

Abstract
Through alternative splicing, most human genes express multiple isoforms that often differ in function. To infer isoform regulation from high-throughput sequencing of cDNA fragments (RNA-seq), we developed the mixture-of-isoforms (MISO) model, a statistical model that estimates expression of alternatively spliced exons and isoforms and assesses confidence in these estimates. Incorporation of mRNA fragment length distribution in paired-end RNA-seq greatly improved estimation of alternative-splicing levels. MISO also detects differentially regulated exons or isoforms. Application of MISO implicated the RNA splicing factor hnRNP H1 in the regulation of alternative cleavage and polyadenylation, a role that was supported by UV cross-linking–immunoprecipitation sequencing (CLIP-seq) analysis in human cells. Our results provide a probabilistic framework for RNA-seq analysis, give functional insights into pre-mRNA processing and yield guidelines for the optimal design of RNA-seq experiments for studies of gene and isoform expression.
The distinct isoforms expressed from metazoan genes through alternative splicing can be important in development, differentiation and disease1. For example, the pyruvate kinase gene produces two distinct tissue-specific spliced isoforms that differ in their enzymatic activity, allosteric regulation and ability to support tumor growth2. Conservative estimates predict 2–12 mRNA isoforms for most mammalian genes (Supplementary Fig. 1), though some genes, including neurexins, may express more than 1,000 isoforms each3.
Recently, high-throughput sequencing of short cDNA fragments, RNA-seq, has emerged as a powerful approach to characterizing the transcriptome. RNA-seq data have recently been used to show that the vast majority of human genes are alternatively spliced and that most alternative exons show tissue-specific regulation4. To date, RNA-seq analysis methods have focused mostly on estimation of gene expression levels and discovery of novel exons and genes4-6, assembly and annotation of mRNA transcripts5,7, and estimation of the expression levels of alternative exons4. Two recent methods, Cufflinks and Scripture, can produce de novo annotations of transcripts in metazoan genomes using RNA-seq data alone8-10.
Accurate quantification of alternative-exon abundance and detection of differentially regulated exons and isoforms remain challenging. Paired-end RNA-seq protocols, in which both ends of a cDNA fragment are sequenced, are paving the way for isoform-centric rather than exon-centric analyses. Here we have developed the MISO model, a probabilistic framework that uses information in single-end or paired-end RNA-seq data to enable more comprehensive and accurate analysis of alternative splicing, at either the exon or isoform level. MISO provides confidence intervals (CIs) for estimates of exon and isoform abundance, detects differential expression and uses latent information to improve accuracy. We applied MISO to analyze isoform regulation by the splicing factor hnRNP H. Using MISO, we showed how the mean and variance of the library insert length affects the information obtained about splicing events in paired-end RNA-seq data, yielding guidelines for the design of RNA-seq experiments.
RESULTS
Quantifying alternative splicing with MISO
To detect alternative splicing using RNA-seq data, MISO and other methods use sequence reads aligned to splice-junction sequences that are either precomputed from known or predicted exon-intron boundaries, or discovered de novo by spliced alignment to the genome (Online Methods). In the most common type of alternative splicing in mammals, an exon is included or excluded from the mature mRNA; ‘percentage spliced in’ (PSI or Ψ)11 denotes the fraction of mRNAs that represent the inclusion isoform. Reads aligning to the alternative exon or to its junctions with adjacent constitutive exons provide support for the inclusion isoform, whereas reads aligning to the junction between the adjacent constitutive exons support the exclusion isoform; the relative read density of these two sets forms the standard estimate of Ψ, denoted Ψ̂SJ (Fig. 1 and Supplementary Fig. 2)4.
Figure 1.
