

Abstract
Most recent evidence suggests that the process of tissue repair is driven by stem-like cells that reside in multiple tissues but are replenished by precursor cells from bone marrow. Among the candidates for the reparative cells are the adult stem cells from bone marrow referred to as either mesenchymal stem cells or marrow stromal cells (MSCs). We recently found that after MSCs were replated at very low densities to generate single-cell-derived colonies, they did not exit a prolonged lag period until they synthesized and secreted considerable quantities of Dickkopf-1, an inhibitor of the canonical Wnt signaling pathway. We also found that when the cells were cocultured with heat-shocked pulmonary epithelial cells, they differentiated into epithelial cells. Most of the MSCs differentiated without evidence of cell fusion but up to one-quarter underwent cell fusion with the epithelial cells. A few also underwent nuclear fusion. The results are consistent with the interesting possibility that MSCs and similar cells repair tissue injury by three different mechanisms: creation of a milieu that enhances regeneration of endogenous cells, transdifferentiation, and perhaps cell fusion.
One of the most intriguing questions in both biology and medicine is: How do complex organisms repair injured tissues? The repair of tissues in mammals is not as efficient as the dramatic regeneration of whole body parts that is observed in simple organisms such as planaria, hydra, or even vertebrates such as newts (see ref. 1). At the same time, it is apparent that tissue repair is an ongoing process throughout the lives of the most complex organisms.
How Are Tissues Repaired? The Cohnheim Hypothesis
One hypothesis about tissue repair in mammals was advanced in the middle of the 19th century by Cohnheim (2), who suggested that all of the cells come from the bloodstream and therefore, in light of subsequent observations, from the bone marrow. Cohnheim's hypothesis has been the subject of much debate and experimentation. Some early experiments in animal models appeared to rule out the possibility that any of the noninflammatory cells involved in healing tissues and organ originated from the bloodstream and bone marrow (see ref. 3). Therefore, most studies on wound repair focused on cells resident in the tissues such as pericytes that are seen to proliferate during the repair in most tissues (see refs. 4 and 5). The interest in resident, reparative cells has been heightened by the recent observations that small, stem-like cells with considerable plasticity are found in a variety of tissues including muscle (6, 7), fat (8), liver (9, 10), synovial membranes (11), and brain (12). At the same time, recent observations have largely validated Cohnheim's hypothesis and indicated that the stem cells found in most tissues are replenished by stem cells for nonhematopoietic tissues found in the marrow.
Reparative Cells from Bone Marrow: Data from Experimental Animals
A large series of reports have indicated that marrow-derived cells were found to engraft as differentiated cells into multiple tissues after infusion into experimental animals of either whole bone marrow or one or more subpopulations from marrow. The number of such publications is now too large to summarize in the present context. (For a partial list of the publications, see refs. 4 and 13-27.) Also, several reports have described isolation of precursor cells for nonhematopoietic tissues from peripheral blood (28-30). However, several recent publications have challenged some of the evidence that bone marrow contains cells that can repair nonhematopoietic tissues.
Wagers et al. (31) transplanted single hematopoietic cells that expressed GFP into lethally irradiated mice and examined parabiotic mice in which the cells of one partner expressed GFP. They concluded that there was “little evidence for developmental plasticity of adult hematopoietic stem cells.” Their results therefore seemed to challenge several publications reporting that some hematopoietic stem cells can also differentiate in vivo into nonhematopoietic cells (see refs. 23-25). As indicated by the subsequent exchange of comments (32, 33), the apparent discrepancies are probably explained by several subtle differences in the experimental conditions. In particular, there were differences in the subpopulations of the cells used for the experiments and in the sensitivity and the stability of the genes used to mark the donor cells. A similar challenge came from the report by Castro et al. (34), who followed LacZ gene label in hematopoietic stem cells, referred to as SP cells, after i.v. infusion into mice that were lethally irradiated. They examined brains from the recipient mice and interpreted their results as a “failure of bone marrow cells to transdifferentiate into neural cells in vivo.” Therefore the results were apparently inconsistent with several previous reports that identified donor-derived neural cells in mice after i.v. infusion of bone marrow cells (15, 16, 21). As again reflected in an exchange of comments (35, 36), the apparent discrepancies can probably be explained by a number of critical experimental variables.
A further challenge to some of the conclusions about reparative cells in bone marrow came from reports indicating that at least some of the plasticity of adult stem cells can be attributed to cell fusion and not direct differentiation (37-39). Cell fusion was observed when adult stem cells from the CNS (37) or unfractionated mononucleated cells from marrow (38) were added to cultured embryonic stem cells, and the added cells were selected for by the presence of a selectable marker gene. The resulting tetraploid cells exhibited full pluripotentiality, including multilineage contribution to chimeras (37). In unrelated experiments, bone marrow-derived hepatocytes were serially transplanted into transgenic mice that are a model for tyrosinemia (40). The results indicated that the marrow-derived hepatocytes that rescued the mice arose by cell fusion and not by differentiation.
At the moment it is difficult to resolve all of the apparent discrepancies in observations on the presence of reparative cells in bone marrow, but several generalizations can be made. One is that many of the apparent discrepancies are explained by the surprisingly difficult problem of devising markers for donor cells that are sensitive enough and that remain stable after the cells engraft into diverse tissues. Another is that the level of engraftment of marrow-derived cells into nonhematopoietic tissues is low unless the recipient animal is a rapidly growing embryo or newborn animal or has injury to some organ or tissue. Unfortunately, it has been extremely difficult to define the kind and degree of tissue injury that promotes engraftment. Still another generalization is that many of the apparent discrepancies can be explained by subtle but critical differences in the donor cells used for the experiments. For example, the inbred strain of mice used for donor cells (41) or even the cell cycle of the infused cells (42) can critically affect the results. Finally, the observations indicating that cell fusion is unexpectedly common in experiments with adult stem cells raise a new series of unanswered questions as to the fate of such cells during repair of tissue damage. Cell fusion cannot explain the ability of clonally derived marrow cells to differentiate into multiple cell phenotypes in culture (for examples, see refs. 43 and 44). The possible contribution of cell fusion to the engraftment and differentiation of marrow-derived cells observed in in vivo experiments has not been defined. The available data do not resolve questions such as: Are the fused cells dead-end products that disappear with time, or are they intermediates in the normal process of tissue repair?
