Metalloporphyrins [M(TPP)Cl] Inorg Chem. 2006;45:2835–2856. doi: 10.1021/ic0510866. [DOI] [PubMed] [Google Scholar]
- 63.Spolitak T, Dawson JH, Ballou DP. Reaction of ferric Cytochrome P450cam with peracids. J Biol Chem. 2005;280:20300–20309. doi: 10.1074/jbc.M501761200. [DOI] [PubMed] [Google Scholar]
- 64.Lehnert N, DeBeer George S, Solomon EI. Recent advances in bioinorganic spectroscopy. Curr Opin Chem Biol. 2001;5:176–187. doi: 10.1016/s1367-5931(00)00188-5. [DOI] [PubMed] [Google Scholar]
- 65.Becke AD. Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 66.Lee C, Yang W, Parr RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B (Condensed Matter) 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 67.Wadt WR, Hay PJ. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J Chem Phys. 1985;82:270–283. [Google Scholar]
- 68.Wadt WR, Hay PJ. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J Chem Phys. 1985;82:284–298. [Google Scholar]
- 69.Wadt WR, Hay PJ. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J Chem Phys. 1985;82:299–311. [Google Scholar]
- 70.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Jr, M JA, Vreven T, Kudin KN, Burant JC, Milliam JM, Lyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Danneberg JJ, Zakrewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03. Gaussian, Inc.; Pittsburgh, PA: 2003. [Google Scholar]
- 71.Nemykin VN, Hadt RG, Belosludov RN, Mizuseki H, Kawazoe Y. Influence of molecular geometry, exchange-correlation functional, and solvent effects in the modeling of vertical excitation energies in phthalocyanines using time-dependent density functional theory (TDDFT) and polarized continuum model TDDFT methods: Can modern computational chemistry methods explain experimental controversies? J Phys Chem A. 2007;111:12901–12913. doi: 10.1021/jp0759731. [DOI] [PubMed] [Google Scholar]
- 72.Peralta GA, Seth M, Ziegler T. Magnetic circular dichroism of porphyrins containing M = Ca, Ni, and Zn. A computational study based on time-dependent density functional theory. Inorg Chem. 2007;46:9111–9125. doi: 10.1021/ic7008545. [DOI] [PubMed] [Google Scholar]
- 73.De Luca G, Romeo A, Scolaro LM, Ricciardi G, R A. Sitting-Atop Metallo-Porphyrin Complexes: Experimental and Theoretical Investigations on Such Elusive Species. Inorg Chem. 2009;48:8493–8507. doi: 10.1021/ic9012153. [DOI] [PubMed] [Google Scholar]
- 74.Becke AD. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 75.Perdew JP. Density-Functional Approximation for the Correlation-Energy of the. Inhomogeneous Electron-Gas. Phys Rev B (Condensed Matter) 1986;33:8822–8824. doi: 10.1103/physrevb.33.8822. [DOI] [PubMed] [Google Scholar]
- 76.Schaefer A, Horn H, Ahrichs R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J Chem Phys. 1992;97:2571–2577. [Google Scholar]
- 77.Schaefer A, Huber C, Ahrichs R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J Chem Phys. 1994;100:5829–5835. [Google Scholar]
- 78.Neese F. ORCA, ORCA 2.4 ed. Universität Bonn Bonn; Germany: 2004. [Google Scholar]
- 79.Neese F, Solomon EI. MCD C-Term Signs, Saturation Behavior, and Determination of Band Polarizations in Randomly Oriented Systems with Spin S ≥ 1/2. Applications to S = 1/2 and S = 5/2. Inorg Chem. 1999;38:1847–1865. doi: 10.1021/ic981264d. [DOI] [PubMed] [Google Scholar]
- 80.Spolitak T, Ballou DP. Personal Communication. 2008. [Google Scholar]
