

Abstract
Various interaction modes between a group of six ruthenium polypyridyl complexes and DNA have been studied using a number of spectroscopic techniques. Five mononuclear species were selected with formula [Ru(tpy)L1L2](2−n)+, and one closely related dinuclear cation of formula [{Ru(apy)(tpy)}2{μ-H2N(CH2)6NH2}]4+. The ligand tpy is 2,2′:6′,2″-terpyridine and the ligand L1 is a bidentate ligand, namely, apy (2,2′-azobispyridine), 2-phenylazopyridine, or 2-phenylpyridinylmethylene amine. The ligand L2 is a labile monodentate ligand, being Cl−, H2O, or CH3CN. All six species containing a labile L2 were found to be able to coordinate to the DNA model base 9-ethylguanine by 1H NMR and mass spectrometry. The dinuclear cationic species, which has no positions available for coordination to a DNA base, was studied for comparison purposes. The interactions between a selection of four representative complexes and calf-thymus DNA were studied by circular and linear dichroism. To explore a possible relation between DNA-binding ability and toxicity, all compounds were screened for anticancer activity in a variety of cancer cell lines, showing in some cases an activity which is comparable to that of cisplatin. Comparison of the details of the compound structures, their DNA binding, and their toxicity allows the exploration of structure–activity relationships that might be used to guide optimization of the activity of agents of this class of compounds.
Electronic supplementary material
The online version of this article (doi:10.1007/s00775-008-0460-x) contains supplementary material, which is available to authorized users.
Keywords: Ruthenium, Polypyridyl, DNA binding, Linear dichroism, Cytotoxicity
Introduction
Since the introduction of cisplatin in medical protocols for treatment of certain cancers in 1978 [1], anticancer metallopharmaceuticals [2–4] have attracted significant attention. The clinical drawbacks of cisplatin therapy soon became apparent, including the limited applicability of the medicine, the acquired resistance displayed by certain tumors, and the serious side effects [5]. To design improved antitumor platinum drugs, research has developed and focused on understanding the mechanisms of the action of cisplatin both in the living cell and in the body. To date, DNA is generally accepted to be the main target of cisplatin, which has been demonstrated to bind most frequently to two adjacent guanine residues via their N7 position, thereby generating a kink in the DNA structure [5, 6].
Initially anticancer research was guided by a few structure–activity relationship (SAR) “rules” [7], which dictated, for example, the structure that a platinum complex should have for it to display anticancer activity, and showed the importance of the lability of its ligands. However, a number of compounds were reported later that violated these rules, but that still displayed anticancer activity [8–18]; it is now recognized that different molecular-level drug actions are most readily achieved by deliberately breaking the platinum drug SARs.
A relatively new line of investigation focuses on ruthenium chemistry as an alternative metallopharmaceutical approach to platinum [19, 20]. The higher coordination number of ruthenium compared with platinum provides additional coordination sites, which can potentially be used to fine-tune the properties of the complex, for example, by influencing the way the complex interacts with DNA [19]. The different redox properties of ruthenium can also play an important role in the transport mechanisms of the drug in the body, as well as in the interaction between the drug and several different biologically relevant proteins [19]. Other important factors are the differences in ligand substitution kinetics and in the water solubility of the compounds. Ruthenium chemistry may also allow for photodynamic approaches to therapy [21–24].
Ruthenium anticancer chemistry has already yielded many promising results. Several compounds have been described which display an activity comparable to that of cisplatin, and in some cases activities are even better [25–30]. Indeed, two ruthenium dimethyl sulfoxide (DMSO) compounds are currently in clinical trials [31, 32]. For one of these, NAMI-A, the compound does not show dramatic cytotoxicity in tumor cell lines in vitro, but it displays a very high activity against metastases [31, 32]. However, the mechanism of action of these compounds is not well established and SARs are not yet known which might provide starting points to optimize the design of any ruthenium anticancer drug.
Nevertheless a large variety of potential ruthenium drugs have been synthesized, with ligands such as amines, imines, DMSO, polypyridyl compounds, and arenes [19, 33, 34]. The diversity of the active structures in fact suggests that different mechanisms of action may be involved for different types of ruthenium complex [35].
The present investigation focuses on ruthenium polypyridyl coordination compounds which contain only one available coordination site, in an attempt to see whether this is a significant variable in ruthenium complex cytotoxicity. The selected series of ruthenium(II) complexes contains the chelating polypyridyl ligand 2,2′:6′,2″-terpyridine (tpy), a bidentate arylazopyridine or aryliminopyridine ligand, and also a labile monodentate ligand L2 [36–38] (Fig. 1). The choice of tpy as a coligand is based on the known anticancer activity of some Ru(tpy)-containing complexes [39, 40]. The bidentate ligand has selected variations in the structure, for example, by substituting a pyridine ring for a phenyl ring and an imino group for an azo group. These variations, together with the fact that three different labile ligands were used, would hopefully allow for the proposal of a possible SAR. For comparison reasons a symmetric, homodinuclear cationic species [{Ru(apy)(tpy)}2{μ-H2N(CH2)6NH2}]4+ (1f), where apy is 2,2′-azobispyridine, was also synthesized (Fig. 2), which, unlike complexes 1a, 1b, 1c, 1d, and 1e, has no free positions available for coordination to a DNA base. This compound might still interact with DNA through a noncoordinative mechanism.
Fig. 1.
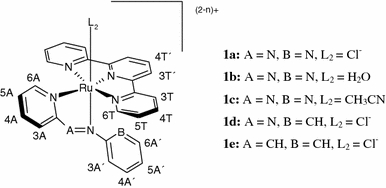
Structure of [Ru(tpy)L1L2](2−n)+ compounds (1a, 1b, 1c, 1d, 1e), where tpy is 2,2′:6′,2″-terpyridine. The proton numbering scheme for use in 1H NMR spectra is indicated as well
Fig. 2.
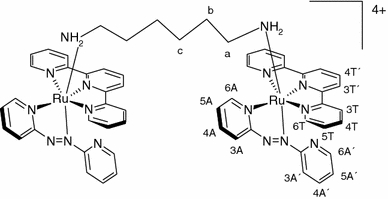
Structure of the dinuclear cation [{Ru(apy)(tpy)}2{μ-H2N(CH2)6NH2}]4+ (1f), where apy is 2,2′-azobispyridine. The proton numbering scheme for use in 1H NMR spectra is indicated as well
The DNA binding and cytotoxicity of these six new agents were explored. These new complexes show a significant cytotoxicity in several cell lines, and, equally excitingly, the results obtained suggest that the mechanism of action of this kind of ruthenium complex may be quite different from that of the classic platinum anticancer agents.
Materials and methods
Starting reagents and coordination compounds
LiCl, NaClO4 (both from Merck), NaClO, AgNO3 (both from Acros), tpy (Aldrich), RuCl3·3H2O (Johnson Matthey), 9-ethylguanine (9-EtGua; Sigma), and H2N(CH2)6NH2 (Fluka) were used as supplied. Ultrapure water (18.2 MΩ; Aldrich) was used for the mass spectrometry (MS), circular dichroism (CD), and linear dichroism (LD) experiments. All other chemicals and solvents were reagent grade, commercial materials and were used as received.
