

Abstract
Summary: Accurate and complete mapping of short-read sequencing to a reference genome greatly enhances the discovery of biological results and improves statistical predictions. We recently presented RNA-MATE, a pipeline for the recursive mapping of RNA-Seq datasets. With the rapid increase in genome re-sequencing projects, progression of available mapping software and the evolution of file formats, we now present X-MATE, an updated version of RNA-MATE, capable of mapping both RNA-Seq and DNA datasets and with improved performance, output file formats, configuration files, and flexibility in core mapping software.
Availability: Executables, source code, junction libraries, test data and results and the user manual are available from http://grimmond.imb.uq.edu.au/X-MATE/.
Contact: n.cloonan@uq.edu.au; s.grimmond@uq.edu.au
Supplementary information: Supplementary data are available at Bioinformatics Online.
A major step in most deep-sequencing data analysis is mapping of the many million short reads to a reference genome, a process which is both computationally and strategically challenging (Trapnell and Salzberg, 2009). Effective downstream analysis relies heavily on the quality of this mapping (Pepke et al., 2009). Incomplete mapping discards precious data, and may limit the identification of biologically relevant information. Problems associated with inaccurate mapping can be more difficult to identify, but include bias in the quantification of differential gene expression, poor statistical power when detecting sequence variants, or greater noise and complexity when de-convoluting genomic re-arrangements.
We have previously described the recursive mapping strategy which combats the typical decrease in tag quality towards the 3′ end, and allows the alignment of short inserts with 3′ trailing adaptor sequence (Cloonan et al., 2009). In this strategy, tags are first mapped at full length, and then if unaligned, truncated then mapped again. Quality profiles for mapped tags indicate that those mapping at a shorter lengths do so because the truncated section contains poorer quality sequence (Fig. 1; Supplementary Fig. 1). The recursive approach increases the mapped data yield while maintaining exon mapping specificity (Supplementary Fig. 2), extracting as much value as possible from the experiment.
Fig. 1.
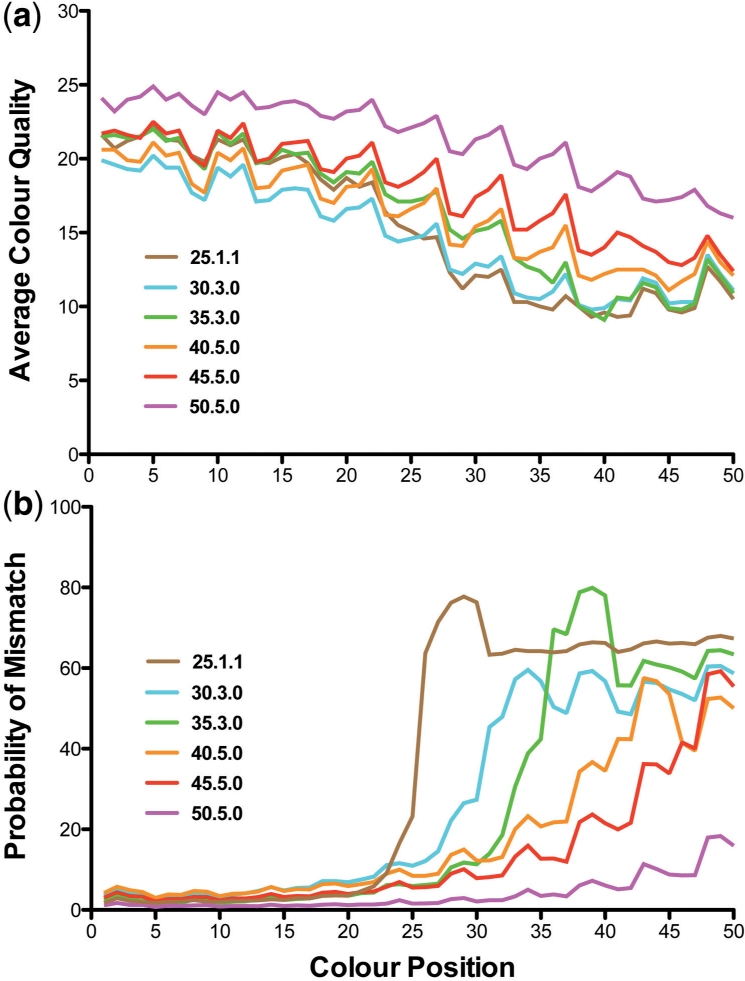
Average quality of each di-nucleotide (colour) over 50 nt SOLiD gDNA tags for five recursive runs, denoted using the code length.mismatches.mode. Mode 1 treats valid adjacent mismatches as a single mismatch (optimal for 25mer tags), and mode 0 as two mismatches. Although reads are truncated when mapped, the data shown represents the original length and quality. Truncated reads show a steeper decline in quality (a), and an increased probability of a mismatch in the truncated portion (b). The wobble particularly noticeable in (a) and also evident in (b) is a quality profile characteristic of the SOLiD five primer ligation cycle. The general trend is consistent in base-space data (Supplementary Fig. 1).
Although RNA-MATE was specifically designed for (and effectively limited to) colour-space stranded RNA-Seq data, the recursive approach works equally well on all sequencing datasets. To increase the utility of this software, and provide further enhancements, we have significantly revised and updated the RNA-MATE pipeline. Here, we present X-MATE, a system for flexible and comprehensive mapping of all deep-sequencing datasets. A comparison of features between RNA-MATE and X-MATE can be found in Table 1.
Table 1.