Reparative Cells from Bone Marrow: Data from Patients with Marrow or Organ Transplants
In view of the questions raised by some of the observations in experimental animals, it is surprising that the most convincing evidence for Cohnheim's hypothesis appears to come from assays on tissues of patients who have received either bone marrow transplants or organ transplants. In female patients who received bone marrow transplants from male donors, male cells were found in the liver as hepatocytes and cholangiocytes (45), in kidney as tubular epithelial cells (46), in lung as epithelial and endothelial cells (47), in heart as cardiomyocytes (48), and in brain as neurons (49) and Purkinje cells (50). In most of the reports, the number of differentiated cells detected was small. In patients who received allografts of organ transplants, the number of differentiated recipient cells detected in the organ was low in many patients, but high in some instances in which pathological changes occurred in the organ. A value of 38% of cholangiocytes and 43% of hepatocytes were male in a male who received a female liver and then developed fibrosing cholestatic recurrent hepatitis C (45). In males who received cardiac transplants from female donors, the Y chromosome was detected by cardiac biopsies in a mean of 0.04% of cardiomyocytes in one study (51) and 0.16% in a second study (52). However, 29% of the cardiomyocytes contained the Y chromosome in “hot spots” of pathological changes in the heart of the only 1 of the 5 patients in the first study who died of cardiac rejection (51). In patients who received lung allografts (53), recipient-derived cells were detected as bronchial epithelial cells, type II pneumocytes, and seromucous glands lying adjacent to large bronchi in the transplanted lungs from all seven patients. The chimerism was as high as 24% in epithelial structures displaying chronic injury. Taken as a whole, the results indicate that although many tissues contain stem-like cells that can participate in tissue repair, the resident cells in tissues are replenished by cells from the bloodstream and the bone marrow during some forms of tissue repair.
Which Cells from Bone Marrow?
A more difficult question to resolve has been: Which cells from bone marrow repair injured tissues? A number of candidate cell types are currently being explored, including marrow cells that can serve as hematopoietic precursors (23-25, 43). One class of cells that has received attention for many years are the cells currently referred to as mesenchymal stem cells or marrow stromal cells (MSCs).
MSCs were discovered by Friedenstein and his associates (54, 55) >30 years ago. They demonstrated that a small fraction of cells from bone marrow adhere to tissue culture surfaces and that the adherent cells can be differentiated both in culture and in vivo into osteoblasts, chondrocytes, and adipocytes. Friedenstein et al.'s observations were confirmed by a large number of subsequent investigators (4, 56-64), who demonstrated that the cells can also differentiate in culture into muscle (65), early precursors of neural cells (66, 67), and cardiomyocytes (68). In parallel experiments, adherent cells from bone marrow with most of the properties of MSCs were shown to provide effective feeder layers for the expansion of hematopoietic stem cells (see refs. 69 and 70). The cells have attracted considerable attention in efforts to develop cell and gene therapies (4, 14, 18, 71), because they are readily obtained from the patient to be treated. Therefore their use can avoid any immune responses. Also, extensive experiments over several decades have not shown any evidence of the tumorgenicity that is prominently observed with embryonic stem cells. Promising results have been reported with use of MSCs or closely related cells from bone marrow in animal models for a number of diseases, including osteogenesis imperfecta (14), parkinsonism (72, 73), spinal cord injury (74-77), stroke (78, 79), myelin deficiency (80), cardiac disorders (81-84), and lung diseases (85, 86). Also, several clinical trials have been initiated and encouraging results have been reported in using administration of MSCs for osteogenesis imperfecta (87, 88) and Hurler's syndrome and metachromatic leukodystrophy (89) and to enhance engraftment of heterologous bone marrow transplants (90).
Properties of MSCs
Despite the great interest in MSCs, the cells are still poorly characterized. Part of the difficulty lies in the subtle changes the cells undergo as they are expanded in culture. The adherent cells initially isolated from whole bone marrow readily can be cloned as single-cell-derived colonies if they are plated at very low densities of 1-10 cells per cm2 (43, 62, 91-93). The single-cell-derived colonies can be differentiated into either osteoblasts, adipocytes, or chondrocytes in culture or in vivo. Therefore, they are multipotential and can differentiate without cell fusion. However, the cells within the single-cell-derived colonies are morphologically heterogeneous (57) in that they contain both small, rapidly self-renewing cells (RS cells) and larger, more slowly replicating cells (44, 57, 91-93). The cultures can be expanded rapidly and over one billion-fold in ≈8 weeks. The cultures remain relatively rich in RS cells through four or five passages if they are maintained at low cell density. If allowed to reach confluency, or if expanded beyond the Hayflick limit of ≈50 population doublings, the cultures are dominated by larger, more mature cells that gradually cease to proliferate (44, 63). Cultures enriched for RS cells have a greater potential to differentiate than cultures of the large, mature cells. However, confluent cultures of the large, mature cells continue to secrete a number of growth factors, an observation consistent with their ability to serve as feeder layers for hematopoietic stem cells.
By analogy with the hematopoietic stem cell system (69, 70), the RS cells seen in early-passage, low-density cultures (Fig. 1) are comparable to transitory amplifying cells that also replicate rapidly in culture. Therefore, RS cells probably do not constitute the earliest progenitors in the pathway. Also, they may not account for all of the marrow-derived cells that are seen to engraft and differentiate in many of the animal models that have been studied. Some of the results obtained with experiments in vivo may be explained by other subfractions of marrow cells. For example, Verfaillie and her associates (24, 43) isolated marrow cells referred to as multipotential adult progenitor cells (MAPCs) that are more multipotential than MSCs in that they can differentiate into hematopoietic cells. They also may more readily differentiate into other cell phenotypes and are incorporated into many tissues after injection into mouse embryos. MAPCs differ from MSCs in that they appear in cultures after extensive incubation in medium containing low concentrations of serum and a mixture of growth factors, express telomerase, and are apparently immortal in culture. Another possible candidate is the rare hematopoietic stem cell isolated by Krause et al. (23) that can engraft in vivo as epithelial cells. The current data, however, do not exclude a number of other possibilities as to which cells in marrow can most effectively home to and repair injured tissues. The most likely possibility is that some subfractions of cells more effective in repairing one type of tissue injury but other subfractions are more effective in repairing other types of tissue injury.