Calf-thymus DNA (ct-DNA) was purchased from Sigma-Aldrich and used without further purification. The solid DNA salt was dissolved in ultrapure water (18.2 MΩ; Aldrich) and left at 278 K for 24 h to fully hydrate. The resulting stock DNA solution was kept frozen and it was thawed when needed. The concentration of the DNA stock solution was determined spectroscopically, using the known molar extinction coefficient of ct-DNA at 258 nm: ε 258 = 6,600 molar base−1 cm−1 dm3 [41].
A 100 mM stock solution of sodium cacodylate buffer (pH 6.8) was prepared, as well as a 1 M sodium chloride stock solution, using in both cases ultrapure water (18.2 MΩ; Aldrich).
The ligands apy, 2-phenylazopyridine (azpy), and 2-phenylpyridinylmethylene amine (impy) and the metal compounds Ru(tpy)Cl3, [Ru(apy)(tpy)Cl](ClO4), [Ru(apy)(tpy)(H2O)](ClO4)2·2H2O, [Ru(apy)(tpy)(CH3CN)](ClO4)2, [Ru(azpy)(tpy)Cl]Cl·5H2O, and [Ru(impy)(tpy)Cl](ClO4) were synthesized as described in the literature [42, 43] and in our earlier reports [36–38].
Synthesis and characterization of [{Ru(apy)(tpy)}2{μ-H2N(CH2)6NH2}](ClO4)4
[Ru(apy)(tpy)(H2O)](ClO4)2·2H2O (26 mg, 0.034 mmol) and H2N(CH2)6NH2 (2 mg, 0.016 mmol) were dissolved in 12 mL 5:1 absolute EtOH/MeOH. The solution was vigorously refluxed for 15 h. The pH remained constant around 7. The product was collected by filtration, washed with a little ethanol and diethyl ether, and dried in vacuo over silica. Yield: 20 mg (76%). Anal. calc. for C56H54N16O16Cl4Ru2: C, 43.4; H, 3.5; N, 14.4%. Found: C, 43.8; H, 3.8; N, 14.5%. m/z (electrospray ionization, ESI, MS) 634.1 ([Ru(apy)(tpy) H2N(CH2)6NH2]+); 576.1 ([Ru(apy)(tpy)]2[μ-H2N(CH2)6NH2]2+); 317.3 ([Ru(apy)(tpy)] H2N(CH2)6NH2]2+). 1H NMR (DMSO-d 6, 298 K): δ (ppm) 9.34 (2H, d, 4.81 Hz); 9.00 (2H, d, 8.05 Hz); 8.62 (6H, m); 8.52 (2H, t, 6.84 Hz); 8.30 (4H, m); 8.14 (4H, t, 7.24 Hz); 7.78 (2H, d, 4.83 Hz); 7.73 (2H, t, 7.76 Hz); 7.46 (4H, t, 6.12 Hz); 7.30 (6H, m); 6.98 (2H, d, 7.98 Hz); 4.92 (4H, m); 1.64 (4H, m); 1.10 (4H, m); 0.66 (4H, m).
Physical measurements
Carbon, hydrogen, and nitrogen determinations were performed with a PerkinElmer 2400 series II analyzer. Mass spectra were obtained with a Finnigan AQA mass spectrometer equipped with an ESI source. NMR spectra were recorded using a Bruker DPX-300 spectrometer operating at a frequency of 300 MHz, at a temperature of 310 K, unless otherwise stated. Chemical shifts were calibrated against tetramethylsilane. CD spectra were collected in 2 mm path length quartz cuvettes using a JASCO J-810 spectropolarimeter. Flow LD spectra were collected using a flow Couette cell in the abovementioned spectropolarimeter which has been adapted for LD spectroscopy. All CD and LD spectra were recorded at room temperature.
In vitro cytotoxicity assays
Prior to the experiments, a mycoplasma test was carried out on all cell lines and the test was negative. All cell lines were maintained in a continuous logarithmic culture in RPMI 1640 medium with 4-(2-hydroxyethyl)-1-piperazineethanesulfonate and phenol red. The medium was supplemented with 10% fetal calf serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. The cells were mildly trypsinized for passage and for use in the experiments. Cytotoxicity was estimated by the microculture sulforhodamine B test [44].
A2780 (human ovarian carcinoma) and A2780R cisplatin-resistant cell lines were maintained in continuous logarithmic culture in Dulbecco’s modified Eagle’s medium (Gibco BRL™, Invitrogen, The Netherlands) supplemented with 10% fetal calf serum (Perbio Science, Belgium), 100 U/mL penicillin G sodium (Duchefa Biochemie, The Netherlands), 100 μg/mL streptomycin (Duchefa Biochemie, The Netherlands), and GlutaMAX (100×; Gibco BRL™, The Netherlands) in a humidified 5% CO2, 95% air atmosphere at 310 K. Cisplatin-sensitive and cisplatin-resistant mouse leukemia L1210/0 and L1210/2 cells were grown under the abovementioned conditions. The cells were harvested from confluent monolayers. Cell viability was determined by the trypan blue dye exclusion test.
For the cytotoxicity evaluation in the cell lines WIDR, IGROV, M19 MEL, A498, H226, MCF7, and EVSA-T, the compounds were dissolved to a concentration of 250 μg/mL in full medium by 20-fold dilution of a stock solution which contained 1 mg of compound per 200 μL DMSO. Trypsinized tumor cells (about 150 μL, 1,500–2,000 cells per well) were plated in 96-well flat-bottomed microtiter plates (Falcon 3072, BD). The plates were preincubated for 48 h at 310 K, 5.5% CO2. A threefold dilution sequence of ten steps was then made in full medium, starting with the 250.000 μg/mL stock solution. Every dilution was used in quadruplicate by adding 50 μL to a column of four wells, resulting in a highest concentration of 62.500 mg/mL. The plates were incubated for 5 days, after which the cells were fixed with 10% trichloroacetic acid in phosphate-buffered saline and placed at 277 K for 1 h. After three washings with water, the cells were stained for at least 15 min with 0.4% sulforhodamine B dissolved in 1% acetic acid. The cells were washed with 1% acetic acid to remove the unbound stain. The plates were air-dried and the bound stain was dissolved in 150 μL of 10 mM tris(hydroxymethyl)aminomethane. The absorbance was read at 540 nm using an automated microplate reader (Labsystems Multiskan MS). Data were used for construction of concentration–response curves and determination of the concentration that induces 50% growth inhibition of cells compared with untreated cells (IC50) by use of the Deltasoft 3 software program.
