


Abstract
Cyclin D1 is expressed at abnormally high levels in many cancers and has been specifically implicated in the development of breast cancer. In this report we have extensively analyzed the cyclin D1 promoter in a variety of cancer cell lines that overexpress the protein and identified two critical regulatory elements (CREs), a previously identified CRE at –52 and a novel site at –30. In vivo footprinting experiments demonstrated factors binding at both sites. We have used a novel DNA-binding ligand, GL020924, to target the site at –30 (–30–21) of the cyclin D1 promoter in MCF7 breast cancer cells. A binding site for this novel molecule was constructed by mutating 2 bp of the wild-type cyclin D1 promoter at the –30–21 site. Treatment with GL020924 specifically inhibited expression of the targeted cyclin D1 promoter construct in MCF7 cells in a concentration-dependent manner, thus validating the –30–21 site as a target for minor groove-binding ligands. In addition, this result validates our approach to regulating the expression of genes implicated in disease by targeting small DNA-binding ligands to key regulatory elements in the promoters of those genes.
INTRODUCTION
Aberrant expression of specific genes contributes to the development of many disease states. In particular, misregulation of gene expression is involved in numerous cancers. A number of methods have been employed to correct expression of such misregulated genes, including gene therapy, antisense techniques and novel zinc finger transcription factors (1,2). These techniques all suffer from difficulties in delivering the therapeutic agent.
A potentially powerful way to re-establish proper regulation and hence impact the disease state is to target critical regulatory regions in the promoters of therapeutically relevant genes with small, cell permeable ligands that interfere with transcription factor binding. By targeting the relatively non-conserved sequences flanking these critical regulatory regions, both gene-specific regulation and a reduction in side-effects can be attained. Recent examples of such an approach using linked polyamides (3,4) have been published. However, neither study demonstrated a direct effect on the expression of an RNA polymerase II-regulated gene. For example, one case looked at a polymerase III-regulated gene, 5S RNA, while another examined HIV replication rather than expression of mRNA.
A therapeutically relevant gene that we have pursued in our approach to targeting gene expression with DNA-binding ligands is cyclin D1. Inappropriately high levels of cyclin D1 gene expression are characteristic of a range of human tumors, in some instances as a consequence of chromosomal inversion, translocation or amplification (5). In addition, overexpression of cyclin D1 at the level of gene transcription, without gene amplification or rearrangement, is common in primary breast carcinoma, colon adenocarcinoma (5), familial adenomatous polyposis (6) and pancreatic carcinoma (7). In human esophageal, colon, squamous or pancreatic cancer cells with abnormally high cyclin D1 levels, expression of cyclin D1 antisense molecules slowed the cell cycle, restored anchorage-dependent growth and reversed the tumorigenicity of cells injected into nude mice (8–15), thus validating cyclin D1 as a target for therapeutic intervention.
A number of previously identified transcription factor-binding sites in the human cyclin D1 promoter have been implicated as critical for cyclin D1 expression in cancer cells. Potential Sp1, E2F, CRE, Oct1, Myc/Max, AP-1, Egr, NFκB, STAT5, Ets and TCF/LEF sites have been previously noted in the cyclin D1 promoter (16–23). Several of these sites have been demonstrated to play a role in cyclin D1 regulation in various human and non-human cell lines (18–29). However, as most of the sites have not been confirmed in therapeutically relevant cell lines, we analyzed the cyclin D1 promoter in several human carcinoma cell lines to identify critical transcription factor-binding sites as targets of small DNA-binding ligands.
We have identified two critical transcription factor-binding sites in the cyclin D1 promoter and have used a novel DNA-binding ligand, GL020924, to validate one site as appropriate to target with a sequence-specific DNA-binding molecule. Treatment of MCF7 breast cancer cells with GL020924 specifically decreased the activity of cyclin D1 promoters in which a GL020924-binding site overlapped the critical regulatory site, –30–21. Significantly, gene expression was reduced to levels equivalent to those observed with mutated regulatory sites, but there was no effect on promoters lacking a ligand-binding site. These studies validate the –30–21 site as a potential target for regulating cyclin D1 expression. In addition, they demonstrate the feasibility of modulating gene expression by directing small DNA-binding ligands to critical regulatory regions of genes implicated in the progression of cancer and other diseases.
MATERIALS AND METHODS
Tissue culture
The human breast carcinoma cell lines MCF7 and ZR75-1 were maintained in DMEM/F12 medium with 10% fetal bovine serum, 10 µg/ml bovine insulin and antibiotics (penicillin and streptomycin). The human colon carcinoma cell line HCT116 was maintained in McCoy’s medium with 10% fetal bovine serum, penicillin and streptomycin. The human pancreatic cell line PANC-1 was maintained in DMEM/F12 with 10% fetal bovine serum, penicillin and streptomycin. Human mammary epithelial cells (HMEC) were maintained in Epithelial Growth Medium supplemented with bovine pituitary extract (50 µg/ml), hydrocortisone (500 ng/ml), hEGF (10 ng/ml) and insulin (5 µg/ml). All lines were maintained at 37°C, 5% CO2. MCF7, ZR75-1, HCT116 and PANC-1 cells were purchased from the American Type Culture Collection. HMEC were purchased from Clonetics Corp.
Construction of luciferase plasmids
A 1900 bp fragment of the human cyclin D1 promoter was PCR amplified from genomic DNA using the following oligonucleotides: 5′-GCA CGC GTG CTA GCC AGC TGG GCC GCC CTT GT-3′ and 5′-ATC CAT GGA AGC TTT GGG GCT CTT CCT GGG CA-3′. This purified fragment, representing nucleotides –1745 to +155 relative to the transcription start site of the cyclin D1 promoter, was subcloned into the vector pGL3-Basic (Promega) at the MluI and HindIII sites to form the reporter –1745D1/LUC. A series of 5′ deletions were cloned using PCR amplification of the native promoter plasmid and the following oligonucleotides: –1590, 5′-GCA CGC GTG CTA GCC CCC CCC AGG ACC CGG ATT AT-3′; –1440, 5′-GCA CGC GTG CTA GCG AGC TTT TAC TGT TAA GAG-3′; –690, 5′-GCA CGC GTG CTA GCA AAT CCC TTT AAC TTT TAG GG-3′; –545, 5′-GCA CGC GTG CTA GCA AAT GAA AGA AGA TGC AGT CG-3′; –390, 5′-GCA CGC GTG CTA GCT GCT GTG CCG GCC TTC CTA G-3′; –245, 5′-GCA CGC GTG CTA GCT ATG AAA ACC GGA CTA CA-3′; –90, 5′-GCA CGC GTG CTA GCT GGA GCC TCC AGA GGG CTG T-3′.
Site-directed mutagenesis of the AP1, critical regulatory element (CRE), E2F, SP1 and Oct1 sites and linker-scanning mutagenesis of the proximal promoter were done using the QuikChange mutagenesis system (Stratagene) using the –1745D1/LUC plasmid as template. The –310 reporters were constructed by subcloning the XhoI–HindIII fragments from the full-length reporter containing the desired mutations into pGL3-Basic. Restriction enzyme analysis and DNA sequencing confirmed the integrity of all constructs.