Feature matrix summarizing the major improvements between RNA-MATE and X-MATE
| Feature | RNA-MATE | X-MATE |
|---|---|---|
| Mapping Engine | Fixed | Flexible |
| Data type | Stranded RNA-Seq only | Stranded or unstranded RNA-Seq, gDNA, CAGE, ChIP-Seq, etc |
| Data Encoding | Colour-space only | Colour-space or base-space |
| Map to junctions | Required | Optional |
| Output formats | BED, Wiggle | BED, Wiggle, SAM |
| Multi-map rescue | MuMRescue | MuMRescueLite |
| Configuration | Cumbersome | Improved |
1 SUPPORT FOR MULTIPLE DATATYPES
X-MATE provides full support for both unstranded and stranded RNA- and DNA-derived datasets, including CAGE and ChIP-Seq.
The expected strand for a dataset can be easily configured, and when mapping stranded datasets, output files summarizing mapping results are created for each strand, allowing analysis and visualization of strand mapping specificity. When mapping unstranded data these results are combined into single output files. In both cases, libraries of known exon–exon junctions can be optionally used during alignment, either simultaneously with chromosome mapping, or after chromosome mapping.
2 ALTERNATIVE MAPPING ENGINES SUPPORTING BASE- AND COLOUR-SPACE
By default mapreads is used, but if licensed, ISAS can be chosen. ISAS is an extremely fast alignment system capable of performing exhaustive searches, or using a heuristic algorithm to greatly increase search speeds (see Supplementary File 1 for notes on performance). ISAS also implements a variable-length mapping mode and can be chosen in place of the X-MATE recursive mapping. The modular redesign of X-MATE allows for further alignment programs to be incorporated into the package if required. As both mapreads and ISAS can map in base-space, we have passed this additional capability to X-MATE, and it now maps base-space data natively (fasta format for mapreads, and fastq format for ISAS).
3 SAM FORMAT OUTPUT NOW SUPPORTED
To assist with interrogating the mapped data, X-MATE provides multiple data output formats (bed, and wiggle files) consistent with commonly used visualization tools such as UCSC genome browser (Kent et al., 2002) and powerful bioinformatic middleware platforms like Galaxy (Goecks et al., 2010). In addition, to provide for backward compatibility of previously mapped data (output in ‘collated’ format), and to assist in tertiary analysis, we have included a Java utility for data transformation of colour-space mapped data into Sequence Alignment/Map (SAM) format. SAM files can be converted to Binary Alignment/Map (BAM) files using Samtools (Li et al., 2009), and are becoming the default input file format for many genome browsers (e.g. the Integrative Genomics Viewer, IGV, http://www.broadinstitute.org/igv). Using SAM and BAM files, mapped data can be viewed in single nucleotide resolution, and can also be passed directly to downstream software for tertiary analysis such as transcript assembly, SNP calling, structural re-arrangement detection and more.
4 RESOURCE USAGE AND UTILITIES
X-MATE requires modest memory (4 GB is sufficient), and although running on desktop computers is possible, the use of a distributed cluster is recommended. We have updated the multi-map rescue strategy module to use the more memory efficient MuM Rescue Lite (Hashimoto et al., 2009), and included a utility to restart a mapping run from the ‘rescue’ stage. Utilities tailored to RNA-Seq data for the creation of junction libraries and for the quantification of reads mapping to the expected strand when library protocols generate stranded data are added, the latter useful in determining the quality of a sequencing library. Finally, utilities for analysis of mapping yield and for cleaning up residual files after a mapping run have been included (see descriptions in Supplementary File 1).
5 SIMPLIFIED CONFIGURATION FILES
Setting up an X-MATE run is now straightforward using our new configuration format. Example configuration files for many possible mapping strategies and dataset types have been provided with the X-MATE distribution. The system contains improved code quality, commenting and modularization, facilitating further enhancements and ongoing maintenance.
6 FUTURE DIRECTIONS
Future enhancements planned are the addition of more mapping engines, utilities to compare mapping runs and provide information to optimize mapping parameters and upkeep with file input and output formats. All source code and documentation, test datasets with results and junction libraries are freely available from http://grimmond.imb.uq.edu.au/X-MATE/.
Supplementary Material
ACKNOWLEDGEMENTS
We thank D.F. Taylor and S. Wood for web and system administrator support, R. Yip and G. Kolle for SAM file conversion software components and advice and H. Isaac for advice on ISAS.
Funding: National Health and Medical Research Council (455857 to S.M.G. 456140, 631701); Australian Research Council (DP1093164 to N.C., DP0988754). D.L.A.W receives an Australian Postgraduate Award from the Australian Federal Government.
Conflict of Interest: None declared.
REFERENCES
- Cloonan N., et al. RNA-MATE: a recursive mapping strategy for high-throughput RNA-sequencing data. Bioinformatics. 2009;25:2615–2616. doi: 10.1093/bioinformatics/btp459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goecks J., et al. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T., et al. Probabilistic resolution of multi-mapping reads in massively parallel sequencing data using MuMRescueLite. Bioinformatics. 2009;25:2613–2614. doi: 10.1093/bioinformatics/btp438. [DOI] [PubMed] [Google Scholar]
- Kent W.J., et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepke S., et al. Computation for ChIP-seq and RNA-seq studies. Nature. Methods. 2009;6:S22–S32. doi: 10.1038/nmeth.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., Salzberg S.L. How to map billions of short reads onto genomes. Nat. Biotechnol. 2009;27:455–457. doi: 10.1038/nbt0509-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.