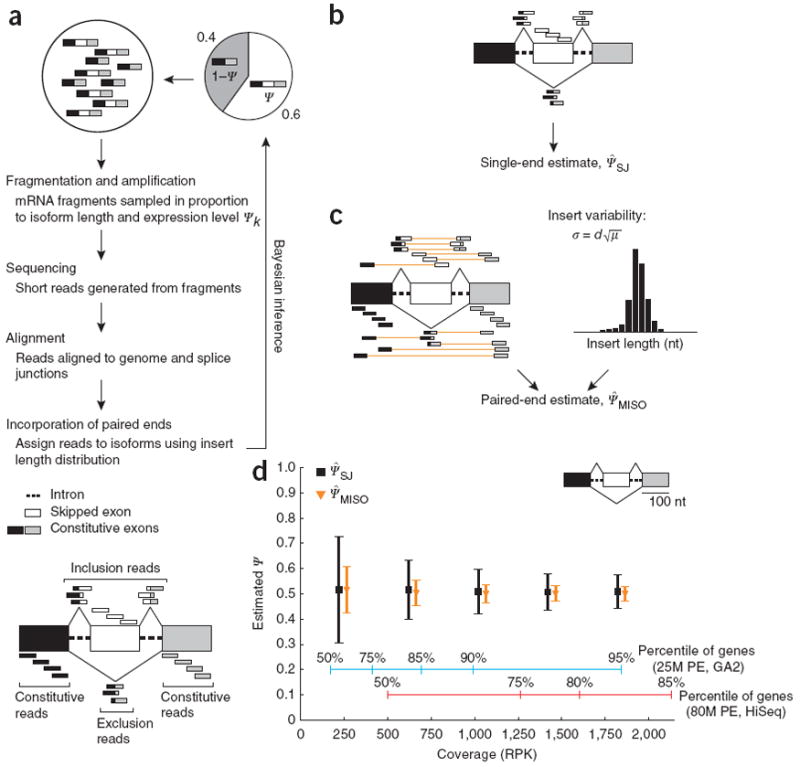
More accurate inference of splicing levels using MISO. (a) Generative process for MISO model. White, alternatively spliced exon; gray and black, flanking constitutive exons. RNA-seq reads aligning to the alternative exon body (white) or to splice junctions involving this exon support the inclusive isoform, whereas reads joining the two constitutive exons (black-gray exon junction) support the exclusive isoform. Reads aligning to the constitutive exons are common to both isoforms. (b) The Ψ̂SJ estimate uses splice-junction and alternative exon–body reads only. (c) The MISO estimate, Ψ̂MISO (derived here analytically), also uses constitutive reads and paired-end read information; orange lines connect reads in a pair; the insert length distribution is shown at right. (d) Comparison of Ψ̂SJ and Ψ̂MISO estimates from simulated data. Reads were sampled at varying coverage, measured in RPK, from the gene structure shown at top right, with underlying true Ψ = 0.5. Mean values from 3,000 simulations are shown (±s.d.) for each coverage value. Percentiles of gene expression values are shown for a data set assuming 25 million mapped paired-end (PE) read pairs (25M PE; blue, extrapolating from an Illumina GA2 run that yielded 15 million mapped read pairs) and for a data set of 78 million mapped read pairs from an Illumina HiSeq 2000 instrument (78M PE; red), both obtained from human heart tissue.
This estimate ignores reads that align to the bodies of the flanking constitutive exons, which could have derived from either isoform. Nevertheless, these constitutive reads contain latent information about the splicing of the alternative exon, as higher expression of the exclusion isoform will generally increase the density of reads in the flanking exons relative to the alternative exon, and lower expression of the exclusion isoform will decrease this ratio of densities. MISO captures this, as well as the information in the lengths of library inserts in paired-end data, by recasting the analysis of isoforms as a Bayesian inference problem. Our approach is related to the alternative-splicing quantification method12, which does not use paired-end information.
MISO samples reads uniformly from the chosen isoform, then recovers the underlying abundances of isoforms (Ψ and 1 − Ψ in the case of a single alternative exon) using the short read data (Fig. 1a and Supplementary Fig. 3). As a result of mRNA fragmentation in library preparation, mRNA abundance and length contribute roughly linearly to read sampling in RNA-seq. This effect is treated by rescaling the abundances Ψ and 1 − Ψ of the two isoforms by the number of possible reads that could be generated from each isoform, respectively. In the model, reads from a gene locus are produced by a generative process in which an isoform is first chosen according to its rescaled abundance, and a sequence read is then sampled uniformly from possible read positions along the mRNA (Online Methods). For the exon-centric analyses involving a single alternative exon we derived an analytic solution to the inference problem, whereas for isoform-centric analyses and estimation using CIs we developed an efficient inference technique based on Monte Carlo sampling (Online Methods). Our new estimator, Ψ̂MISO, uses all of the read positions used in Ψ̂SJ, plus reads aligning to the adjacent exons (Fig. 1b,c) and information about the library insert length distribution in paired-end RNA-seq. Both Ψ̂SJ and Ψ̂MISO are unbiased estimators of Ψ.
An improved measure of exon expression
Simulating read generation from an alternatively spliced gene, we observed that the Ψ̂MISO estimate had consistently much lower variance and error than Ψ̂SJ (Fig. 1d). For reference, the distribution of read-coverage values at depths typically obtained from one lane of sequencing on an Illumina Genome Analyzer 2 (GA2) and on a HiSeq 2000 are shown, in units of reads per kilobase of exon model (RPK). For a gene with median coverage in the GA2 data set (~220 RPK), the s.d. of the estimated Ψ value was reduced more than twofold, from 0.21 for Ψ̂SJ to 0.09 for Ψ̂MISO.