Transient transfections
Cells were transiently transfected with LipofectAMINE PLUS reagent (Gibco Life Sciences) in triplicate in 6-well tissue culture plates (Corning, NY). Equal numbers of cells (3 × 105/well) were seeded in each well 24 h prior to transfection. Prior to transfection, cells were equilibrated in 800 µl of fresh medium (OptiMEM with 5% fetal bovine serum, penicillin and streptomycin). Cells were transfected with 5 µg reporter plasmid containing the different cyclin D1 promoter constructs in 200 µl of transfection buffer. Corrections were made for transfection efficiency and variations in harvesting by co-transfecting a Renilla luciferase reporter gene driven either by no (Fig. 5) or the SV40 promoter. After a 4 h incubation with the transfection solution, cells were fed with 4 ml of OptiMEM with 5% fetal bovine serum, penicillin and streptomycin. Cells were harvested 48 h after transfection. The activities of the cyclin D1 firefly reporter gene and the Renilla luciferase internal control were determined in a Dual Luciferase assay (Promega). When GL020924 was used, the procedure was modified in that after 4 h incubation the transfection solution was removed and 4 ml of OptiMEM with 1% fetal bovine serum with 0, 1 or 10 µM GL020924 were added. The parental plasmid, pGL3-Basic, was unaffected by treatment with GL020924 at these levels (data not shown).
Figure 5.

The effect of GL020924 on cyclin D1 promoter reporter constructs in MCF7 cells. Various cyclin D1 promoter derivatives within the –310 to +155 context driving firefly luciferase in pGL3 basic were transfected into MCF7 cells. The constructs were wild-type (black), the –30 to –21 mutant (white), the 10 bp AT-rich promoter derivative (gray) and the 8 bp AT-rich promoter derivative (hatched). Four hours post-transfection, cells were incubated with or without GL020924 for 48 h, at which time promoter activities were assayed. Promoter activities were normalized relative to the co-transfected SV40 control driving Renilla luciferase and are expressed as a percentage of the untreated wild-type promoter construct. All samples were in triplicate; the error bars represent SEM for three separate experiments.
Luciferase assays
Cells were washed once with phosphate-buffered saline (PBS), harvested in 1 ml of PBS, pelleted and lysed with 100 µl of passive lysis buffer (Promega) at room temperature for 15–20 min. The cell lysate was centrifuged for 5 min. Then 10 µl of lysate was assayed for firefly and Renilla luciferase activity using the Dual Luciferase assay (Promega). Assays were carried out in an EG&G Berthold luminometer. After standardization with Renilla luciferase activity, a relative luciferase activity was obtained and the mean and standard deviation from triplicate wells was calculated. Transfections were repeated and reproduced in at least two independent experiments.
In vivo footprinting
In vivo footprinting with DMS of various regions of the cyclin D1 promoter in HCT116 cells was performed as described previously (17) using the following sets of oligonucleotides: 5′-GCCTGGAGACTCTTCGG-3′; 5′-TCGGGCTGCCTTCCTACCTTGACCA-3′; 5′-CCTCGACCAGTCGGTCCTTGCGGGGGT-3′; 5′-GCTCTCGCTTCTGCTGC-3′; 5′-GCTCTTCTGCCCCTCGCCGGAG-3′; 5′-CCTCGCCGGAGCGTGCGGACTCTGCT-3′.
UV mutagenesis was done essentially as described (30). In brief, cells were washed twice with cold PBS and irradiated with 1500 µJ in a Stratagene Stratalinker. Cells were then harvested and DNA was isolated, treated with piperidine and assayed by LMPCR as previously described (17).
Quantitative DNase I footprinting reactions
All reactions were done in a 15 µl final volume. The DNA sequence used, with the footprinted area in bold, was: CGTGAATTCTGCAGATGAGGTACCGTATTAATACCGTTCG-CACTTTCTAGAGCTCTCC. 5′-32P-labeled DNA was incubated with either netropsin or GL02094 at the final concentrations indicated in Figure 4C at room temperature for 30 min in 20 mM Tris–HCl, pH 7.5, 50 mM KCl, 10 mM EDTA and 1% glycerol. DNase I was added in 15 µl of 20 mM Tris–HCl, pH 7.5, 50 mM KCl, 5 mM MgCl2 and 1% glycerol and the mixture incubated for 1 min. The reactions were stopped by addition of 30 µl of 80% formamide load buffer/1× TBE. Reactions were then heated to 90°C for 2 min, placed on ice and electrophoresed on a 20% polyacrylamide denaturing gel in 1× TBE. The gels were exposed to a storage phosphor screen (Molecular Dynamics) and the resulting image was quantitated using ImageQuant software (Molecular Dynamics).
Figure 4.

Biochemical analysis of the DNA-binding properties of GL020924. (A) The chemical structure of GL020924. (B) DNase I footprint of GL020924 on DNA. Increasing concentrations of either netropsin or GL020924 (amounts are indicated in nM above their respective lanes) were assayed by DNase I footprinting as described in Materials and Methods. The DNA sequence of the oligonucleotide is as described in Materials and Methods and the footprinted region, TATTAATA, is indicated by the arrow. (C) Various cyclin D1 promoter sequences were used to compete with the fluorescent/DABCYL indicator DNA duplex (5′-fluorescein-CTTTATTATTTT and 3′-DABCYL-AAAATAATAAAG) for GL020924 binding. Competitor duplex DNAs were: wild-type cyclin D1 sequence, 5′-GGGAGTTTTGTTGAAGTTG-3′ (solid circles); –30 to –21 mutant sequence, 5′-GGTCTGGGATCCGAAGTTG-3′ (open circles); 10 bp AT-rich site-containing sequence, 5′-GGGAGTTTTTTTTAAGTTG-3′ (solid triangles); 8 bp AT-rich site-containing sequence, 5′-GGGAGTTTTAAAAGAGTTG-3′ (open triangles); mutagenized bases are underlined, GL020924 concentration was 1000 nM in all samples.
Hybridization stabilization assay
Hybridization stabilization assays were performed in 96-well plates on a CytoFluor Series 4000 Fluorescence Multi-Well Plate Reader from PerSeptives Biosystems (MA). Emission was set at 530 nm, excitation at 485 nm and gain at 70. Samples consisted of 25 nM 5′-fluorescently labeled oligonucleotide (CTTTATTATTTT) mixed with 30 nM 3′-DABCYL complementary quenching oligonucleotide (both oligonucleotides were purchased from Sigma-Genosys, The Woodlands, TX) in 10 mM HEPES, pH 7.2, 50 mM NaCl and 0.1 mM EDTA. To this was added 1000 nM GL020924 to stabilize the fluorescent- and DABCYL-labeled strands and the mixture was equilibrated at room temperature. The signal was then calculated as %F of the –drug control/+ drug duplex.
For the competiton analysis described in Figure 4C the ligand-bound quenched duplex described above was titrated with the various unlabeled competitor duplexes (see Fig. 4C legend) in 10 mM HEPES, pH 7.2, 50 mM NaCl and 0.1 mM EDTA. The ligand-bound duplex and competitors were equilibrated overnight at room temperature (as above) and the various sequences were quantitatively ranked and assessed as %F. %F is calculated using the following formula:
%F = (Fi – FQ)/(FM – FQ) × 100
where Fi is the relative fluorescence observed for each competitor in the presence of the drug, FQ the relative fluorescence observed in the presence of the drug without competitor and FM the value observed in the absence of drug and competitor. %F was calculated for each individual competitor duplex and that value plotted.