Fig. 1.
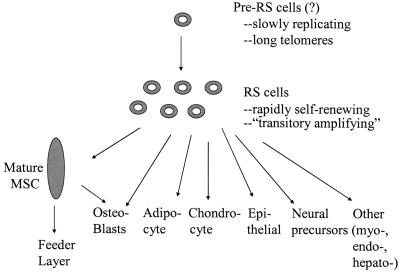
Schematic summarizing relationship between subpopulations of MSCs and their differentiation to specific cell phenotypes.
Recent Results
Conditions for Culture and Expansion of MSCs. If marrow cells are to be used for cell and gene therapy, it will be important to define the conditions for isolation and expansion of the cells. As demonstrated by Friedenstein and colleagues (54, 55), MSCs are relatively easy to isolate from marrow from most species by their adherence to tissue culture plates and flasks. However, the cells display several unusual features as they expand in culture. One of the intriguing observations (63, 91) made with early-passage MSCs is that they display a prolonged lag phase of 3-4 days whenever they are plated at low density in tissue culture (Fig. 2). The lag phase is followed by a phase of rapid exponential growth during which the cells have doubling times as short as 10 h (93). The cultures then pass into a stationary phase in which single-cell-derived colonies markedly decease their rate of proliferation without colony-to-colony contact (44, 63, 91). If the stationary-phase cultures are replated at low density, they replicate the lag phase, exponential growth phase, and stationary phase through four or five passages. In initial experiments, we observed that conditioned medium from stationary cultures increased expansion of MSCs when added to newly initiated cultures (94). One of the active components in the conditioned medium had a molecular mass of ≈30 kDa. Recently, we (95) demonstrated that the active principal in the conditioned medium was Dickkopf-1 (Dkk-1), an inhibitor of the canonical Wnt signaling pathway. The addition of recombinant Dkk-1 decreased the lag period (Fig. 2), and antibodies to Dkk-1 prolonged the lag period. In the early log phase, MSCs synthesized and secreted considerable quantities of Dkk-1, but expression of the Dkk-1 gene and its receptor LRP6 ceased as the cells approached the stationary phase. As the cultures expanded, the gene for Wnt5a demonstrated a reverse pattern of expression in that the gene was not expressed in early log-phase cultures, but was expressed in stationary cultures (Fig. 3). The expression of Wnt5a by stationary-phase cultures was consistent with their role as feeder layers for hematopoietic stem cells, because recent observations indicate that several Wnt ligands enhance hematopoiesis (96-99).
Fig. 2.
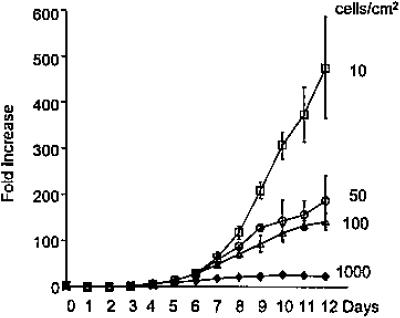
Lag period and rapid expansion of early-passage human MSCs plated at low densities. [Reproduced with permission from ref. 93 (Copyright 2002, AlphaMed Press).]
Fig. 3.
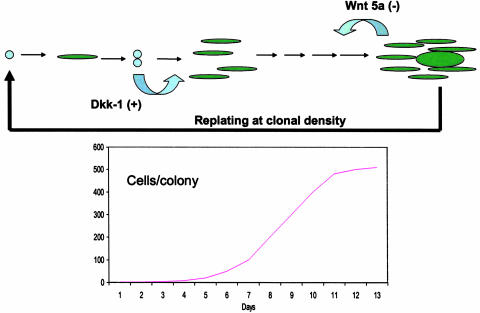
Scheme summarizing transitions of colonies of MSCs from lag period to log phase and stationary phase of growth. As indicated, Dkk-1 is synthesized and secreted as late log/early log phase and stimulates expansion of cells. Wnt 5a in expressed at the end of log phase and in stationary phase. As the cells expand in the colonies, spindle-shaped RS cells give rise to larger, more mature cells. For four or five passages of preparation of MSCs the sequence is repeated after the cells are lifted and replated at low density.
A Simplified System for Studying Repair by MSCs in Culture. The difficulty of carrying out experiments in animal models with MSCs and other marrow cells has prompted us to develop a coculture system to study the repair of injured cells and tissues by MSCs. In initial experiments, we (100) cocultured MSCs with heat-shocked human small airway epithelial cells (SAECs).
In culture, SAECs formed a continuous monolayer of broad, flat cells with a raised perinuclear region (Fig. 4). In the same serum-free medium used to culture SAECs, MSCs became thin and elongated. Therefore, as a first approximation, it was possible to follow differentiation of the MSCs by light microscopy. After heat shock of the SAEC cultures, the epithelial monolayer was disrupted as many of the cells became necrotic and apoptotic. After addition of MSCs labeled with GFP to the heat-shocked SAECs, some of the GFP+ MSCs entered the monolayer and assumed the broad, flat morphology of the epithelial cells. Assays by immunohistochemistry and cDNA microarrays demonstrated that the GFP+ MSCs began to express many proteins characteristic of epithelial cells and they formed adherens junctions (data not shown). Time-lapse photomicroscopy (Fig. 5) demonstrated that many of the GFP+ MSCs assumed the characteristic broad morphology of epithelial cells directly as they entered the monolayer (data not shown), but some of the cells fused with epithelial cells to form multinuclear cells (Fig. 5). Reverse-tracking of 381 GFP+ cells that had assumed an epithelial morphology indicated that most of the MSCs differentiated without any evidence of cell fusion. However, ≈14% had undergone a distinct cell fusion event.
Fig. 4.