Validation of MISO estimates
To assess the uncertainty in the splicing estimates for each exon, we calculated CIs for Ψ (Online Methods) from moderate-depth breast cancer RNA-seq data (Supplementary Table 1; examples are shown in Fig. 2a,b). Comparing Ψ̂MISO estimates for 52 alternative exons to corresponding quantitative reverse-transcription PCR (qRT-PCR) values11,13 yielded a Pearson correlation r = 0.87 (Fig. 2c and Supplementary Table 2; a bias in the RT-PCR data was analyzed in Supplementary Figs. 4–6). Restricting the analysis to exons with 95% CI width <0.25 increased the correlation with qRT-PCR data considerably, to r = 0.96 (Fig. 2d). Thus, MISO CIs identify exons whose RNA-seq–based Ψ-value estimates are more reliable.
Figure 2.
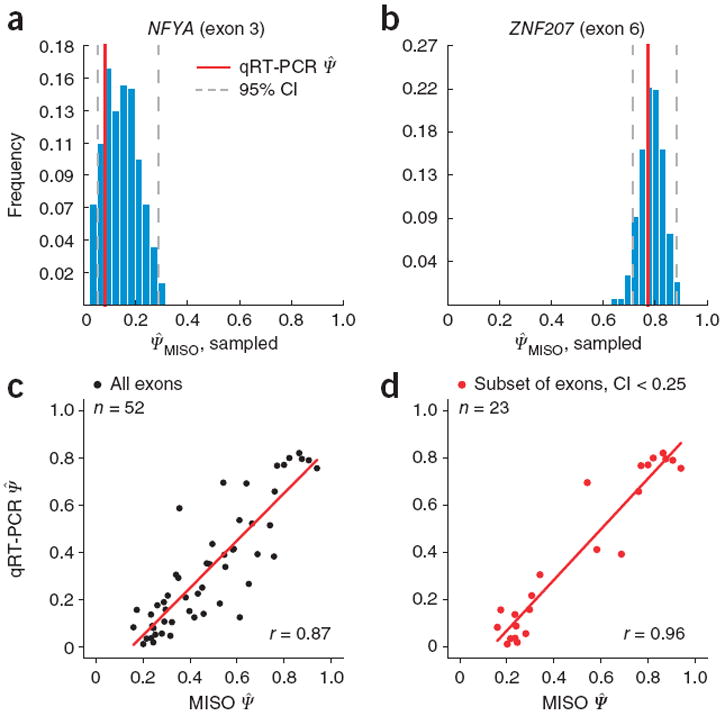
MISO CIs for Ψ values and qRT-PCR validation. qRT-PCR measurements from ref. 13 for a set of 52 alternatively skipped exons were compared to MISO posterior mean estimates of Ψ, denoted Ψ̂MISO. Full listing of events is given in Supplementary Table 1. (a,b) The Ψ posterior distributions obtained by sampling and 95% CIs are shown for two representative exons, one with a wide (NFYA, exon 3) and one with a narrower (ZNF207, exon 6) CI. qRT-PCR Ψ measurements are indicated in red. (c) Scatterplot of MISO and qRT-PCR Ψ estimates for the full set of 52 events. (d) Scatterplot of MISO and qRT-PCR estimates for the subset of 23 high-confidence events, for which CI width <0.25. One exon was excluded from this plot because of expressed sequence tag (EST) evidence of an alternative isoform expected to confound the qRT-PCR analysis (Supplementary Fig. 6).
Detection of differentially expressed isoforms
Differential splicing of alternative exons entails a difference in Ψ values, ΔΨ, and can be evaluated statistically using the Bayes factor (BF), which quantifies the odds of differential regulation occurring. MISO is used to calculate the posterior probability distributions of Ψ and ΔΨ for the two samples. The latter distribution is used to calculate the BF, defined as the ratio of the posterior probability of the alternative hypothesis, ΔΨ ≠ 0, to that of the null hypothesis, ΔΨ = 0 (Online Methods); thus, higher values of the BF indicate increased confidence in differential regulation.
In a recent study we used RNA-seq to characterize transcriptome changes after RNA-interference knockdown of the splicing factor hnRNP H in cultured human cells14. This factor is known to bind polyguanine (poly(G)) runs, typically activating splicing when binding in introns flanking an exon and repressing splicing when binding in exons (Fig. 3a,b). An example of BF calculation for a gene with moderately high read coverage is shown in Figure 3c. When we compared RNA-seq to qRT-PCR data, we found that 100% of exons (6 of 6) with BF ≥ 20 were detected as differentially regulated by qRT-PCR, compared to 21% of exons (4 of 19) with BF < 20 (P < 0.0004, Fisher’s exact test), and the magnitude of ΔΨ showed good agreement (Supplementary Fig. 7). Overall, 15% of alternative exons changed with BF ≥ 20 (Fig. 3d); similarly widespread changes in splicing have been observed by all-exon microarray analysis14.