In the case of cell lines A2780, A2780R, L1210/0, and L1210/2, 2,000 cells per well were seeded in 150 μL of complete medium in 96-multiwell flat-bottomed microtiter plates (Corning Costar®). The plates were incubated at 310 K, 5% CO2 for 48 h prior to drug testing to allow cell adhesion. The stock solutions of all compounds tested were freshly prepared and directly used for the dilutions. Both 1a and α-[Ru(azpy)2Cl2] are poorly water soluble, so for the sake of comparison with the water-soluble compounds, a DMSO/H2O stock solution was chosen for all the compounds tested, except compound 1f. The latter was dissolved directly in water, to avoid decomposition. The nondecomposition was proved by the CD and LD experiment. The dilutions (eight-step dilutions) were prepared in complete medium. The final tested concentrations were 0.019, 0.012, 0.0015, 0.0009, 0.0005, 0.0001, 0.00005, and 0.00001 mM in the case of α-[Ru(azpy)2Cl2] and 0.17, 0.11, 0.06, 0.04, 0.01, 0.003, 0.001, and 0.0003 mM for the other compounds. Each concentration was tested in quadruplicate, using 45 μL per well added to the 150 μL of complete medium. In the control group only 45 μL of complete medium was added containing the corresponding percentages of H2O and DMSO. The maximum content of DMSO in the wells was 0.96%. Parallel experiments showed that no difference in cell proliferation was observed in control groups with or without 1% DMSO. The plates were incubated for 48 h and the evaluation of cell proliferation was performed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide colorimetric assay [45–47]. About 50 μL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide solution (5 mg/mL in phosphate-buffered saline, Sigma Chemical) was added to each well and incubated for 3 h. Formazan crystals were dissolved in 100 μL DMSO. Optical density was measured using a microplate reader (Bio-Rad) at 590 nm. IC50 values were obtained by GraphPad Prism, version 3.02.
Interaction between ruthenium polypyridyl complexes and 9-ethylguanine
Aqueous solutions with a 1.3 mM concentration of the ruthenium compound and a 2.6 mM concentration of the DNA model base 9-EtGua were incubated at 310 K for 24 h. Subsequently the mass spectrum of each of the mixtures was recorded. m/z (ESI-MS) of the mixture 1a + 9-EtGua: 618.1 [Ru(apy)(tpy)](ClO4)+; 554.2 ([Ru(apy)(tpy)Cl]+); 536.3 ([Ru(apy)(tpy)(H2O)]+); 348.9 ([Ru(apy)(tpy)(9-EtGua)]2+). m/z (ESI-MS) of the mixture 1b + 9-EtGua: 696.7 ([Ru(apy)(tpy)(9-EtGua)]+); 617.6 [Ru(apy)(tpy)](ClO4)+; 535.7 ([Ru(apy)(tpy)(H2O)]+); 517.7 ([Ru(apy)(tpy)]+); 348.9 ([Ru(apy)(tpy)(9-EtGua)]2+). m/z (ESI-MS) of the mixture 1d + 9-EtGua: 695.8 ([Ru(azpy)(tpy)(9-EtGua)]+); 552.7 ([Ru(azpy)(tpy)Cl)]+); 534.8 ([Ru(azpy)(tpy)(H2O)]+); 348.3 ([Ru(azpy)(tpy)(9-EtGua)]2+). m/z (ESI-MS) of the mixture 1e + 9-EtGua: 695 ([Ru(impy)(tpy)(9-EtGua)]+); 616 [Ru(impy)(tpy)](ClO4)+; 552 ([Ru(impy)(tpy)Cl]+); 534 ([Ru(impy)(tpy)(H2O)]+); 516 ([Ru(impy)(tpy)]+); 348 ([Ru(impy)(tpy)(9-EtGua)]2+).
Each ruthenium compound was dissolved in 600 μL D2O and the appropriate amount of 9-EtGua was added to prepare solutions with a 1.3 mM concentration of the ruthenium compound and 2.6 mM 9-EtGua. The interaction between each ruthenium complex, H2O, and 9-EtGua was followed by 1H NMR for 24 h at 310 K.
Interaction between ruthenium polypyridyl complexes and calf-thymus DNA
Fresh samples were made with constant concentrations of DNA (300 μM in ultrapure water for the experiments involving complexes 1b, 1d, and 1e and 100 μM concentration for the experiment with complex 1f), NaCl (20 mM), and sodium cacodylate buffer (1 mM), and a range of the metal concentration using a concentrated stock solution of each complex (500 μM 1b, 1d, and 1e and 300 μM 1f in in ultrapure water). The ratio of DNA to metal complex was decreased from 50:1 to 1.5:1 in the various samples. The CD spectra of these solutions were measured after 24 h of incubation at 310 K. The solutions prepared with complex 1f were also measured fresh.
For the LD measurements, a 300 μM solution of DNA in ultrapure water containing NaCl (20 mM) and sodium cacodylate buffer (1 mM) was prepared. This solution was titrated with two stock solutions. The first solution contained each of the complexes 1b, 1d, and 1e in 1,000 μM concentration in ultrapure water or complex 1f in 500 μM concentration. The second stock solution contained DNA (600 μM), NaCl (40 mM), and sodium cacodylate buffer (2 mM). The DNA, NaCl, and sodium cacodylate concentrations were kept constant, while the ratio of DNA to metal complex was decreased from 20:1 to 3:1 for complexes 1b, 1d, and 1e and from 40:1 to 6:1 for complex 1f.
Results and discussion
Synthesis and characterization of the coordination compounds
The anticancer activity of compounds analogous to 1a, 1b, 1c, 1d, and 1e is easily hypothesized to be related to their ability to bind to DNA model bases. To prove this relation and eliminate other possible mechanisms of action of the ruthenium complexes, the compounds in Fig. 1 and an additional new compound 1f (Fig. 2) were synthesized. The design and choice of 1f was based on the following arguments. Its inability to bind to DNA by metal coordination, due to the blockage of the six coordination positions of ruthenium by nonlabile ligands, would allow the DNA-binding–cytotoxicity relationship to be proved. On the other hand, the compound was chosen to be symmetrical and analogous to the mononuclear parent compounds 1a, 1b, and 1c to make the comparison amongst all these complexes as valid as possible. Finally a chain was added that was long enough to allow complex 1f to act as two units of the parent compound.
Compound 1f was found to be pure by 1H NMR and elemental analysis and was characterized by ESI-MS. The mass spectrum showed a peak corresponding to the dinuclear species and also peaks corresponding to the mononuclear fragment arising from fragmentation under the conditions used. The 1H NMR spectrum of 1f was recorded in DMSO-d 6 because although the solubility of this compound in water was adequate for cell testing, it was not sufficient for 1H NMR. Full spectral assignment was made using correlation spectroscopy and nuclear Overhauser effect spectroscopy experiments (Table 1). The stability of 1f in water was studied by dissolving it in water, incubating the solution at 310 K for 2 weeks, evaporating the water, and subsequently recording the 1H NMR spectrum in DMSO-d 6. The compound was found to remain unchanged after this time.
Table 1.
