

Respiratory syncytial virus infection of lung epithelial cells represses glucocorticoid receptor-mediated gene activation by reducing glucocorticoid receptor binding to promoter genes of responsive genes.
Abstract
Respiratory syncytial virus (RSV) is a common cause of bronchiolitis in infants. Although antiinflammatory in nature, glucocorticoids have been shown to be ineffective in the treatment of RSV-induced bronchiolitis and wheezing. In addition, the effectiveness of glucocorticoids at inhibiting RSV-induced proinflammatory cytokine production in cell culture has been questioned. In this study, we have investigated the effect of RSV infection on glucocorticoid-induced gene activation in lung epithelium-derived cells. We show that RSV infection inhibits dexamethasone induction of three glucocorticoid receptor (GR)-regulated genes (glucocorticoid-inducible leucine zipper, FK506 binding protein, and MAPK phosphatase 1) in A549, BEAS-2B cells, and primary small airway epithelial cells. UV irradiation of the virus prevents this repression, suggesting that viral replication is required. RSV is known to activate the nuclear factor κB (NFκB) pathway, which is mutually antagonistic towards the GR pathway. However, specific inhibition of NFκB had no effect on the repression of GR-induced genes by RSV infection, indicating that RSV repression of GR is independent of NFκB. RSV infection of A549 cells does not alter GR protein levels or GR nuclear translocation but does reduce GR binding to the promoters of the glucocorticoid responsive genes analyzed in this study. Repression of GR by RSV infection may account for the apparent clinical ineffectiveness of glucocorticoids in RSV bronchiolitis therapy. In addition, this data adds to our previously published data suggesting that GR may be a general target for infectious agents. Identifying the mechanisms through which this suppression occurs may lead to the development of novel therapeutics.
Respiratory syncytial virus (RSV), a negative single-stranded RNA pneumovirus of the Paramyxoviridae family, is the major cause of severe lower respiratory tract infection in children worldwide and results in 70,000–126,000 infant hospitalizations per year for bronchiolitis in the United States alone, depending on the study (1). RSV plays a considerable role in acute respiratory infections in children under 5 yr of age. It has been estimated to be involved in 20% of hospitalizations, 18% of emergency room visits, and 15% of primary care physician office visits (2). Although much research has focused on the production of a vaccine, these efforts have so far been unsuccessful (3). In addition to children, immunocompromised adults, persons with cardiopulmonary disease, and the elderly are also at risk from RSV (4–6).
Glucocorticoids act through the glucocorticoid receptor (GR), playing an important role in regulation of many systems, including the cardiovascular system, central nervous system, metabolism, and homeostasis, as well as their well-known role in regulation of the immune system (7). Glucocorticoid resistance is a major problem in the treatment of many diseases with approximately 30% of asthma (8), ulcerative colitis (9), systemic lupus erythematosus (10), and rheumatoid arthritis (11) patients exhibiting glucocorticoid resistance. In addition, it is estimated that 30% of a normal healthy population would be glucocorticoid nonresponsive if they should require treatment with glucocorticoids (12). One area that has been neglected is the role of infectious agents in glucocorticoid resistance. We have recently established that a bacterial protein, the anthrax lethal toxin, can repress GR function by interfering with GR-DNA binding (13, 14). There is additional fragmentary evidence in the literature that other bacterial and viral infections might alter GR function (15).
Glucocorticoid therapy for RSV-related respiratory diseases remains controversial. Most studies have found no beneficial effect (16, 17), although a few did find some improvements with glucocorticoids (18, 19). In cell culture, some reports show that glucocorticoids inhibit RSV-induced cytokine production (20, 21), whereas others do not (22, 23). These data, although controversial, suggest that RSV may have a deleterious effect on glucocorticoid signaling to suppress inflammation.
Direct effects of viral infections on GR in general are not well studied, although there is some evidence that such an interaction between GR and a viral infection may exist (15). In this study, we show that infection of lung epithelium-derived cells by RSV results in reduced glucocorticoid induction of GR-regulated genes. This effect is dependent on viral replication but is independent of the nuclear factor κB (NFκB) pathway. In addition, RSV infection does not alter GR nuclear translocation but does reduce GR binding to DNA.
Materials and Methods
Materials
Dexamethasone was purchased from Sigma-Aldrich (St. Louis, MO) and was dissolved in 99.5% ethanol. Pyrrolidine dithiocarbamate (PDTC) and BAY 11-7082 were purchased from EMD Chemicals, Inc. (Gibbstown, NJ) and were dissolved in dimethylsulfoxide.
Cell culture
A549 cells, an alveolar cell carcinoma-derived cell line that retains features of the type II alveolar epithelial cells, and BEAS-2B, an SV40-transformed nontumorigenic human bronchial epithelial cell line are routinely used as a model for RSV infection of epithelial cells (24). A549 cells (American Type Culture Collection, Manassas, VA) were grown in DMEM/F12 (50/50) media with 10% fetal bovine serum (FBS), 100 IU penicillin and 100 μg/ml streptomycin at 37 C, and 5% CO2. BEAS-2B cells (American Type Culture Collection) were grown in DMEM with 10% FBS, 100 IU penicillin, and 100 μg/ml streptomycin. Human small airway epithelial cells (SAECs) (Lonza Walkersville, Inc., Walkersville, MD) were grown in small airway growth media with SingleQuot growth factors and supplements according to manufacturer's instructions; 24 h before experimental stimuli, they were transferred into small airway growth media without growth factors or supplements.
Virus preparation and infection
Recombinant green fluorescent protein (GFP)-expressing RSV (rgRSV) was grown in HeLa cells, separated from debris by low-speed centrifugation, and further purified by pelleting in a high-speed centrifuge (25). Mock-infected condition media were produced by treating the cells in the same manner, but no RSV was used to infect the cells. Viral titer was determined by a fluorescent titration assay. Briefly, a serial dilution of viral stocks was used to infect HeLa cells, and the number of GFP expressing cells was counted 24 h after infection. Viral pools were aliquoted and frozen on dry ice and stored at −80 C until used. For viral adsorption, media were removed from cells, and fresh media without FBS containing RSV at the required multiplicity of infection (MOI) were added to the cells for 1 h. Media were then replaced with fresh media containing 10% charcoal-stripped serum (CSS) to reduce background levels from endogenous hormones. Successful infection was confirmed by the presence of GFP viewed under a fluorescent microscope.
UV irradiation of RSV
Viral replication was inhibited by UV irradiation. The virus, rgRSV, was diluted in media without FBS, distributed in 800 μl aliquots in an open 12-well cell culture plate, and irradiated in a Stratagene UV Crosslinker at 70,000 μJ/cm2 for 1 min (Agilent Technologies, Santa Clara, CA). Success of UV irradiation was confirmed by the failure to detect GFP in cells inoculated with the UV-rgRSV.
Transient transfections
A549 cells were plated in 24-well cell culture plates in DMEM/F12 containing 10% CSS at a concentration of 5 × 104 cells/well. The next day, cells were transfected overnight with 160 ng of the GR-responsive promoter, mouse mammary tumor virus (MMTV) luc, 20 ng of the constitutively active renilla internal control, pRL TK (Promega Corp., Madison, WI), and 20 ng pSG5 using Effectene (QIAGEN, Inc., Valencia, CA) according to the manufacturer's instructions. The next day, the cells were inoculated with rgRSV at an MOI of 3 as described above. After 1 h, the inoculum was replaced with media containing dexamethasone at various concentrations. After 24 h, the cells were lysed and luciferase measured using the Dual Luciferase Assay (Promega Corp.).
Real-time PCR
Total RNA was extracted using acid guanidium phenol extraction (TRIzol; Invitrogen Corp., Carlsbad, CA); 500 ng RNA was reverse transcribed using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Inc., Foster City, CA) in a 20 μl reaction. cDNA was then diluted by addition of 100 μl diethylpyrocarbonate-treated H2O. Real-time PCR was performed on 8 μl diluted cDNA in a 20 μl reaction containing 1× Power SYBR Green PCR Master Mix (Applied Biosystems, Inc.) and 0.125 μm of each primer (Table 1) using the following protocol on an ABI 7300 Real-Time PCR system. Plates were heated at 50 C from 2 min, denatured for 10 min at 50 C, and then subjected to 40 cycles of 95 C for 15 sec and 60 C for 1 min. Duplicate cycle threshold (CT) values of duplicate samples were analyzed using the comparative CT (ΔΔCT) method (Applied Biosystems, Inc.). The fold induction (2−ΔΔCT) by dexamethasone was obtained by normalizing to two endogenous genes, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and cAMP-accessory protein (CAP-1) (26) and expressed relative to the amount in nontreated cells.
