

Summary
The VAMP-associated proteins termed VAP are a small gene family of proteins characterised by the presence of an N-terminal major sperm protein (MSP) domain. The P56S mutation of the B isoform (VAPB) has been linked to late-onset amyotrophic lateral sclerosis (ALS8) and its expression causes formation of large ER aggregates. Overexpression of the wild-type A isoform (VAPA) but not the B isoform (VAPB), inhibited ER-to-Golgi transport of membrane proteins. This transport block by VAPA was primarily due to decreased segregation of membrane cargo into ER vesicles. We also found that VAPA inhibited lateral diffusion of membrane proteins, most likely through its stable association with microtubules. The MSP domain of VAP is known to interact with the FFAT motif (two phenylalanines in an acidic tract) of proteins involved in sterol regulation. Overexpression of FFAT restored ER-to-Golgi transport and lateral diffusion of membrane proteins, and resolved the large ER aggregates in VAPB-P56S. Application of a FFAT peptide restored in vitro ER vesicle budding and disrupted VAP-microtubule association. Thus, overexpression of the two VAP isoforms causes retention of ER membrane proteins by impeding lateral diffusion and their incorporation into transport vesicles. This inhibitory effect can be relieved by expression of the FFAT motif.
Keywords: VAPA, VAPB, FFAT motif, Endoplasmic reticulum, ER budding, Lateral diffusion, Microtubule
Introduction
The endoplasmic reticulum (ER) is the site of a diverse array of cellular processes. It is an entry point for lipid biosynthesis as well as for secretory and membrane proteins, which are translocated through the ER membrane where they undergo further post-translational modification. They are also subjected to ER quality control mechanisms to ensure proper folding and assembly into higher order complexes. Proteins destined for other intracellular organelles are sequestered into ER exit sites through a passive or selective process before their eventual incorporation as cargoes of coated transport vesicles. Because membrane proteins are confined within the two-dimensional lattice of the membrane, their lateral diffusion to designated ER exit sites can influence the efficiency of the sorting process. In this context, interconnectivity of ER tubules is vital to the movement and sorting of cargo molecules. The characteristic reticular tubular pattern of the ER is highly dependent on association with the microtubule network, which provides stable anchor points for motor and non-motor proteins. Motor proteins such as kinesin and dynein provide the mechanical force for ER tubule extension along microtubules whereas non-motor proteins, such as cytoskeleton-associated protein 4 (CKAP4; also known as CLIMP63), p22 and vesicle-associated membrane protein (VAMP)-associated protein (VAP), anchor the ER membranes to microtubules for stability (Vedrenne and Hauri, 2006). Disruption of these anchor points can lead to collapse of peripheral ER tubules into disorganised membrane sheets near the centre of the cell as well as reorganisation of the microtubule network because of the close relationship between the ER membrane and microtubules (Andrade et al., 2004). It remains unclear whether the non-motor proteins are confined to specific ER subdomains or whether they play a similar role with respect to their interaction with the microtubule network and maintenance of ER morphology.
There are two VAP genes in humans, designated VAPA and VAPB. VAPA was initially identified as a VAMP-binding protein (Skehel et al., 1995), and VAPB was identified as ERG30, a protein involved in COPI-coated vesicle transport (Soussan et al., 1999). The VAPA and VAPB isoforms are type II membrane proteins and are structurally similar with an N-terminal major sperm protein (MSP) domain, a central coiled-coil region and a C-terminal transmembrane domain that anchors the protein to the ER membrane. A P56S mutation within the highly conserved MSP domain of VAPB has been linked to a late-onset form of amyotrophic lateral sclerosis (Nishimura et al., 2004). This mutation is predicted to introduce a kink between two short stretches of β strands, and causes insolubility and accumulation of large ER aggregates in cells (Kanekura et al., 2006). The VAPA and VAPB genes are broadly expressed (Nishimura et al., 1999; Weir et al., 1998), suggesting they perform a function required by all cells. VAPA is primarily localised to the early secretory pathway where it colocalises with ER markers and microtubules (Skehel et al., 2000), but it is also found in tight junctions, late secretory and synaptic vesicles (Lapierre et al., 1999; Nishimura et al., 1999; Pennetta et al., 2002; Weir et al., 1998). Although studies have implicated VAPA in late secretory events, its localisation in the ER suggests a more prominent role in early secretory compartments.
VAPA is conserved through evolution. Its yeast orthologue, SCS2, is a suppressor of ire15 inositol auxotrophic phenotype (Nikawa et al., 1995) and choline-sensitivity of cse1 (Hosaka et al., 1992). However, mammalian VAPA can only partially complement SCS2, suggesting that functional conservation of the two proteins may be limited (Loewen and Levine, 2005). This may reflect the lack of Opi1 in mammals, which in yeast interacts with Scs2 to regulate expression of a number of phospholipid biosynthetic genes (Daum, 2004). Mammalian VAPA interacts with a number of proteins with the largest category of interacting proteins involved in regulation of sterol and lipid biosynthesis or trafficking. This group comprises the oxysterol-binding protein (OSBP) and the oxysterol-binding protein-related protein (ORP) families (Wyles et al., 2002), the human orthologues of Drosophila retinal degeneration type B (rdgB) also known as NIR (Amarilio et al., 2005), and ceramide transport protein CERT (Kawano et al., 2006). They share a common motif known as FFAT (two phenylalanines in an acidic tract) (Loewen et al., 2003), which interacts with the MSP domain of VAP and is partially responsible for targeting the lipid binding proteins to the cytosolic surface of the ER (Wyles and Ridgway, 2004). Binding to the FFAT motif is tightly linked to function such that mutations disrupting FFAT interaction result in complete loss of function (Loewen and Levine, 2005). In addition to FFAT-containing proteins, VAPA also interacts with viral proteins (Ettayebi and Hardy, 2003; Hamamoto et al., 2005) whereby VAP modulates their ER-to-Golgi transport. Thus, VAPA is likely to function in trafficking of membrane proteins from the ER to Golgi complex in mammals.
Here we show that overexpression of VAPA but not VAPB blocks ER-to-Golgi transport by inhibiting incorporation of the anterograde membrane cargo VSVG into ER vesicles. The P56S mutation in VAPB but not in VAPA caused accumulation of large ER aggregates and loss of the characteristic reticular pattern of the ER. VAPA also inhibits lateral diffusion of membrane proteins in the ER. It associates with the polymerised microtubule network to stably anchor the ER. This stable interaction may inadvertently create immobile obstacles that inhibit lateral diffusion and sequestration of membrane proteins to ER exit sites. Finally, overexpression or application of the FFAT motif relieved the inhibitory effect of the wild-type protein and most of the effect of the P56S mutation.
Results
FFAT rescues the ER aggregation effect of VAPB-P56S
When epitope-tagged wild-type VAPA and VAPB were expressed in CHO fibroblasts, they exhibited a reticular pattern that colocalised with the ER marker Calreticulin (Fig. 1; supplementary material Fig. S1). Colocalisation analysis with other intracellular markers indicated that overexpression did not alter the overall morphology of other intracellular organelles, in particular, the Golgi complex remained unaffected (data not shown). Neither VAP isoform colocalised with Sar1 (supplementary material Fig. S2), an Arf-like GTPase involved in COPII coat formation, suggesting that VAPA and VAPB are not localised to ER exit sites. There also was no detectable change in the organisation of the microtubule network by any of the VAP constructs (supplementary material Fig. S3), in contrast to previous studies on VAPB (Amarilio et al., 2005) and Drosophila VAP (Pennetta et al., 2002). Interestingly, overexpression of the P56S mutant in VAPA showed no significant change in distribution of Calreticulin or overall ER morphology, although small, Calreticulin-positive aggregates were occasionally observed in <1% of the transfected cells (Table 1) in contrast to a previous study (Teuling et al., 2007). Overexpression of VAPB-P56S caused accumulation of large membranous aggregates with ~70% of the transfected cells exhibiting large aggregates, consistent with previous studies (Kanekura et al., 2006; Teuling et al., 2007). We also noticed a reduction in the characteristic reticular ER pattern in these cells. Calreticulin colocalised with these large membranous aggregates, suggesting that they originated in the ER. Since interaction with the FFAT motif is essential for VAPA function, we examined whether coexpression of FFAT altered VAP localisation and ER morphology. A FFAT-containing fragment of oxysterol-binding protein 1 (OSBP1) spanning residues 347–468 was subcloned into the mammalian pcDNA3.1(+) vector with a Myc tag to directly test the effect of FFAT without any potential influence from other OSBP1-related functions. Expression of this FFAT motif alone produced no detectable changes in ER morphology and microtubule organisation (data not shown), nor did it alter the phenotype of wild-type VAPA, VAPB and VAPA-P56S (Fig. 1). However, its coexpression with VAPB-P56S resolved most of the large membranous aggregates and partially restored the reticular ER pattern. We quantified the percentage of transfected cells exhibiting VAP-containing large aggregates (≥300 nm diameter) and found that coexpression of FFAT significantly reduced the number of cells with large membranous aggregates from 70.2±1.2% to 37.7±6.0% (Table 2). Thus, coexpression of a FFAT motif partially prevented or resolved the accumulation of large membranous aggregates and restored the reticular ER pattern in VAPB-P56S cells.