RESULTS
Promoter analysis identifies two elements critical for basal expression of the cyclin D1 gene in MCF7 cells
In order to identify promoter elements critical for cyclin D1 gene expression in human cancer cells, an extensive promoter analysis study was performed in several different cell types. A 1900 bp fragment of the human cyclin D1 promoter from –1745 to +155 was PCR amplified and cloned into the firefly luciferase reporter plasmid pGL3-Basic. The full-length construct (–1745) was 50-fold more active than pGL3-Basic (data not shown). A series of cyclin D1 5′ promoter deletions were similarly constructed and cloned into pGL3-Basic. Promoter activities for the 5′ deletion constructs were compared to that of the full-length (–1745) cyclin D1 promoter following transfection into asynchronous MCF7 human breast carcinoma cells. MCF7 cells overexpress cyclin D1 through amplification of the 11q13 chromosomal region that contains the cyclin D1 gene (31). In addition, overexpression occurs at the level of transcription regulation as the increased amount of mRNA cannot be fully explained by amplification alone in a number of cell lines (31). Deletion of cyclin D1 promoter regions between –1745 and –245 had little effect on basal promoter activity in MCF7 cells (Fig. 1A), even though several potential transcription factor-binding sites have been previously noted in this region. Further deletion of the promoter to –90 reduced activity 2-fold, suggesting the presence of an activator site between –245 and –90 (Fig. 1A). Deletion of the region between –90 and –10 reduced activity to 11% of the full-length promoter, suggesting that much of the basal promoter activity is dependent on this region (Fig. 1A).
Figure 1.
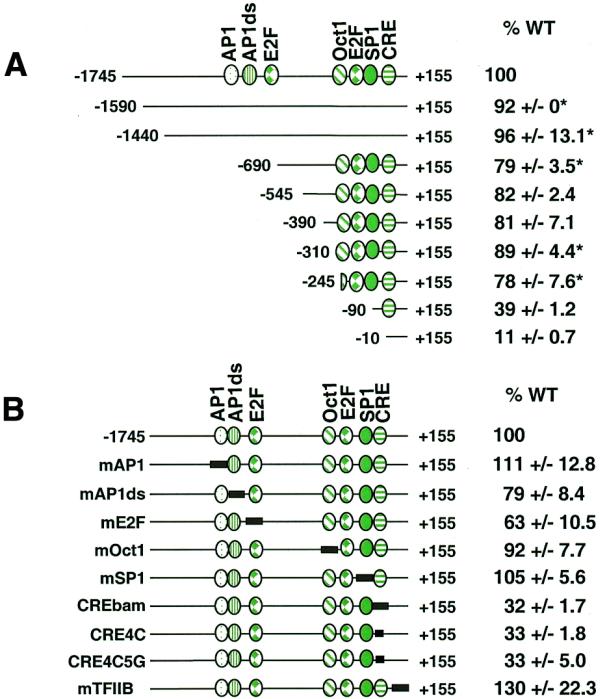
Cyclin D1 promoter analysis in MCF7 cells. Various 5′ deletions or site-directed mutations of the cyclin D1 promoter were inserted into the promoterless firefly luciferase plasmid (pGL3-Basic) and co-transfected into MCF7 cells together with a SV40 promoter-driven Renilla luciferase control plasmid. The length of each construct is indicated relative to the transcriptional start site (+1) and known transcription factor-binding sites are indicated. Firefly luciferase activity for each construct was normalized to Renilla luciferase activity and is shown relative to that of the full-length wild-type promoter (–1745). The data are presented as means ± SEM for a minimum of two independent transfections done in triplicate unless otherwise indicated. Error values denoted with * are standard deviations. (A) 5′ Deletion analysis. (B) Site-specific mutational analysis of known transcription factor-binding sites; the mutagenized region is indicated by a solid box.
Several potential transcription factor-binding sites in the cyclin D1 promoter have been previously noted by homology or by their role in controlling cyclin D1 expression in a variety of human and non-human cell lines. A series of site-specific mutations in these transcription factor-binding sites was constructed in the context of the full-length promoter (–1745) in order to determine whether any of these sites played a role in regulating cyclin D1 gene expression in human cancer cells. Mutation of either the AP-1 site (17), a site immediately downstream of AP-1 (AP1ds) which had been previously footprinted in vivo (17), the Oct1 site (17) or the Sp1 site (16) had little or no effect on basal promoter activity in MCF7 cells (Fig. 1B). Effects that ranged from 63 to 120% of promoter activity were seen in response to mutation of the distal E2F site (Fig. 1B). This site has been shown to be necessary for E2F-1-mediated repression of cyclin D1 in JEG-3 cells (27), suggesting that this E2F site may be a negative or positive regulatory element in different cell types or under different cell growth conditions. The cyclin D1 promoter does not contain a canonical TATA box although it does contain a potential TFIIB-binding site (32). Mutation of this sequence (construct mTFIIB) had no effect on basal promoter activity in MCF7 cells (Fig. 1B).
Mutation of the CRE (TAACGTCA→TGGATCCA, construct CREbam) resulted in a 3-fold down-regulation of promoter activity, suggesting that this element is important for basal cyclin D1 expression in MCF7 cells (Fig. 1B). The CRE has previously been identified as a critical element in the control of cyclin D1 basal expression in both rat chondrocytes (28) and HeLa cells (23). In addition, the CRE has been shown to be involved in activated cyclin D1 expression in response to serum stimulation in NIH 3T3 mouse cells (18), in p60v-src-induced expression in MCF7 cells (29) and in Ras-induced expression in HeLa cells (23). The sequence of the CRE element in the cyclin D1 promoter most closely fits the CRE consensus site but is also similar to the consensus for AP-1 family member binding. The C at position 4 in the CRE consensus (TGACGTc/aA) has been shown to be critical for CREB/CREM/ATF binding but would not be expected to significantly impact AP-1 binding (33). For this reason this base was specifically mutated both alone and in conjunction with the G at position 5 (constructs CRE4C and CRE4C5G, respectively). When either mutant was transfected into MCF7 cells, promoter activity was 33% compared to that of the wild-type promoter, suggesting that a member of the CREB/CREM/ATF family of transcription factors is likely responsible for transcriptional activation from this site. In addition, this DNA sequence was shown to specifically bind purified CREB protein in vitro using a probe from –81 to –23 in an electrophoretic mobility shift assay (data not shown).
A series of linker scanner mutations were made in 10 bp segments from –62 to +20 in the context of the full-length promoter (–1745) in pGL3-Basic to examine the proximal promoter region in more detail. These constructs were assayed for luciferase activity following transfection into MCF7 cells. Mutation of the 10 bp immediately 3′ to the CRE (construct 3′CREm) or bases –40 to –31 or –10 to –1 had little effect on promoter activity in MCF7 cells (Fig. 2). Mutation of either the 10 bp immediately 5′ of the CRE (construct 5′CREm) or of bases –20 to –11 increased promoter activity mildly, suggesting the presence of negative transcriptional regulation sites in these regions (Fig. 2). Mutation of bases –30 to –21 reduced basal promoter activity to 33%, revealing a second important activator site for cyclin D1 expression in MCF7 cells (Fig. 2). Mutation of bases +1 to +9 or +10 to +19 reduced basal promoter activity to 37 and 62%, respectively (Fig. 2). Since these sites are both downstream of the transcriptional start site, they may play a role in cyclin D1 regulation at either the transcriptional or translational level. As both the CRE and the –30–21 sites appear critical to basal promoter activity, a double mutant containing both the CRE4C mutation and the –30–21 mutation was constructed and transfected into MCF7 cells. This double mutant retained only 11% of the activity of the full-length wild-type promoter, activity equal to that of the –10 deletion construct (Fig. 2). A double mutant of the CRE in combination with the +1 to +9 site reduced activity to 14% (Fig. 2).