Table 1.
Real-time PCR primers
| Gene name | Accession no. | Direction | Primer sequence (5′-3′) | Ref. |
|---|---|---|---|---|
| CAP-1 | NM_006367.3 | F | ATTCCCTGGATTGTGAAATAGTC | 26 |
| R | ATTAAAGTCACCGCCTTCTGTAG | |||
| FKBP51 | NM_004117 | F | TCCCTCGAATGCAACTCTCT | |
| R | AAACATCCTTCCACCACAGC | |||
| GAPDH | NM_002046.3 | F | ACTTTGGTATCGTGGAAGGACT | 26 |
| R | GTAGAGGCAGGGATGATGTTCT | |||
| GILZ | NM_004089 | F | GCACAATTTCTCCATCTCCTTCTT | 69 |
| R | TCAGCATGATTCTTCACCAGATCCA | |||
| IL-6 | NM_000600.2 | F | CACAGACAGCCACTCACCTC | |
| R | TTTTCTGCCAGTGCCTCTTT | |||
| IL-8 | NM_000584.2 | F | AGTTTTTGAAGAGGGCTGAGAAT | |
| R | CAACAGACCCACACAATACATGA | |||
| MKP-1 | NM_004417 | F | CTGCCTTGATCAACGTCTCA | |
| R | ACCCTTCCTCCAGCATTCTT |
F, Forward; R, reverse.
Cell viability assay
A549 and BEAS-2B cells were plated in 96-well opaque-walled cell culture plates at a concentration of 1 × 104 cells/well in growth media containing 10% CSS. The next day, cells were inoculated as described above with rgRSV at an MOI of 3. After 1 h, the inoculum was replaced with fresh growth media containing 10% CSS plus increasing concentrations of dexamethasone and the cells incubated for a further 5 h. Cell viability was then assayed using the CellTiter-Glo Luminescent Cell Viability Assay (Promega Corp.) according to the manufacturer's instructions.
Enzyme-linked immunosorbent assays
Cell culture supernatants were harvested and centrifuged at 14,000 × g for 5 min to remove cell debris. IL-6 and IL-8 protein was determined by ELISA using the human IL-6 and IL-8 ELISA Max Delux kit (BioLegend, San Diego, CA) according to manufacturer's instructions.
Small interfering RNA (siRNA)-mediated gene silencing
SignalSilence NFκB p65 siRNA I (Cell Signaling Technology, Inc., Danvers, MA) or Alexa Fluor 488 AllStars Negative Control siRNA (QIAGEN, Inc.) was transfected into A549 cells using DharmaFECT 2 (Thermo Fisher Scientific, Inc., Pittsburgh, PA) according to the manufacturer's instructions; 48 h after transfection, the cells were inoculated with rgRSV at an MOI of 3 as described above. After 1 h, the inoculum was removed, cells treated with increasing concentrations of dexamethasone, and the expression of GR-regulated genes determined by real-time PCR. The efficiency of siRNA silencing was determined by Western blotting.
Western blotting
Total cellular protein was isolated using mammalian protein extraction reagent (Thermo Fisher Scientific, Inc.) or nuclear and cytosolic extracts isolated using nuclear protein extraction kit (Thermo Fisher Scientific, Inc.) according to Manufacturer's instructions; 10 μg protein were subjected to SDS-PAGE on NuPAGE Novex 4–12% bis-Tris precast gels (Invitrogen Corp.). Proteins were then transferred to nitrocellulose membranes by semidry blotting and blocked in 5% nonfat dry milk in Tris-buffered saline with 0.05% Tween 20 (TBST). They were then incubated with antibodies raised against NFκB p65 (3034, 1:1000; Cell Signaling Technology, Inc.), GR (H-300) (sc-8992; 1:1000; Santa Cruz Biotechnology, Inc., Santa Cruz, CA), RSV (AB1128, 1:5000; Millipore, Billerica, MA), or FK506 binding protein 51 (FKBP51) (sc-11514, 1:200; Santa Cruz Biotechnology, Inc.) in 5% milk in TBST overnight at 4 C. Membranes were then washed three times with TBST and then incubated with goat antirabbit IgG-horseradish peroxidase (HRP) (sc-2004, 1:2000; Santa Cruz Biotechnology, Inc.) or bovine antigoat IgG-HRP (sc-2350, 1:2000; Santa Cruz Biotechnology, Inc.) in TBST for 1 h at room temperature. Membranes were again washed with TBST and chemiluminescence detected using SuperSignal West Pico Chemiluminscence Substrate (Thermo Fisher Scientific, Inc.) according to the manufacturer's instructions and exposed to autoradiographic film or visualized using the Kodak Image Station 4000MM Pro and quantified by densiometry. Membranes were then stripped for 15 min at room temperature in Restore Stripping buffer (Thermo Fisher Scientific, Inc.) and reprobed with β-actin antibody (sc-47778, 1:2000; Santa Cruz Biotechnology, Inc.) in 5% milk in TBST for 1 h at room temperature. Membranes were again washed with TBST and then incubated with goat antimouse IgG-HRP (sc-2005, 1:2000; Santa Cruz Biotechnology, Inc.) in TBST for 1 h at room temperature. Membranes were washed and developed as previously.
Immunocytochemistry
A549 cells were plated on coverslips in DMEM/F12 media containing 10% CSS at a concentration of 2 × 105 cells/well. The next day, cells were inoculated with rgRSV at an MOI of 3 as described above. After 1 h, the inoculum was replaced with media containing 100 nm dexamethasone. After 5 h, the cells were fixed with 4% paraformaldehyde for 1 h at room temperature and then permeabilized with 100% methanol for 15 min. The coverslips were blocked in 3% BSA (Sigma-Aldrich) in PBS containing 0.05% Tween 20 (PBST) for 1 h. Rabbit polyclonal GR antibody (sc-8992, 4 μg/ml; Santa Cruz Biotechnology, Inc.) or normal rabbit IgG (sc-2027; Santa Cruz Biotechnology, Inc.) diluted in blocking buffer was applied to the coverslips for 1 h in a humidified chamber. Coverslips were washed three times for 5 min per wash in PBST. Goat antirabbit IgG-TR (sc-2780, 2 μg/ml; Santa Cruz Biotechnology, Inc.) was added to each of the coverslips for 1 h in a dark humidified chamber. The coverslips were again washed three times with PBST for 5 min. The coverslips were mounted onto microscope slides with 10 μl ProLong Gold antifade with 4′,6-diamidino-2-phenylindole (Invitrogen Corp.). This was incubated overnight in the dark. The coverslips were viewed and photographed on an upright fluorescent microscope and camera (Leica Microsystems, Wetzlar, Germany).
Chromatin immunoprecipitation (ChIP)
A549 cells were plated in 150-mm cell culture dishes in DMEM/F12 containing 10% CSS at a concentration of 7 × 105 cells/dish. The cells were serum starved for 24 h before inoculation. Cells were inoculated with rgRSV at an MOI of 3 as described above. After 1 h, the inoculum was replaced and the cells stimulated with 100 nm dexamethasone for 5 h. ChIP was performed as described by Nissen and Yamamoto (27) and Wang et al. (28) with the following modifications. Nuclei were sonicated (power setting 6) with a Misonix microson with a microtip as described. Chromatin was precleared by rotating for 1 h at 4 C with 50 μl 50% slurry of protein-A/G plus agarose beads (sc-2003; Santa Cruz Biotechnology, Inc.) (preblocked with 0.2 mg/ml sonicated salmon sperm DNA and 0.5 mg/ml BSA). The cleared chromatin was incubated with rotation overnight at 4 C with 6 μg antibody raised against GR (sc-8992; Santa Cruz Biotechnology, Inc.) or normal rabbit IgG (sc-2027; Santa Cruz Biotechnology, Inc.). Preblocked agarose-A/G plus was added (50 μl) and chromatin rotated for a further 2 h at 4 C. Beads were pelleted by centrifugation at 1000 × g for 1 min and washed twice with RIPA, twice with RIPA containing 500 nm NaCl, twice with LiCl buffer [20 mm Tris (pH 8), 1 mm EDTA, 250 mm LiCl, 0.05% Nonidet P-40, and 0.01% Na-deoxycholate]), and once with Tris EDTA buffer; 5 μl ChIP product was analyzed by real-time PCR in a 20 μl reaction containing 1 × Power SYBR Green PCR Master Mix and 1 μm of each primer. The glucocorticoid-inducible leucine zipper (GILZ) primers corresponded to the −1919/−1794 region of the GILZ promoter as described previously (28). The FKBP51 primers (forward, 5′ GCATGGTTTAGGGGTTCTTG 3′ and reverse, 5′ TGCCCTAGAGCAATTTTGTTT 3′) spanned the pIE2 region that has been described (29). The MAPK phosphatase 1 (MKP-1) primers corresponded to the −1464/−1412 region of the MKP-1 promoter as described previously (30). Data were analyzed by the ΔΔCT method and the fold change in occupancy calculated.