- 81.Gouterman M. In: The Porphyrins. Dolphin DH, editor. Academic; New York: 1979. pp. 1–156. [Google Scholar]
- 82.Gouterman M. Spectra of porphyrins. J Mol Spectrosc. 1961;6:138–163. [Google Scholar]
- 83.Gouterman M. Study of the effects of substitution on the absorption spectra of porphyrin. J Chem Phys. 1959;30:1139–1161. [Google Scholar]
- 84.Spiro TG, Li XY. Resonance Raman Spectra of Heme and Metalloproteins. Wiley; New York: 1988. [Google Scholar]
- 85.Rush TS, III, Kozlowski PM, Piffat CA, Kumble R, Zgierski MZ, Spiro TG. Computational Modeling of Metalloporphyrin Structure and Vibrational Spectra: Porphyrin Ruffling in NiTPP. J Phys Chem B. 2000;104:5020–5034. [Google Scholar]
- 86.Praneeth VKK, Näther C, Peters G, Lehnert N. Spectroscopic Properties and Electronic Structure of Five- and Six-Coordinate Iron(II) Porphyrin NO Complexes: Effect of the Axial N-Donor Ligand. Inorg Chem. 2006;45:2795–2811. doi: 10.1021/ic050865j. [DOI] [PubMed] [Google Scholar]
- 87.Lehnert N. Electron Paramagnetic Resonance and Low-Temperature Magnetic Circular Dichroism Spectroscopy of Ferrous Heme Nitrosyls. In: Ghosh A, editor. The Smallest Biomolecules: Diatomics and their Interactions with Heme Proteins. Elsevier; Amsterdam: 2008. pp. 147–171. [Google Scholar]
- 88.Cheesman MR, Greenwood C, Thomson AJ. Magnetic Circular Dichroism of Hemoproteins. Adv Inorg Chem. 1991;36:201–255. [Google Scholar]
- 89.Hanson L, Sligar SG, Gunsalus IC. Electronic Structure of Cytochrome P450. Croat Chem Acta. 1977;49:237–250. [Google Scholar]
- 90.Bangcharoenpaurpong O, Champion PM, Martinis SA, Sligar SG. Investigations of the resonance Raman excitation profiles of cytochrome P450cam. J Chem Phys. 1987;87:4273–4284. [Google Scholar]
- 91.Sono M, Roach MP, Coulter ED, Dawson JH. Heme-Containing Oxygenases. Chem Rev. 1996;96:2841–2887. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 92.Shokhireva TK, Berry RE, Uno E, Balfour CA, Zhang H, Walker FA. Electrochemical and NMR spectroscopic studies of distal pocket mutants of nitrophorin 2: Stability, structure, and dynamics of axial ligand complexes. PNAS. 2003;100:3778–3783. doi: 10.1073/pnas.0536641100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Paulat F, Lehnert N. Electronic Structure of Ferric Heme Nitrosyl Complexes with Thiolate Coordination. Inorg Chem. 2007;46:1547–1549. doi: 10.1021/ic070023f. [DOI] [PubMed] [Google Scholar]
- 94.Thomson AJ, Johnson AK. Magnetization curves of haemoproteins measured by low-temperature magnetic-circular-dichroism spectroscopy. Biochem J. 1980;191:411–420. doi: 10.1042/bj1910411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Thomson AJ, Johnson MK, Greenwood C, Gooding PE. A study of the magnetic properties of haem a3 in cytochrome c oxidase by using magnetic circular dichroism spectroscopy. Biochem J. 1981;193:687–697. doi: 10.1042/bj1930687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nagano S, Tosha T, Ishimori K, Morishima I, Poulos TL. Crystal structure of the cytochrome P450cam mutant that exhibits the same spectral perturbations induced by putidaredoxin bindin. J Biochem Chem. 2004;279:42844–42849. doi: 10.1074/jbc.M404217200. [DOI] [PubMed] [Google Scholar]
- 97.Poulos TL, Cupp-Vickery JLH. Cytochrome P450 Structure, Mechanism, and Biochemistry. Plenum Press; New York: 1995. [Google Scholar]
- 98.Poulos TL. The role of the proximal ligand in heme enzymes. J Biol Inorg Chem. 1996;1:356–359. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.