Figure 2.

Linker-scanning mutagenesis of the –62+19 region of the cyclin D1 promoter. Various linker-scanning mutations of the cyclin D1 promoter from –62 to +19 were co-transfected into MCF7 cells together with a SV40 promoter-driven Renilla luciferase control plasmid. Mutated sequences are indicated by a solid black rectangle. Firefly luciferase activity for each construct was normalized to Renilla luciferase activity and is shown relative to that of the full-length wild-type promoter (–1745). The data are presented as means ± SEM for a minimum of two independent transfections done in triplicate unless otherwise indicated.
To examine the –30–21 site in more detail, a series of smaller mutations were made within this region and constructs were assayed in MCF7 cells. Mutation of bases –30 to –26, –25 to –21, –30 to –28 or –28 to –23 resulted in compromised cyclin D1 promoter activity in MCF7 cells while mutation of bases –23 to –21 increased promoter activity slightly (Table 1). These results indicate that bases between –30 and –24 are the most important for transcriptional activation from this site. This sequence (GAGTTTT) overlaps a recently identified NFκB-binding site between –33 and –24 (GGGGAGTTTT) (20) as well as a potential TCF/LEF site at –27 to –21 (TTTTGTT) (23). However, the identity of the factor responsible for activation of cyclin D1 expression through the –30–21 site in MCF7 cells remains to be determined.
Table 1. Activities of linker-scanning mutations of the –30–18 region of the cyclin D1 promoter in MCF-7 cells.
| Promoter construct |
Sequence (–31 to –18) |
Percent wild-type activity |
| Wild-type | GAGTTTTGTTGAA | 100 |
| –30–21 | tctgggatccGAA | 33 ± 2.2 |
| –30–26 | tctggTTGTTGAA | 43 ± 3.5 |
| –25–21 | GAGTTggcggGAA | 34 ± 4.7 |
| –30–28 | tctTTTTGTTGAA | 33 ± 6.3a |
| –28–23 | GAtgggatTTGAA | 46 ± 5.1 |
| –23–21 | GAGTTTTtccGAA | 138 ± 16.4 |
| 10 bp site | GAGTTTTtTTtAA | 87 ± 11.4 |
| 8 bp site | GAGTTTTaaaagAG | 85 ± 7.8 |
The –1745 wild-type and various linker-scanning mutants of the cyclin D1 promoter were co-transfected into MCF7 cells together with a SV40 promoter-driven Renilla luciferase control plasmid. Firefly luciferase activity for each construct was normalized to Renilla luciferase activity and is shown relative to that of the full-length wild-type promoter (–1745). The data are presented as means ± SEM for a minimum of two independent transfections performed in triplicate. Error values denoted with a are standard deviations. The mutated sequence is in lower case.
The CRE and –30–21 sites are required for basal cyclin D1 expression in several cancer cell lines
As a therapeutic strategy wherein expression of cyclin D1 would be regulated via critical promoter elements, it is important to establish whether the critical transcription factor-binding sites identified above are common between carcinoma cell lines generated from tumors of varying tissue origin. As a first step toward this goal, we assayed mutant promoter constructs in a second breast carcinoma cell line, ZR75-1, that overexpress cyclin D1 through the same chromosomal amplification of the cyclin D1-containing region as found in MCF7 cells. Mutational analysis was also done in a breast cell line that expresses cyclin D1 normally (HMEC). Cyclin D1 is also overexpressed without amplification or chromosomal rearrangement in many colon and pancreatic cancers. Consequently, the promoter constructs were also assayed in a cyclin D1-overexpressing colon cancer cell line (HCT116) and an overexpressing pancreatic cancer cell line (PANC-1). In all cell lines tested mutation of the CRE (constructs CREbam and CRE4C5G) reduced basal promoter activity considerably, although the strongest effect was seen in MCF7 cells (Table 2). Indeed, the –10 deletion construct also retained more basal promoter activity in these other cell lines, ∼20% in HCT116 and ZR75-1 cells and 50% in PANC-1 and HMEC cells, compared to 10% in MCF7 cells (Table 2).
Table 2. Activities of cyclin D1 promoter mutant constructs in MCF7, HCT116, ZR75, PANC-1 and HMEC cells.
| Promoter construct |
MCF7 cells
(% wild-type) |
HCT116 cells
(% wild-type) |
ZR75 cells
(% wild-type) |
PANC-1 cells
(% wild-type) |
HMEC cells
(% wild-type) |
| –1745 (wild-type) | 100 | 100 | 100 | 100 | 100 |
| –10 | 11 ± 0.7 | 22 ± 1.6 | 21 ± 1.1 | 45 ± 1.9 | 50 ± 4.8 |
| CREbam | 32 ± 1.7 | 46 ± 3.3 | 64 ± 6.8 | 52 ± 7.5 | 50 ± 2.1 |
| 3′CREm | 102 ± 7.1 | 86 ± 8.8 | 92 ± 6.4 | 89 ± 4.3 | 74 ± 2.1 |
| 5′CREm | 160 ± 3.6 | 120 ± 15.6 | N/D | 99 ± 6.3 | N/D |
| CRE4C5G | 33 ± 5.0 | 69 ± 5.1 | 54 ± 8.3 | 52 ± 4.9 | N/D |
| –30–21 | 33 ± 2.2 | 91 ± 12.2 | 77 ± 7.0 | 46 ± 4.8 | 78 ± 4.8 |
| +1+9 | 37 ± 4.0 | 46 ± 4.1 | 92 ± 12.5 | 53 ± 8.3 | 74 ± 5.0 |
| CRE4C/–30–21 | 11 ± 1.3 | 30 ± 4.5 | 38 ± 11.4 | 40 ± 6.9 | 26 ± 1.7 |
| CRE4C/+1+9 | 14 ± 0.8 | 32 ± 4.2 | 43 ± 4.0 | 17 ± 3.4 | N/D |
The –1745 wild-type, the –10 deletion or various site-directed mutants of the cyclin D1 promoter were inserted into the promoterless firefly luciferase plasmid (pGL3-basic) and co-transfected into MCF7, HCT116, ZR75, PANC-1 or HMEC cells together with a SV40 promoter-driven Renilla luciferase control plasmid. Firefly luciferase activity for each construct was normalized to Renilla luciferase activity and is shown relative to that of the full-length wild-type promoter (–1745). The data are presented as means ± SEM for a minimum of two independent transfections performed in triplicate.
While mutation of the –30–21 site had little or no effect on basal promoter activity in HCT116 cells and only a small effect in ZR75-1 and HMEC cells, this mutation reduced basal cyclin D1 promoter activity to 46% of wild-type in PANC-1 cells (Table 2). Importantly, however, in all cell lines tested mutation of this region in combination with mutation of the CRE (construct CRE4C/–30–21) reduced basal promoter activity considerably and to a greater extent than did mutation of either site alone (Table 1). This suggests that both the CRE and the –30–21 sites are involved in transcriptional regulation of cyclin D1 basal expression in all of the cyclin D1-overexpressing cancer cell lines tested, as well as in HMEC cells which express normal levels of cyclin D1.