Statistical analysis
Experiments were performed in duplicate, and the average was treated as one value. Curve fitting was performed using the log(agonist) vs. response (three parameter) function with Prism 5 software (GraphPad Software, San Diego, CA). Maximal activity (Amax) was calculated as the highest activity at the peak of the dose-response curve. The average values of n independent experiments were analyzed for statistical significance using the two-tailed unpaired Student's t test using Prism 5 software. Significance is determined by P values less than 0.05 (one asterisk indicates P = 0.05–0.01, two asterisks indicate P = 0.01–0.001, and three asterisks indicate P < 0.001).
Results
Dexamethasone suppression of RSV and IL-1β-induced cytokines
The efficacy of glucocorticoids in suppressing RSV-induced cytokine production in lung epithelium-derived cell lines has been controversial (20–23). To determine the suppression of RSV-induced cytokines by dexamethasone in our system, A549 cells were inoculated with RSV (MOI = 3) and then treated with 100 nm dexamethasone for a further 5 h. Expression of IL-6 and IL-8 was determined by real-time PCR and normalized to two house keeping genes, GAPDH and CAP-1. As a positive control, A549 cells were also treated with 1 ng/ml IL-1β in the presence or absence of 100 nm dexamethasone. RSV infection or treatment with IL-1β induced expression of IL-6 and IL-8 in A549 cells. The IL-1β-induced IL-6 and IL-8 was reduced by 95 and 90% by cotreatment with 100 nm dexamethasone, whereas the RSV-induced IL-6 and IL-8 was only slightly reduced (20–30%) by dexamethasone treatment (Fig. 1). These data suggest that RSV-induced cytokine production is resistant to suppression by glucocorticoids.
Fig. 1.
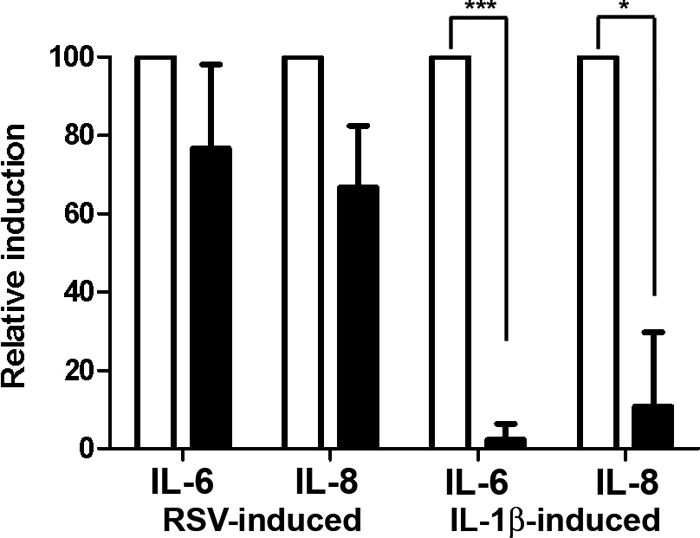
Dexamethasone represses IL-1β-induced cytokines but not RSV-induced cytokines. A549 cells were inoculated with live rgRSV (MOI = 3) for 1 h followed by treatment with 100 nm dexamethasone for 5 h or were treated with 1 ng/ml IL-1β in the presence or absence of 100 nm dexamethasone for 6 h. RSV or IL-1β induction of IL-6 and IL-8 was determined by real-time PCR and normalized to GAPDH and CAP-1. Induction by RSV or IL-1β alone was normalized to 100% (white bars) and induction in the presence of dexamethasone (black bars) calculated. Means and sd are shown (n = 3).
Repression of dexamethasone activation of a GR-responsive promoter by RSV infection
To determine the effect of RSV infection on GR function, A549 cells were transiently transfected with the GR responsive MMTV promoter driving expression of the firefly luciferase and a constitutively active renilla luciferase control for normalization. Cells were subsequently inoculated with rgRSV (MOI = 3) or treated with media only for 1 h followed by dexamethasone treatment for 24 h. An MOI of 3 was chosen as preliminary studies determined that this was optimal for detecting RSV effects on GR-regulated genes at 6 h after infection (data not shown). In addition, efficient infection was confirmed by detection of GFP under a fluorescent microscope. In the absence of RSV infection, dexamethasone induced a ligand-dependent activation of the MMTV promoter in A549 cells with a maximum 23-fold induction (EC50 = 11.5 ± 2.8 nm). Infection of the cells with RSV reduced the maximum induction to only 7-fold (EC50 = 13.3 ± 6.2 nm). The reduced induction of the MMTV promoter was seen at all dexamethasone concentrations (Fig. 2A). Such repression is indicative of a noncompetitive inhibitor of GR. RSV infection significantly repressed dexamethasone induction of the MMTV promoter compared with no infection in A549 cells (Fig. 2B).
Fig. 2.

RSV infection represses dexamethasone induction of MMTV promoter in A549 cells. A549 cells were transfected with the GR-responsive MMTV promoter and a constitutively active control renilla vector. They were then inoculated with rgRSV (MOI = 3) (open squares and white bar) or uninfected (closed squares and black bar) for 1 h followed by dexamethasone treatment for 24 h. MMTV-controlled firefly luciferase was normalized to renilla luciferase, and fold increase after dexamethasone treatment was calculated (A). In addition, Amax was also calculated (B). Means and sd are shown (n = 4).
Effect of RSV infection on cell viability
One possibility that could account for the observed reduction in GR function in RSV-infected cells is that the cells are simply dying in response to viral infection. To test this, cell viability assays were performed using the CellTiter-glo assay to quantify ATP. Neither RSV infection (MOI = 3) nor mock infection (with HeLa-conditioned media) (data not shown) in the presence or absence of increasing concentrations of dexamethasone in either A549 or BEAS-2B (Supplemental Fig. 1, published on The Endocrine Society's Journals Online web site at http://endo.endojournals.org) cells had any significant effect on cell viability compared with no infection at 6 h after inoculation. In addition, RSV infection at an MOI of 3 also had no significant effect on cell viability 24 h after infection (data not shown). These data suggest that RSV infection has no effect on cell viability under the experimental conditions used in our studies.