Proton chemical shift values (given in parts per million) for [{Ru(apy)(tpy)}2{μ-H2N(CH2)6NH2}](ClO4)4 (1f), where apy is 2,2′-azobispyridine and tpy is 2,2′:6′,2″-terpyridine, recorded in dimethyl sulfoxide-d 6 at 298 K
| 6A | 3A | 3T 3T′ | 4A | 5A 4T′ | 4T | 6A′ | 4A′ | 5T | 5A′ 6T | 3A′ | NH 2 | (CH 2)a | (CH 2)b | (CH 2)c |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 9.34 | 9.00 | 8.62 | 8.52 | 8.30 | 8.14 | 7.78 | 7.73 | 7.46 | 7.30 | 6.98 | 4.92 | 1.64 | 1.10 | 0.66 |
In vitro cytotoxicity assays
The cytotoxicity of compounds 1a, 1b, 1c, 1d, 1e, and 1f was tested in vitro in a series of selected cell lines belonging to the currently used anticancer screening panel of the National Cancer Institute, USA [48]: WIDR (human colon cancer), IGROV (human ovarian cancer), M19 MEL (human melanoma), A498 (human renal cancer), and H226 (non-small human cell lung cancer). The human breast cancer cell lines MCF7 and EVSA-T [estrogen receptor (ER)+/progesterone receptor (PgR)+ and (ER)−/(PgR)−, respectively] were also included as were the cisplatin-sensitive and cisplatin-resistant mouse leukemia L1210/0 and L1210/2 cells and A2780 (human ovarian carcinoma) and A2780R cisplatin-resistant cell lines.
The cytotoxicity of the mononuclear [Ru(apy)(tpy)L2 n−](2−n)+ complexes (1a, 1b, 1c) and that of their dinuclear analogue [Ru(apy)(tpy)]2[μ-H2N(CH2)6NH2](ClO4)4 (1f) against several selected cell lines were compared to see the differences that might arise from their structural differences. All compounds showed good activity in the EVSA-T cell line and moderate activity in H226, M19 MEL and MCF7 cell lines (Table 2), suggesting the different axial ligands have little effect on their activity. A minor difference was also noted between these compounds in A2780 normal and resistant cell lines and the L1210 cells lines, though the compounds were generally found to be ineffective in the latter.
Table 2.
Concentration that induces 50% growth inhibition of cells compared with untreated cells (IC50) (μM) of the [Ru(apy)(tpy)L2 n−](2−n)+ complexes (1a, 1b, 1c) and their dinuclear analogue [Ru(apy)(tpy)]2[μ-H2N(CH2)6NH2](ClO4)4 (1f) after a 5-day treatment with selected cell lines
| Compound tested | A498 | EVSA-T | H226 | IGROV | M19 MEL | MCF7 | WIDR |
|---|---|---|---|---|---|---|---|
| [Ru(apy)(tpy)Cl](ClO4) (1a) | >96 | 7 | 17 | >96 | 25 | 13 | 66 |
| [Ru(apy)(tpy)(H2O)](ClO4)2·2H2O (1b) | >81 | 6 | 17 | 44 | 26 | 18 | 50 |
| [Ru(apy)(tpy)(CH3CN)] (ClO4)2 (1c) | >82 | 6 | 26 | 78 | 30 | 21 | 73 |
| [Ru(azpy)(tpy)Cl]Cl·5H2O (1d) | 39 | 11 | 34 | 65 | 15 | 30 | 51 |
| [Ru(apy)(tpy)]2[μ-H2 N(CH2)6NH2] (ClO4)4 (1f) | >40 | 17 | 28 | >40 | 33 | >40 | >40 |
| α-[Ru(azpy)2Cl2] | 0.3 | 0.1 | 0.5 | 0.3 | 0.1 | 0.3 | 0.3 |
| Cisplatin | 2 | 1 | 2 | 0.2 | 3 | 2 | 2 |
α-[Ru(azpy)2Cl2] and cisplatin have been included as reference compounds
The non-azo complex 1e showed very low or no activity at all in the cell lines tested (Table 3), indicating that the azo group is important for activity. The most active drug in the case of the nonresistant cell line, A2780, was found to be 1b, but all the drugs were significantly less active than cisplatin, whereas in the resistant cell lines all the activities are comparable to that of cisplatin and similar to that of the nonresistant cell line, suggesting a mechanism of action different from that of cisplatin. In the case of the murine leukemia cell lines L1210 1b was found by way of contrast one of the least effective compounds, and 1a, 1c, and 1d show somewhat better activity in the resistant cell line than in the nonresistant one. Neither the non-azo complex (1e) nor the homodinuclear complex (1f) shows any activity in the L1210 cell lines.
Table 3.
IC50 values (μM) of the [Ru(tpy)L1L2 n−](2−n)+ complexes (1a, 1b, 1c, 1d, 1e) and the dinuclear complex [Ru(apy)(tpy)]2[μ-H2N(CH2)6NH2](ClO4)4 (1f) after a 48-h treatment in some selected cell lines
| Compound tested | A2780 | A2780R | L1210/0 | L1210/2 |
|---|---|---|---|---|
| [Ru(apy)(tpy)Cl](ClO4) (1a) | 23 | 25 | 100 | 56 |
| [Ru(apy)(tpy)(H2O)](ClO4)2·2H2O (1b) | 11 | 30 | 80 | 97 |
| [Ru(apy)(tpy)(CH3CN)] (ClO4)2 (1c) | 31 | 28 | 70 | 40 |
| [Ru(azpy)(tpy)Cl]Cl·5H2O (1d) | 19 | 42 | 42 | 26 |
| [Ru(impy)(tpy)Cl] (ClO4) (1e) | >100 | 62 | >100 | >100 |
| [Ru(apy)(tpy)]2[μ-H2N(CH2)6NH2] (ClO4)4 (1f) | 33 | 28 | >100 | >100 |
| α-[Ru(azpy)2Cl2] | 0.1 | 0.2 | 0.1 | 0.2 |
| Cisplatin | 6 | 25 | 2 | 24 |
α-[Ru(azpy)2Cl2] and cisplatin have been included as reference compounds
Interaction between ruthenium polypyridyl complexes and 9-ethylguanine
It is generally accepted that the target for cisplatin is DNA, as discussed earlier. The compounds of this study were also designed to target DNA. However, it is clear from the cytotoxicity studies that the resistance mechanisms developed for cisplatin do not affect the compounds of this work. The first question was whether they could coordinatively bind to guanine by displacement of the labile ligand. In this respect it should be mentioned that a previous 1H NMR study of the interaction between each of the complexes 1a, 1b, and 1c ([Ru(apy)(tpy)L2](2−n)+, where L is Cl−, H2O, and CH3CN, respectively) and 9-EtGua [49] demonstrated that these three complexes are capable of binding to the DNA model base in water at 310 K and pH 7, albeit with different kinetics in each case. We therefore carried out an analogous study involving 9-EtGua and the new cationic complexes [Ru(azpy)(tpy)Cl]+ (1d) and [Ru(impy)(tpy)Cl]+ (1e), respectively. The hydrolysis of these complexes in the absence of the DNA model base was also investigated by 1H NMR under the same experimental conditions. Comparison of the spectra (data shown in the electronic supplementary information) indicated that both compounds 1d and 1e undergo two reactions, similar to the previously reported ones for 1c [49], i.e., hydrolysis followed by the formation of a ruthenium–base adduct. The reaction between 1d and 9-EtGua is estimated to reach its maximum in about 2 h, with an approximate conversion of 25%, while complex 1e yields as much as a 60% conversion after 9 h (Fig. S1, Table S1). The maximum conversions observed in the cases of complexes 1b and 1c were reported to be 20% in 5 h and 30% in 18 h, respectively [49]. ESI-MS confirmed the 9-EtGua binding for each of the chlorido complexes 1a, 1d, and 1e after incubation for 24 h at 310 K. The spectrum of 1a and 9-EtGua showed a peak at m/z 348.9, corresponding to the species [Ru(apy)(tpy)(9-EtGua)]2+. Two peaks appeared in the spectrum of 1d + 9-EtGua at m/z 695.8 and 348.3, corresponding to the species [Ru(azpy)(tpy)(9-EtGua)]+ and [Ru(azpy)(tpy)(9-EtGua)]2+, respectively. The mass spectrum of 1e + 9-EtGua similarly showed two peaks at m/z 695 ([Ru(impy)(tpy)(9-EtGua)]+) and 348 ([Ru(impy)(tpy)(9-EtGua)]2+). Peaks corresponding to hydrolysis products were also observed for each complex.