While mutation of the +1 to +9 site had the greatest effect in MCF7 cells, it also reduced promoter activity in HCT116 and PANC-1 cells and had a modest effect on activity in HMEC cells. Mutation of this region had little or no effect on activity in ZR75-1 cells (Table 2). When double mutant constructs containing both this mutation and the CRE4C mutation were tested in MCF7, HCT116, ZR75-1 or PANC-1 cells, luciferase activities were lower than for the –30–21, the CRE4C5G or the CRE4C mutation alone (Table 2 and data not shown). From these data the best regulatory sites to target are the CRE at –52 and the –30–21 element.
The CRE and –30–21 sites are footprinted in vivo in HCT116 cells
In vivo footprinting was done at the CRE and –30–21 sites to verify the importance of these two sites by showing transcription factor binding in vivo. Transcription factor binding at the CRE and the –30–21 site was visualized by in vivo footprinting using DMS or UV light in HCT116 cells in the presence or absence of serum (serum induces cyclin D1 expression; 17,34). Experiments using DMS to probe the reactivity of bases in the CRE region revealed that the G on the top strand of the CRE site is readily methylated both in vitro (Fig. 3A, lanes 1 and 2) and in T cells, which do not express cyclin D1 (Fig. 3A, lanes 5 and 6). However, in DMS-treated serum-starved HCT116 cells (Fig. 3A, lanes 7 and 8) and also in cells released from starvation by serum addition (Fig. 3A, lanes 9 and 10) this G residue is protected from methylation, suggesting transcription factor binding at this site. This is in agreement with previous in vivo footprints of the CRE in quiescent, serum-stimulated and normally cycling human WI-38 lung fibroblasts (17). The finding that the CRE is protected in both serum-starved and serum-stimulated cells is in accordance with current understanding of the mode of action of CREB. CREB has been shown to bind to the CRE in both its active and inactive states. Transcriptional activation by CREB occurs upon phosphorylation of CREB and subsequent recruitment of the transcriptional co-activator CBP to promoter-bound CREB (35).
Figure 3.

In vivo footprinting of the cyclin D1 promoter. (A) Visualization of factor binding in the CRE region using DMS. In vitro treated DNA control (lanes 1–4, lanes 1 and 2 are a darker exposure), T cells (lanes 5 and 6), serum-starved HCT116 cells (lanes 7 and 8) or HCT116 cells with serum (lanes 9 and 10). The sequence of the CRE-containing region with the hypomethylated G residue is indicated. (B) Visualization of factor-binding in the –75 to –62 and the –30 to –21 regions using UV. In vitro treated DNA control (lane 1), resting T cells (lanes 2 and 3), activated T cells (lanes 4 and 5), non-irradiated HCT116 cells (lanes 6 and 7), serum-starved HCT116 cells (lanes 8 and 9), serum-released HCT116 cells (lanes 10 and 11) DMS ladder (lanes 12 and 13), molecular weight markers (lane M). The sequences of the –75 to –62 and the –30 to –21 regions are shown with the hyperreactive T residues indicated.
The –30–21 site was probed using UV light as the reactive agent, which can produce 6–4 photoproducts in dipyrimidine residues. In UV-irradiated serum-starved or serum-stimulated HCT116 cells two adjacent reactive T residues within the –30–21 site appear as sites of enhanced cleavage when the extracted DNA is treated with piperidine (Fig. 3B, lanes 8–11). These cleavage products are only weakly detectable in the in vitro treated control DNA (lane 1) or in DNA from resting (lanes 2 and 3) or activated T cells (lanes 4 and 5), which do not express cyclin D1, and are absent from non-irradiated HCT116 cells (lanes 6 and 7). This suggests that a protein is binding to the –30–21 site in HCT116 cells causing localized conformational changes that lead to increased photoreactivity of these two T residues. As with the CRE footprint, binding is equivalent in serum-starved and serum-stimulated cells. As indicated previously, the identity of the factor responsible for binding to the –30–21 site remains to be determined.
A third in vivo footprint in HCT116 cells was identified at –74/–73 (Fig. 3B) using UV light. This footprint overlaps a TCF/LEF-binding site at –76 to –64 (TTTGATCTTT) shown to be required for β-catenin activation of the cyclin D1 promoter in HeLa, 293T and cyclin D1-overexpressing SW480 colon cancer cells (22,23). Indeed, mutation of residues –85 to –70 or –75 to –60 results in a 3-fold reduction in basal promoter activity in transiently transfected HCT116 but not MCF7 cells (data not shown). Nuclear β-catenin accumulation can occur as a result of mutation of the adenomatous polyposis coli tumor suppressor (APC) gene, a mutation that occurs in most colon cancers (36) leading to increased transactivation of target genes by β-catenin/LEF complexes. HCT116 cells are wild-type for APC but express mutant β-catenin that also hyper-accumulates in the nucleus (37,38). The in vivo footprint visualized in the –74/–73 region in HCT116 cells may be due to β-catenin/LEF binding.
Targeting of the –30–21 site with a DNA-binding ligand results in inhibition of cyclin D1 promoter activity in MCF7 cells
The previous data clearly demonstrate that the CRE at –52 is critical in a variety of cancer cell lines and that the –30–21 element, while critical in a number of cancer cell lines in conjunction with other mutations, is important by itself only in the breast cancer cell line MCF-7. The next step was to validate each as a target for minor groove-binding ligands by testing the effect of these ligands on protein binding and/or promoter activity.
In order to efficiently compete with transcription factors for binding to a specific DNA site in vivo, DNA-binding ligands need to have considerable specificity and affinity for that site. In addition, in order to attain gene specificity and thus ameliorate potential side-effects, such a ligand must recognize a relatively large DNA-binding site. Known small, organic DNA-binding ligands do not meet either criterion: they bind with relatively low affinity and to short DNA sequences of 2–5 bp. Netropsin, for example, binds to the minor groove of DNA with a preference for four to five A/T base pairs and with an EC50 of 86–1250 nM, depending upon sequence (39), making it inappropriate for specific displacement of transcription factors. To circumvent netropsin’s limitations, GL020924 (structure shown in Fig. 4A) has been chemically synthesized (manuscript in preparation). As would be predicted from its structure, GL020924 footprints a longer DNA site than does netropsin, as measured by DNase footprinting, ∼10 bp compared to 5 bp for netropsin (Fig. 4B). In addition, GL020924 binds with 15-fold increased affinity as compared to netropsin, 0.37 versus 5.7 nM (Fig. 4B). In terms of specificity, GL020924 has been shown to have high affinity and specificity for A/T-rich DNA sequences (data not shown).
Due to its affinity for A/T-rich DNA sequences, GL020924 would not be predicted to bind strongly to either the CRE or the –30–21 site of the cyclin D1 promoter. Therefore, GL020924-binding sites were engineered into cyclin D1 reporters overlapping either the CRE or the –30–21 site. For the CRE the three ligand-binding sites all overlapped the final A of the CRE, TAACGTCA, and including this A were A10, AT2A4T2A2 or A5T5. GL020924 was unable to displace CREB in in vitro electrophoretic mobility shift assay experiments and had no effect in transient transfection assays utilizing the engineered reporters (data not shown). However, GL020924 was successful when targeted to the –30–21 site.