Repression of GR-mediated gene activation by RSV infection is inhibited by UV irradiation
The effect of RSV infection on dexamethasone induction of endogenous GR-regulated genes in A549 cells was determined by real-time PCR. Primers were designed across exon boundaries to prevent amplification of any contaminating genomic DNA. The endogenous genes chosen had previously been shown to be up-regulated by dexamethasone in A549 cells and the role of GR in their induction shown by the use of RNAi to knock down GR (28). To determine the effect of RSV infection on dexamethasone induction of GR-regulated genes, cells were inoculated with RSV (MOI = 3) and then treated with varying concentrations of dexamethasone for a further 5 h. Expression of three GR-regulated genes was determined by real-time PCR and normalized to two house keeping genes, GAPDH and CAP-1. To confirm infection, the presence of GFP was detected 24 h after infection using a fluorescent microscope. Loss of viral replication of UV-irradiated RSV was detected by loss of GFP in the infected cells (Supplemental Fig. 2). Compared with mock infection, live, but not UV irradiated, RSV repressed dexamethasone induction of the antiinflammatory gene GILZ (Fig. 3A) (EC50 for mock infected = 4.8 ± 2.4 nm; RSV infected = 4.2 ± 1.6 nm; UV-irradiated RSV = 4.5 ± 1.1 nm). Comparison of the Amax showed that RSV repressed dexamethasone induction of GILZ compared with either mock infection or UV-irradiated RSV infection (18-fold increase for mock, 9-fold for RSV, and 18-fold for UV-irradiated RSV) (Fig. 3B). Similar results were seen for dexamethasone induction of the immunophilin FKBP51 (14-fold increase and EC50 2.8 ± 0.7 nm for mock, 10-fold increase and EC50 2.6 ± 1.6 nm for RSV, and 15-fold increase and EC50 2.9 ± 1.6 nm for UV-irradiated RSV) (Fig. 3, C and D) and the MAPK suppressor MKP-1 (4-fold increase and EC50 2.9 ± 1.3 nm for mock, 2.5-fold increase and EC50 4.2 ± 3.4 nm for RSV, and 4-fold increase and EC50 2.2 ± 0.3 nm for UV-irradiated RSV) (Fig. 3, E and F). In addition, RSV infection also inhibited dexamethasone induction of FKBP51 protein levels in A549 cells (Fig. 3, G and H).
Fig. 3.

RSV infection repression of dexamethasone induction of endogenous GR-regulated genes and proteins in A549 cells. A549 cells were mock inoculated with HeLa-conditioned media (closed square and black bar) or inoculated with live rgRSV (open square and white bar) or UV-irradiated rgRSV (open circle with dashed line and hatched bar) (MOI = 3) for 1 h followed by treatment with dexamethasone for 5 h. Dexamethasone (Dex) induction of GILZ (A and B), FKBP51 (C and D), and MKP-1 (E and F) was determined by real-time PCR and normalized to GAPDH and CAP-1. Means and sd are shown (n = 4). G and H, A549 cells were either inoculated with HeLa-conditioned media (mock infected) (black bar) or rgRSV (MOI = 3) (white bar) for 1 h followed by incubation with increasing concentrations of dexamethasone for 24 h. Western blotting of 10 μg total protein was performed for FKBP51 and stripped and reprobed for β-actin. A representative Western blotting is shown (G) and quantification using the Kodak Image Station 4000MM Pro (H). Means and sd are shown (n = 3).
Next, the effect of RSV infection on the noncarcinoma BEAS-2B bronchial epithelial cell line was determined. RSV infection (MOI = 3) repressed dexamethasone induction of GILZ compared with mock infection. In these cells, the RSV repression of dexamethasone-induced GILZ was not quite significant when compared with mock infected, although it followed a similar trend (33-fold increase and EC50 3.1 ± 1 nm for mock and 25-fold increase and EC50 2.3 ± 1.8 nm for RSV infected). Again, UV irradiation of RSV before infection inhibited its effects on dexamethasone induction of GILZ (35-fold increase and EC50 2.8 ± 2.1 nm for UV-irradiated) (Fig. 4, A and B). Similar effects were seen for dexamethasone induction of MKP-1 (24-fold increase and EC50 2.1 ± 0.6 nm for mock, 8.5-fold increase and EC50 1.6 ± 0.6 nm for RSV, and 20-fold increase and EC50 2.3 ± 0.5 nm for UV irradiated) (Fig. 4, C and D). Interestingly, no effect of RSV infection on dexamethasone induction of FKBP51 was observed in these cells (data not shown), suggesting some cell-specific effects.
Fig. 4.

RSV infection repression of dexamethasone induction of endogenous GR-regulated genes in BEAS-2B cells is dependent on viral replication. BEAS-2B cells were mock inoculated with HeLa-conditioned media (closed square and black bar) or inoculated with live rgRSV (open square and white bar) or UV-irradiated rgRSV (open circle with dashed line and hatched bar) (MOI = 3) for 1 h followed by treatment with dexamethasone for 5 h. Dexamethasone induction of GILZ (A and B) and MKP-1 (C and D) was determined by real-time PCR and normalized to GAPDH and CAP-1. Means and sd are shown (n = 5).
Finally, the effect of RSV infection on primary SAECs was determined. Three different levels of RSV infection were tested. Infection with an MOI of 3 for 5 h, an MOI of 1 for 24 h, and an MOI of 48 for 48 h. Dexamethasone was added for the last 5 h only. RSV infection was confirmed by detection of GFP by fluorescence microscopy (data not shown). Dexamethasone induced GILZ mRNA by about 20- to 30-fold, which was repressed by RSV infection at all three MOIs to about 10-fold or less (Fig. 5, A, C, and E). In addition, dexamethasone induced FKBP51 mRNA about 15- to 30-fold, which was also repressed by RSV infection to 5- to 10-fold (Fig. 5, B, D, and F).
Fig. 5.

RSV infection repression of dexamethasone induction of endogenous GR-regulated genes in SAECs. SAECs were mock inoculated with HeLa-conditioned media (closed square) or inoculated with live rgRSV (open square) at an MOI of 3 (A and B), 1 (C and D), or 0.5 (E and F) for 1 h. After 1 h, the media were replaced and cells incubated for a further 5 h (A and B), 24 h (C and D), or 48 h (E and F). Increasing concentrations of dexamethasone were added for the last 5 h. RSV infection was determined by detected of GFP by fluorescent microscope (data not shown). Dexamethasone induction of GILZ (A, C, and E) and FKBP51 (B, D, and F) was determined by real-time PCR and normalized to GAPDH and CAP-1. Means of duplicate samples and EC50 values are shown.
Involvement of NFκB in repression of GR-regulated genes by RSV infection in A549 cells
A mutual antagonism exists between GR and NFκB, and RSV infection is known to activate NFκB, thereby inducing proinflammatory cytokines, such as IL-6 and IL-8 (31–36). Therefore, NFκB induction in infected cells may inhibit GR activity. To examine the role of NFκB in the repression of dexamethasone-induced genes during RSV infection, we used the specific NFκB inhibitors PDTC and BAY 11-7082. To confirm that RSV infection induced NFκB under our conditions and that the inhibitors used repressed NFκB, induction of IL-6 and IL-8 mRNA and protein was measured in A549 cells at 6 h after infection by real-time PCR and ELISA. Although there were experimental variations, RSV infection induced expression of IL-6 and IL-8 mRNA and protein in A549 cells compared with mock infection. Pretreatment with the NFκB inhibitors for 1 h before RSV inoculation prevented induction of IL-6 and IL-8 confirming the induction of NFκB by RSV (Supplemental Fig. 3). Interestingly, neither PDTC nor BAY 11-7082 reversed the RSV mediated reduction in dexamethasone induction of GR-regulated genes, GILZ, FKBP51, and MKP-1 (Fig. 6, A–F). These results suggest that NFκB is not required for the suppressive effects of RSV infection on GR-regulated gene expression.
Fig. 6.

Inhibitors of NFκB do not reverse the effects of RSV infection on GR-mediated gene activation. A549 cells were pretreated for 1 h with 50 μm PDTC (open circle and hatched bar) or 10 μm BAY 11-7082 (closed circle and stripped bar) and then either mock inoculated with HeLa-conditioned media (close square and black bar) or inoculated with rgRSV (open square and white bar, open circle and hatched bar, closed circle and stripped bar) (MOI = 3) for 1 h followed by treatment with dexamethasone for 5 h. Dexamethasone induction of GILZ (A and B), FKBP51 (C and D), and MKP-1 (E and F) was determined by real-time PCR and normalized to GAPDH and CAP-1. Means and sd are shown (n = 4).
To confirm the lack of involvement of NFκB in the RSV repression of GR, siRNA was employed to reduce the protein levels of the p65 subunit of NFκB. Transfection of p65 siRNA reduced the protein levels of the p65 NFκB protein almost completely (Fig. 7A). A control siRNA had no effect. In cells transfected with the p65 NFκB siRNA, RSV infection reduced dexamethasone induction of GILZ to a similar extent as for cells transfected with the control siRNA (Fig. 7, B and C). A similar result was seen for dexamethasone induction of FKBP51, except cells transfected with p65 NFκB siRNA had a slightly reduced dexamethasone induction of FKBP51 compared with cells transfected with the control siRNA (Fig. 7, D and E). Taken together, these data indicate that NFκB is not required for the repressive effects of RSV infection on GR-mediated gene activation.