Interaction between ruthenium polypyridyl complexes and calf-thymus DNA
That coordination binding of the compounds to guanine is possible does not necessarily imply that such binding can also happen in the more constrained environment of duplex DNA. The addition of the metal complexes to ct-DNA resulted in small changes in the UV–vis absorption spectra. This behavior is not necessarily indicative of a noncovalent interaction, since previous reports on [Ru(bpy)(tpy)X]n+ systems, where bpy is 2,2′-bipyridine, showed that replacement of the chloride by an aqua ligand, or by a guanine DNA base, leads to only very small changes in the metal-to-ligand charge transfer spectra [50, 51].
To probe the binding of the complexes to polymeric ct-DNA in more detail we also recorded CD spectra. CD is a well-established analytical tool for the study of conformational changes in chiral systems [52, 53] and has often been used for the study of DNA–metal complex systems [54–58], including ruthenium metallodrug complexes [30, 59–63] where an induced CD signal is indicative of binding to the chiral DNA. The ruthenium complexes 1b, 1d, 1e, and 1f were therefore mixed with ct-DNA in a range of ratios and left to incubate for 24 h at 310 K. Complex 1b is the aqua analogue of complex 1a, and was used for this study because of its much higher water solubility. The CD spectra of these samples after incubation are shown in Fig. 3. The DNA region of the spectrum shown from 220 to 300 nm is characteristic of retention of a B-DNA conformation, whereas the induction of a positive CD signal at approximately 330 nm when 1e is incubated with ct-DNA shows it binds strongly. This transition is associated with the pyridylimine unit [38]; a similar CD band is not observed for the azopyridine-containing complexes. Nevertheless, changes in the DNA region of the spectrum are indicative of interactions between the DNA and these other compounds.
Fig. 3.
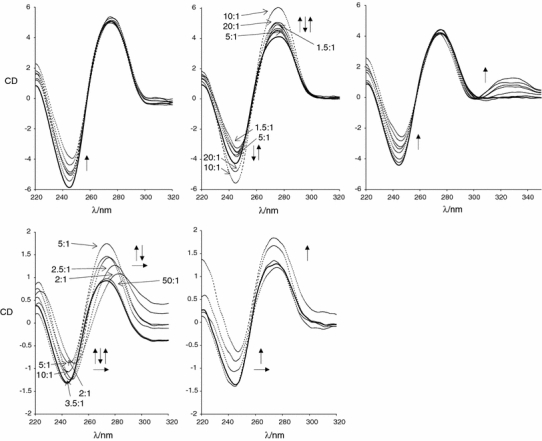
Top circular dichroism (CD) spectra of 300 μM calf-thymus DNA (ct-DNA) incubated for 24 h with increasing concentrations of the mononuclear ruthenium complexes 1b (left), 1d (center), and 1e (right). The DNA base pairs to ruthenium complex molecules ratios are 20:1, 10:1, 5:1, 3:1, 2.5:1, 2:1, and 1.5:1. Bottom CD spectra of 100 μM ct-DNA with increasing concentrations of the dinuclear complex 1f, from freshly prepared samples (left) and from samples incubated for 24 h (right). The DNA base pairs to ruthenium complex molecules ratios are 50:1, 10:1, 5:1, 3.5:1, 2.5:1, and 2:1; the last two ratios were eliminated in the incubated sample, because of precipitation. The solid line represents the ct-DNA; some of the curves are labeled with the base pairs to ruthenium complex molecules ratios. The bold arrows indicate an increase in ruthenium concentration
While the B-DNA conformation is clearly retained, small changes in the bands in the DNA region of the spectrum are difficult to interpret, as induced CD signals from the ligand-based spectroscopic transitions of the compounds also fall in this region. In fact for 1b and 1e the changes are similar to those reported for the ruthenium(II) compound [(η6-p-cymene)Ru(en)(Cl)]+, where en is ethylenediamine [64]. The solutions were prepared and allowed to stand for 24 h, to obtain full hydrolysis. Solutions with complex 1f were also measured fresh for comparison.
Compound 1f is a tetracation and therefore DNA precipitation was observed at high loading. In this case the B-DNA conformation is also clearly retained.
Flow LD (defined as the difference in absorption of light polarized parallel and perpendicular to an orientation axis [52, 65] when the sample is subject to flow) is also sensitive to interactions between DNA and ligands. In this study we used a Couette cell, in which one cylinder rotates inside another, with the sample in the gap between the cylinders [66]. Since the base pairs are oriented perpendicular to the DNA helix axis, a negative LD signal is expected for B-DNA (Fig. 4, band at 258 nm). The LD signal of bound metal complexes indicates the orientations they adopt on the DNA [64, 67–69]. In addition, LD is a useful tool for assessing DNA coiling [58, 65, 70, 71]. When reacted with DNA, 1b, 1d, and 1f show a positive LD signal at 330 nm (whereas 1e shows none), confirming the specific binding of these complexes to DNA. The absence of the band in 1e could simply reflect different polarization of transitions in the pyridylimine and azopyridine complexes. This means the azopyridine ligand based transition [38] lies more parallel than perpendicular to the DNA helix axis.
Fig. 4.