Fig. 1.
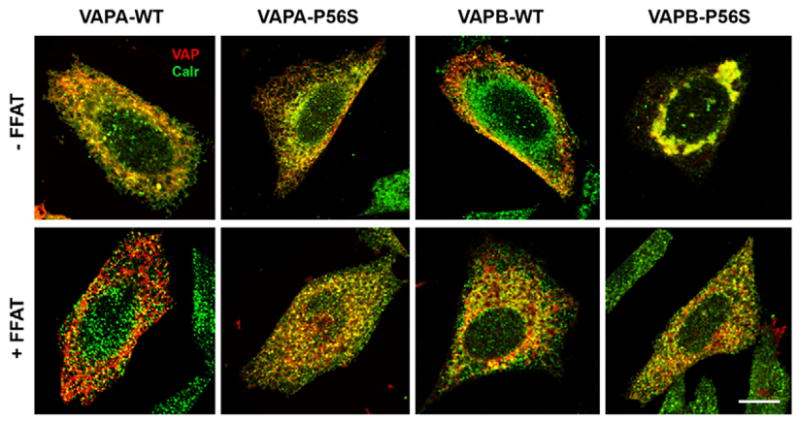
Subcellular localisation of WT and P56S VAPA and VAPB. CHO cells were transfected with either FLAG-tagged WT or P56S mutant in the absence of FFAT (top row) or with FFAT (bottom row). The proteins were visualised with anti-FLAG (red) and anti-Calreticulin (green). Scale bar: 10 μm.
Table 1.
Quantitative analysis of the effect of FFAT expression on ER aggregation
| Condition | % cells with ER aggregates |
|---|---|
| VAPA-WT | 0.7±0.4 |
| VAPA-WT + FFAT | 1.1±0.6 |
| VAPA-P56S | 0.9±0.4 |
| VAPA-P56S + FFAT | 1.9±1.2 |
| VAPB-WT | 4.3±2.8 |
| VAPB-WT + FFAT | 4.5±0.3 |
| VAPB-P56S | 70.2±1.2 |
| VAPB-P56S + FFAT | 37.7±6.0* |
Structures with a diameter of ≥300 nm were considered to be an ER aggregate. An average of 200 cells was counted for each condition and the values represent mean ± s.e.m. from three independent experiments.
P<0.05 when compared with VAPB-P56S without FFAT.
Table 2.
Effect of FFAT expression on fluorescence recovery rate
| Condition | Recovery rate (minute−1) |
|---|---|
| Control | 0.181±0.012 |
| Control + FFAT | 0.185±0.024 |
| VAPA(WT) | 0.091±0.013† |
| VAPA(WT) + FFAT | 0.198±0.011* |
| VAPA(P56S) | 0.046±0.008† |
| VAPA(P56S) + FFAT | 0.133±0.035 |
| VAPB(WT) | 0.210±0.005 |
| VAPB(WT) + FFAT | 0.156±0.014 |
| VAPB(P56S) | 0.014±0.011† |
| VAPB(P56S) + FFAT | 0.186±0.017* |
The recovery rate was calculated by linear regression from fluorescence values within the first 2 minutes of recovery.
P<0.05 compared with values in the absence of FFAT;
P<0.01 compared with control.
FFAT motif rescues VAPA inhibition of ER-to-Golgi transport
We used the temperature-sensitive VSVGts045-GFP as a marker to monitor the effect of VAP on anterograde intracellular transport. The folding-defective protein was trapped in the ER at 42°C for 5–6 hours and trafficking was restored upon temperature shift to 32°C. In control untransfected cells, VSVG was efficiently transported to the Golgi complex 30 minutes after release, and reached the plasma membrane in 60–90 minutes (Fig. 2; see supplementary material Fig. S4 for quantitative summary). Overexpression of VAPA-WT inhibited ER-to-Golgi transport of VSVG such that the protein largely remained in the ER at the 90 minute chase period, with only a small fraction evident at the plasma membrane after digital enhancement. VAPA-P56S similarly inhibited progression of VSVG through the secretory pathway, although larger vesicular structures were more prominent at the later time points. By contrast, overexpression of wild-type VAPB (VAPB-WT) had no effect on VSVG trafficking. VSVG was transported to the Golgi complex within 30 minutes after release and reached the plasma membrane with the same kinetics as untransfected cells, consistent with a previous study (Amarilio et al., 2005). VSVG was trapped in the large membranous aggregates after incubation at 42°C instead of the normal reticular, tubular ER pattern observed in VAPB-P56S-expressing cells. VSVG remained in these ER aggregates throughout the 90 minute chase period, suggesting that these ER aggregates are very stable structures with no significant turnover of trapped VSVG. Together, the data indicate that the two VAP isoforms differentially affect anterograde transport of VSVG and that anterograde membrane cargoes, such as VSVG, are retained in the large membranous aggregates in VAPB-P56S-expressing cells for an extended period of time.
Fig. 2.
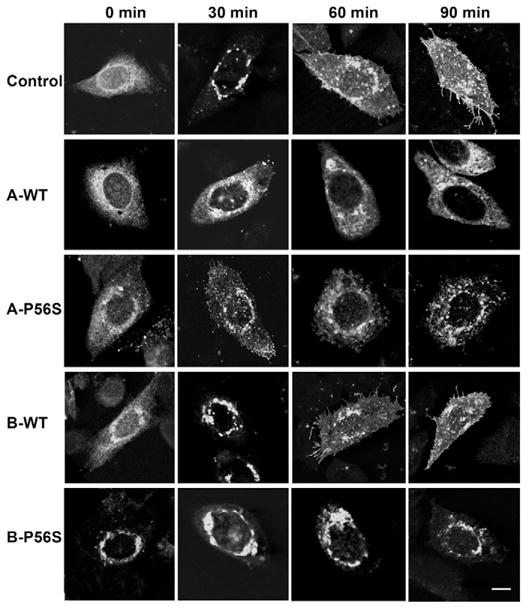
Time course of VSVGts045-GFP trafficking in CHO cells transfected with WT or mutant VAPA and VAPB in the absence of FFAT. VSVGts045-GFP was trapped in the ER at 42°C for 6 hours and shifted to 32°C. The cells were fixed with 4% paraformaldehyde at the indicated time (minutes). Scale bar: 10 μm.
Because coexpression of the FFAT motif restored the aggregation phenotype of VAPB-P56S, we next examined the effect of its coexpression on VSVG trafficking. Expression of FFAT alone had no effect on VSVG trafficking from the ER to plasma membrane (Fig. 3; supplementary material Fig. S4). However, its expression relieved the inhibition of ER-to-Golgi transport by VAPA-WT and VAPA-P56S mutant. In both cases, VSVG was transported to the Golgi complex within 30 minutes after release and reached the plasma membrane in roughly the same time course as control cells transfected with FFAT alone. Together, this indicates that ER-to-Golgi transport was the rate-limiting step affected by VAPA and interaction with FFAT effectively relieved this inhibition. Transport of VSVG was unaffected by FFAT in VAPB-WT-expressing cells. Interestingly, coexpression of FFAT resolved the ER aggregation phenotype and partially restored the reticular pattern of ER-trapped VSVG after incubation at 42°C in VAPB-P56S-expressing cells. Intracellular transport was restored upon shift to the permissive temperature with VSVG transported to the Golgi complex and plasma membrane in the same time course as control cells. Thus, FFAT expression not only resolved the ER aggregation phenotype but also functionally restored intracellular transport of VSVG to the plasma membrane in VAPB-P56S cells. This effect was observed only with FFAT, and coexpression of the corresponding AAAT sequence, whereby the two phenylalanines were substituted with alanines, had no effect.
Fig. 3.