Fig. 7.

p65 NFκB siRNA does not reverse the effects of RSV infection on GR-mediated gene activation. A549 cells were transfected with 10 nm NFκB p65 siRNA (open circle and open square) or with the Alexa Fluor 488 AllStars Negative Control siRNA (closed circle and closed square) and then either mock inoculated with HeLa-conditioned media (closed circle, open circle, and black bar) or inoculated with rgRSV (closed square, open square, and white bar) (MOI = 3) for 1 h followed by treatment with dexamethasone for 5 h. A, Western blotting showing loss of p65 protein with the NFκB p65 siRNA. B–E, Dexamethasone induction of GILZ (B and C) and FKBP51 (D and E) was determined by real-time PCR and normalized to GAPDH and CAP-1. Means and sd are shown (n = 3).
RSV reduces GR-DNA binding
To determine the mechanism by which RSV represses GR function, we sought to analyze various steps in the GR signaling pathway. Total GR protein was determined by Western blotting of whole-cell extracts. RSV infection had no effect on GR protein levels (Supplemental Fig. 4). GR nuclear translocation was determined by analysis of GR protein levels in nuclear and cytosolic extracts and by immunocytochemistry of noninfected and RSV-infected A549 cells. RSV infection did not alter GR nuclear translocation (Supplemental Fig. 5). Interestingly, RSV proteins were detected in both the cytoplasmic and nuclear compartments of RSV-infected cells (Supplemental Fig. 5). GR-DNA binding was analyzed using ChIP. Treatment with 100 nm dexamethasone increased binding of GR to the GILZ promoter 5-fold, to the FKBP51 promoter 4-fold, and to the MKP-1 promoter 3.5-fold. Infection of the cells with RSV before dexamethasone reduced the binding of GR to the GILZ promoter to 3-fold (although not significant) and significantly abolished binding to the FKBP51 and MKP-1 promoters (Fig. 8). There was minimal pull-down of both these promoters with nonspecific immune serum, and this was not altered by RSV infection. A single product from the PCR was confirmed using a dissociation curve (Supplemental Fig. 6). Interestingly, no difference was seen in GR-DNA binding using EMSAs using nuclear extracts from RSV-infected or mock-infected cells (data not shown).
Fig. 8.
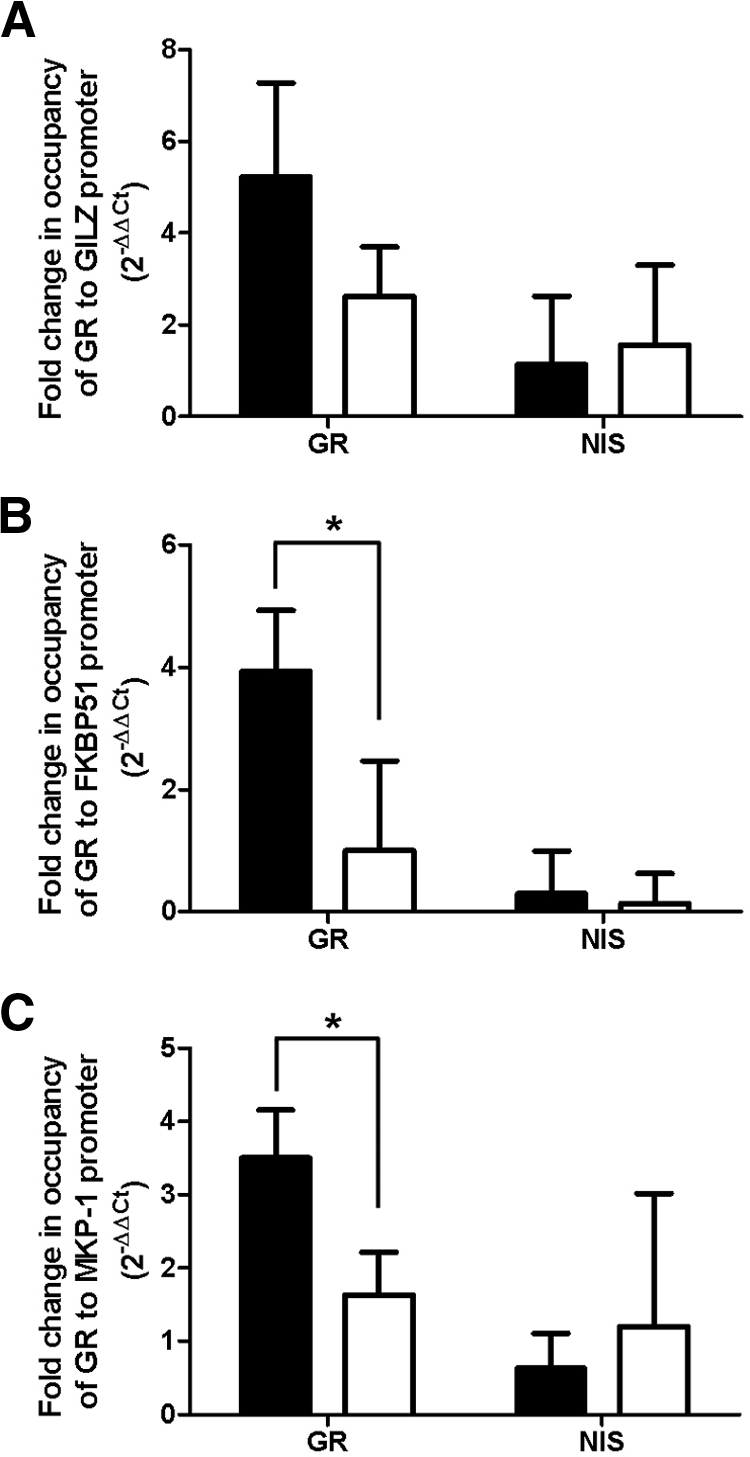
RSV infection reduces GR-DNA binding. A549 cells were mock inoculated with HeLa-conditioned media (black bar) or inoculated with live rgRSV (white bar) (MOI = 3) for 1 h followed by treatment with 100 nm dexamethasone for 5 h. Cells were fixed and ChIP performed using an antibody specific to GR or nonspecific immune serum (NIS). GR binding to the GILZ (A), FKBP51 (B), and MKP-1 (C) promoters was determined by real-time PCR. A threshold of 0.1 was used. Means and sd are shown (n = 3).
Discussion
Glucocorticoids are produced in response to physical and psychological insults through activation of the hypothalamic-pituitary-adrenal axis. This axis and resultant glucocorticoid action is essential to survival from a number of bacterial, viral, and inflammatory insults. Interruption of this axis or inhibition of GR function is associated with increased mortality from these agents, which can be reversed by administration of glucocorticoids (reviewed in Ref. 15). Defects in the GR signaling pathway resulting in glucocorticoid resistance/insensitivity exist in a number of inflammatory diseases (8–11), and there is clinical evidence to suggest that RSV may similarly induce glucocorticoid resistance. Indeed, with few exceptions (18), the majority of clinical studies has shown that glucocorticoids are ineffective against RSV-induced bronchiolitis (16, 17).
The ability of glucocorticoids to repress RSV-induced cytokine production in cell culture has been investigated, although with conflicting results. Several groups have shown that the glucocorticoids dexamethasone and fluticasone propionate can repress RSV induction of IL-11 mRNA, as well as IL-8, Regulated upon Activation, Normal T-cell Expressed and Secreted, and macrophage inflammatory protein 1α protein levels in a variety of cell types including A549 cells, MRC-5 cells (lung fibroblasts), and neutrophils (20, 21). However, others have shown that glucocorticoids do not repress RSV induction of IL-8, monocyte chemotactic protein-1, macrophage inflammatory protein 1α, and Regulated upon Activation, Normal T-cell Expressed, and Secreted in A549 cells or normal small airway human bronchial epithelial cells (22, 23). The reasons for the apparent discrepancies in the literature are unknown. However, our findings tend to support those of Bonville et al. (22) and Carpenter et al. (23), because we also found that RSV induced insensitivity to glucocorticoids (Fig. 1).