Two potential binding sites for GL020924, a 10 bp AT-rich and an 8 bp AT-rich sequence were engineered into the cyclin D1 promoter overlapping the –30–21 site. Since the 3′-end of the –30–21 site is A/T rich, it was possible to introduce a potential GL020924 binding site that overlapped the –30–21 site by changing 2 bp (10 bp site, Table 1). The 8 bp site was constructed by mutating 5 bp of the wild-type promoter sequence to produce an uninterrupted eight A/T stretch (8 bp site, Table 1). Binding of GL020924 to these sites and not to the wild-type or mutant sites was confirmed using a hybridization stabilization assay (Fig. 4C) as described in Materials and Methods. When this assay was run using the various cyclin D1 sequences as competitors for GL020924 binding, both the 8 and 10 bp ligand-binding site sequences resulted in markedly increased fluorescence compared to the wild-type or –30–21 mutant sequences (Fig. 4C). This indicates a higher affinity interaction of GL020924 with both the 8 and 10 bp sites and a weak interaction with the –30–21 wild-type and mutant sequences.
Both ligand-binding sites were cloned separately into the context of the –310 cyclin D1 promoter in pGL3-Basic to address whether binding of GL020924 to the 8 and 10 bp sites would affect gene expression in cell-based assays. These constructs were transfected into MCF7 cells and shown to retain 85 and 87% of wild-type promoter activity, respectively (Fig. 5, 0 µM) in the absence of GL020924, indicating that introduction of these ligand-binding sites overlapping the –30–21 site did not adversely affect transcriptional activation. This is consistent with results demonstrating that mutation of the –20–11 and the –23–21 regions of the cyclin D1 promoter did not markedly affect promoter activity in MCF7 cells (Fig. 2 and Table 1). When transiently transfected MCF7 cells were treated with 1 µM GL020924 and assayed after 48 h, promoters with either the 8 or 10 bp ligand-binding site showed reduced promoter activity. At 10 µM GL020924, activity of these promoters was reduced to a level of activity equivalent to mutation of the –30–21 site (25% of wild-type, Fig. 5). Activity of the wild-type cyclin D1 promoter construct remained high and was unaffected by GL020924. The activity of the –30–21 mutant construct was ∼25% of wild-type and was also unaffected by GL020924 treatment (Fig. 5). Thus, GL020924 specifically reduced cyclin D1 promoter activity 4-fold, to the level of activity achieved by mutation of this region, when a GL020924-binding site was present overlapping the –30–21 site. The observation that GL020924 had no effect on the transcriptional activity of promoters that lacked a ligand-binding site in the –30–21 region demonstrates that this DNA-binding ligand can regulate gene expression in a sequence-specific manner in mammalian cells.
DISCUSSION
Expression of cyclin D1 is a tightly regulated step in the normal mammalian cell cycle and is abnormally high in many human cancers, suggesting that interfering with cyclin D1 gene expression in cancer cells would have therapeutic efficacy. The design and use of ligands that will regulate the expression of target genes by recognizing specific DNA sequences critical for gene expression provides a novel method for controlling gene expression, as well as an innovative approach to drug development and therapeutic intervention. Toward this end, we are developing DNA-binding ligands to target specific promoter sequences in disease-associated genes such as cyclin D1. In order to utilize such ligands to control gene expression, a critical regulatory site in the promoter must first be identified, the interaction between the transcription factor and its binding site in DNA must be influenced by a minor groove-binding ligand and, finally, a sequence-specific molecule must be synthesized that can target the native promoter sequence. The studies performed here on the –30–21 site of the cyclin D1 promoter meet the first two of these criteria and, most importantly, validate the –30–21 site as a potential target site.
The first criterion in this strategy was met through a thorough promoter analysis that identified two sites critical for expression of cyclin D1 in a variety of carcinoma cell lines. Our analysis of the cyclin D1 promoter indicates that the CRE at –52 is critical for cyclin D1 promoter expression in two different breast cancer cell lines (MCF7 and ZR75-1), as well as in HCT116 colon cancer cells and PANC-1 pancreatic cancer cells (Table 2). This element was also found to be critical for cyclin D1 promoter expression in HMEC breast cells that express cyclin D1 at normal levels. Thus the CRE may be a regulatory element that could be targeted in many types of human cancer in which cyclin D1 is regulated incorrectly. An in vivo footprint was clearly visible in the CRE region in HCT116 cells (Fig. 3A). Mutational analysis of the CRE in a variety of cell lines (Table 2), together with in vitro binding data (not shown), strongly suggests that a member of the CREB family of transcription factors is binding at this site. Mutation of the regions flanking the CRE had no effect on gene expression, suggesting that this region would be an appropriate target for a DNA-binding ligand. However, attempts to disrupt the binding of CREB family members to the cyclin D1 CRE in vitro and in cell-based assays with GL020924 by engineering a ligand-binding site overlapping the CRE were unsuccessful (data not shown), making this promoter target site refractive to regulation by conventional minor groove-binding ligands. This result is not surprising, as evidence from the crystal structure of bZIP–DNA complexes indicates that bZIP proteins bind to their consensus sites by contacting DNA almost exclusively in the major groove (40). As has been shown previously, displacement of this class of proteins with minor groove-binding ligands such as GL020924 may be problematical (41).
A second element between –30 and –21 (–30–21) was identified as important for basal cyclin D1 promoter activity in MCF7 breast cancer cells and in PANC-1 pancreatic cancer cells (Table 2). Mutation of the –30–21 site alone had less of an effect in ZR75-1, HCT116 or HMEC cells, although it did reduce promoter activity. In all of the cell lines tested activity of cyclin D1 promoter constructs containing a mutation of the CRE in combination with a mutation of the –30–21 site was severely compromised beyond the level of the CRE mutation itself (Table 2). Thus it seems that both of these elements play a role in basal transcriptional regulation of cyclin D1 in a number of different cyclin D1-overexpressing cancer cells as well as in a transformed cell line with normal cyclin D1 levels. The demonstration that expression of cyclin D1 in diverse cell types is controlled by common regulatory regions of the promoter suggests that it may be possible to develop a single DNA-binding ligand with efficacy in cancers arising from a variety of tissues.
The identity of the transcription factor bound at the –30–21 site remains to be determined. A detailed mutational analysis of the –30 to –21 region indicated that bases between –30 and –24 are the most important for transcriptional activation from this site (Table 1). This sequence (GAGTTTT) overlaps a recently identified NFκB-binding site between –33 and –24 (GGGGAGTTTT) that was found to be required for NFκB-mediated cyclin D1 expression in COS-7 cells and for serum-induced expression in NIH 3T3 cells (20). It is possible that the reduced promoter activity observed with the –30–21 mutation is a consequence of mutating the NFκB site. However, it should be noted that in MCF7 cells mutation of the –40 to –31 region, which includes the first three G residues of the NFκB site, did not reduce cyclin D1 promoter activity (Fig. 2). Hence, the transcription factor that binds the –30–21 site and regulates cyclin D1 basal promoter activity in MCF7 cells may be distinct from NFκB.