Figure 3.
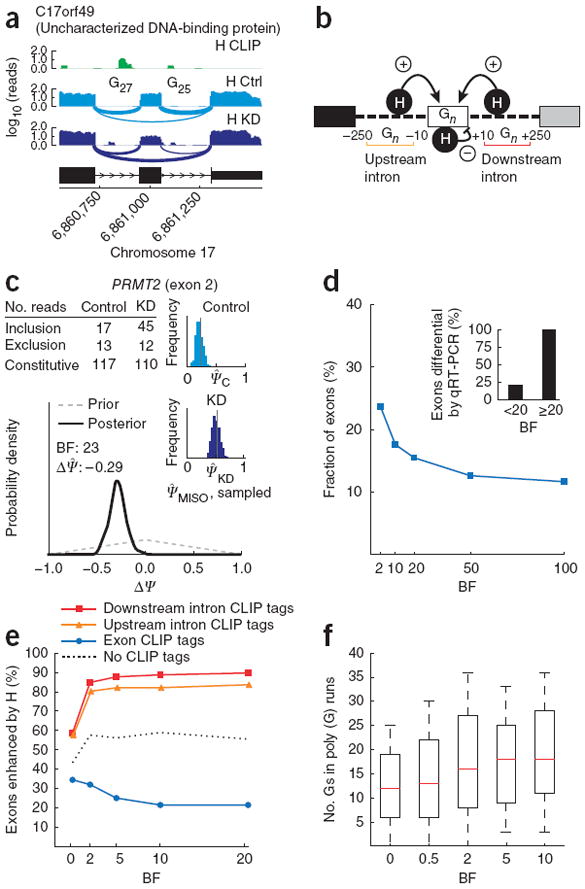
Bayes factor analysis of hnRNP H regulation of exon splicing. (a) CLIP tag density (H CLIP; green) and RNA-seq read densities in hnRNP H–knockdown and control conditions (H KD and H Ctrl; light and dark blue, respectively) for an alternative exon in human C17orf49. Number of guanines in poly(G) runs in upstream and downstream introns is shown. (b) Model of hnRNP H function in splicing regulation: binding of poly(G) runs (Gn) adjacent to an exon enhances the exon’s splicing (+ arrows); binding in exon body represses splicing (− arrow). A 250-nt window in flanking introns was used to count CLIP tags in analyses. (c) BF for exon 2 of PRMT2 gene. Gray dashed line, distribution over ΔΨ under the null hypothesis; black solid line, posterior distribution. (d) Cumulative distribution of BFs using hnRNP H RNA-seq data for exons with sufficiently high read coverage. Inset, fraction of differentially regulated exons (ΔΨ ≥ 0.15 by qRT-PCR), grouping exons by BF (n = 25 exons). (e) Percentage of exons enhanced by hnRNP H (ΔΨ > 0), plotted against increasing BF thresholds, for exons with CLIP tags in downstream or upstream introns but not in exon body (red and orange curves), for exons with CLIP tags in exon body but not in flanking introns (blue curve) and for exons with no CLIP tags (dotted black line). (f) Guanines in poly(G) runs in downstream intron, plotted against increasing BFs.
Genome-wide validation of isoform regulation by CLIP-seq
To identify events directly regulated by hnRNP H and further validate the BF analysis, we performed CLIP-seq analysis of hnRNP H1 under the same conditions as in ref. 14 to identify RNA binding sites of hnRNP H transcriptome-wide. Notably, the percentage of exons with CLIP tags in their flanking introns whose splicing was enhanced by hnRNP H (ΔΨ > 0 between control and knockdown conditions) increased from 60% to over 90% as the BF threshold was increased, approaching a plateau at a BF = 5 (Fig. 3e), corresponding to 5:1 odds of regulation. This effect was stronger for hnRNP H binding in the downstream intron and was reversed for events with exonic CLIP tags, consistent with previous studies (for example, ref. 14 and references therein); virtually no bias was detected, on average, for exons not associated with CLIP tags. Further evidence that BF values reflect regulated exons came from the observation that exons with larger BFs had more guanines in poly(G) runs in their downstream introns (Fig. 3f).
A possible role for hnRNP H in alternative polyadenylation
We used a similar approach to examine whether hnRNP H also has a role in regulating tandem alternative cleavage and polyadenylation (APA), in which cleavage at distinct polyadenylation sites (PASs), without intervening splicing, results in mRNAs with longer or shorter 3′ untranslated regions (UTRs), often affecting mRNA stability, localization or translation15. Evidence that hnRNP H1 and its paralogs hnRNPs F and H2 affect the efficiency of constitutive cleavage and polyadenylation has been described16,17, but regulation of alternative 3′ UTR events by this factor has not previously been reported. Notably, we observed that increased density of CLIP tags just upstream of the core (5′) PAS correlated with greater use of this site in control conditions than in the hnRNP H knockdown, suggesting a role for hnRNP H in promoting core PAS use.