Although glucocorticoids mediate many of their antiinflammatory actions through inhibition of the NFκB and activator protein-1 signaling pathways resulting in down-regulation of inflammatory mediators (7), glucocorticoids also activate target genes through GR (7, 37, 38). Some of these target genes also play a role in the regulation of inflammation (38). There are multiple examples of inflammation-related genes whose expression is activated through GR. These include GILZ, which has been shown to interact with NFκB and impair its function (39–41) and also impair activator protein-1 (39, 42); MKP-1 (also known as dual specific phosphatase 1), which dephosphorylates and inactivates members of the MAPK family, thereby inhibiting the expression of several inflammatory genes (43–46); and recently has been shown to play a role in NFκB signaling (47). We have investigated the effect of RSV infection on glucocorticoid induction of GR-regulated genes and have shown that RSV infection represses dexamethasone induction of the GR-activated genes, GILZ, FKBP51, and MKP-1, in A549 and BEAS-2B cells and SAECs, without altering the EC50 values and that this repression is dependent on viral replication (Figs. 3–5). Interestingly, there is some evidence of cell specific effects, i.e. dexamethasone induction of FKBP51 is repressed by RSV infection in A549 cells but not in BEAS-2B cells. The mechanisms behind these effects are unknown and are currently under investigation.
RSV is known to activate NFκB (31–36). A mutual antagonism has been described between GR and NFκB, whereby NFκB represses GR and vice versa (48, 49). The simplest mechanism explaining the repressive effects of RSV on GR-mediated gene activation would be that RSV infection results in activation of NFκB, which, in turn, would repress GR function. If this were the case, then inhibition of NFκB would alleviate the repressive effects of RSV infection on GR. However, as shown in Figs. 6 and 7, inhibition of NFκB either by specific inhibitors or siRNA does not alter RSV repression of dexamethasone-induced genes, suggesting that the effect of RSV infection on GR is independent of NFκB. Analysis of the pathway involved in GR-mediated gene activation shows that RSV infection does not alter GR protein levels (Supplemental Fig. 4) or nuclear translocation (Supplemental Fig. 5) but does reduce GR binding to the FKBP51 and MKP-1 promoter and to a small extent the GILZ promoter (Fig. 8). Interestingly, RSV infection does not reduce GR-DNA binding in vitro, as determined using EMSA, which suggests that chromatin context of the promoter may play a role in these effects. Taken together, these data suggest that RSV infection inhibits GR-mediated gene activation by reducing GR-DNA binding independent of the NFκB pathway.
Very little is known about the interaction of infectious agents and the GR signaling pathway. In some cases, an interaction has been shown but not fully investigated [for a review, see Webster and Sternberg (15)]. Interestingly, the effect of RSV infection on GR-DNA is remarkably similar to our previous data for the anthrax lethal toxin (13, 14). It is possible that a similar pathway is targeted by both these agents, but further investigation is needed to determine the exact mechanism. One intriguing possibility is that these agents are not targeting GR directly but may be altering chromatin structure, thereby reducing GR accessibility to the DNA. Some virally encoded enzymes are known to have chromatin remodeling actions (50, 51). However, RSV replicates in the cytoplasm and the effects observed on GR-DNA binding occur in the nucleus. There are several possibilities that should be considered. Firstly, although we have shown that the effects of RSV infection on GR-mediated gene activation are independent of NFκB, RSV infection also induces other signaling pathways, such as MAPK, phosphatidylinositol 3 kinase, protein kinase C, and signal transducer and activator of transcription (52–60). It is possible that an interaction between these signaling pathways and GR could result in RSV-mediated repression of GR function. Secondly, GR is subject to posttranslational modifications (61), and it is feasible that such modifications could reduce GR-DNA binding but not affect nuclear translocation. Finally, the matrix (M) protein is the only RSV protein that has been shown to be present in the host cell nucleus (62, 63), and it has been described to prevent global transcription (63). Interestingly, the M proteins of the related cytoplasmic replicating viruses, vesicular stomatitis virus, and Newcastle disease virus also are found in the nucleus of infected cells (64–66). The vesicular stomatitis virus M protein inhibits host cell RNA transcription (67, 68). Thus, there is the possibility that there is an interaction between viral M proteins and the host transcriptional machinery. These potential mechanisms are currently under investigation.
In summary, we have shown that RSV infection inhibits GR-mediated gene activation in lung epithelial cells through a mechanism that requires viral replication, reduces GR-DNA binding, but is independent of the classical NFκB signaling pathway. This inhibition of GR function may explain why glucocorticoids are generally ineffective in the treatment of RSV-induced bronchiolitis in infants. Together with our previously published data (13, 14), these data suggest that GR may be a target for multiple infectious agents.
Supplementary Material
Acknowledgments
Recombinant GFP-expressing RSV was kindly provided by Mark Peeples (The Research Institute at Nationwide Children's Hospital, Department of Pediatrics, The Ohio State University) and by Peter Collins (National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD). Primer sequences for GAPDH, CAP-1, IL-6, and IL-8 were kindly provided by Mikhail Gavrilin (The Ohio State University Medical Center).
Present address for A.M.C.: Booz Allen Hamilton, Suite 750, 4001 Fairfax Drive, Arlington, Virginia 22203.
Present address for J.A.: Lake Erie College of Osteopathic Medicine, 5000 Lakewood Ranch Boulevard, Bradenton, Florida 34211.
This work was supported by the National Institutes of Health Grant T32 AI554411 (to A.M.C.) and the National Center for Research Resources Grant RR-17626 (to I.C.D.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- Amax
- Maximal activity
- CAP-1
- cAMP-accessory protein
- ChIP
- chromatin immunoprecipitation
- CSS
- charcoal-stripped serum
- CT
- cycle threshold
- FBS
- fetal bovine serum
- FKBP51
- FK506 binding protein 51
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- GFP
- green fluorescent protein
- GILZ
- glucocorticoid-inducible leucine zipper
- GR
- glucocorticoid receptor
- HRP
- horseradish peroxidase
- M
- matrix
- MKP-1
- MAPK phosphatase 1
- MMTV
- mouse mammary tumor virus
- MOI
- multiplicity of infection
- NFκB
- nuclear factor κB
- PBST
- PBS containing 0.05% Tween 20
- PDTC
- pyrrolidine dithiocarbamate
- rgRSV
- recombinant GFP-expressing RSV
- RSV
- respiratory syncytial virus
- SAEC
- small airway epithelial cell
- siRNA
- small interfering RNA
- TBST
- Tris-buffered saline with 0.05% Tween 20.