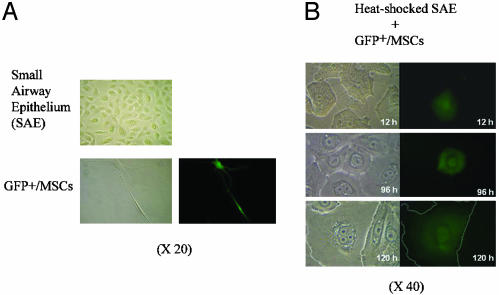
Cocultures of GFP+ MSCs and heat-shocked SAECs. (A) The SAECs form a continuous monolayer of epithelial cells that are broad and thin with a raised perinuclear region. In the serum-free medium used to culture SAECs, GFP+ MSCs become thin and elongated (epifluorescence frame, Lower Right). (B) Differential interference (Left) and epifluorescence (Right) of cocultures. After heat shock, the monolayer of SAECs is disrupted as some of the cells undergo necrosis and apoptosis. Over 12-120 h, some GFP+ MSCs enter the monolayer and become broad, flat cells that reform the monolayer. (Outline of GFP+ cell in Bottom is enhanced.) [Reproduced with permission from ref. 100 (Copyright 2003, National Academy of Sciences).]
Fig. 5.
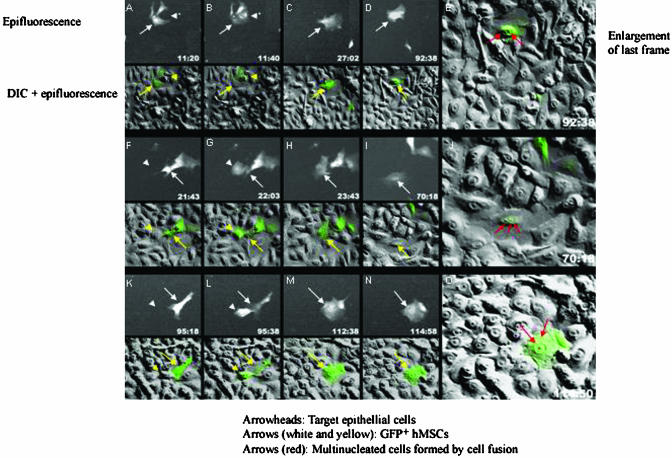
Time-lapse photomicroscopy of cocultures of GFP-labeled human MSCs added to heat-shocked SAECs demonstrating cell fusion. Epifluorescence and differential interference (DIC) photomicrographs were taken every 20 min for >114 h, and selective frames for three sequences are shown. In alternate rows, the epifluorescence and DIC frames are superimposed. [Reproduced with permission from ref. 100 (Copyright 2003, National Academy of Sciences).]
Differentiation of the MCSs was also examined by sorting the cocultures for cells that were positive both for GFP and CD24, an epitope found on the epithelial cells but not on the MSCs. After coculture for 1 wk, assays by flow cytometry demonstrated that ≈4% of the added GFP+ cells were positive for both GFP and CD24. About three-quarters of the isolated doubly positive cells were mononucleated. One-quarter were binucleated, an observation consistent with cell fusion.
In further experiments, male GFP+ MSCs were cocultured with female heat-shocked SAECS, GFP+/CD24+ cells were isolated from the cocultures, and the double-labeled cells were assayed for the X and Y chromosome by in situ hybridization (Fig. 6). GFP+/CD24+ cells were seen that contained a single nucleus with one Y and one X chromosome, an observation consistent with direct differentiation of a male MSC. A number of cells contained a single nucleus with one Y and three X chromosomes, indicating that a male MSC and a female SAEC had undergone both cell fusion and nuclear fusion. A rare cell was found with a single nucleus that contained one Y and five X chromosomes, indicating fusion of three nuclei, one from a male GFP+ MSC cell, and two from female SAECs.
Fig. 6.
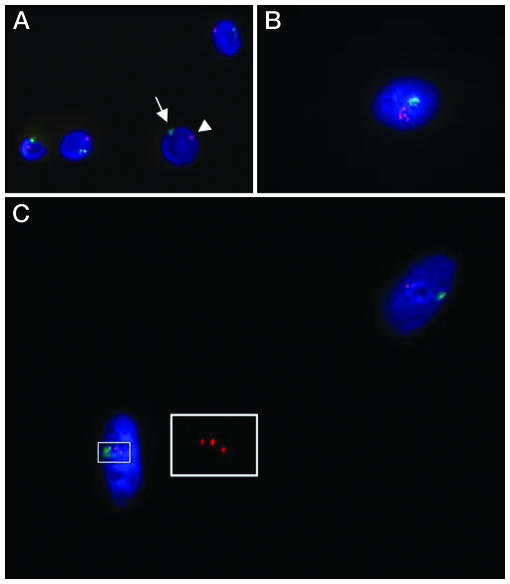
Evidence for nuclear fusion in cocultures of GFP-labeled male MSCs and heat-shocked female SAECs. After coculture, fluorescence-activated cell sorting was used to isolate cells positive both for GFP and CD24, a surface epitope expressed on the epithelial cells but not on MSCs. The isolated cells were then assayed by in situ hybridization for the X and Y chromosome. (A) Control male cell (arrows) containing one X and one Y chromosome. (B) A rare cell with a single nucleus containing one Y chromosome and five X chromosomes. (C) A highlighted cell with a single nucleus containing one Y and three X chromosomes. (Inset) Enlargement to demonstrate the three X chromosomes. The second nucleus has one X and one Y chromosome, an observation consistent with differentiation without cell fusion of a male MSC.
Conclusions
The cyclical expression of Dkk-1 and Wnt5a by MSCs in culture provides a simple explanation for why the cells repeatedly display a lag phase, an exponential growth phase, and a stationary phase as they are passed in culture (63, 91). Within bone marrow, the expression of Wnt5a instead of Dkk-1 may prevent MSCs from expanding at the extremely rapid rate seen in low-density cultures (Fig. 1) and thereby prevent them from overpopulating the marrow. Also, the expression of Wnt5a is consistent with effectiveness of MSCs in serving as feeder layers for hematopoietic stem cells, because recent observations indicate that several Wnt ligands enhance hematopoieses (96-99). The results also raise the possibility that recombinant Dkk-1 or analogues can be used to enhance the expansion of RS cells in culture and perhaps in vivo. In related experiments (95), we observed that Dkk-1 was secreted by two lines of osteosarcoma cells as they exited a lag phase similar to the lag phase seen with MSCs. Therefore, it is possible that antagonists of Dkk-1 may be useful to limit the growth of some malignancies.