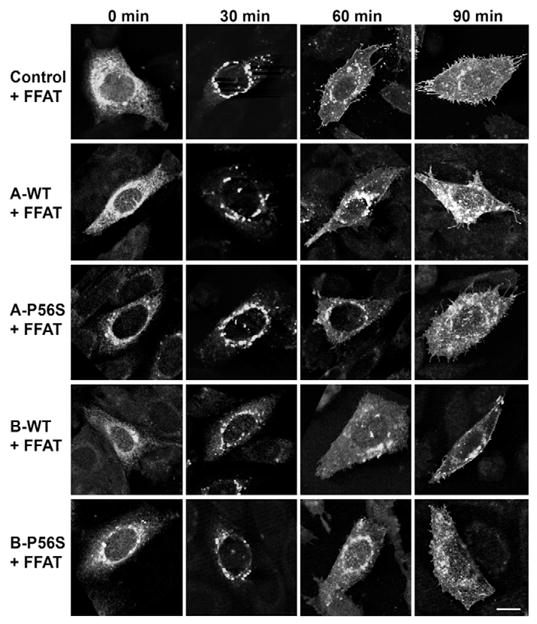
Time course of VSVGts045-GFP trafficking in CHO cells transfected with WT or mutant VAPA and VAPB in the presence of FFAT. The conditions were as described in Fig. 2. Scale bar: 10 μm.
VAPA and VAPB do not affect transport of soluble chromogranin B cargo
Inhibition of anterograde vesicle trafficking with the fungal metabolite brefeldin A causes disassembly of the Golgi complex (Fujiwara et al., 1988). However, the Golgi complex remained intact in our VAPA-transfected cells, suggesting that not all anterograde membrane trafficking was inhibited. To determine whether trafficking of a soluble ER cargo was also affected, we examined the transport of chromogranin B (CgB) fused to the temperature-sensitive GFP(S65T) variant, which shows low fluorescence at 37°C but is brightly fluorescent at 20°C or below (Wacker et al., 1997). Cells were incubated at 15°C for 2 hours to enhance fluorescence as well as to trap any cargo that has exited the ER in the ER-Golgi intermediate compartment (ERGIC) (Schweizer et al., 1991). Transport to the Golgi complex was then assessed by rapidly shifting to 37°C for 30 minutes. If VAPA also blocks ER exit of CgB-GFP(S65T), the activated fluorescent protein will remain in the ER during the 15°C treatment. Conversely, if ER exit is not affected, CgB-GFP(S65T) will be trapped in the ERGIC and subsequent release of the 15°C block will allow the trapped cargo to progress to the Golgi complex. Since a cargo cannot reach the ERGIC if its ER exit was blocked, only cargo that has exited the ER but trapped in the ERGIC will reach the Golgi complex. We found that CgB-GFP(S65T) was efficiently transported to the Golgi complex at 30 minutes in control CHO and VAP-transfected cells (Fig. 4; supplementary material Fig. S5). Strikingly, CgB-GFP(S65T) was also transported to the Golgi complex in VAPB-P56S mutant cells within the same time course as control cells. Under identical conditions, VSVG remains trapped in the ER in VAPA-transfected cells (data not shown). Thus, VAPA selectively inhibits anterograde transport of VSVG while transport of luminal CgB remains largely unaffected.
Fig. 4.
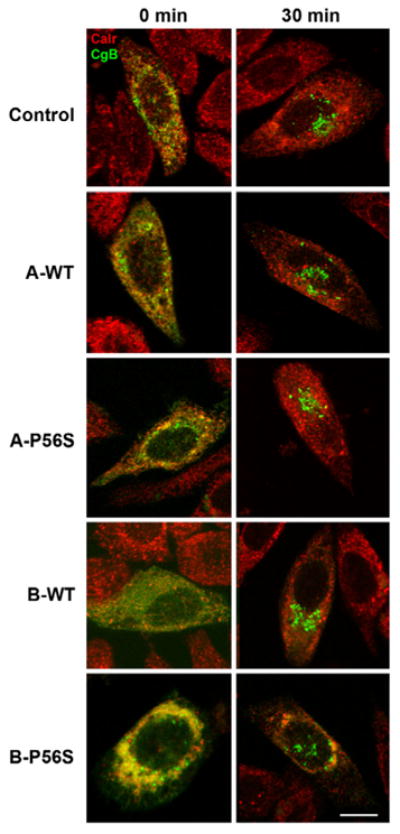
Effect of VAPA on transport of CgB-GFP(S65T). CHO cells were cotransfected with CgB-GFP(S65T) and control or VAPA (A) or VAPB (B) wild-type (WT) and mutant (P56S) vectors. The cells were incubated at 15°C for 2 hours and rapidly shifted to 37°C for 30 minutes before fixation and staining with anti-Calreticulin (Red). Scale bar: 10 μm.
Properly folded VSVG remains trapped in the ER
Proper folding and assembly is a key factor in ER exit of VSVG. To determine whether this might be a contributing factor in the inhibition of ER-to-Golgi transport by VAPA, we used the conformation-specific I14 antibody that only recognises properly folded VSVG (Lefrançois and Lyles, 1983) to determine whether VSVG folding or assembly was compromised. VSVGts045-GFP was clearly retained in the ER in the unfolded state after incubation at 42°C in control, VAPA-WT- and VAPB-WT-transfected cells as indicated by the prominent reticular GFP signal and absence of I14 antibody immunoreactivity (Fig. 5). VSVG was localised to the perinuclear Golgi complex and readily detected by the I14 antibody 30 minutes after release in control and VAPB-transfected cells. The extensive overlap between the GFP and I14 signals indicates effective anterograde transport of properly folded and assembled VSVG in these cells. By contrast, VSVGts045-GFP was retained in the ER compartment in a characteristic reticular pattern 30 minutes after release in VAPA-expressing cells. More importantly, the GFP signal overlapped with I14 immunoreactivity, indicating that VSVG was properly folded but retained in the ER. Thus, defective VSVG folding or assembly can be ruled out as a causative factor in VAPA-mediated inhibition of ER-to-Golgi transport.
Fig. 5.
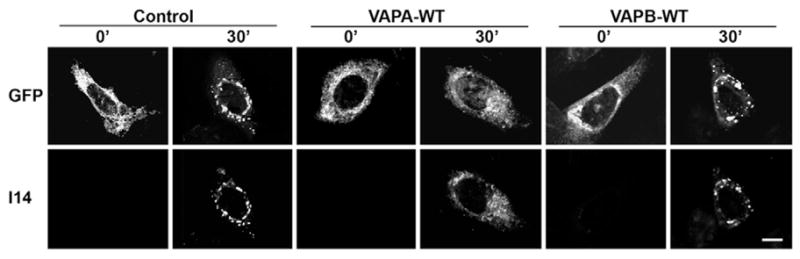
Effect of VAPA and VAPB on VSVG folding. VSVGts045-GFP was trapped in the ER at 42°C for 6 hours in transfected CHO cells and shifted to 32°C for 0 or 30 minutes before paraformaldehyde fixation. VSVG was detected by its GFP signal (top panel) or with the conformation-specific I14 antibody, which recognises properly folded VSVG (lower panel). Scale bar: 10 μm.
VAPA and VAPB have an inhibitory effect on ER vesicle budding
We next determined whether defective ER vesicle budding might be the underlying cause of VAPA-mediated inhibition of ER-to-Golgi transport. We used a modified in vitro reconstituted assay (Xu and Hay, 2004) to directly assess whether VSVG incorporation into ER budded vesicles was compromised. Cells cotransfected with VSVGts045-Myc were perforated and ER vesicle budding was reconstituted with rat liver cytosol and an energy regenerating system. Budded vesicles released into the medium were recovered and the VSVG content analysed by quantitative western immunoblot. Under control conditions, a fraction of the total ER-trapped VSVG (5.9±0.3%) was consistently recovered in the budded vesicle fraction (Fig. 6). Coexpression of VAPA-WT and P56S mutant resulted in near complete loss of VSVG from budded vesicles (to 0.8±0.3% and 0.3±0.1%, respectively). VSVG recovered in the ER-budded vesicles was also reduced in VAPB-WT and VAPB-P56S cells, although not as robustly as in VAPA-WT transfected cells. Thus, the primary cause of the inhibition in ER-to-Golgi transport by VAPA is due to a defect in incorporation of membrane-associated cargo into budding ER vesicles. The milder inhibitory effect of VAPB-WT on ER vesicle formation is consistent with its lack of inhibition on VSVG ER-to-Golgi trafficking. It remains unclear whether the vesicles recovered from the VAPB-P56S-expressing cells were from residual functional ER or from the large ER aggregates.
Fig. 6.