References
- 1. Henrickson KJ, Hoover S, Kehl KS, Hua W. 2004. National disease burden of respiratory viruses detected in children by polymerase chain reaction. Pediatr Infect Dis J 23:S11–S18 [DOI] [PubMed] [Google Scholar]
- 2. Hall CB, Weinberg GA, Iwane MK, Blumkin AK, Edwards KM, Staat MA, Auinger P, Griffin MR, Poehling KA, Erdman D, Grijalva CG, Zhu Y, Szilagyi P. 2009. The burden of respiratory syncytial virus infection in young children. N Engl J Med 360:588–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Power UF. 2008. Respiratory syncytial virus (RSV) vaccines—two steps back for one leap forward. J Clin Virol 41:38–44 [DOI] [PubMed] [Google Scholar]
- 4. Walsh EE, Falsey AR, Hennessey PA. 1999. Respiratory syncytial and other virus infections in persons with chronic cardiopulmonary disease. Am J Respir Crit Care Med 160:791–795 [DOI] [PubMed] [Google Scholar]
- 5. Raboni SM, Nogueira MB, Tsuchiya LR, Takahashi GA, Pereira LA, Pasquini R, Siqueira MM. 2003. Respiratory tract viral infections in bone marrow transplant patients. Transplantation 76:142–146 [DOI] [PubMed] [Google Scholar]
- 6. Falsey AR, Walsh EE. 2005. Respiratory syncytial virus infection in elderly adults. Drugs Aging 22:577–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Webster JI, Tonelli L, Sternberg EM. 2002. Neuroendocrine regulation of immunity. Annu Rev Immunol 20:125–163 [DOI] [PubMed] [Google Scholar]
- 8. Leung DY, de Castro M, Szefler SJ, Chrousos GP. 1998. Mechanisms of glucocorticoid-resistant asthma. Ann NY Acad Sci 840:735–746 [DOI] [PubMed] [Google Scholar]
- 9. Hearing SD, Norman M, Probert CS, Haslam N, Dayan CM. 1999. Predicting therapeutic outcome in severe ulcerative colitis by measuring in vitro steroid sensitivity of proliferating peripheral blood lymphocytes. Gut 45:382–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Seki M, Ushiyama C, Seta N, Abe K, Fukazawa T, Asakawa J, Takasaki Y, Hashimoto H. 1998. Apoptosis of lymphocytes induced by glucocorticoids and relationship to therapeutic efficacy in patients with systemic lupus erythematosus. Arthritis Rheum 41:823–830 [DOI] [PubMed] [Google Scholar]
- 11. van Schaardenburg D, Valkema R, Dijkmans BA, Papapoulos S, Zwinderman AH, Han KH, Pauwels EK, Breedveld FC. 1995. Prednisone treatment of elderly-onset rheumatoid arthritis. Disease activity and bone mass in comparison with chloroquine treatment. Arthritis Rheum 38:334–342 [DOI] [PubMed] [Google Scholar]
- 12. Hearing SD, Norman M, Smyth C, Foy C, Dayan CM. 1999. Wide variation in lymphocyte steroid sensitivity among healthy human volunteers. J Clin Endocrinol Metab 84:4149–4154 [DOI] [PubMed] [Google Scholar]
- 13. Webster JI, Sternberg EM. 2005. Anthrax lethal toxin represses glucocorticoid receptor (GR) transactivation by inhibiting GR-DNA binding in vivo. Mol Cell Endocrinol 241:21–31 [DOI] [PubMed] [Google Scholar]
- 14. Webster JI, Tonelli LH, Moayeri M, Simons SS, Jr, Leppla SH, Sternberg EM. 2003. Anthrax lethal factor represses glucocorticoid and progesterone receptor activity. Proc Natl Acad Sci USA 100:5706–5711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Webster JI, Sternberg EM. 2004. Role of the hypothalamic-pituitary-adrenal axis, glucocorticoids and glucocorticoid receptors in toxic sequelae of exposure to bacterial and viral products. J Endocrinol 181:207–221 [DOI] [PubMed] [Google Scholar]
- 16. Panickar J, Lakhanpaul M, Lambert PC, Kenia P, Stephenson T, Smyth A, Grigg J. 2009. Oral prednisolone for preschool children with acute virus-induced wheezing. N Engl J Med 360:329–338 [DOI] [PubMed] [Google Scholar]
- 17. Ermers MJ, Rovers MM, van Woensel JB, Kimpen JL, Bont LJ. 2009. The effect of high dose inhaled corticosteroids on wheeze in infants after respiratory syncytial virus infection: randomised double blind placebo controlled trial. BMJ 338:b897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. van Woensel JB, Wolfs TF, van Aalderen WM, Brand PL, Kimpen JL. 1997. Randomised double blind placebo controlled trial of prednisolone in children admitted to hospital with respiratory syncytial virus bronchiolitis. Thorax 52:634–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. van Woensel JB, Lutter R, Biezeveld MH, Dekker T, Nijhuis M, van Aalderen WM, Kuijpers TW. 2003. Effect of dexamethasone on tracheal viral load and interleukin-8 tracheal concentration in children with respiratory syncytial virus infection. Pediatr Infect Dis J 22:721–726 [DOI] [PubMed] [Google Scholar]
- 20. Wang J, Zhu Z, Nolfo R, Elias JA. 1999. Dexamethasone regulation of lung epithelial cell and fibroblast interleukin-11 production. Am J Physiol 276:L175–L185 [DOI] [PubMed] [Google Scholar]
- 21. Jaovisidha P, Peeples ME, Brees AA, Carpenter LR, Moy JN. 1999. Respiratory syncytial virus stimulates neutrophil degranulation and chemokine release. J Immunol 163:2816–2820 [PubMed] [Google Scholar]
- 22. Bonville CA, Mehta PA, Krilov LR, Rosenberg HF, Domachowske JB. 2001. Epithelial cells infected with respiratory syncytial virus are resistant to the anti-inflammatory effects of hydrocortisone. Cell Immunol 213:134–140 [DOI] [PubMed] [Google Scholar]
- 23. Carpenter LR, Moy JN, Roebuck KA. 2002. Respiratory syncytial virus and TNF α induction of chemokine gene expression involves differential activation of Rel A and NF-κB1. BMC Infect Dis 2:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huang YC, Li Z, Hyseni X, Schmitt M, Devlin RB, Karoly ED, Soukup JM. 2008. Identification of gene biomarkers for respiratory syncytial virus infection in a bronchial epithelial cell line. Genomic Med 2:113–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hallak LK, Spillmann D, Collins PL, Peeples ME. 2000. Glycosaminoglycan sulfation requirements for respiratory syncytial virus infection. J Virol 74:10508–10513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gavrilin MA, Bouakl IJ, Knatz NL, Duncan MD, Hall MW, Gunn JS, Wewers MD. 2006. Internalization and phagosome escape required for Francisella to induce human monocyte IL-1β processing and release. Proc Natl Acad Sci USA 103:141–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nissen RM, Yamamoto KR. 2000. The glucocorticoid receptor inhibits NFκB by interfering with serine-2 phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes Dev 14:2314–2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang JC, Derynck MK, Nonaka DF, Khodabakhsh DB, Haqq C, Yamamoto KR. 2004. Chromatin immunoprecipitation (ChIP) scanning identifies primary glucocorticoid receptor target genes. Proc Natl Acad Sci USA 101:15603–15608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hubler TR, Scammell JG. 2004. Intronic hormone response elements mediate regulation of FKBP5 by progestins and glucocorticoids. Cell Stress Chaperones 9:243–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Johansson-Haque K, Palanichamy E, Okret S. 2008. Stimulation of MAPK-phosphatase 1 gene expression by glucocorticoids occurs through a tethering mechanism involving C/EBP. J Mol Endocrinol 41:239–249 [DOI] [PubMed] [Google Scholar]
- 31. Bitko V, Barik S. 1998. Persistent activation of RelA by respiratory syncytial virus involves protein kinase C, underphosphorylated IκBβ, and sequestration of protein phosphatase 2A by the viral phosphoprotein. J Virol 72:5610–5618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chini BA, Fiedler MA, Milligan L, Hopkins T, Stark JM. 1998. Essential roles of NF-κB and C/EBP in the regulation of intercellular adhesion molecule-1 after respiratory syncytial virus infection of human respiratory epithelial cell cultures. J Virol 72:1623–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Choudhary S, Boldogh S, Garofalo R, Jamaluddin M, Brasier AR. 2005. Respiratory syncytial virus influences NF-κB-dependent gene expression through a novel pathway involving MAP3K14/NIK expression and nuclear complex formation with NF-κB2. J Virol 79:8948–8959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fiedler MA, Wernke-Dollries K, Stark JM. 1996. Inhibition of viral replication reverses respiratory syncytial virus-induced NF-κB activation and interleukin-8 gene expression in A549 cells. J Virol 70:9079–9082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Garofalo R, Sabry M, Jamaluddin M, Yu RK, Casola A, Ogra PL, Brasier AR. 1996. Transcriptional activation of the interleukin-8 gene by respiratory syncytial virus infection in alveolar epithelial cells: nuclear translocation of the RelA transcription factor as a mechanism producing airway mucosal inflammation. J Virol 70:8773–8781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Haeberle HA, Takizawa R, Casola A, Brasier AR, Dieterich HJ, Van Rooijen N, Gatalica Z, Garofalo RP. 2002. Respiratory syncytial virus-induced activation of nuclear factor-κB in the lung involves alveolar macrophages and toll-like receptor 4-dependent pathways. J Infect Dis 186:1199–1206 [DOI] [PubMed] [Google Scholar]