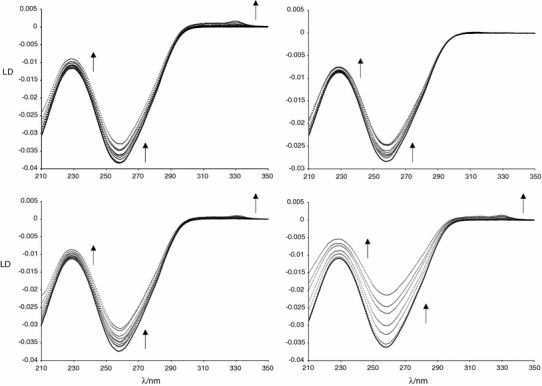
Linear dichroism spectra of 300 μM ct-DNA with increasing concentrations of the ruthenium complexes 1b (top left), 1d (bottom left), 1e (top right), and 1f (bottom right). The DNA base pairs to ruthenium complex molecules ratios are 20:1, 15:1, 10:1, 8:1, 5:1, 3.5:1, 3:1, 2.5:1, and 2:1 in the case of the mononuclear complexes (1b, 1d and 1e) and 40:1, 20:1, 15:1, 10:1, 8:1, and 6:1 for the dinuclear complex (1f). The solid line represents the ct-DNA. The arrows indicate the direction of an increase in ruthenium concentration
For each complex the DNA LD signal at 258 nm decreases in magnitude upon addition of the ruthenium complex, though the effect is small compared with that induced by the metallocylinders that have been reported to coil DNA [58, 65].
Concluding remarks
The IC50 values found for the apy complexes 1a, 1b, and 1c suggest no correlations exist between the lability of the axial leaving group and the cytotoxicity of the compound. From the results with 9-EtGua binding, the most rapidly reacting is the azpy complex 1d, the slowest reacting is 1c, whereas the impy complex 1e yields the maximal conversion. From the IC50 values for 1c, 1d, and 1e it is evident that, the ability of the compounds to bind to 9-EtGua and their anticancer activity are uncorrelated. The mechanisms of activity of the dinuclear coordinatively saturated compound 1f must be different from the mechanisms of other compounds, as it cannot bind to a DNA base; however, its cytotoxic activity is comparable to that of the mononuclear compounds. A small effect on the DNA band in LD is observed with non-covalent-binding dinuclear complex 1f that may indicate some DNA bending or coiling, The effect is small compared with the dramatic effects caused by the non-coordinative-binding dinuclear cylinder compounds which can interact in the major groove of DNA [65, 70, 71], as well as in three-way junctions [72, 73].
In conclusion, a set of ruthenium compounds with differing axial ligands and internal functional groups was synthesized. The cytotoxicity of the compounds appears to be unaffected by the nature of axial ligand leaving group and the dinuclear compound which lacks a leaving group shows activity similar to that of the other compounds. Nevertheless, the presence of the azo group is required for anticancer activity. Interestingly [Ru(bpy)(tpy)Cl]Cl, which is to some extent analogous to the compounds described herein, and which also lacks an azo group, has also been reported to be inactive [40]. In general, the activity of compounds is not affected by the cisplatin resistance mechanisms, suggesting their modes of action differ. Their efficacy is in some cases better than that of cisplatin in resistant cells. The mononuclear compounds can all bind to the isolated model base 9-EtGua, but their DNA binding neither results in kinking like with cisplatin nor in the coiling as known for the dimetallo helicates [65]. At this stage we cannot exclude the possibility that the target of these compounds is DNA, since they all bind to DNA in a non-cisplatin mode.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supporting Information: Figure with NMR spectra of the reactions of 1d and 1e with 9-ethylguanine in D2O; Table with the chemical shifts of the proton H8 and 6A in the several complexes. (PDF 107 kb)
Acknowledgments
The Council for Chemical Sciences of the Netherlands Organization for Scientific Research (CW-NWO) and the EU (A.H., Marie Curie fellowship MEIF-CT-2005-024818; A.R. DNAREC training site, MEST-CT-2005-020842) are thanked for support. This work was performed under the auspices of the Graduate Research School HRSMC, a joint activity of Leiden University and the two universities in Amsterdam and in the context of COST D20 (WG D20/0001/00, D20/0002/00, and D20/0003/01) and of COST D39, which supported an exchange of researchers (E.C.). We kindly acknowledge Dick de Vos (PCM, Haarlem) for some of the cell testing and Fons Lefeber and Kees Erkelens for NMR technical assistance and helpful discussions. Finally Johnson Matthey (Reading, UK) is thanked for the generous loan of RuCl3·3H2O (J.R.).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- apy
2,2′-Azobispyridine
- azpy
2-Phenylazopyridine
- CD
Circular dichrosim
- ct-DNA
Calf-thymus DNA
- DMSO
Dimethyl sulfoxide
- 9-EtGua
9-Ethylguanine
- IC50
Concentration that induces 50% growth inhibition of cells compared with untreated cells
- impy
2-Phenylpyridinylmethylene amine
- LD
Linear dichroism
- MS
Mass spectrometry
- SAR
Structure–activity relationship
- tpy
2,2′:6′,2″-Terpyridine
Contributor Information
Michael J. Hannon, FAX: +44-121-4147871, Email: m.j.hannon@bham.ac.uk
Jan Reedijk, FAX: +31-71-5274671, Email: reedijk@chem.leidenuniv.nl.