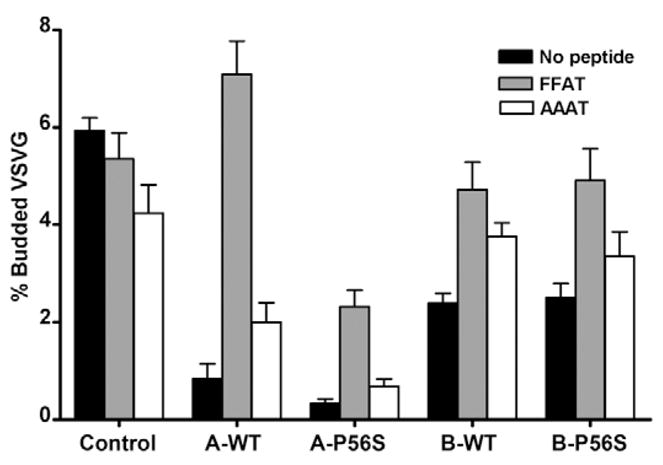
Effect of VAPA and VAPB on ER-vesicle budding. VSVGts045-Myc was trapped in the ER and ER-vesicle budding was reconstituted in vitro in perforated cells. Released VSVG-containing vesicles were collected by centrifugation and subjected to quantitative western immunoblot. The amount of VSVGts045 in vesicles released from semi-intact cells incubated at 4°C was subtracted from that in cells incubated at 32°C and the values were normalised as a percentage of the total cellular VSVG signal. Vehicle (DMSO) control (solid bars), FFAT (grey bars) and AAAT peptides (white bars) were added at 62.5 μM final concentration to the budding assay where indicated. The values represent mean ± s.e.m.
Since expression of FFAT rescued the VAPA-induced inhibition of ER-to-Golgi transport and the morphological effect of VAPB-P56S, we tested whether addition of a FFAT-containing peptide could also restore cargo sorting into ER vesicles. We synthesised a short 10-residue peptide based on OSBP1, spanning the FFAT motif and a control AAAT peptide in which the two phenylalanines were substituted by alanines. Addition of FFAT or AAAT peptide (at 62.5 μM) had little effect on ER-vesicle budding in control cells, although a small but statistically insignificant decrease (to 4.2±0.6; P>0.05) was observed with the AAAT peptide (Fig. 6). Interestingly, addition of the FFAT peptide to VAPA-WT-transfected cells resulted in a greater than eightfold increase in vesicle budding (to 7.1±0.7%) with the amount of VSVG-containing budded vesicles exceeding that of the control. Although the FFAT peptide completely nullified the inhibitory effect of VAPA-WT, only a modest increase (to 2.0±0.4%) was observed with an identical dose of the AAAT peptide. Thus, the FFAT peptide effectively relieved the VAPA-meditated inhibition of ER-vesicle budding, with the two phenylalanines being indispensable. The effect was also dosage-dependent, because a lower concentration of 31 μM of the FFAT peptide was ineffective in relieving the inhibitory effect of VAPA (data not shown). The FFAT peptide also relieved the inhibition of vesicle budding by VAPA-P56S with the absolute amount of vesicle budding increasing fourfold to 1.4±0.6%. However, this relief was clearly not as effective when compared with VAPA-WT. The AAAT peptide was completely ineffective in relieving the inhibitory effect of the VAPA-P56S mutant, producing only a small but insignificant increase in vesicle budding (to 0.5±0.2%; P>0.05). The FFAT peptide also restored ER-vesicle budding in VAPB-WT and VAPB-P56S cells to levels slightly below that of the control. However, the lack of statistical significance between the FFAT and AAAT peptide (P>0.05) suggests that the effect cannot be attributed to the two Phe residues alone. Thus, restoration of ER budding by FFAT supports a role for FFAT-containing proteins in modulating ER vesicle formation or VSVG cargo incorporation into ER vesicles. The partial relief in VAPA-P56S mutants implicates an adverse effect of the mutation on other FFAT-independent processes.
FFAT rescues VAPA-mediated inhibition of lateral diffusion of membrane proteins in the ER
Proteins are sequestered either passively or by a molecular sorting process to ER exit sites prior to vesicle budding. Because membrane proteins are confined within the two-dimensional lattice of the ER membrane, they must diffuse laterally to reach these exit sites. We reasoned that stable association of VAPA with the underlying microtubule network could create immobile obstacles impeding lateral diffusion of integral membrane cargoes. This, in turn, could decrease sorting efficiency and sequestration of properly assembled protein cargoes into budding vesicles. To test this interpretation, we performed fluorescence recovery after photobleaching (FRAP) on ER-trapped VSVGts045-GFP using the rate of fluorescence signal recovery as an indication of lateral diffusion and ER exit of the VSVG cargo (see supplementary material Movies 1–10). The photobleached fluorescent signal rapidly recovered in control cells reaching a plateau within 10 minutes after release (Fig. 7A). The recovery rate was greatest within the first 2 minutes, followed by a slower rate of recovery up to 10 minutes. A gradual loss of fluorescence signal was observed after 10 minutes, because of transport of VSVGts045-GFP to the Golgi complex (data not shown). We used the rate of recovery within the first 2 minutes, where the fluorescence recovery was greatest, as a measure of lateral diffusion immediately upon release. The rate of recovery in control cells was nearly twofold higher than in VAPA-WT-expressing cells (Table 2). A further decrease in the rate of recovery relative to VAPA-WT was observed in VAPA-P56S. Thus, expression of VAPA-WT or P56S significantly decreased the overall lateral mobility of VSVG in the ER. Overexpression of VAPB had no detectable effect on lateral diffusion of VSVG relative to control cells. We were unable to assess lateral diffusion in VAPB-P56S-expressing cells because of the lack of tubular ER structures, which was due to sequestration of VSVG-GFP to the large membranous aggregates in these cells. However, FRAP analysis on VSVG-GFP trapped in large membranous aggregates indicated that these structures are highly immobile with very little signal recovery over 10 minutes. Thus, anterograde membrane cargoes trapped in these large aggregates are unlikely to be transported to the Golgi compartment.
Fig. 7.
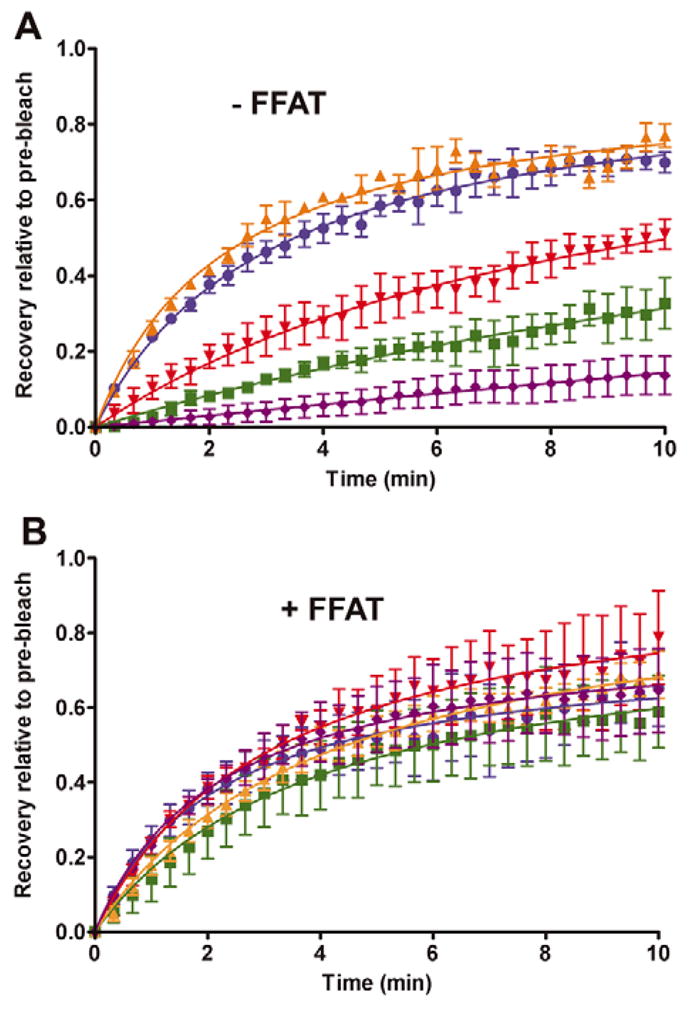
FRAP analysis in the absence or presence of coexpressed FFAT motif. VSVGts045-GFP was trapped in the ER and shifted to 32°C immediately before a small area was photobleached. Fluorescence signal recovery was measured at 20 second intervals over a 10 minute period and values were normalised to the pre-bleach value. Cells were transfected with control vector (blue), VAPA-WT (red), VAPA-P56S (green), VAPB-WT (orange) or VAPB-P56S (purple) (A) or cotransfected with FFAT-expressing vector (B).