For example, a high density of hnRNP H CLIP tags was observed upstream of the core PAS of the NFATC4 gene, and RNA-seq data indicated greater use of this site in control conditions than in knockdown conditions (Fig. 4a). Because MISO encodes isoforms in a general way as lists of exon coordinates, APA events can be analyzed similarly to alternative splicing events (Online Methods). Applying MISO to RNA-seq data from control and hnRNP H knockdown cells, we observed that genes with higher expression of the shorter 3′ UTR isoform in the presence of hnRNP H—particularly those with large BF values—had higher CLIP tag density near the core PAS (Fig. 4b). Together, these analyses implicate hnRNP H1 in widespread regulation of APA in human genes by activation of the core PAS when bound nearby. Elevated levels of hnRNP H1 have been observed in certain cancers18, and it would be of interest to determine whether hnRNP H1 contributes to the widespread ‘3′ UTR shortening’ (preferential expression of upstream PASs) that occurs in cancer cells19,20.
Figure 4.
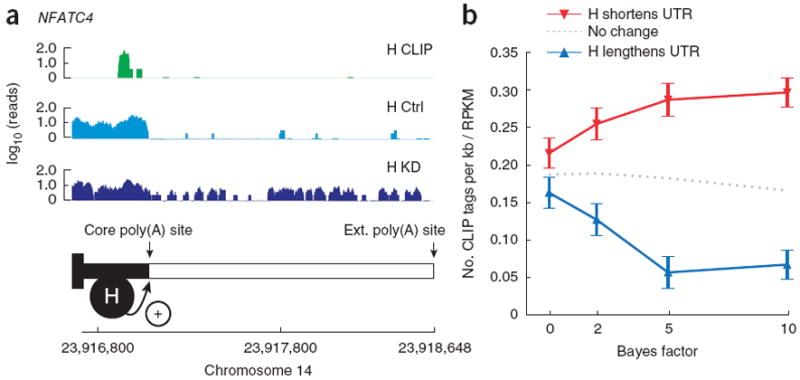
Bayes factor analysis implicates hnRNP H in alternative cleavage and polyadenylation. (a) CLIP tag density (H CLIP; green) and RNA-seq read densities in hnRNP H control and knockdown conditions (H Ctrl and H KD; light and dark blue, respectively) along the 3′ UTR of the NFATC4 gene. Core and extension poly(A) sites for NFATC4 are shown, with a model illustrating the effect of hnRNP H effect on poly(A) site selection. (b) Number of CLIP tags per kilobase normalized by expression (RPKM) for exons with shortened and lengthened UTRs between hnRNP H control and knockdown conditions (red and blue curves, respectively). Values plotted are averages of subsampled mean densities (n = 100 subsamplings) where exons were matched for expression (RPKM). Error bars show s.e.m. CLIP tag density for UTRs not differentially regulated (BF < 1), as shown by dotted gray line.
RNA-seq design: paired-end reads and insert length
A size-selection step is used in RNA-seq library preparation to control the mean length of inserted cDNA fragments. In paired-end sequencing, the full distribution of the lengths of these inserts can be measured precisely from read pairs that map to large constitutive regions such as 3′ UTRs, which are typically intronless. This length distribution can then be used to make qualitatively new types of inferences about alternative isoforms. For example, when the reads in a pair map upstream and downstream of an alternatively spliced exon, the inclusion and exclusion isoforms will typically imply different intervening insert lengths, often enabling the isoform from which the read was generated to be inferred with high confidence.
These considerations led us to compare the fraction of reads that are ‘assignable’—that is, consistent with only one of the two isoforms—in simulations of paired-end and single-end sequencing, varying the mean, μ, of the insert length distribution (Fig. 5). To assess the amount of splicing information present in the length distribution, we considered read pairs that were 20 times more likely to have derived from one isoform than the other under the insert length distribution to be ‘probabilistically assignable’, with a ‘false read assignment’ (FRA) frequency of 1/20 = 5%. In Figure 5d, the insert length distribution has a mean ~260 ± 10 nucleotides (nt), making it far more likely that the read pair shown derived from the inclusion isoform.
Figure 5.