References
- 1.Loehrer PJ, Einhorn LH. Ann Intern Med. 1984;100:704–713. doi: 10.7326/0003-4819-100-5-704. [DOI] [PubMed] [Google Scholar]
- 2.Dyson PJ, Sava G (2006) Dalton Trans 1929–1933 [DOI] [PubMed]
- 3.Reedijk J. Curr Opin Chem Biol. 1999;3:236–240. doi: 10.1016/S1367-5931(99)80037-4. [DOI] [PubMed] [Google Scholar]
- 4.Reedijk J. Chem Rev. 1999;99:2499–2510. doi: 10.1021/cr980422f. [DOI] [PubMed] [Google Scholar]
- 5.Reedijk J (1996) Chem Commun 801–806
- 6.Jamieson ER, Lippard SJ. Chem Rev. 1999;99:2467–2498. doi: 10.1021/cr980421n. [DOI] [PubMed] [Google Scholar]
- 7.Reedijk J. Pure Appl Chem. 1987;59:181–192. doi: 10.1351/pac198759020181. [DOI] [Google Scholar]
- 8.Bierbach U, Qu Y, Hambley TW, Peroutka J, Nguyen HL, Doedee M, Farrell N. Inorg Chem. 1999;38:3535–3542. doi: 10.1021/ic981181x. [DOI] [PubMed] [Google Scholar]
- 9.Farrell N, Ha TTB, Souchard JP, Wimmer FL, Cros S, Johnson NP. J Med Chem. 1989;32:2240–2241. doi: 10.1021/jm00130a002. [DOI] [PubMed] [Google Scholar]
- 10.van Beusichem M, Farrell N. Inorg Chem. 1992;31:634–639. doi: 10.1021/ic00030a021. [DOI] [Google Scholar]
- 11.Farrell N, Kelland LR, Roberts JD, van Beusichem M. Cancer Res. 1992;52:5065–5072. [PubMed] [Google Scholar]
- 12.Coluccia M, Sava G, Loseto F, Nassi A, Boccarelli A, Giordano D, Alessio E, Mestroni G. Eur J Cancer. 1993;29A:1873–1879. doi: 10.1016/0959-8049(93)90541-M. [DOI] [PubMed] [Google Scholar]
- 13.Zou Y, van Houten B, Farrell N. Biochemistry. 1993;32:9632–9638. doi: 10.1021/bi00088a015. [DOI] [PubMed] [Google Scholar]
- 14.Montero EI, Díaz S, González-Vadillo AM, Pérez JM, Alonso C, Navarro-Ranninger C. J Med Chem. 1999;42:4264–4268. doi: 10.1021/jm991015e. [DOI] [PubMed] [Google Scholar]
- 15.Montero EI, Pérez JM, Schwartz A, Fuertes MA, Malinge JM, Alonso C, Leng M, Navarro-Ranninger C. Chembiochem. 2002;3:61–67. doi: 10.1002/1439-7633(20020104)3:1<61::AID-CBIC61>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 16.Boccarelli A, Intini FP, Sasanelli R, Sivo MF, Coluccia M, Natile G. J Med Chem. 2006;49:829–837. doi: 10.1021/jm050986t. [DOI] [PubMed] [Google Scholar]
- 17.Kasparkova J, Marini V, Najajreh Y, Gibson D, Brabec V. Biochemistry. 2003;42:6321–6332. doi: 10.1021/bi0342315. [DOI] [PubMed] [Google Scholar]
- 18.Kasparkova J, Nováková O, Marini V, Najajreh Y, Gibson D, Pérez JM, Brabec V. J Biol Chem. 2003;278:47516–47525. doi: 10.1074/jbc.M304720200. [DOI] [PubMed] [Google Scholar]
- 19.Clarke MJ. Coord Chem Rev. 2003;236:209–233. doi: 10.1016/S0010-8545(02)00312-0. [DOI] [Google Scholar]
- 20.Kostova I. Curr Med Chem. 2006;13:1085–1107. doi: 10.2174/092986706776360941. [DOI] [PubMed] [Google Scholar]
- 21.Armitage B. Chem Rev. 1998;98:1171–1200. doi: 10.1021/cr960428+. [DOI] [PubMed] [Google Scholar]
- 22.Davia K, King D, Hong YL, Swavey S. Inorg Chem Commun. 2008;11:584–586. doi: 10.1016/j.inoche.2008.02.023. [DOI] [Google Scholar]
- 23.Elias B, Kirsch-De Mesmaeker A. Coord Chem Rev. 2006;250:1627–1641. doi: 10.1016/j.ccr.2005.11.011. [DOI] [Google Scholar]
- 24.Lang K, Mosinger J, Wagnerova DM. Coord Chem Rev. 2004;248:321–350. doi: 10.1016/j.ccr.2004.02.004. [DOI] [Google Scholar]
- 25.Reedijk J. Proc Natl Acad Sci USA. 2003;100:3611–3616. doi: 10.1073/pnas.0737293100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hotze ACG, Caspers SE, de Vos D, Kooijman H, Spek AL, Flamigni A, Bacac M, Sava G, Haasnoot JG, Reedijk J. J Biol Inorg Chem. 2004;9:354–364. doi: 10.1007/s00775-004-0531-6. [DOI] [PubMed] [Google Scholar]
- 27.Velders AH, Kooijman H, Spek AL, Haasnoot JG, de Vos D, Reedijk J. Inorg Chem. 2000;39:2966–2967. doi: 10.1021/ic000167t. [DOI] [PubMed] [Google Scholar]
- 28.Habtemariam A, Melchart M, Fernandez R, Parsons S, Oswald IDH, Parkin A, Fabbiani FPA, Davidson JE, Dawson A, Aird RE, Jodrell DI, Sadler PJ. J Med Chem. 2006;49:6858–6868. doi: 10.1021/jm060596m. [DOI] [PubMed] [Google Scholar]
- 29.Vilaplana RA, González-Vilchez F, Gutiérrez-Puebla E, Ruiz-Valero C. Inorg Chim Acta. 1994;224:15–18. doi: 10.1016/0020-1693(94)04159-8. [DOI] [Google Scholar]
- 30.Vilaplana RA, Delmani F, Manteca C, Torreblanca J, Moreno J, García-Herdugo G, González-Vilchez F. J Inorg Biochem. 2006;100:1834–1841. doi: 10.1016/j.jinorgbio.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 31.Sava G, Pacor S, Bergamo A, Cocchietto M, Mestroni G, Alessio E. Chem Biol Interact. 1995;95:109–126. doi: 10.1016/0009-2797(94)03350-1. [DOI] [PubMed] [Google Scholar]
- 32.Hartinger CG, Zorbas-Seifried S, Jakupec MA, Kynast B, Zorbas H, Keppler BK. J Inorg Biochem. 2006;100:891–904. doi: 10.1016/j.jinorgbio.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 33.Allardyce CS, Dyson PJ. Platinum Met Rev. 2001;45:62–69. [Google Scholar]
- 34.Ang WH, Dyson PJ (2006) Eur J Inorg Chem 4003–4018
- 35.Brabec V, Nováková O. Drug Resist Update. 2006;9:111–122. doi: 10.1016/j.drup.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 36.Corral E, Hotze ACG, Tooke DM, Spek AL, Reedijk J. Inorg Chim Acta. 2006;359:830–838. doi: 10.1016/j.ica.2005.05.017. [DOI] [Google Scholar]
- 37.Boelrijk AEM, Jorna AMJ, Reedijk J. J Mol Catal A Chem. 1995;103:73–85. doi: 10.1016/1381-1169(95)00112-3. [DOI] [Google Scholar]
- 38.Hotze ACG, Faiz JA, Mourtzis N, Pascu GI, Webber PRA, Clarkson GJ, Yannakopoulou K, Pikramenou Z, Hannon MJ (2006) Dalton Trans 3025–3034 [DOI] [PubMed]