The coculture system with MSCs and epithelial cells made it possible to demonstrate directly the response of MSCs to cell injury (100). Up to 4% of the added MSCs differentiated into epithelial cells. Therefore it should be possible to use the system to isolate the signals released by the heat-shocked epithelial cells that attract MSCs and initiate their differentiation. Most of the MSCs that differentiated did so without any evidence of a cell fusion event. However, cell fusion occurred with up to one-quarter of the cells that acquired an epithelial phenotype (Fig. 7). Fusion of the MSCs and the epithelial cells was confirmed by the observation that some of the cells also underwent nuclear fusion. The rate of cell fusion was unexpectedly high for a system in which the process was not driven by membrane damage or amplified by selective pressure (see refs. 37-39). At the moment it is not clear whether the high rate of cell fusion is a unique property of MSCs or some fortuitous combination of the MSCs and the heat-shocked epithelial cells. Also, because proliferation of cells in the system ceased after ≈48 h under the conditions used here, it was not possible to define the fate of the fused cells. Further refinement of the coculture system should make it possible to resolve these and other issues.
Fig. 7.
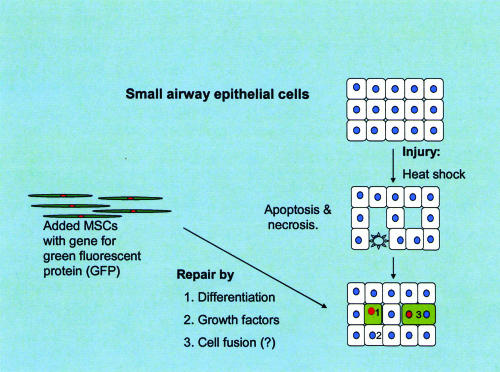
Schematic showing that in the coculture system some MSCs differentiated directly into SAECs and others fused. In addition, the MSCs probably synthesize and secrete growth factors that enhance regeneration of epithelial and other cells.
Acknowledgments
This work was supported by National Institutes of Health Grants AR47796 and AR44210, the Oberkotter Foundation, the Louisiana Gene Therapy Research Consortium, and HCA, The Healthcare Company.
This paper results from the Arthur M. Sackler Colloquium of the National Academy of Sciences, “Regenerative Medicine,” held October 18-22, 2002, at the Arnold and Mabel Beckman Center of the National Academies of Science and Engineering in Irvine, CA.
Abbreviations: MSC, mesenchymal stem cell or marrow stromal cell; RS cells, rapidly self-renewing cells; Dkk-1, Dickkopf-1; SAEC, small airway epithelial cell.
References
- 1.Tanaka, E. M. (2003) Cell 113, 559-562. [DOI] [PubMed] [Google Scholar]
- 2.Cohnheim, J. (1867) Pathol. Anat. Physiol. Klin. Med. 40, 1-79. [Google Scholar]
- 3.Ross, R., Everett, N. B. & Tyler, R. (1970) J. Cell Biol. 44, 645-654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prockop, D. J. (1997) Science 276, 71-74. [DOI] [PubMed] [Google Scholar]
- 5.Doherty, M. J. & Canfield, A. E. (1999) Crit. Rev. Eukaryotic Gene Expression 9, 1-17. [PubMed] [Google Scholar]
- 6.Majka, S. M., Jackson, D. A., Kienstra, K. A., Majesky, M. W., Goodell, M. A. & Hirschi, K. K. (2003) J. Clin. Invest. 111, 29-30.12511584 [Google Scholar]
- 7.LaBarge, M. A. & Blau, H. M. (2002) Cell 111, 589-601. [DOI] [PubMed] [Google Scholar]
- 8.Zuk, P. A., Zhu, M., Mizuno, H., Huang, J., Futrell, J. W., Katz, A. J., Benhaim, P., Lorenz, H. P. & Hedrick, M. H. (2001) Tissue Eng. 7, 211-228. [DOI] [PubMed] [Google Scholar]
- 9.Campagnoli, C., Roberts, I. A., Kumar, S., Bennett, P. R., Bellantuono, I. & Fisk, N. M. (2001) Blood 98, 2396-2402. [DOI] [PubMed] [Google Scholar]
- 10.Wulf, G. G., Luo, K. L., Jackson, K. A., Brenner, M. K. & Goodell, M. A. (2003) Haematologica 88, 368-378. [PubMed] [Google Scholar]
- 11.De Bari, C., Dell'Accio, F., Vandenabeele, F., Vermeesch, J. R., Raymackers, J. M. & Luyten, F. P. (2003) J. Cell Biol. 160, 909-918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taupin, P. & Gage, F. H. (2002) J. Neurosci. Res. 69, 745-749. [DOI] [PubMed] [Google Scholar]
- 13.Pereira, R. F., Halford, K. W., O'Hara, M. D., Leeper, D. B., Sokolov, B. P., Pollard, M. D., Bagasra, O. & Prockop, D. J. (1995) Proc. Natl. Acad. Sci. USA 92, 4857-4861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pereira, R. F., O'Hara, M. D., Laptev, A. V., Halford, K. W., Pollard, M. D., Class, R., Simon, D., Livezey, K. & Prockop, D. J. (1998) Proc. Natl. Acad. Sci. USA 95, 1142-1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eglitis, M. A. & Mezey, E. (1997) Proc. Natl. Acad. Sci. USA 94, 4080-4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brazelton, T. R., Rossi, F. M., Keshet, G. I. & Blau, H. M. (2000) Science 290, 1672-1674. [DOI] [PubMed] [Google Scholar]