Since FFAT relieved the VAPA-mediated inhibition of ER budding, we anticipated that coexpression of the FFAT motif in vivo would also relieve the VAPA-induced decrease in lateral diffusion. Indeed, coexpression of FFAT restored lateral diffusion to control levels for both VAPA-WT and VAPA-P56S mutant (Fig. 7B). It caused a twofold increase in recovery rate in VAPA-WT cells such that it is statistically similar to control cells expressing FFAT (Table 2). FFAT also relieved the inhibitory effect of VAPA-P56S, although the rate of recovery was slightly lower but not statistically significant when compared with VAPA-WT cells. Coexpression of FFAT had no effect on lateral diffusion of VSVG in VAPB-WT-expressing cells, which remained comparable to control cells. Restoration of the reticular ER pattern by FFAT coexpression in VAPB-P56S cells allowed us to monitor its effect on lateral diffusion of VSVG-GFP. In this case, lateral diffusion of VSVG-GFP was also completely restored to the control level. Together, overexpression of VAPA-WT and VAPA-P56S but not VAPB-WT decreased lateral diffusion of VSVG. Coexpression of FFAT effectively restored lateral diffusion, supporting a modulatory role for this motif in VAPA-dependent function. VSVG trapped in large membranous aggregates in VAPB-P56S-expressing cells was highly immobile, and FFAT coexpression effectively restored VSVG mobility to control levels.
FFAT disrupts microtubule association of VAP
We next examined whether creation of immobile obstacles might underlie the decrease in lateral diffusion by VAPA. To create an immobile obstacle that could impede lateral diffusion, VAPA must possess at least two anchor points and disrupting either one would release the immobile obstacle. The C-terminal transmembrane domain is clearly one anchor point by virtue of spanning the ER membrane. The close association of VAPA with the microtubule network (Skehel et al., 2000) makes this a plausible second anchor point. If correct, disrupting microtubule association would relieve the inhibitory effect of VAPA. We first assessed whether the MSP domain is indeed essential for association with polymerised microtubules by performing a pull-down assay. Detergent-solubilised extracts from transfected cells were incubated with purified polymerised microtubules, and proteins recovered with the polymerised microtubule pellet after centrifugation through a sucrose cushion were analysed by western immunoblot. VAPA-WT, VAPA-P56S and VAPB-WT were recovered with polymerised microtubules (Fig. 8A). Unfortunately, we were unable to assess microtubule association of VAPB-P56S because the mutant protein was largely insoluble in Triton X-100 (data not shown). The small fraction that was recovered after detergent solubilisation migrated through the sucrose cushion and was recovered in the pellet fraction even in the absence of exogenous microtubules. Microtubule association is clearly mediated by the N-terminal MSP domain, because deletion of this domain abolished polymerised microtubule association. Its microtubule association is probably mediated by other VAP-interacting proteins because purified recombinant GST-VAPA showed no direct binding to polymerised microtubules (data not shown).
Fig. 8.
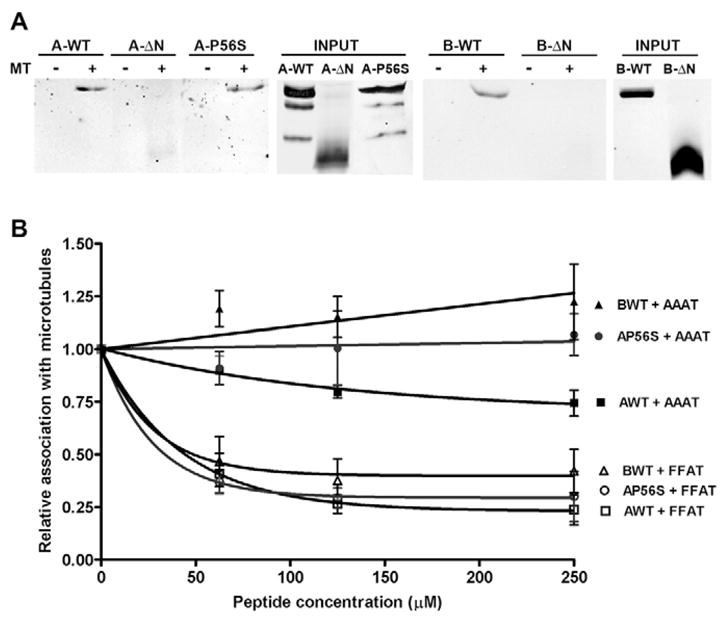
Association of VAPA and VAPB with polymerised microtubules. (A) Detergent-solubilised extracts of transfected cells were pulled down with polymerised microtubules. The ‘input’ panels show 25% of the input signal. A-WT, wild-type VAPA; A-ΔN, N-terminal deletion VAPA; A-P56S, mutant P56S VAPA; B-WT, wild-type VAPB; B-ΔN, N-terminal deletion VAPB. (B) Quantitative analysis of VAPA and VAPB recovered with polymerised microtubules in the presence of AAAT or FFAT peptide at the indicated concentrations. Values were normalised to the amount of VAPA or VAPB recovered in the absence of either peptide. Filled symbols represent microtubule association of VAPA-WT, VAPA-P56S and VAPB-WT in the presence of AAAT peptide whereas open symbols represent association of VAPA-WT, VAPA-P56S and VAPB-WT in the presence of FFAT peptide.
If stable association of VAPA with microtubules is a factor in lateral mobility of VSVG, then disruption of this interaction by FFAT might contribute to the restoration of lateral diffusion in the VAPA-WT- and VAPA-P56S-expressing cells. To test this interpretation, we added varying amounts of FFAT or AAAT peptide to the extracts and quantified the amount of VAPA or VAPB recovered with polymerised microtubules. The values were normalised to vehicle (DMSO) control. Addition of the FFAT peptide equally disrupted microtubule association of VAPA-WT and VAPA-P56S mutant (Fig. 8B). In both cases, a >60% decrease in microtubule association was observed in the presence of 62.5 μM FFAT, a concentration that effectively restored in vitro ER budding. The AAAT peptide produced a modest decrease in microtubule association of VAPA-WT, but had no effect on microtubule association of VAPA-P56S. Microtubule association of VAPB-WT was equally inhibited by the FFAT peptide but not by the AAAT peptide. Thus, the FFAT motif clearly disrupted association of VAPA and VAPB with the microtubule network. This could result in enhanced lateral diffusion of VSVG by virtue of creating a more mobile VAPA-containing complex.
Discussion
Although we and others (Amarilio et al., 2005; Kanekura et al., 2006; Teuling et al., 2007) have found that the two VAP isoforms showed extensive overlap with the ER marker Calreticulin, there are striking differences in their cellular effects on anterograde transport and ER morphology. With overexpression of the transgene approximately fourfold higher than endogenous proteins (data not shown), VAPA significantly inhibited transport of VSVG to the Golgi complex while VSVG trafficking remained largely unaffected by VAPB, consistent with a previous study (Amarilio et al., 2005). We found that the trapped VSVG in the VAPA-transfected cells colocalised extensively with Calreticulin, suggesting that VSVG was not efficiently transported out of the ER. This differential effect of the two VAP isoforms on ER-to-Golgi transport suggests that they might act on different ER subdomains or transport steps, with VAPA exerting a stronger effect than VAPB on early stages of anterograde transport. VAPB has been proposed to predominantly affect COPI-mediated transport (Soussan et al., 1999), which preferentially affects retrograde transport processes, but might indirectly affect anterograde transport by limiting recycling of key secretory components to the ER. There is also a striking difference in the morphological effect of the P56S mutation, despite the fact that the P56S mutant VAPA and VAPB were expressed at levels comparable to the endogenous proteins (data not shown). Surprisingly, the P56S mutation had limited effect on VAPA with only a small fraction (~1–2%) of the transfected cells containing visible aggregates. In general, the aggregates were relatively small and randomly distributed throughout the cell. By contrast, the P56S mutation in VAPB induced clustering and aggregation of ER membranes in ~70% of transfected cells, as also seen by others (Amarilio et al., 2005; Kanekura et al., 2006; Teuling et al., 2007). This lack of defect in VAPA could reflect differences in intrinsic properties of the protein, its interaction with other proteins, or may simply be due to a wider membrane distribution of VAPA throughout the ER tubules, which could accommodate protein aggregates without significant loss of overall function. We also noted that the large membranous aggregates contained Calreticulin, which is in contrast to a previous observation (Teuling et al., 2007). This may be due to differences in cell line or expression levels, but the Calreticulin colocalisation along with the collapse of the reticular ER pattern strongly suggests that the aggregates originated from the ER.