- 39.van Vliet PM, Toekimin SMS, Haasnoot JG, Reedijk J, Novakova O, Vrana O, Brabec V. Inorg Chim Acta. 1995;231:57–64. doi: 10.1016/0020-1693(94)04320-U. [DOI] [Google Scholar]
- 40.Nováková O, Kasparková J, Vrána O, van Vliet PM, Reedijk J, Brabec V. Biochemistry. 1995;34:12369–12378. doi: 10.1021/bi00038a034. [DOI] [PubMed] [Google Scholar]
- 41.Reichmann ME, Rice SA, Thomas CA, Doty P. J Am Chem Soc. 1954;76:3047–3053. doi: 10.1021/ja01640a067. [DOI] [Google Scholar]
- 42.Kirpal A, Reiter E. Ber Dtsch Chem Ges. 1927;60:664–666. doi: 10.1002/cber.19270600312. [DOI] [Google Scholar]
- 43.Adcock PA, Keene FR, Smythe RS, Snow MR. Inorg Chem. 1984;23:2336–2343. doi: 10.1021/ic00183a025. [DOI] [Google Scholar]
- 44.Keepers YP, Pizao PE, Peters GJ, Vanarkotte J, Winograd B, Pinedo HM. Eur J Cancer. 1991;27:897–900. doi: 10.1016/0277-5379(91)90142-Z. [DOI] [PubMed] [Google Scholar]
- 45.Alley MC, Scudiero DA, Monks A, Hursey ML, Czerwinski MJ, Fine DL, Abbott BJ, Mayo JG, Shoemaker RH, Boyd MR. Cancer Res. 1988;48:589–601. [PubMed] [Google Scholar]
- 46.Mosmann T. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 47.Tada H, Shiho O, Kuroshima K, Koyama M, Tsukamoto K. J Immunol Methods. 1986;93:157–165. doi: 10.1016/0022-1759(86)90183-3. [DOI] [PubMed] [Google Scholar]
- 48.Boyd MR. Princ Pract Oncol. 1989;3:1–12. [Google Scholar]
- 49.Corral E, Hotze ACG, Magistrato A, Reedijk J. Inorg Chem. 2007;46:6715–6722. doi: 10.1021/ic070092u. [DOI] [PubMed] [Google Scholar]
- 50.Grover N, Gupta N, Thorp HH. J Am Chem Soc. 1992;114:3390–3393. doi: 10.1021/ja00035a034. [DOI] [Google Scholar]
- 51.Smith SR, Neyhart GA, Kalsbeck WA, Thorp HH. New J Chem. 1994;18:397–406. [Google Scholar]
- 52.Rodger A, Nordén B. Circular dichroism and linear dichroism. Oxford: Oxford University Press; 1997. [Google Scholar]
- 53.Kelly SM, Jess TJ, Price NC. Biochim Biophys Acta Proteins Proteomics. 2005;1751:119–139. doi: 10.1016/j.bbapap.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 54.Macquet JP, Butour JL. Eur J Biochem. 1978;83:375–387. doi: 10.1111/j.1432-1033.1978.tb12103.x. [DOI] [PubMed] [Google Scholar]
- 55.Howegrant M, Lippard SJ. Biochemistry. 1979;18:5762–5769. doi: 10.1021/bi00593a003. [DOI] [PubMed] [Google Scholar]
- 56.Brabec V, Kleinwachter V, Butour JL, Johnson NP. Biophys Chem. 1990;35:129–141. doi: 10.1016/0301-4622(90)80003-P. [DOI] [PubMed] [Google Scholar]
- 57.Natile G, Marzilli LG. Coord Chem Rev. 2006;250:1315–1331. doi: 10.1016/j.ccr.2005.12.004. [DOI] [Google Scholar]
- 58.Pascu GI, Hotze ACG, Sánchez-Cano C, Kariuki BM, Hannon MJ. Angew Chem Int Ed. 2007;46:4374–4378. doi: 10.1002/anie.200700656. [DOI] [PubMed] [Google Scholar]
- 59.Chen HM, Parkinson JA, Nováková O, Bella J, Wang FY, Dawson A, Gould R, Parsons S, Brabec V, Sadler PJ. Proc Natl Acad Sci USA. 2003;100:14623–14628. doi: 10.1073/pnas.2434016100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Karidi K, Garoufis A, Hadjiliadis N, Lutz M, Spek AL, Reedijk J. Inorg Chem. 2006;45:10282–10292. doi: 10.1021/ic0608039. [DOI] [PubMed] [Google Scholar]
- 61.Chao H, Yuan YX, Zhou F, Ji LN, Zhang J. Transit Met Chem. 2006;31:465–469. doi: 10.1007/s11243-006-0013-5. [DOI] [Google Scholar]
- 62.Grguric-Sipka SR, Vilaplana RA, Pérez JM, Fuertes MA, Alonso C, Álvarez Y, Sabo TJ, González-Vilchez F. J Inorg Biochem. 2003;97:215–220. doi: 10.1016/S0162-0134(03)00281-2. [DOI] [PubMed] [Google Scholar]
- 63.Hiort C, Nordén B, Rodger A. J Am Chem Soc. 1990;112:1971–1982. doi: 10.1021/ja00161a050. [DOI] [Google Scholar]
- 64.Nováková O, Chen HM, Vrána O, Rodger A, Sadler PJ, Brabec V. Biochemistry. 2003;42:11544–11554. doi: 10.1021/bi034933u. [DOI] [PubMed] [Google Scholar]
- 65.Hannon MJ, Moreno V, Prieto MJ, Moldrheim E, Sletten E, Meistermann I, Isaac CJ, Sanders KJ, Rodger A. Angew Chem Int Ed. 2001;40:880–884. doi: 10.1002/1521-3773(20010302)40:5<879::AID-ANIE879>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 66.Rodger A, Marrington R, Geeves MA, Hicks M, de Alwis L, Halsall DJ, Dafforn TR. Phys Chem Chem Phys. 2006;8:3161–3171. doi: 10.1039/b604810m. [DOI] [PubMed] [Google Scholar]
- 67.Nordén B. Inorg Chim Acta. 1978;31:83–95. doi: 10.1016/S0020-1693(00)94983-1. [DOI] [Google Scholar]
- 68.Nordén B. FEBS Lett. 1978;94:204–206. doi: 10.1016/0014-5793(78)80938-7. [DOI] [PubMed] [Google Scholar]
- 69.Coggan DZM, Haworth IS, Bates PJ, Robinson A, Rodger A. Inorg Chem. 1999;38:4486–4497. doi: 10.1021/ic990654c. [DOI] [PubMed] [Google Scholar]
- 70.Meistermann I, Moreno V, Prieto MJ, Moldrheim E, Sletten E, Khalid S, Rodger PM, Peberdy JC, Isaac CJ, Rodger A, Hannon MJ. Proc Natl Acad Sci USA. 2002;99:5069–5074. doi: 10.1073/pnas.062634499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Uerpmann C, Malina J, Pascu M, Clarkson GJ, Moreno V, Rodger A, Grandas A, Hannon MJ. Chem Eur J. 2005;11:1750–1756. doi: 10.1002/chem.200401054. [DOI] [PubMed] [Google Scholar]
- 72.Hannon MJ. Chem Soc Rev. 2007;36:280–295. doi: 10.1039/b606046n. [DOI] [PubMed] [Google Scholar]
- 73.Oleksi A, Blanco AG, Boer R, Uson I, Aymami J, Rodger A, Hannon MJ, Coll M. Angew Chem Int Ed. 2006;45:1227–1231. doi: 10.1002/anie.200503822. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information: Figure with NMR spectra of the reactions of 1d and 1e with 9-ethylguanine in D2O; Table with the chemical shifts of the proton H8 and 6A in the several complexes. (PDF 107 kb)