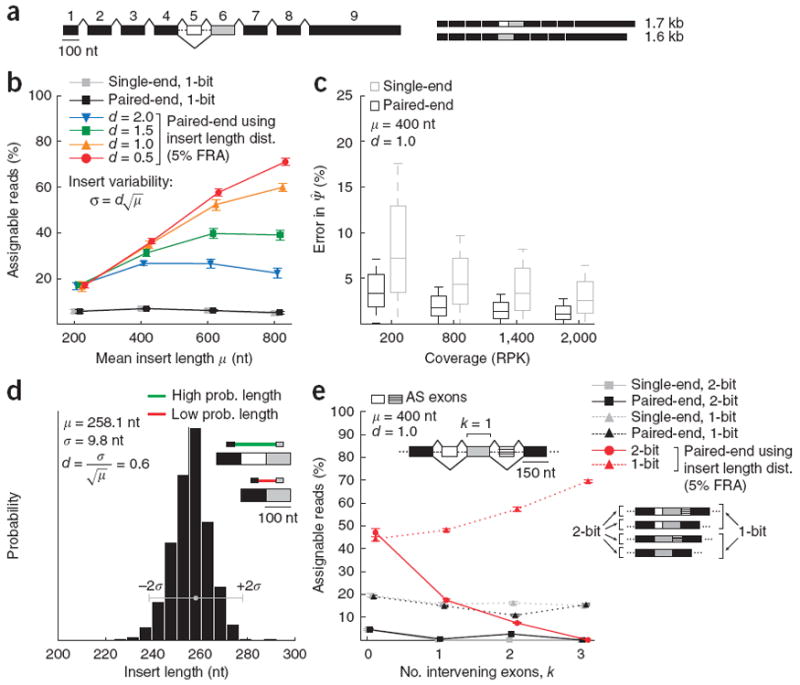
Improved estimation of isoform abundance using paired-end reads. (a) Representative gene model with 100-nt first exon, 100-nt skipped exon (exon 5, in white), 150-nt constitutive exons and 600-nt last exon. (b) We simulated reads from the two-isoform gene model shown in a while varying the mean, μ, of the insert length distribution, setting the s.d. to adjust for the higher variability expected in the size selection for longer fragments. Fraction of 1-bit (assignable to only one isoform) paired and single-end reads is plotted (±s.d.). (c) Distribution of errors for paired-end and single-end estimation as coverage increases (measured in RPK). (d) Histogram shows library insert length distribution computed from read pairs mapped to long constitutive 3′ UTRs in a human testes RNA-seq data set. In the example exon trio shown (similar to that in Fig. 1d), the insert length distribution assigns a higher probability to the top (inclusion) isoform than to the bottom (exclusion) isoform, for which the inferred insert length is improbably small. (e) Fraction of assignable 2-bit and 1-bit reads (±s.d.) for paired-end and single-end reads as a function of the number of intervening constitutive exons, k.
Variability in the insert length distribution influences the confidence with which read pairs can be assigned to isoforms. Varying the s.d., σ, of the insert length distribution by a dispersion factor, d (where σ = d√μ), we observed that even for a relatively broad insert length distribution (d = 2), inclusion of the 5% FRA reads substantially increased the fraction of assignable reads for a gene containing a (typically sized) 100-nt alternative exon (Fig. 5b). For tighter length distributions (d = 1 or d = 0.5), the fraction of assignable reads increased markedly, from ~15% when ignoring insert length information to >50% when considering insert length for large mean lengths, indicating that paired-end data with low-dispersion length distributions can potentially increase the yield of information about splicing by threefold or more at a given sequencing depth. Obtaining a length distribution with d near 1 requires care in library preparation but is achievable in practice (the libraries used in this study had d values between 0.6 and 1.5). For d < 1.5, the proportion of assignable reads increased steadily with insert length (Fig. 5a), as larger inserts make it more likely that reads from a pair will fall on opposite sides of an alternative exon and be probabilistically assignable. Thus, if dispersion is kept near or below 1, use of longer insert lengths should yield more information about splicing. However, changing mRNA fragment size can have other effects on RNA-seq experiments, potentially affecting the priming and reverse-transcription steps and the sampling of mRNAs of different lengths.
To assess the nature and extent of these effects, we generated libraries with mean insert lengths of ~100 nt and ~280 nt from the same RNA sample, derived from control mouse myoblasts, and generated similar libraries from myoblasts depleted of the splicing factor CUGBP1 (Supplementary Fig. 8a). Gene expression estimates were relatively unaffected by insert length for mRNAs 1 kilobase (kb) or longer, but, as expected, read coverage of very short mRNAs only a few hundred bases in length was reduced by ~20–40% in the longer-insert libraries (Supplementary Fig. 8b). The precise pattern of fluctuations in read coverage along constitutive regions differed between libraries with different insert sizes but was highly correlated between libraries generated with similar insert sizes (Supplementary Fig. 8c). The reproducibility of the patterns of local fluctuations indicated that they are primarily determined by fragment size21—which could affect RNA secondary structure and therefore the priming and reverse-transcription steps—rather than by technical noise. Because such fluctuations could affect analysis of alternative splicing, comparisons made between RNA-seq data sets prepared using similar library insert lengths will be most accurate. Changes in gene expression resulting from the knockdown of CUGBP1 were detected highly reproducibly at the two different library insert sizes (r ≈ 0.9; Supplementary Fig. 8d), indicating that library insert size can be varied at least over this range without affecting the ability to detect changes in expression. The overall magnitude of read-coverage fluctuations was only modestly greater for the 100-nt-insert library than for the library with 280-nt inserts (Supplementary Fig. 8e), but further tests of longer insert libraries will be needed to determine the magnitude and impact of the expected increases in local read-coverage fluctuations. Overall, the optimal insert size to use in an RNA-seq experiment will depend on the importance one places on outputs such as detection of splicing changes relative to efficient capture of short mRNAs.