The domain structure of VAPA is ideally suited to link the ER membrane to the microtubule network, where a minimum of two anchor points is required, and disruption of either of these releases or compromises this tight association. Deletion of the C-terminal transmembrane domain (TMD) is likely to result in a soluble truncated protein, and we found that its expression had no inhibitory effect on ER-to-Golgi transport (data not shown). The TMD, along with the coiled-coil domain, has been proposed to play a critical role in dimerisation (Weir et al., 2001). Although some MSP domains are known to form a dimer, the critical residues at the dimerisation interface are not conserved in VAPA and VAPB (Kaiser et al., 2005), suggesting that their MSP domain in isolation dimerises poorly, if at all, and requires the coiled-coil and TMD for stable dimer formation. Our observations are consistent with this view. We noticed a delay in transport of VSVG-GFP out of the ER in VAPA-ΔN-expressing cells, with a larger fraction of cells exhibiting VSVG-GFP retention in the ER at the 30 minute chase period (~30% in VAPA-ΔN compared with 13% in control cells); however, this was clearly not as severe as with VAPA-WT, where over 90% of the cells exhibited ER retention at this time period. VSVG-GFP was eventually evident at the plasma membrane in 28% and 98% of VAPA-ΔN cells at the 60 and 90 minute chase periods, respectively, suggesting a significant fraction was able to overcome the delay in ER-to-Golgi transport. Likewise, VAPA-ΔN only partially inhibited incorporation of VSVG into ER vesicles in the in vitro reconstituted budding assay (2.2±0.5%, n=4) when compared with VAPA-WT (0.8±0.3%). Thus, deletion of the N-terminal MSP domain is expected to retain residual ability to form a heterodimer with endogenous VAP, and could potentially interfere with endogenous VAP.
The MSP domain of VAPA is known to interact with the FFAT motif present in OSBP and ORP protein families (Loewen and Levine, 2005; Wyles et al., 2002; Wyles and Ridgway, 2004). We found that this simple interaction is sufficient to relieve the VAPA-mediated inhibition of ER-to-Golgi transport. The impaired anterograde transport of VSVG-GFP (Fig. 2) and near complete loss of in vitro ER budding (Fig. 6) independently implicates defective incorporation of membrane cargo into budding vesicles as the primary cause of ER retention of VSVG. We considered the possibility that budded vesicles might be trapped inside the perforated cells by tethering onto residual microtubule network. However, treatment with the microtubule-depolymerising drug Nocodazole after the 40 minute budding period showed no further increase in the amount of recoverable VSVG (data not shown) arguing against the existence of a microtubule-trapped fraction. Furthermore, the ability of the luminal cargo protein CgB to efficiently transit from ER to Golgi (Fig. 4) suggests that budded vesicles are not statically tethered to microtubules, but can traffic to, and fuse with, the appropriate target compartment. This argues that VAPA primarily inhibits sequestration of membrane-bound VSVG and the decrease in VSVG-containing vesicles is not due to microtubule attachment of budded vesicles. Viewed in light of the indirect association of VAP with polymerised microtubules, it might be predicted that depolymerising microtubules could relieve the inhibitory effect of VAP on budding of VSVG from the ER. However, treatment with Nocodazole, either before or during the budding assay, significantly interfered with vesicle budding, reducing ER budding in control cells from 5.9±0.3% to 1.4±0.3%. The latter value was slightly higher but not statistically significant from that in VAPA-WT-expressing cells (0.8±0.3%; P>0.1) or VAPA-WT cells treated with Nocodazole during the budding assay (1.6±0.2%; P>0.05). Although treatment with Nocodazole appeared to enhance vesicle budding in VAPA-WT-expressing cells, the increase was not statistically significant when compared with untreated VAPA-WT cells. Thus, an intact microtubule network clearly plays a critical role in ER-vesicle budding, and the adverse effect of depolymerising agents such as Nocodazole on vesicle budding negates their possible use to relieve the inhibitory effect of VAPA.
VAPA-P56S similarly inhibited ER-to-Golgi transport, ER budding and lateral diffusion of VSVG. Coexpression of FFAT rescued the inhibition of lateral diffusion, which probably contributes to the restoration of ER exit and eventual plasma membrane localisation of VSVG. Taken together with the disruption of microtubule association by the FFAT peptide, it suggests that the primary effect of the FFAT motif is to disrupt VAPA-mediated anchoring of ER membrane to the microtubule network. Because the Pro56 residue is not located within the hydrophobic FFAT-binding pocket (Kaiser et al., 2005; Loewen and Levine, 2005), it may not have a direct effect on FFAT binding, such that FFAT could still bind to the P56S mutant protein, causing detachment of the ER membrane and restoration of the defect in lateral diffusion and fluorescence recovery rate. Although it also enhanced ER budding, it did not completely restore ER budding to control levels, suggesting that additional steps are involved. The effect of FFAT was more dramatic on the VAPB-P56S mutation. Its coexpression not only resolved most of the ER aggregates, but it also restored lateral diffusion and transport of VSVG to the plasma membrane. This significantly differed from the lack of effect of FFAT reported in a previous study (Teuling et al., 2007) and may reflect differences in the FFAT constructs. We used the FFAT sequence from OSBP1 with a small Myc tag at the N-terminus as opposed to the FFAT sequence from Nir2 with an N-terminal GFP moiety used by Teuling et al. (Teuling et al., 2007). It is possible that differences in the FFAT sequence might affect efficacy of the motif. Alternatively, the presence of the large N-terminal GFP tag might affect accessibility or binding of the FFAT motif. In either case, it raises the possibility that a FFAT-containing peptide might be used to alleviate the effect of VAPB-P56S on protein transport through the secretory pathway.
Finally, we offer a possible explanation of the effect of VAPA and VAPB on ER-to-Golgi transport, which appears to be limited to integral membrane proteins, such as VSVG. Membrane proteins are sorted and sequestered prior to incorporation as cargoes of ER-transport vesicles. This process is thought to drive COPII-dependent coat formation and vesicle budding. Because membrane proteins are confined within the lipid bilayer, their access to these exit sites is highly dependent on lateral diffusion within the two-dimensional lattice of the membrane. Membrane proteins are known to diffuse anomalously, and a number of models have been proposed over the years to explain this property. These include motion impedance by immobile obstacles (Saxton, 1987), anchored picket posts (Kusumi et al., 2005) and interactions with membrane microdomains or corrals (Schutz et al., 1997). After excluding changes in membrane viscosity, lateral diffusion of integral ER membrane proteins is most sensitive to obstacles within the membrane (Nicolau et al., 2007; Saxton, 1987). Interaction with the cytoskeleton provides the attachment points for these immobile obstacles, although proteins that could generate these relatively immobile obstacles either directly through microtubule association or indirectly through interaction with other proteins or lipids have never been identified.
With its membrane-embedded C-terminus and interaction of its MSP domain with membrane-associated microtubules on the cytoplasmic face, we believe that VAPA could readily form immobile membrane domains that serve as folding and assembly sites for membrane proteins. Since the MSP domain of VAPA does not directly interact with polymerised microtubules (Kaiser et al., 2005), other accessory proteins must be involved in stabilising its association with polymerised microtubules. Binding of the FFAT motif to the MSP domain could disrupt this microtubule association, destabilising association of the VAPA-containing complex with the underlying microtubule network. This could lead to enhanced mobility of membrane proteins. Physiologically, recruitment of FFAT-containing proteins could also create regions of increased fluidity that are conducive to membrane-remodelling events, such as incorporation of membrane lipids or their removal through vesicle budding. Thus, a VAPA-containing protein complex could physically link the ER membrane to the microtubule network. This not only stabilises the ER membrane, but also provides an anchor point for outward ER membrane expansion driven by motor proteins. It may also serve as a stationary site for protein folding and assembly. Overexpression of VAPA may have inadvertently created an excess amount of anchor sites such that they form a sea of immobile obstacles that physically restricted lateral diffusion of membrane proteins. This could affect ER trafficking to the extent that even properly folded VSVG was unable to exit the ER. Addition of the FFAT motif then relieves this inhibition of lateral diffusion by disrupting the microtubule association of VAPA leading to disengagement of these relatively immobile anchor points. Thus, FFAT not only serves to recruit lipid-binding proteins to the ER, but also creates membrane regions that are physically more fluid and conducive to remodelling.