Recently, the cyclin D1 promoter has been shown to be inducible by β-catenin through a number of TCF/LEF-binding sites (22,23). The –30–21 site overlaps a potential TCF/LEF site at –27 to –21 (TTTTGTT), although mutation of this site did not seem to affect β-catenin-dependent activation of the cyclin D1 promoter or basal promoter activity in HeLa cells (23). In MCF7 cells bases –23 to –21 (GTT of the TCF/LEF site) appear to be dispensable for transcriptional activation mediated through the –30–21 site while bases –28 to –30, which are outside the core TCF/LEF consensus sequence, are required for this activity (Table 1). However, the possibility that TCF/LEF binding is responsible for activation of cyclin D1 basal activity through the –30 to –21 site cannot be ruled out. Interestingly, mutation of the –30–21 site had very little effect on cyclin D1 promoter activity in HCT116 colon cancer cells which contain mutant β-catenin and in which TCF/LEF has been shown to play an important role in regulating cyclin D1 expression (23).
Upon identification of a critical site in the cyclin D1 promoter at –30–21, overlapping ligand-binding sites were engineered around this region with the specific ligand GL020924 in mind. GL020924 binds to the minor groove of DNA and has a preference for A/T-rich stretches of 8–10 bp. Thus, cyclin D1 promoter constructs were engineered to contain an 8 and a 10 bp AT-rich ligand-binding site overlapping the critical –30–21 region. These promoter constructs retained high levels of promoter activity in MCF7 cells in the absence of GL020924 (Table 1 and Fig. 5). Treatment of MCF7 cells transiently transfected with engineered promoters with GL020924 resulted in dose-dependent down-regulation of cyclin D1 promoter activity to levels equivalent to direct mutation of the –30–21 site. Promoter constructs lacking the GL020924 ligand-binding sites were unaffected, demonstrating regulation of gene expression in a sequence-specific manner in mammalian cells. These results suggest that this strategy may be used to specifically regulate improperly expressed endogenous cyclin D1 in tumor cells by developing a DNA-binding ligand with specificity for the –30–21 promoter region. Indeed, antisense data suggests that a 3- to 4-fold down-regulation of cyclin D1 is sufficient to impact the disease state in breast cancer, suggesting that the level of down-regulation observed with the –30–21 mutation may be sufficient for a therapeutic effect.
The final criterion, synthesis and testing of ligands specific to the –30–21 region, has not yet been met, but efforts are currently underway to generate such compounds. Once identified, the assays are in place to directly assess the effect of these ligands on cyclin D1 expression. Given the results with GL020924 and the –30–21 regulatory region, once such –30–21-specific ligands have been synthesized, regulation of cyclin D1 expression should be possible in breast cancer cells and, perhaps, in breast cancer itself.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Laurakay Bruhn and Lillian Lou for careful reading of this manuscript.
References
- 1.Beerli R.R., Dreier,B. and Barbas,C.F. (2000) Positive and negative regulation of endogenous genes by designed transcription factors. Proc. Natl Acad. Sci. USA, 97, 1495–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kang J.S. and Kim,J.-S. (2000) Zinc finger proteins as designer transcription factors. J. Biol. Chem., 275, 8742–8748. [DOI] [PubMed] [Google Scholar]
- 3.Dickinson L.A., Gulizia,R.J., Trauger,J.W., Baird,E.E., Mosier,D.E. and Gottesfeld,J.M. (1998) Inhibition of RNA polymerase II transcription in human cells by synthetic DNA-binding ligands. Proc. Natl Acad. Sci. USA, 95, 12890–12895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dickinson L.A., Trauger,J.W., Baird,E.E., Dervan,P.B., Graves,B.J. and Gottesfeld,J.M. (1999) Inhibition of Ets-1 DNA binding and ternary complex formation between Ets-1, NF-kappaB and DNA by a designed DNA-binding ligand. J. Biol. Chem., 274, 12765–12773. [DOI] [PubMed] [Google Scholar]
- 5.Hall M. and Peters,G. (1996) Genetic alterations of cyclins, cyclin-dependent kinases and Cdk inhibitors in human cancer. Adv. Cancer Res., 68, 67–108. [DOI] [PubMed] [Google Scholar]
- 6.Zhang T., Nanney,L.B., Luongo,C., Lamps,L., Heppner,K.J., DuBois,R.N. and Beauchamp,R.D. (1997) Concurrent overexpression of cyclin D1 and cyclin-dependent kinase 4 (Cdk4) in intestinal adenomas from multiple intestinal neoplasia (Min) mice and human familial adenomatous polyposis patients. Cancer Res., 57, 169–175. [PubMed] [Google Scholar]
- 7.Gansauge S., Gansauge,F., Ramadani,M., Stobbe,H., Rau,B., Harada,N. and Beger,H.G. (1997) Overexpression of cyclin D1 in human pancreatic carcinoma is associated with poor prognosis. Cancer Res., 57, 1634–1637. [PubMed] [Google Scholar]
- 8.Zhou P., Jiang,W., Zhang,Y.-J., Kahn,S.M., Schieren,I., Santella,R.M. and Weinstein,I.B. (1995) Antisense to cyclin D1 inhibits growth and reverses the transformed phenotype of human esophageal cancer cells. Oncogene, 11, 571–580. [PubMed] [Google Scholar]
- 9.Arber N., Doki,Y., Han,E.K.-H., Sgambato,A., Zhou,P., Kim,N.-H., Delohery,T., Klein,M.G., Holt,P.R. and Weinstein,I.B. (1997) Antisense to cyclin D1 inhibits the growth and tumorigenicity of human colon cancer cells. Cancer Res., 57, 1569–1574. [PubMed] [Google Scholar]
- 10.Kornmann M., Arber,N. and Korc,M. (1998) Inhibition of basal and mitogen-stimulated pancreatic cancer cell growth by cyclin D1 antisense is associated with loss of tumorigenicity and potentiation of cytotoxicity to cisplatinum. J. Clin. Invest., 101, 344–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cagnoli M., Barbieri,F., Bruzzo,C. and Alama,A. (1998) Control of cyclin D1 expression by antisense oligonucleotides in three ovarian cancer cell lines. Gynecol. Oncol., 70, 372–377. [DOI] [PubMed] [Google Scholar]
- 12.Wang M.B., Billings,K.R., Venkatesan,N., Hall,F.L. and Srivatsan,E.S. (1998) Inhibition of cell proliferation in head and neck squamous cell carcinoma cell lines with antisense cyclin D1. Otolaryngol. Head Neck Surg., 119, 593–599. [DOI] [PubMed] [Google Scholar]
- 13.Kornmann M., Danenberg,K.D., Arber,N., Beger,H.G., Danenberg,P.V. and Korc,M. (1999) Inhibition of cyclin D1 expression in human pancreatic cancer cells is associated with increased chemosensitivity and decreased expression of multiple chemoresistance genes. Cancer Res., 59, 3505–3511. [PubMed] [Google Scholar]