- 17.Ferrari, G., Cusella-De Angelis, G., Coletta, M., Paolucci, E., Stornaiuolo, A., Cossu, G. & Mavilio, F. (1998) Science 279, 1528-1530. [DOI] [PubMed] [Google Scholar]
- 18.Azizi, S. A., Stokes, D., Augelli, B. J., DiGirolamo, C. & Prockop, D. J. (1998) Proc. Natl. Acad. Sci. USA 95, 3908-3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kopen, G. C., Prockop, D. J. & Phinney, D. G. (1999) Proc. Natl. Acad. Sci. USA 96, 10711-10716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hou, Z., Nguyen, Q., Frenkel, B., Nilsson, S. K., Milne, M., van Wijnen, A. J., Stein, J. L., Quesenberry, P., Lian, J. B. & Stein, G. S. (1999) Proc. Natl. Acad. Sci. USA 96, 7294-7299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eglitis, M. A., Dawson, D., Park, K. W. & Mouradian, M. M. (1999) NeuroReport 10, 1289-1292. [DOI] [PubMed] [Google Scholar]
- 22.Liechty, K. W., MacKenzie, T. C., Shaaban, A. F., Radu, A., Moseley, A. M., Deans, R., Marshak, D. R. & Flake, A. W. (2000) Nat. Med. 6, 1282-1286. [DOI] [PubMed] [Google Scholar]
- 23.Krause, D. S., Thiese, N. D., Collector, M. I., Henegariu, O., Hwang, S., Gardner, R., Neutzel, S. & Sharkis, S. J. (2001) Cell 105, 369-377. [DOI] [PubMed] [Google Scholar]
- 24.Jiang, Y., Jahagirdar, B. N., Reinhardt, R. L., Schwartz, R. E, Keene, C. D., Ortiz-Gonzalez, X. R., Reyes, M., Lenvik, T., Lund, T., Blackstad, M., et al. (2002) Nature 418, 41-49. [DOI] [PubMed] [Google Scholar]
- 25.Kale, S., Karihaloo, A., Clark, P. R., Kashgarian, M., Krause, D. S. & Cantley, L. G. (2003) J. Clin. Invest. 112, 42-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Orlic, D. (2002) Int. J. Hematol. 76, 144-145. [DOI] [PubMed] [Google Scholar]
- 27.Badiavas, E. V., Abedi, M., Butmarc, J., Galanga, V. & Quesenberry, P. (2003) J. Cell Physiol. 196, 245-250. [DOI] [PubMed] [Google Scholar]
- 28.Chesney, J. & Bucala, R. (2000) Curr. Rheumatol. Rep. 2, 501-505. [DOI] [PubMed] [Google Scholar]
- 29.Kuznetsov, S. A., Mankani, M. H., Gronthos, S., Satomura, K., Bianco, P. & Robey, P. G. (2001) J. Cell Biol. 153, 1133-1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zvaifler, N. J., Marinova-Mutafchieva, L., Adams, G., Edwards, C. J., Moss, J., Burger, J. A. & Maini, R. N. (2000) Arthritis Res. 2, 477-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wagers, A. J., Sherwood, R. I., Christensen, J. L. & Weissman, I. L. (2002) Science 297, 2256-2259. [DOI] [PubMed] [Google Scholar]
- 32.Theise, N. D., Krause, D. S. & Sharkis, S. (2003) Science 299, 1317a. [DOI] [PubMed] [Google Scholar]
- 33.Wagers, A. J., Sherwood, R. I., Christensen, J. L. & Weissman, I. L. (2003) Science 299, 1317b.12610282 [Google Scholar]
- 34.Castro, R. F., Jackson, K. A., Goodell, M. A., Robertson, C. S., Liu, H. & Shine, H. D. (2002) Science 297, 1299. [DOI] [PubMed] [Google Scholar]
- 35.Blau, H., Brazelton, T., Keshet, G. & Rossi, F. (2002) Science 298, 361-362. [DOI] [PubMed] [Google Scholar]
- 36.Castro, F. F., Jackson, K. A., Goodell, M. A., Robertson, C. S., Liu, H. & Shine, H. D. (2002) Science 298, 362. [DOI] [PubMed] [Google Scholar]
- 37.Ying, Q.-L., Nichols, J., Evans, E. P. & Smith, A. G. (2002) Nature 416, 545-547. [DOI] [PubMed] [Google Scholar]
- 38.Terada, N., Hamazaki, T., Oka, M., Hoki, M., Mastalerz, D. M., Nakano, Y., Meyer, E. M., Morel, L., Petersen, B. E. & Scott, E. W. (2002) Nature 416, 542-545. [DOI] [PubMed] [Google Scholar]
- 39.Wang, X., Willenbring, H., Akkari, Y., Torimaru, Y., Foster, M., Al-Dhalimy, M., Lagasse, E., Finegold, M., Olson, S. & Grompe, M. (2003) Nature 422, 897-901. [DOI] [PubMed] [Google Scholar]
- 40.Vassilopoulos, G., Wang, P. R. & Russell, D. W. (2003) Nature 422, 901-904. [DOI] [PubMed] [Google Scholar]
- 41.Phinney, D. G., Kopen, G., Isaacson, R. L. & Prockop, D. J. (1999) J. Cell Biochem. 72, 570-585. [PubMed] [Google Scholar]
- 42.Lambert, J. F., Liu, M., Colvin, G. A., Dooner, M., McAuliffe, C. I., Becker, P. S., Forget, B. G., Weissman, S. M. & Quesenberry, P. J. (2003) J. Exp. Med. 197, 1563-1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reyes, M., Lund, T., Lenvik, T., Aguiar, D., Koodie, L. & Verfaillie, C. M. (2001) Blood 98, 2615-2625. [DOI] [PubMed] [Google Scholar]
- 44.DiGirolamo, C. M., Stokes, D., Colter, D., Phinney, D. G., Class, R. & Prockop, D. J. (1999) Br. J. Haematol. 107, 275-281. [DOI] [PubMed] [Google Scholar]