- 37. Aranda A, Pascual A. 2001. Nuclear hormone receptors and gene expression. Physiol Rev 81:1269–1304 [DOI] [PubMed] [Google Scholar]
- 38. Clark AR. 2007. Anti-inflammatory functions of glucocorticoid-induced genes. Mol Cell Endocrinol 275:79–97 [DOI] [PubMed] [Google Scholar]
- 39. Ayroldi E, Riccardi C. 2009. Glucocorticoid-induced leucine zipper (GILZ): a new important mediator of glucocorticoid action. FASEB J 23:3649–3658 [DOI] [PubMed] [Google Scholar]
- 40. Yang N, Zhang W, Shi XM. 2008. Glucocorticoid-induced leucine zipper (GILZ) mediates glucocorticoid action and inhibits inflammatory cytokine-induced COX-2 expression. J Cell Biochem 103:1760–1771 [DOI] [PubMed] [Google Scholar]
- 41. Eddleston J, Herschbach J, Wagelie-Steffen AL, Christiansen SC, Zuraw BL. 2007. The anti-inflammatory effect of glucocorticoids is mediated by glucocorticoid-induced leucine zipper in epithelial cells. J Allergy Clin Immunol 119:115–122 [DOI] [PubMed] [Google Scholar]
- 42. Mittelstadt PR, Ashwell JD. 2001. Inhibition of AP-1 by the glucocorticoid-inducible protein GILZ. J Biol Chem 276:29603–29610 [DOI] [PubMed] [Google Scholar]
- 43. Shepherd EG, Zhao Q, Welty SE, Hansen TN, Smith CV, Liu Y. 2004. The function of mitogen-activated protein kinase phosphatase-1 in peptidoglycan-stimulated macrophages. J Biol Chem 279:54023–54031 [DOI] [PubMed] [Google Scholar]
- 44. Chen P, Li J, Barnes J, Kokkonen GC, Lee JC, Liu Y. 2002. Restraint of proinflammatory cytokine biosynthesis by mitogen-activated protein kinase phosphatase-1 in lipopolysaccharide-stimulated macrophages. J Immunol 169:6408–6416 [DOI] [PubMed] [Google Scholar]
- 45. Li L, Chen SF, Liu Y. 2009. MAP kinase phosphatase-1, a critical negative regulator of the innate immune response. Int J Clin Exp Med 2:48–67 [PMC free article] [PubMed] [Google Scholar]
- 46. Zhao Q, Shepherd EG, Manson ME, Nelin LD, Sorokin A, Liu Y. 2005. The role of mitogen-activated protein kinase phosphatase-1 in the response of alveolar macrophages to lipopolysaccharide: attenuation of proinflammatory cytokine biosynthesis via feedback control of p38. J Biol Chem 280:8101–8108 [DOI] [PubMed] [Google Scholar]
- 47. King EM, Holden NS, Gong W, Rider CF, Newton R. 2009. Inhibition of NF-κB-dependent transcription by MKP-1: transcriptional repression by glucocorticoids occurring via p38 MAPK. J Biol Chem 284:26803–26815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Caldenhoven E, Liden J, Wissink S, Van de Stolpe A, Raaijmakers J, Koenderman L, Okret S, Gustafsson JA, Van der Saag PT. 1995. Negative cross-talk between RelA and the glucocorticoid receptor: a possible mechanism for the antiinflammatory action of glucocorticoids. Mol Endocrinol 9:401–412 [DOI] [PubMed] [Google Scholar]
- 49. McKay LI, Cidlowski JA. 1998. Cross-talk between nuclear factor-κB and the steroid hormone receptors: mechanisms of mutual antagonism. Mol Endocrinol 12:45–56 [DOI] [PubMed] [Google Scholar]
- 50. Lilley CE, Chaurushiya MS, Weitzman MD. 2010. Chromatin at the intersection of viral infection and DNA damage. Biochim Biophys Acta 1799:319–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wei H, Zhou MM. 2010. Viral-encoded enzymes that target host chromatin functions. Biochim Biophys Acta 1799:296–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Arnold R, König W. 2005. Respiratory syncytial virus infection of human lung endothelial cells enhances selectively intercellular adhesion molecule-1 expression. J Immunol 174:7359–7367 [DOI] [PubMed] [Google Scholar]
- 53. Chen W, Monick MM, Carter AB, Hunninghake GW. 2000. Activation of ERK2 by respiratory syncytial virus in A549 cells is linked to the production of interleukin 8. Exp Lung Res 26:13–26 [DOI] [PubMed] [Google Scholar]
- 54. Marchant D, Singhera GK, Utokaparch S, Hackett TL, Boyd JH, Luo Z, Si X, Dorscheid DR, McManus BM, Hegele RG. 2010. Toll-like receptor 4-mediated activation of p38 mitogen-activated protein kinase is a determinant of respiratory virus entry and tropism. J Virol 84:11359–11373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Meusel TR, Imani F. 2003. Viral induction of inflammatory cytokines in human epithelial cells follows a p38 mitogen-activated protein kinase-dependent but NF-κB-independent pathway. J Immunol 171:3768–3774 [DOI] [PubMed] [Google Scholar]
- 56. Singh D, McCann KL, Imani F. 2007. MAPK and heat shock protein 27 activation are associated with respiratory syncytial virus induction of human bronchial epithelial monolayer disruption. Am J Physiol Lung Cell Mol Physiol 293:L436–L445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ennaciri J, Ahmad R, Menezes J. 2007. Interaction of monocytic cells with respiratory syncytial virus results in activation of NF-κB and PKC-α/β leading to up-regulation of IL-15 gene expression. J Leukoc Biol 81:625–631 [DOI] [PubMed] [Google Scholar]
- 58. Kong X, San Juan H, Kumar M, Behera AK, Mohapatra A, Hellermann GR, Mane S, Lockey RF, Mohapatra SS. 2003. Respiratory syncytial virus infection activates STAT signaling in human epithelial cells. Biochem Biophys Res Commun 306:616–622 [DOI] [PubMed] [Google Scholar]
- 59. Lindemans CA, Coffer PJ, Schellens IM, de Graaff PM, Kimpen JL, Koenderman L. 2006. Respiratory syncytial virus inhibits granulocyte apoptosis through a phosphatidylinositol 3-kinase and NF-κB-dependent mechanism. J Immunol 176:5529–5537 [DOI] [PubMed] [Google Scholar]
- 60. Monick M, Staber J, Thomas K, Hunninghake G. 2001. Respiratory syncytial virus infection results in activation of multiple protein kinase C isoforms leading to activation of mitogen-activated protein kinase. J Immunol 166:2681–2687 [DOI] [PubMed] [Google Scholar]
- 61. Duma D, Jewell CM, Cidlowski JA. 2006. Multiple glucocorticoid receptor isoforms and mechanisms of post-translational modification. J Steroid Biochem Mol Biol 102:11–21 [DOI] [PubMed] [Google Scholar]
- 62. Ghildyal R, Mills J, Murray M, Vardaxis N, Meanger J. 2002. Respiratory syncytial virus matrix protein associates with nucleocapsids in infected cells. J Gen Virol 83:753–757 [DOI] [PubMed] [Google Scholar]
- 63. Ghildyal R, Baulch-Brown C, Mills J, Meanger J. 2003. The matrix protein of human respiratory syncytial virus localises to the nucleus of infected cells and inhibits transcription. Arch Virol 148:1419–1429 [DOI] [PubMed] [Google Scholar]
- 64. Lyles DS, Puddington L, McCreedy BJ., Jr 1988. Vesicular stomatitis virus M protein in the nuclei of infected cells. J Virol 62:4387–4392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Peeples ME. 1988. Differential detergent treatment allows immunofluorescent localization of the Newcastle disease virus matrix protein within the nucleus of infected cells. Virology 162:255–259 [DOI] [PubMed] [Google Scholar]
- 66. Peeples ME, Wang C, Gupta KC, Coleman N. 1992. Nuclear entry and nucleolar localization of the Newcastle disease virus (NDV) matrix protein occur early in infection and do not require other NDV proteins. J Virol 66:3263–3269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Black BL, Lyles DS. 1992. Vesicular stomatitis virus matrix protein inhibits host cell-directed transcription of target genes in vivo. J Virol 66:4058–4064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ferran MC, Lucas-Lenard JM. 1997. The vesicular stomatitis virus matrix protein inhibits transcription from the human β interferon promoter. J Virol 71:371–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Smit P, Russcher H, de Jong FH, Brinkmann AO, Lamberts SW, Koper JW. 2005. Differential regulation of synthetic glucocorticoids on gene expression levels of glucocorticoid-induced leucine zipper and interleukin-2. J Clin Endocrinol Metab 90:2994–3000 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.