Materials and Methods
Generation of cDNA constructs
For all experiments, the cDNAs encoding human VAPA and VAPB were subcloned into pFlag-CMV2 at the NotI and BamHI sites. The ΔN constructs were also subcloned into pFlag-CMV2 starting with residues 135 and 124 for VAPA and VAPB, respectively. The P56S mutation was generated by site-directed mutagenesis, and was subcloned into pFlag-CMV2 at the NotI and EcoRI sites. The FFAT motif of rabbit OSBP (residues 347–468) was inserted into pcDNA3.1(+)-Myc at the BamHI and EcoRI sites. This was subsequently used to generate a vector with the AAAT motif by site-directed mutagenesis.
Cell culture, transfection and immunocytochemistry
Chinese hamster ovary (CHO) cells were maintained in minimum essential medium α (Invitrogen) supplemented with 5% fetal bovine serum (Invitrogen), 100 U/ml penicillin and 100 μg/ml streptomycin (Invitrogen). Transfection was performed using LipofectAMINE reagent (Invitrogen) according to the manufacturer’s instructions.
For colocalisation analysis, cells were washed with phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde in PBS for 30 minutes. The fixed cells were then permeabilised in blocking solution (PBS containing 0.4% saponin, 1% bovine serum albumin, 2% normal goat serum and 0.01% NaN3) before staining with mouse or rabbit anti-Calreticulin (Stressgen), mouse anti-Flag (Applied Biological Materials), rabbit anti-Flag (Sigma), or rabbit anti-Sar1 (Upstate) antibodies, followed by staining with Alexa Fluor 488- or Texas-Red-conjugated secondary antibodies (Invitrogen and Jackson Laboratories, respectively). Coverslips were mounted in SlowFade Gold reagent (Invitrogen). All images were captured on an Olympus IX70 microscope equipped with a Bio-Rad MRC1024 laser-scanning confocal unit using a 60×, 1.4 NA oil-immersion objective and LaserSharp image acquisition software.
Time course of VSVGts045 and CgB transport
CHO cells were cotransfected with pCDM8.1/VSVGts045-GFP (Presley et al., 1997) and either empty vector, wild-type or P56S mutant VAPA or VAPB. pcDNA3.1(+)-Myc/OSBP-FFAT was cotransfected in an additional set of experiments to determine the effect of the FFAT motif on VAP-mediated transport inhibition. 16 hours after transfection, cells were shifted to 42°C for 6 hours to accumulate VSVGts045-GFP in the ER. Cycloheximide (20 μg/ml) was then added, and cells were chased at 32°C for 0, 30, 60 and 90 minutes. At each time interval, cells were washed with PBS, and fixed with 4% paraformaldehyde. A similar protocol was used to directly visualise VSVG folding. At the 30 minute chase period, cells were fixed and stained with monoclonal I14 antibody, which specifically recognises folded VSVG (Lefrançois and Lyles, 1983), and Texas Red goat anti-mouse antibodies as described above.
Transport of CgB from ER to Golgi was assessed by cotransfection of CHO cells with pCDM8/CgB-GFP(S65T) (Wacker et al., 1997) and either empty vector, wild-type or P56S mutant VAPA or VAPB. 16 hours after transfection, cells were shifted to 15°C for 2 hours to enhance the fluorescent signal and to prevent transport to the Golgi complex by trapping the cargo in the ERGIC. Cycloheximide was then added to 20 μg/ml for 10 minutes before shifting to 37°C. Cells were then chased for 0 or 30 minutes, at which time coverslips were fixed with 4% paraformaldehyde and stained with mouse anti-Calreticulin antibodies as described above.
In vitro ER-budding assay
A modified in vitro budding assay (Xu and Hay, 2004) based on transfected CHO was used to assess ER vesicle budding. Briefly, perforated CHO cells cotransfected with VSVG-Myc3 and various VAP vectors were incubated with rat liver cytosol and energy regenerating system at 32°C for 40 minutes. Identical samples were incubated at 4°C as a measure of nonspecific ER fragmentation. The cells were removed by low speed centrifugation at 4000 g for 1 minute, followed by 15,000 g for 1 minute, and budded vesicles in the resulting supernatant were recovered by centrifugation at 100,000 g for 1 hour. The amount of VSVG cargo in the budded vesicle fraction was subjected to quantitative western immunoblot with anti-Myc antibody (Stressgen). The values for budded vesicles were normalised as a percentage of the input and corrected for energy-independent nonspecific ER fragmentation.
Fluorescence recovery after photobleaching (FRAP)
For live imaging experiments, CHO cells were seeded onto 0.17 mm glass-bottomed Delta T dishes (Bioptechs), and cotransfected with pCDM8.1/VSVGts045-GFP and either empty vector, wild-type or P56S mutant VAPA or VAPB. For experiments to determine the effect of the FFAT motif on diffusion of VSVGts045 through the ER, cells were additionally cotransfected with pcDNA3.1(+)-myc/OSBP-FFAT. VSVGts045-GFP was accumulated in the ER at 42°C for 6 hours. Cycloheximide was then added to a concentration of 20 μg/ml, and cells were imaged on a confocal microscope equipped with a Delta T stage adapter (Bioptechs) maintained at a constant temperature of 32°C. A portion of the ER in the periphery of the cell was photobleached, and recovery was monitored by imaging every 20 seconds for 10 minutes. The extent of recovery into the bleached area was determined using Medical Image Processing, Analysis and Visualisation (MIPAV) software from NIH.
Microtubule pull-down experiments
CHO cells were transfected with wild-type or mutant VAPA and VAPB and homogenised by sonication in buffer H (5 mM HEPES-NaOH pH 7.4, 100 mM NaCl, 1× protease inhibitor cocktail). After clearing the lysate at 5000 g for 5 minutes, Triton X-100 was added to a final concentration of 0.25%, and insoluble proteins were removed by centrifugation at 130,000 g for 20 minutes. FFAT (DENEFFDAPE) or AAAT (DENEAADAPE) peptide (GL Biochem) was then added to equivalent amounts of supernatant to give final concentrations of 0, 62.5, 125 or 250 μM. Bovine brain tubulin (Cytoskeleton) was prepolymerised according to the manufacturer’s instructions. Polymerised microtubules were added to the samples for 20 minutes at 25°C. The mixture was then layered onto a cushion of 20% sucrose in 80 mM PIPES pH 6.9, 1 mM EGTA, 1 mM MgCl2 and 10 μM Taxol, and polymerised microtubules were isolated by centrifugation at 100,000 g for 40 minutes. The high-speed pellet was solubilised in SDS sample buffer, and associated proteins were resolved by SDS-PAGE followed by western blotting with mouse anti-Flag and 12G10 mouse anti-α-tubulin antibodies (a gift from J. Frankel and E. M. Nelsen, DSHB, University of Iowa, IA).