- 14.Sauter E.R., Nesbit,M., Litwin,S., Klein-Szanto,A.J., Cheffetz,S. and Herlyn,M. (1999) Antisense cyclin D1 induces apoptosis and tumor shrinkage in human squamous carcinomas. Cancer Res., 59, 4876–4881. [PubMed] [Google Scholar]
- 15.Sauter E.R., Herlyn,M., Liu,S.C., Litwin,S. and Ridge,J.A. (2000) Prolonged response to antisense cyclin D1 in a human squamous cancer xenograft model. Clin. Cancer Res., 6, 654–660. [PubMed] [Google Scholar]
- 16.Motokura T. and Arnold,A. (1993) PRAD1/cyclin D1 proto-oncogene: genomic organization, 5′ DNA sequence and sequence of a tumor-specific rearrangement breakpoint. Genes Chromosom. Cancer, 7, 89–95. [DOI] [PubMed] [Google Scholar]
- 17.Herber B., Truss,M., Beato,M. and Müller,R. (1994) Inducible regulatory elements in the human cyclin D1 promoter. Oncogene, 9, 1295–1304. [PubMed] [Google Scholar]
- 18.Philipp A., Schneider,A., Vasrik,I., Finke,K., Xiong,Y., Beach,D., Alitalo,K. and Eilers,M. (1994) Repression of cyclin D1: a novel function of MYC. Mol. Cell. Biol., 14, 4032–4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Watanabe G., Howe,A., Lee,R.J., Albanese,C., Shu,I.-W., Karnezis,A.N., Zon,L., Kyriakis,J., Rundell,K. and Pestell,R.G. (1996) Induction of cyclin D1 by simian virus 40 small tumor antigen. Proc. Natl Acad. Sci. USA, 93, 12861–12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hinz M., Krappmann,D., Eichten,A., Heder,A., Scheidereit,C. and Strauss,M. (1999) NF-kappaB function in growth control: regulation of cyclin D1 expression and G0/G1-to-S-phase transition. Mol. Cell. Biol., 19, 2690–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsumura I., Kitamura,T., Wakao,H., Tanaka,H., Hashimoto,K., Albanese,C., Downward,J., Pestell,R.G. and Kanakura,Y. (1999) Transcriptional regulation of the cyclin D1 promoter by STAT5: its involvement in cytokine-dependent growth of hematopoietic cells. EMBO J., 18, 1367–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shtutman M., Zhurinsky,J., Simcha,I., Albanese,C., D’Amico,M., Pestell,R.G. and Ben-Ze’ev,A. (1999) The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc. Natl Acad. Sci. USA, 96, 5522–5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tetsu O. and McCormick,F. (1999) Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature, 398, 422–426. [DOI] [PubMed] [Google Scholar]
- 24.Albanese C., Johnson,J., Watanabe,G., Eklund,N., Vu,D., Arnold,A. and Pestell,R.G. (1995) Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J. Biol. Chem., 270, 23589–23597. [DOI] [PubMed] [Google Scholar]
- 25.Watanabe G., Lee,R.J., Albanese,C., Rainey,W.E., Batlle,D. and Pestell,R.G. (1996) Angiotensin II activation of cyclin D1-dependent kinase activity. J. Biol. Chem., 271, 22570–22577. [DOI] [PubMed] [Google Scholar]
- 26.Yan Y.-X., Nakagawa,H., Lees,M.-H. and Rustig,A.K. (1997) Transforming growth factor-alpha enhances cyclin D1 transcription through the binding of early growth response protein to a cis-regulatory element in the cyclin D1 promoter. J. Biol. Chem., 272, 33181–33190. [DOI] [PubMed] [Google Scholar]
- 27.Watanabe G., Albanese,C., Lee,R.J., Reutens,A., Vairo,G., Henglein,B. and Pestell,R.G. (1998) Inhibition of cyclin D1 kinase activity is associated with E2F-mediated inhibition of cyclin D1 promoter activity through E2F and Sp1. Mol. Cell. Biol., 18, 3212–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beier F., Lee,R.J., Taylor,A.C., Pestell,R.G. and LuValle,P. (1999) Identification of the cyclin D1 gene as a target of activating transcription factor 2 in chondrocytes. Proc. Natl Acad. Sci. USA, 96, 1433–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee R.J., Albanese,C., Stenger,R.J., Watanabe,G., Inghirami,G., Haines,G.K.I., Webster,M., Muller,W.J., Brugge,J.S., Davis,R.J. and Pestell,R.G. (1999) pp60(v-src) induction of cyclin D1 requires collaborative interactions between the extracellular signal-regulated kinase, p38 and Jun kinase pathways. A role for cAMP response element-binding protein and activating transcription factor-2 in pp60(v-src) signaling in breast cancer cells. J. Biol. Chem., 274, 7341–7350. [DOI] [PubMed] [Google Scholar]
- 30.Pfeifer G.P. and Tornaletti,S. (1997) Footprinting with UV irradiation and LMPCR. Methods, 11, 189–196. [DOI] [PubMed] [Google Scholar]
- 31.Buckley M.F., Sweeney,K.J., Hamilton,J.A, Sini,R.L., Manning,D.L., Nicholson,R.I., deFazio,A., Watts,C.K., Musgrove,E.A. and Sutherland,R.L. (1993) Expression and amplification of cyclin genes in human breast cancer. Oncogene, 8, 2127–2133. [PubMed] [Google Scholar]
- 32.Lagrange T., Kapanidis,A.N., Tang,H., Reinberg,D. and Ebright,R.H. (1998) New core promoter element in RNA polymerase II-dependent transcription: sequence-specific DNA binding by transcription factor IIB. Genes Dev., 12, 34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meyer T.E. and Habener,J.F. (1993) Cyclic adenosine 3′,5′-monophosphate response element binding protein (CREB) and related transcription-activating deoxyribonucleic acid-binding proteins. Endocr. Rev., 14, 269–290. [DOI] [PubMed] [Google Scholar]
- 34.Sherr C.J. (1993) Mammalian G1 cyclins. Cell, 73, 1059–1065. [DOI] [PubMed] [Google Scholar]
- 35.Kwok R.P., Lundblad,J.R., Chrivia,J.C., Richards,J.P., Bachinger,H.P., Brennan,R.G., Roberts,S.G., Green,M.R., Goodman,R.H. and Kwok (1994) Nuclear protein CBP is a coactivator for the transcription factor CREB. Nature, 370, 223–226. [DOI] [PubMed] [Google Scholar]
- 36.Kinzler K.W. and Vogelstein,B. (1996) Lessons from hereditary colorectal cancer. Cell, 87, 159–170. [DOI] [PubMed] [Google Scholar]
- 37.Morin P.J., Sparks,A.B., Korinek,V., Barker,N., Clevers,H., Vogelstein,B. and Kinzler,K.W. (1997) Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science, 275, 1787–1790. [DOI] [PubMed] [Google Scholar]
- 38.Ilyas M., Tomlinson,I.P., Rowan,A., Pignatelli,M. and Bodmer,W.F. (1997) Beta-catenin mutations in cell lines established from human colorectal cancers. Proc. Natl Acad. Sci. USA, 94, 10330–10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abu-Daya A., Brown,P.M. and Fox,K.R. (1995) DNA sequence preferences of several AT-selective minor groove binding ligands. Nucleic Acids Res., 23, 3385–3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Keller W., Konig,P. and Richmond,T.J. (1995) Crystal structure of a bZIP/DNA complex at 2.2 Å: determinants of DNA specific recognition. J. Mol. Biol., 254, 657–667. [DOI] [PubMed] [Google Scholar]
- 41.Bremer R.E., Baird,E.E. and Dervan,P.B. (1998) Inhibition of major-groove-binding proteins by pyrrole-imidazole polyamides with an Arg-Pro-Arg positive patch. Chem. Biol., 5, 119–133. [DOI] [PubMed] [Google Scholar]