More accurate Ψ values using insert length information
Insert length information is incorporated in MISO by probabilistic assignment of read pairs to isoforms that are consistent with both individual reads, weighting the assignment of read pairs by the relative probability of observing the given insert length, according to the structure of each isoform. To quantify the impact of the increased assignability of reads on accuracy of Ψ estimates, we simulated paired-end reads from a typical gene model containing an alternative exon (Fig. 5a). Use of paired-end reads with insert length information markedly increased the accuracy of estimates of Ψ in simulations, reducing the error by a factor of ~2–5 (Fig. 5c). With a typical gene model containing a typically sized alternative exon, applying the Ψ̂MISO estimation method that makes use of paired-end length information, rather than the standard Ψ̂MISO estimate, reduced the error in estimated Ψ from about 8% to ~4% for a gene with RPK of 200, and the error was further reduced to ~2% at higher coverage values.
Applications to complex alternative splicing
Paired-end data can also be used to make inferences about isoform levels for genes that contain multiple alternative splicing events. To assess how much information can be gained about splicing by paired-end sequencing in these cases, we simulated reads from a gene model containing a pair of alternative exons while varying the number of exons, k, separating the two alternative exons (Fig. 5e). In this gene model, 2 bits of information are required to uniquely specify an isoform: 1 bit to indicate whether the first alternative exon was included or excluded, and 1 bit to describe the splicing of the second alternative exon. Reads that can be uniquely assigned to one of the four isoforms are therefore considered ‘2-bit reads’, whereas reads that are assignable to exactly two of the four isoforms are considered ‘1-bit reads’ (Fig. 5e). When k = 0, a single read may overlap the junction of the two alternative exons or the junction between the flanking constitutive exons, providing 2 bits of information. For k ≥ 1, no 2-bit reads occurred for the typical read and exon lengths used in the simulation, but read pairs can sometimes provide 2 bits of information—for example, if the two reads derive from the two alternative exons or from junctions that are informative about the splicing of these exons, though this is fairly rare. When insert length information is used and probabilistically assignable reads are considered, far more read pairs yield 1 or even 2 bits of information (Fig. 5c and Online Methods), indicating that short-read data has some potential to address more complex alternative splicing events.
The MISO model generalizes to the isoform-centric case in which genes express arbitrarily many isoforms through alternative splicing (Supplementary Note and Supplementary Figs. 9–11); an application of MISO to estimate the abundance of four isoforms from the GRIN1 gene is shown in Supplementary Figure 12. However, sequencing methods involving longer reads, longer library insert lengths or both are needed to quantify isoforms in genes with multiple distant alternative splicing events.
DISCUSSION
Alternative splicing is highly regulated during development and differentiation, and misregulation of RNA processing underlies a variety of human diseases2,22. Because individual alternative exons typically represent only a few percent of the length of the mRNA, analysis of splicing requires greater sequencing depth and more powerful statistical methods than are needed to study gene expression. The MISO model introduced here represents a detailed probabilistic model of RNA-seq, and it has a variety of advantages, including improved accuracy and the ability to analyze all major types of alternative pre-mRNA processing at either the exon level or the isoform level.
This study also has important implications for the design of RNA-seq experiments. Our analyses indicate that paired-end sequencing yields far more information about alternative exons and isoforms than single-end sequencing does. This information derives primarily from cases in which the reads in a pair flank an alternative exon, so that the inclusion and exclusion isoforms imply different intervening mRNA lengths. Use of somewhat longer mRNA fragments, of 300 bases or more, in library preparation should generally enhance isoform inference by increasing the occurrence of such read pairs, with tradeoffs related to the capture of very short mRNAs and changes in the pattern and extent of local fluctuations in read coverage along exons. Our analyses of read-coverage fluctuations strongly imply that RNA-seq–based comparisons of expression and splicing will be most accurate when the insert lengths of the libraries being compared are similar. In some cases a mixed experimental design involving use of different library insert sizes from a single sample may be appropriate—for example, combining one lane of paired-end sequencing from a longer-insert RNA-seq library for inference of mRNA isoform abundance together with a lane of shorter-insert single-end sequencing for analysis of gene expression.