- 45.Theise, N. D., Nimmakayalu, M., Gardner, R., Illei, P. B., Morgan, G., Teperman, L., Henegar, O. & Krause, D. S. (2000) Hepatology 32, 11-16. [DOI] [PubMed] [Google Scholar]
- 46.Poulsom, R., Forbes, S. J., Hodivala-Dilke, K., Ryan, E., Wyles, S., Navaratnarasah, S., Jeffery, R., Hunt, T., Alison, M., Cook, T., et al. (2001) J. Pathol. 195, 229-235. [DOI] [PubMed] [Google Scholar]
- 47.Suratt, B. T., Cool, C. D., Serls, A. E., Chen, I., Varella-Garcia, M., Shpall, E. J., Brown, K. K. & Worthen, G. S. (2003) Am. J. Respir. Crit. Care Med. 168, 318-322. [DOI] [PubMed] [Google Scholar]
- 48.Deb, A., Wang, S., Skelding, K. A., Miller, D., Simper, D. & Caplice, N. M. (2003) Circulation 107, 1247-1249. [DOI] [PubMed] [Google Scholar]
- 49.Mezey, E., Key, S., Vogelsang, G., Szalayova, I., Lange, G. D. & Crain, B. (2003) Proc. Natl. Acad. Sci. USA 100, 1364-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weimann, J. M., Charlton, C. A., Brazelton, T. R., Hackman, R. C. & Blau, H. M. (2003) Proc. Natl. Acad. Sci. USA 100, 2088-2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Laflamme, M. A., Myerson, D., Saffitz, J. E. & Murry, C. E. (2002) Circ. Res. 90, 634-640. [DOI] [PubMed] [Google Scholar]
- 52.Muller, P., Pfeiffer, P., Koglin, J., Schafers, H. J., Seeland, U., Janzen, I., Urbschat, S. & Bohm, M. (2002) Circulation 106, 31-35. [DOI] [PubMed] [Google Scholar]
- 53.Kleeberger, W., Versmold, A., Rothamel, T., Glockner, S., Bredt, M., Haverich, A., Lehman, U. & Kreipe, H. (2003) Am. J. Pathol. 162, 1487-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Friedenstein, A. J., Gorskaja, J. F. & Kulagina, N. N. (1976) Exp. Hematol. 4, 267-274. [PubMed] [Google Scholar]
- 55.Friedenstein, A. J., Chailakhyan, R. K. & Gerasimov, U. V. (1987) Cell Tissue Kinet. 20, 263-272. [DOI] [PubMed] [Google Scholar]
- 56.Castro-Malaspina, H., Gay, R. E., Resnick, G., Kapoor, N., Meyers, P., Chiarieri, D., McKenzie, S., Broxmeyer, H. E. & Moore, M. A. (1980) Blood 56, 289-301. [PubMed] [Google Scholar]
- 57.Mets, T. & Verdonk, G. (1981) Mech. Ageing Dev. 16, 81-89. [DOI] [PubMed] [Google Scholar]
- 58.Piersma, A. H., Brockbank, K. G., Ploemacher, R. E., van Vliet, E., Brakel-van Peer, K. M. & Visser, P. J. (1985) Exp. Hematol. 13, 237-243. [PubMed] [Google Scholar]
- 59.Owen, M. & Friedenstein, A. J. (1988) Ciba Found. Symp. 136, 42-60. [DOI] [PubMed] [Google Scholar]
- 60.Caplan. A. I. (1991) J. Orthop. Res. 9, 641-650. [DOI] [PubMed] [Google Scholar]
- 61.Beresford, J. N., Bennett, J. H., Devlin, C., Leboy, P. S. & Owen, M. E. (1992) J. Cell Sci. 102, 341-351. [DOI] [PubMed] [Google Scholar]
- 62.Kuznetsov, S. A., Krebsbach, P. H., Satomura, K., Kerr, J., Riminucci, M., Benayahu, D. & Robey, P. G. (1997) J. Bone Miner. Res. 12, 1335-1347. [DOI] [PubMed] [Google Scholar]
- 63.Bruder, S. P., Jaiswal, N. & Haynesworth, S. E. (1997) J. Cell Biochem. 64, 278-294. [DOI] [PubMed] [Google Scholar]
- 64.Pittenger, M. F., Mackay, A. M., Beck, S. C., Jaiswal, R. K., Douglas, R., Mosca, J. D., Moorman, M. A., Simonetti, D. W., Craig, S. & Marshak, D. R. (1999) Science 284, 143-147. [DOI] [PubMed] [Google Scholar]
- 65.Wakitani, S., Saito, T. & Caplan, A. I. (1995) Muscle Nerve 18, 1417-1426. [DOI] [PubMed] [Google Scholar]
- 66.Woodbury, D., Schwarz, E. J., Prockop, D. J. & Black, I. B. (2000) J. Neurosci. Res. 61, 364-370. [DOI] [PubMed] [Google Scholar]
- 67.Sanchez-Ramos, J., Song, S., Cardozo-Pelaez, F., Hazzi, C., Stedeford, T., Willing, A., Freeman, T. B., Saporta, S., Janssen, W., Patel, N., et al. (2000) Exp. Neurol. 164, 247-256. [DOI] [PubMed] [Google Scholar]
- 68.Fukuda, K. (2001) Artif. Organs 25, 187-193. [DOI] [PubMed] [Google Scholar]
- 69.Eaves, C., Glimm, H., Eisterer, W., Audet, J., Maguer-Satta, V. & Piret, J. (2001) Ann. N.Y. Acad. Sci. 938, 63-70. [DOI] [PubMed] [Google Scholar]
- 70.Wagers, A. J., Christensen, J. L. & Weissman, I. L. (2002) Gene Ther. 9, 606-612. [DOI] [PubMed] [Google Scholar]
- 71.Caplan, A. I. (1990) Biomaterials 11, 44-46. [PubMed] [Google Scholar]
- 72.Schwarz, E. J., Alexander, G. M., Prockop, D. J. & Azizi, S. A. (1999) Hum. Gene Ther. 10, 2539-2549. [DOI] [PubMed] [Google Scholar]
- 73.Li, Y., Chen, J., Wang, L., Zhang, L., Lu, M. & Chopp, M. (2001) Neurosci. Lett. 316, 67-70. [DOI] [PubMed] [Google Scholar]
- 74.Chopp, M., Zhang, X. H., Li, Y., Wang, L., Chen, J., Lu, D., Lu, M. & Rosenblum, M. (2000) NeuroReport 11, 3001-3005. [DOI] [PubMed] [Google Scholar]