Supplementary Material
Acknowledgments
This work was supported by a CIHR operating grant to J.K.N., and a CGS scholarship to D.C.P.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/121/18/3052/DC1
References
- Amarilio R, Ramachandran S, Sabanay H, Lev S. Differential regulation of endoplasmic reticulum structure through VAP-Nir protein interaction. J Biol Chem. 2005;280:5934–5944. doi: 10.1074/jbc.M409566200. [DOI] [PubMed] [Google Scholar]
- Andrade J, Zhao H, Titus B, Timm Pearce S, Barroso M. The EF-hand Ca2+-binding protein p22 plays a role in microtubule and endoplasmic reticulum organization and dynamics with distinct Ca2+-binding requirements. Mol Biol Cell. 2004;15:481–496. doi: 10.1091/mbc.E03-07-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daum G. Membrane targeting: glued by a lipid to the ER. Curr Biol. 2004;14:R711–R713. doi: 10.1016/j.cub.2004.08.045. [DOI] [PubMed] [Google Scholar]
- Ettayebi K, Hardy ME. Norwalk virus nonstructural protein p48 forms a complex with the SNARE regulator VAP-A and prevents cell surface expression of vesicular stomatitis virus G protein. J Virol. 2003;77:11790–11797. doi: 10.1128/JVI.77.21.11790-11797.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara T, Oda K, Yokota S, Takatsuki A, Ikehara Y. Brefeldin A causes disassembly of the Golgi complex and accumulation of secretory proteins in the endoplasmic reticulum. J Biol Chem. 1988;263:18545–18552. [PubMed] [Google Scholar]
- Hamamoto I, Nishimura Y, Okamoto T, Aizaki H, Liu M, Mori Y, Abe T, Suzuki T, Lai MM, Miyamura T, et al. Human VAP-B is involved in hepatitis C virus replication through interaction with NS5A and NS5B. J Virol. 2005;79:13473–13482. doi: 10.1128/JVI.79.21.13473-13482.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosaka K, Nikawa J, Kodaki T, Yamashita S. A dominant mutation that alters the regulation of INO1 expression in Saccharomyces cerevisiae. J Biochem. 1992;111:352–358. doi: 10.1093/oxfordjournals.jbchem.a123761. [DOI] [PubMed] [Google Scholar]
- Kaiser SE, Brickner JH, Reilein AR, Fenn TD, Walter P, Brunger AT. Structural basis of FFAT motif-mediated ER targeting. Structure. 2005;13:1035–1045. doi: 10.1016/j.str.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Kanekura K, Nishimoto I, Aiso S, Matsuoka M. Characterization of amyotrophic lateral sclerosis-linked P56S mutation of vesicle-associated membrane protein-associated protein B (VAPB/ALS8) J Biol Chem. 2006;281:30223–30233. doi: 10.1074/jbc.M605049200. [DOI] [PubMed] [Google Scholar]
- Kawano M, Kumagai K, Nishijima M, Hanada K. Efficient trafficking of ceramide from the endoplasmic reticulum to the Golgi apparatus requires a VAMP-associated protein-interacting FFAT motif of CERT. J Biol Chem. 2006;281:30279–30288. doi: 10.1074/jbc.M605032200. [DOI] [PubMed] [Google Scholar]
- Kusumi A, Nakada C, Ritchie K, Murase K, Suzuki K, Murakoshi H, Kasai RS, Kondo J, Fujiwara T. Paradigm shift of the plasma membrane concept from the two-dimensional continuum fluid to the partitioned fluid: high-speed single-molecule tracking of membrane molecules. Annu Rev Biophys Biomol Struct. 2005;34:351–378. doi: 10.1146/annurev.biophys.34.040204.144637. [DOI] [PubMed] [Google Scholar]
- Lapierre LA, Tuma PL, Navarre J, Goldenring JR, Anderson JM. VAP-33 localizes to both an intracellular vesicle population and with occludin at the tight junction. J Cell Sci. 1999;112:3723–3732. doi: 10.1242/jcs.112.21.3723. [DOI] [PubMed] [Google Scholar]
- Lefrançois L, Lyles DS. Antigenic determinants of vesicular stomatitis virus: analysis with antigenic variants. J Immunol. 1983;130:394–398. [PubMed] [Google Scholar]
- Loewen CJ, Levine TP. A highly conserved binding site in vesicle-associated membrane protein-associated protein (VAP) for the FFAT motif of lipid-binding proteins. J Biol Chem. 2005;280:14097–14104. doi: 10.1074/jbc.M500147200. [DOI] [PubMed] [Google Scholar]
- Loewen CJ, Roy A, Levine TP. A conserved ER targeting motif in three families of lipid binding proteins and in Opi1p binds VAP. EMBO J. 2003;22:2025–2035. doi: 10.1093/emboj/cdg201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolau DV, Jr, Hancock JF, Burrage K. Sources of anomalous diffusion on cell membranes: a Monte Carlo study. Biophys J. 2007;92:1975–1987. doi: 10.1529/biophysj.105.076869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikawa J, Murakami A, Esumi E, Hosaka K. Cloning and sequence of the SCS2 gene, which can suppress the defect of INO1 expression in an inositol auxotrophic mutant of Saccharomyces cerevisiae. J Biochem. 1995;118:39–45. doi: 10.1093/oxfordjournals.jbchem.a124889. [DOI] [PubMed] [Google Scholar]
- Nishimura Y, Hayashi M, Inada H, Tanaka T. Molecular cloning and characterization of mammalian homologues of vesicle-associated membrane protein-associated (VAMP-associated) proteins. Biochem Biophys Res Commun. 1999;254:21–26. doi: 10.1006/bbrc.1998.9876. [DOI] [PubMed] [Google Scholar]
- Nishimura AL, Mitne-Neto M, Silva HC, Richieri-Costa A, Middleton S, Cascio D, Kok F, Oliveira JR, Gillingwater T, Webb J, et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet. 2004;75:822–831. doi: 10.1086/425287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennetta G, Hiesinger PR, Fabian-Fine R, Meinertzhagen IA, Bellen HJ. Drosophila VAP-33A directs bouton formation at neuromuscular junctions in a dosage-dependent manner. Neuron. 2002;35:291–306. doi: 10.1016/s0896-6273(02)00769-9. [DOI] [PubMed] [Google Scholar]
- Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, Lippincott-Schwartz J. ER-to-Golgi transport visualized in living cells. Nature. 1997;389:81–85. doi: 10.1038/38001. [DOI] [PubMed] [Google Scholar]
- Saxton MJ. Lateral diffusion in an archipelago. The effect of mobile obstacles Biophys J. 1987;52:989–997. doi: 10.1016/S0006-3495(87)83291-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutz GJ, Schindler H, Schmidt T. Single-molecule microscopy on model membranes reveals anomalous diffusion. Biophys J. 1997;73:1073–1080. doi: 10.1016/S0006-3495(97)78139-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer A, Matter K, Ketcham CM, Hauri HP. The isolated ER-Golgi intermediate compartment exhibits properties that are different from ER and cis-Golgi. J Cell Biol. 1991;113:45–54. doi: 10.1083/jcb.113.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skehel PA, Martin KC, Kandel ER, Bartsch D. A VAMP-binding protein from Aplysia required for neurotransmitter release. Science. 1995;269:1580–1583. doi: 10.1126/science.7667638. [DOI] [PubMed] [Google Scholar]
- Skehel PA, Fabian-Fine R, Kandel ER. Mouse VAP33 is associated with the endoplasmic reticulum and microtubules. Proc Natl Acad Sci USA. 2000;97:1101–1106. doi: 10.1073/pnas.97.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soussan L, Burakov D, Daniels MP, Toister-Achituv M, Porat A, Yarden Y, Elazar Z. ERG30, a VAP-33-related protein, functions in protein transport mediated by COPI vesicles. J Cell Biol. 1999;146:301–311. doi: 10.1083/jcb.146.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teuling E, Ahmed S, Haasdijk E, Demmers J, Steinmetz MO, Akhmanova A, Jaarsma D, Hoogenraad CC. Motor neuron disease-associated mutant vesicle-associated membrane protein-associated protein (VAP) B recruits wild-type VAPs into endoplasmic reticulum-derived tubular aggregates. J Neurosci. 2007;27:9801–9815. doi: 10.1523/JNEUROSCI.2661-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedrenne C, Hauri HP. Morphogenesis of the endoplasmic reticulum: beyond active membrane expansion. Traffic. 2006;7:639–646. doi: 10.1111/j.1600-0854.2006.00419.x. [DOI] [PubMed] [Google Scholar]
- Wacker I, Kaether C, Kromer A, Migala A, Almers W, Gerdes HH. Microtubule-dependent transport of secretory vesicles visualized in real time with a GFP-tagged secretory protein. J Cell Sci. 1997;110:1453–1463. doi: 10.1242/jcs.110.13.1453. [DOI] [PubMed] [Google Scholar]
- Weir ML, Klip A, Trimble WS. Identification of a human homologue of the vesicle-associated membrane protein (VAMP)-associated protein of 33 kDa (VAP-33): a broadly expressed protein that binds to VAMP. Biochem J. 1998;333:247–251. doi: 10.1042/bj3330247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir ML, Xie H, Klip A, Trimble WS. VAP-A binds promiscuously to both v- and tSNAREs. Biochem Biophys Res Commun. 2001;286:616–621. doi: 10.1006/bbrc.2001.5437. [DOI] [PubMed] [Google Scholar]
- Wyles JP, Ridgway ND. VAMP-associated protein-A regulates partitioning of oxysterol-binding protein-related protein-9 between the endoplasmic reticulum and Golgi apparatus. Exp Cell Res. 2004;297:533–547. doi: 10.1016/j.yexcr.2004.03.052. [DOI] [PubMed] [Google Scholar]
- Wyles JP, McMaster CR, Ridgway ND. Vesicle-associated membrane protein-associated protein-A (VAP-A) interacts with the oxysterol-binding protein to modify export from the endoplasmic reticulum. J Biol Chem. 2002;277:29908–29918. doi: 10.1074/jbc.M201191200. [DOI] [PubMed] [Google Scholar]
- Xu D, Hay JC. Reconstitution of COPII vesicle fusion to generate a pre-Golgi intermediate compartment. J Cell Biol. 2004;167:997–1003. doi: 10.1083/jcb.200408135. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.