Methods
Methods and any associated references are available in the online version of the paper at http://www.nature.com/naturemethods/.
Supplementary Material
Acknowledgments
We thank C. Wilusz (Colorado State University) for the gift of the CUGBP1-knockdown and control C2C12 cells; R. Darnell for advice regarding CLIP-seq protocols; S. Abou Elela, V. Butty, R. Nutiu and G. Schroth for sharing RNA-seq data; and J. Ernst, D. Gresham, M. Guttman, F. Jäkel, E. Jonas, F. Markowetz, D. Roy, R. Sandberg, T. Velho, X. Xiao and members of the Burge lab for insightful discussions and comments on the manuscript. This work was supported by grants from the US National Science Foundation (E.M.A.) and the US National Institutes of Health (E.M.A. and C.B.B.).
Footnotes
AUTHOR CONTRIBUTIONS Y.K., development of MISO model and software, analyses involving MISO, writing of main text and methods; E.T.W., hnRNP H CLIP-seq experiments and associated computational analyses, CUGBP1 knockdown RNA-seq experiments and associated computational analyses; E.M.A., development of model and statistical analysis, writing of methods; C.B.B., development of MISO model, contributions to computational analyses, writing of main text.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
Accession codes. Gene Expression Omnibus: GSE23694.
Note: Supplementary information is available on the Nature Methods website.
References
- 1.Matlin AJ, Clark F, Smith CWJ. Understanding alternative splicing: towards a cellular code. Nat Rev Mol Cell Biol. 2005;6:386–398. doi: 10.1038/nrm1645. [DOI] [PubMed] [Google Scholar]
- 2.Christofk HR, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- 3.Rowen L, et al. Analysis of the human neurexin genes: alternative splicing and the generation of protein diversity. Genomics. 2002;79:587–597. doi: 10.1006/geno.2002.6734. [DOI] [PubMed] [Google Scholar]
- 4.Wang ET, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mortazavi A, Williams BAA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 6.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 7.Yassour M, et al. Ab initio construction of a eukaryotic transcriptome by massively parallel mRNA sequencing. Proc Natl Acad Sci USA. 2009;106:3264–3269. doi: 10.1073/pnas.0812841106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guttman M, et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol. 2010;28:503–510. doi: 10.1038/nbt.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Griffith M, Griffith OL, Mwenifumbo J, Goya R, Morrissy AS. Alternative expression analysis by RNA sequencing. Nat Methods. 2010;7:843–847. doi: 10.1038/nmeth.1503. [DOI] [PubMed] [Google Scholar]
- 11.Venables JP, et al. Identification of alternative splicing markers for breast cancer. Cancer Res. 2008;68:9525–9531. doi: 10.1158/0008-5472.CAN-08-1769. [DOI] [PubMed] [Google Scholar]
- 12.Jiang H, Wong WH. Statistical inferences for isoform expression in RNA-Seq. Bioinformatics. 2009;25:1026–1032. doi: 10.1093/bioinformatics/btp113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Venables JP, et al. Cancer-associated regulation of alternative splicing. Nat Struct Mol Biol. 2009;16:670–676. doi: 10.1038/nsmb.1608. [DOI] [PubMed] [Google Scholar]
- 14.Xiao X, et al. Splice site strength-dependent activity and genetic buffering by poly-G runs. Nat Struct Mol Biol. 2009;16:1094–1100. doi: 10.1038/nsmb.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Millevoi S, Vagner S. Molecular mechanisms of eukaryotic pre-mRNA 3′ end processing regulation. Nucleic Acids Res. 2010;38:2757–2774. doi: 10.1093/nar/gkp1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alkan SA, Martincic K, Milcarek C. The hnRNPs F and H2 bind to similar sequences to influence gene expression. Biochem J. 2006;393:361–371. doi: 10.1042/BJ20050538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Millevoi S, et al. A physical and functional link between splicing factors promotes pre-mRNA 3′ end processing. Nucleic Acids Res. 2009;37:4672–4683. doi: 10.1093/nar/gkp470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Honoré B, Baandrup U, Vorum H. Heterogeneous nuclear ribonucleoproteins F and H/H’ show differential expression in normal and selected cancer tissues. Exp Cell Res. 2004;294:199–209. doi: 10.1016/j.yexcr.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 19.Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science. 2008;320:1643–1647. doi: 10.1126/science.1155390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mayr C, Bartel DP. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009;138:673–684. doi: 10.1016/j.cell.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J, Jiang H, Wong WH. Modeling non-uniformity in short-read rates in RNA-Seq data. Genome Biol. 2010;11:R50. doi: 10.1186/gb-2010-11-5-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell. 2009;136:777–793. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.