- 75.Hofstetter, C. P., Schwarz, E. J., Hess, D., Widenfalk, J., El Manira, A., Prockop, D. J. & Olson, L. (2002) Proc. Natl. Acad. Sci. USA 99, 2199-2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Akiyama, Y., Radtke, C. & Kocsis, J. D. (2002) J. Neurosci. 22, 6623-6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wu, S., Suzuki, Y., Ejiri, Y., Noda, T., Bai, H., Kitada, M., Kataoka, K., Ohta, M., Chou, H. & Ide, C. (2003) J. Neurosci. Res. 72, 343-351. [DOI] [PubMed] [Google Scholar]
- 78.Li, Y., Chen, J., Chen, X. G., Wang, L., Gautam, S. C., Xu, Y. X., Katakowski, M., Zhang, L. J., Lu, M., Janakiraman, N. & Chopp, M. (2002) Neurology 59, 514-523. [DOI] [PubMed] [Google Scholar]
- 79.Kang, S. K., Lee, D. H., Bae, Y. C., Kim, H. K., Baik, S. Y. & Jung, J. S. (2003) Exp. Neurol., in press. [DOI] [PubMed]
- 80.Jin, H. K., Carter, J. E., Huntley, G. W. & Schuchman, E. H. (2002) J. Clin. Invest. 109, 1183-1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang, J. S., Shum-Tim, D., Galipeau, J., Chedrawy, E., Eliopoulos, N. & Chiu, R. C. (2000) J. Thorac. Cardiovasc. Surg. 120, 999-1005. [DOI] [PubMed] [Google Scholar]
- 82.Toma, C., Pittenger, M. F., Cahill, K. S., Byrne, B. J. & Kessler, P. D. (2002) Circulation 105, 93-98. [DOI] [PubMed] [Google Scholar]
- 83.Tomita, S., Mickle, D. A., Weisel, R. D., Jia, Z. Q., Tumiati, L. C., Allidina, Y., Liu, P. & Li, R. K. (2002) J. Thorac. Cardiovasc. Surg. 123, 1132-1140. [DOI] [PubMed] [Google Scholar]
- 84.Shake, J. G., Gruber, P. J., Baumgartner, W. A., Senechal, G., Meyers, J., Redmond, J. M., Pittenger, M. F. & Martin, B. J. (2002) Ann. Thorac. Surg. 73, 1919-1925. [DOI] [PubMed] [Google Scholar]
- 85.Kotton, D. N., Ma, B. Y., Cardoso, W. V., Sanderson, E. A., Summer, R. S., Williams, M. C. & Fine, A. (2001) Development (Cambridge, U.K.) 128, 5181-5188. [DOI] [PubMed] [Google Scholar]
- 86.Ortiz, L. A., Gambelli, F., McBride, C., Gaupp, D., Baddoo, M., Kaminski, N. & Phinney, D. G. (2003) Proc. Natl. Acad. Sci. USA 100, 8407-8411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Horwitz, E. M., Prockop, D. J., Gordon, P. L., Koo, W. W., Fitzpatrick, L. A., Neel, M. D., McCarville, M. E., Orchard, P. J., Pyeritz, R. E. & Brenner, M. K. (2001) Blood 97, 1227-1231. [DOI] [PubMed] [Google Scholar]
- 88.Horwitz, E. M., Gordon, P. L., Koo, W. K., Marx, J. C., Neel, M. D., McNall, R. Y., Muul, L. & Hofmann, T. (2002) Proc. Natl. Acad. Sci. USA 99, 8932-8937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Koc, O. N., Day, J., Nieder, M., Gerson, S. L., Lazarus, H. M. & Krivit, W. (2002) Bone Marrow Transplant. 30, 215-222. [DOI] [PubMed] [Google Scholar]
- 90.Koc, O. N., Gerson, S. L., Cooper, B. W., Dyhouse, S. M., Haynesworth, S. E., Caplan, A. I. & Lazarus, H. M. (2000) J. Clin. Oncol. 18, 307-316. [DOI] [PubMed] [Google Scholar]
- 91.Colter, D. C., Class, R., DiGirolamo, C. M. & Prockop, D. J. (2000) Proc. Natl. Acad. Sci. USA 97, 3213-3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Colter, D. C., Sekiya, I. & Prockop, D. J. (2001) Proc. Natl. Acad. Sci. USA 98, 7841-7845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sekiya, I., Larson, B. L., Smith, J. R., Pochampally, R., Cui, J. G. & Prockop, D. J. (2002) Stem Cells 20, 530-541. [DOI] [PubMed] [Google Scholar]
- 94.Colter, D. C. (2001) Ph.D. thesis (Tulane Univ., New Orleans).
- 95.Gregory, C. A., Singh, H., Perry, A. S. & Prockop, D. J. (2003) J. Biol. Chem. 278, 28067-28078. [DOI] [PubMed] [Google Scholar]
- 96.Austin, T. W., Solar, G. P., Ziegler, F. C., Liem, L. & Matthews, W. (1997) Blood 89, 3624-3635. [PubMed] [Google Scholar]
- 97.Van Den Berg, D. J., Sharma, A. K., Bruno, E. & Hoffman, R. (1999) Blood 98, 3189-3202. [PubMed] [Google Scholar]
- 98.Murdoch, B., Chadwick, K., Martin, M., Shojaei, F., Shah, K. V., Gallacher, L., Moon, R. T. & Bhatia, M. (2003) Proc. Natl. Acad. Sci. USA 100, 3422-3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Reya, T., Duncan, A. W., Ailles, L., Domen, J., Scherer, D. C., Willert, K., Hintz, I., Nusse, R. & Weissman, I. L. (2003) Nature 423, 409-414. [DOI] [PubMed] [Google Scholar]
- 100.Spees, J. L., Olson, S. D., Ylostalo, J., Lynch, P. J., Smith, J., Perry, A., Peister, A., Wang, M. Y. & Prockop, D. J. (2003) Proc. Natl. Acad. Sci. USA 100, 2397-2402. [DOI] [PMC free article] [PubMed] [Google Scholar]